

Abstract
Reaction of a substituted indole-3-acetyl chloride with N-5-azidopentyl-N′-hydroxyguanidine generated a substituted 3-(5-azidopentylamino)-5-((indol-3-yl)-methyl)-1,2,4-oxadiazole. Reduction of the azide with zinc and ammonium formate afforded the amine, which was elaborated to the guanidine, completing short and efficient syntheses of the cytotoxic natural products phidianidines A and B in 19% overall yield by a convergent route that will make analogues readily available for biological evaluation. Initial screening in the NCI 60 cell line at 10−5 M indicated that the bromine on the indole is necessary for activity and that the amine precursor to phidianidine A is more potent than phidianidine A.
Guo, Gavagnin, and co-workers recently reported the isolation of the guanidine-containing natural products phidianidines A (1a) and B (1b) as the trifluoroacetate salts from the shell-less marine opisthobranch mollusk Phidiana militaris (see Scheme 1).1 Despite their relatively simple structure, the phidianidines are of interest because they appear to be the first natural products that contain an 1,2,4-oxadiazole ring.2 Furthermore, they show significant cytotoxicity at 0.14 to 5.42 μM concentrations against the highly proliferating C6 rat glioma and HeLa epithelial cervical cancer cell lines and the embryonic 3T3-L1 murine embryonic fibroblast and H9c2 rat embryonic cardiac myoblast cell lines and are less toxic to the less rapidly proliferating CaCo-2 epithelial colorectal adenocarcinoma cell line (35 to 100 μM).1 The mode of action is not known, but the closely related dodecylguanidinium acetate (4, dodine) has been in widespread use since 1956 as a fungicide and may act by membrane disruption.3 As part of an ongoing program in the synthesis of structurally novel guanidine-containing natural products,4 we planned to prepare phidianidines A (1a) and B (1b) by coupling of the appropriate ethyl indole-3-acetate (2a or 2b) with hydroxyguanidine 3 containing a suitably protected nitrogen on the other end of the five carbon side chain. There is limited precedent for the synthesis of 3-amino-1,2,4-oxadiazoles by coupling of esters with hydroxyguanidines5 and nature may use a related route for the biosynthesis of the phidianidines.
Scheme 1.
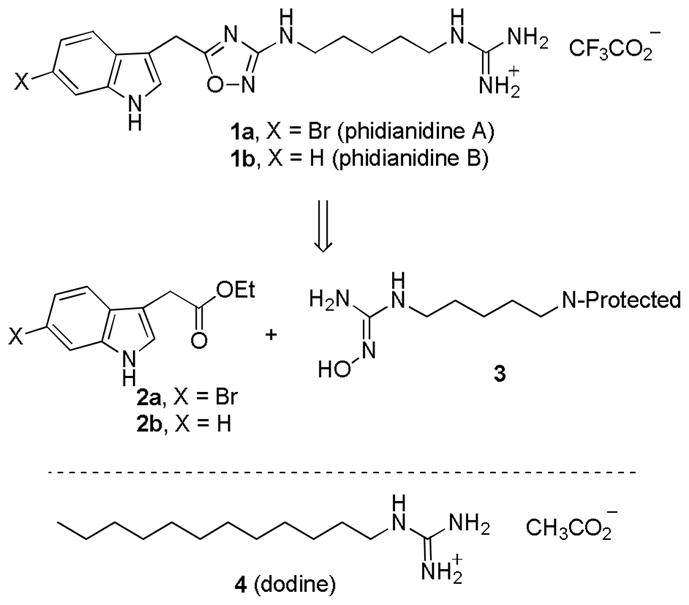
Retrosynthesis of Phidianidines A (1a) and B (1b)
We chose to use hydroxyguanidine 8 with an azidopentyl side chain because the azide group can be easily elaborated into the guanidine of the phidianidines and a wide variety of analogues (see Scheme 2). Furthermore, 5-azido-1-pentanamine (6) is readily available in quantity in 85% yield by selective reduction of diazide 5 by Kim’s procedure with triphenyl-phosphine in a two-phase system.6a After mono-reduction of the diazide in Et2O/EtOAc, the azido amine is protonated and partitions into the 5% HCl solution, thereby preventing reduction of the second azide. Reaction of amine 6 with cyanogen bromide7 in CH2Cl2 and aqueous NaHCO3 solution afforded somewhat unstable cyanamide 7, which was treated with hydroxylamine hydrochloride8 and K2CO3 in EtOH to give the unstable8d hydroxyguanidine 8, which was used immediately for the next step.
Scheme 2.

Synthesis of Phidianidines A (1a) and B (1b)
Our initial attempt at 3-amino-1,2,4-oxadiazole synthesis using models N-butyl-N′-hydroxyguanidine and ethyl phenylacetate (3 equiv) with NaOEt (2–3 equiv) in EtOH at reflux for 5 h proceeded in 50–70% yield based on the hydroxyguanidine. However, the yield dropped to 0–15% without both excess ester and NaOEt and the reaction proceeded in only 30% yield with ethyl indole-3-acetate (3 equiv) and NaOEt (3 equiv). These initial results indicated that an ester is not sufficiently reactive to couple with a hydroxyguanidine unless it is used in large excess. We therefore chose use an acid chloride rather than an ester to form the 3-amino-1,2,4-oxadiazole, a protocol that has been reported in the recent patent literature.9
Indole-3-acetic acid (9b) was reacted with oxalyl chloride and catalytic DMF in CH2Cl2 to give acid chloride 10b, which was treated with 1.5 equiv of freshly prepared 8 and Et3N in CH2Cl2 for 2 h at 25 °C. The solution was concentrated, and a solution of the residue containing the initial coupling product in 1,2-dichloroethane was heated at 80 °C for 2 h to form the 3-amino-1,2,4-oxadiazole giving 11b in 63% yield from 9b and 40% yield from 6. The change of solvents after the initial coupling was necessitated by the high temperature needed for ring closure to form the 1,2,4-oxadiazole and the poor solubility of 8 and 10b in ClCH2CH2Cl.
Reduction of the azide group of 11b was complicated by the sensitivity of the 1,2,4-oxadiazole.2 Attempted hydrogenation over Pd destroyed the 1,2,4-oxadiazole ring. Reduction with Ph3P was successful, but removal of the phosphine oxide byproduct was difficult. Eventually we found that reduction of the azide with activated zinc10 and ammonium formate in MeOH for 7 h at 25 °C proceeded cleanly to give polar amine 12b that was used without purification.11a Coupling of 12b with Boc-protected S-methylisothiourea 13 using Et3N and AgNO34b, 12 in DMF for 2 h at 0 °C and 5 h at 25 °C afforded protected guanidine 14b in 60% yield from 11b. Deprotection of 14b by stirring in 10:1 CH2Cl2/TFA for 8 h at 25 °C removed the Boc protecting groups providing phidianidine B (1b) in 92% yield. The 1H and 13C NMR spectral data of phidianidine B in both CD3OD and DMSO-d6 are identical to those reported for the natural product1 confirming the presence of the 1,2,4-oxadiazole ring.
Phidianidine A (1a) was prepared by an analogous sequence of steps from 6-bromoindole-3-acetic acid (9a).13 Acid chloride 10b was coupled with hydroxyguanidine 8 to give 11a in 61% yield from 9a and 39% yield from 6. Reduction of the azide of 11a with zinc and ammonium formate afforded amine 12a which was coupled with 13 using Et3N and AgNO3 in DMF to give protected guanidine 14a in 61% yield from 11a. Deprotection of 14a in 10:1 CH2Cl2/TFA provided phidianidine A (1a) in 93% yield with 1H and 13C NMR spectral data in both CD3OD and DMSO-d6 identical to those reported for the natural product.1
Initial screening in the NCI 60 cell line screen at 10−5 M showed an average of 33% inhibition of cell growth with phidianidine A (1a). The amine precursor 12a was more potent with an average of 81% inhibition of the 60 cell lines. Phidianidine B (1b) and amine 12b were both much less effective with an average of 5% inhibition of cell growth. These results indicate that the bromine substituent on the indole is important for activity and that the amine of 12a is more effective than the guanidine of 1a. Full details for these four compounds with all 60 cell lines are provided in the Supporting Information.
In conclusion, N-5-azido-1-pentanamine (6) was elaborated to N-5-azidopentyl-N′-hydroxyguanidine (8) in two steps. Reaction of 8 with indole-3-acetyl chloride 10a or 10b afforded 3-(5-azidopentylamino)-5-((indol-3-yl)-methyl)-1,2,4-oxadiazoles 11a and 11b in 61–63% yield. Reduction of the azides with zinc and ammonium formate afforded amines 12a and 12b, which were elaborated to the guanidine, completing short and efficient syntheses of the cytotoxic natural products phidianidines A (1a) and B (1b) in 19% overall yield by a convergent route that will make analogues readily available for biological evaluation.
Experimental Section
General Experimental Methods
Reactions were conducted in flame- or oven-dried glassware under a nitrogen atmosphere and were stirred magnetically. The phrase “concentrated” refers to removal of solvents by means of a rotary-evaporator attached to a diaphragm pump (15–60 Torr) followed by removal of residual solvents at < 1 Torr with an vacuum pump. Flash chromatography was performed on silica gel 60 (230–400 mesh). Analytical thin layer chromatography (TLC) was performed using silica gel 60 F-254 pre-coated glass plates (0.25 mm). TLC Plates were analyzed by short wave UV illumination, or by dipping in CAM stain (40 g of ammonium molybdate, 1.6 g of ceric ammonium molybdate, 80 mL of concentrated sulfuric acid and 720 mL of water) and heating on a hot plate, or by spraying with permanganate solution (5 g KMnO4 in 495 mL water). THF and ether were dried and purified by distillation from sodium/benzophenone. Et3N, pyridine, acetonitrile and benzene were distilled from CaH2. 1H and 13C NMR spectra were obtained on a 400 MHz spectrometer in CDCl3 with tetramethylsilane as internal standard unless specifically indicated. Chemical shifts are reported in δ (ppm downfield from tetramethylsilane). Coupling constants are reported in Hz with multiplicities denoted as s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), m (multiplet) and br (broad). IR spectra were acquired on an FT-IR spectrometer and are reported in wave numbers (cm−1). High resolution mass spectra were obtained using electrospray ionization (ESI) analyzed by quadrupole time of flight (QTof).
5-Azido-1-pentanamine (5)6
To a solution of 56,14 (1.1 g, 7.1 mmol) in 5 mL of Et2O was added 5 mL of EtOAc and 9 mL of 5% HCl aqueous solution successively. To the resulting mixture at 0 °C was added PPh3 (1.9 g, 7.2 mmol, 1.0 equiv) in small portions over 1 h. The mixture was stirred at room temperature for 30 h. 1 M HCl solution (10 mL) was added, the layers were separated, and the organic layer was discarded. The aqueous layer was washed with CH2Cl2 (20 mL × 2), which was discarded, and neutralized with 6 M NaOH until the pH reached 12. The basic aqueous layer was saturated with NaCl and extracted with CH2Cl2 (20 mL × 4). The combined organic layers were dried over Na2SO4 and concentrated to give 780 mg (85% from 5) of 6 as a colorless oil: 1H NMR 3.28 (t, 2, J = 6.8), 2.73 (t, 2, J = 6.8), 2.12 (br, 2, NH2), 1.67-1.58 (m, 2), 1.55-1.45 (m, 2), 1.48-1.38 (m, 2); 13C NMR 51.3, 41.7, 32.7, 28.6, 24.0; IR (neat) 3318 (br), 2933, 2862, 2089, 1558, 1469, 1390, 1300, 1259. The spectral data are identical to those previously reported.6
N-(5-Azidopentyl)-5-[(1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine (11b)
To a suspension of 9b (350 mg, 2.0 mmol) in 10 mL of CH2Cl2 at 0 °C was added DMF (2 drops) and oxalyl chloride (0.51 mL, 5.9 mmol, 3.0 equiv) successively. The mixture was stirred at 0 °C for 1.5 h and concentrated to give 450 mg of crude 1H-indole-3-acetyl chloride (10b), which was used for the synthesis of 11b without further purification.
To a solution of 6 (400 mg, 3.1 mmol) in 6 mL of CH2Cl2 was added NaHCO3 (1.6 g, 19 mmol, 6.0 equiv) and 7 mL of H2O successively. To the resulting mixture was slowly added a solution of BrCN (394 mg, 3.7 mmol, 1.2 equiv) in 2 mL of CH2Cl2 at 0 °C. The mixture was stirred at 0 °C for 0.5 h and at room temperature for 1 h. The reaction was quenched by addition of water (10 mL). The mixture was extracted with CH2Cl2 (15 mL × 3). The combined organic layers were dried over Na2SO4 and concentrated to give 460 mg (96% from 6) of > 90% pure 7 as a somewhat unstable pale yellow oil that can be stored for a few days at 0 °C: 1H NMR 3.73 (br, 1, NH), 3.31 (t, 2, J = 6.8), 3.10 (dt, 2, J = 6.8, 6.8), 1.71-1.59 (m, 4), 1.52-1.42 (m, 2); 13C NMR 116.0, 51.1, 46.0, 29.1, 28.3, 23.4; IR (neat) 3211 (br), 2937, 2865, 2215, 2090, 1454, 1351, 1246, 1159.
To a solution of crude 7 (460 mg, 3.0 mmol) in 10 mL of dry EtOH was added NH2OH•HCl (252 mg, 3.6 mmol, 1.2 equiv) and K2CO3 (1.24 g, 9.0 mmol, 3.0 equiv) successively. The reaction mixture was stirred at room temperature for 5 h, diluted with EtOAc (20 mL) and filtered through Celite. The filtrate was dried over Na2SO4 and concentrated to give 502 mg of crude 8 as an unstable pale yellow sticky oil, which decomposed on storage at 0 °C overnight and should be used immediately in the next step: 1H NMR (DMSO-d6) 9.50 (br, 1, NH or OH), 7.49 (br, 1, NH or OH), 7.32 (br, 2, NH or OH), 3.33 (t, 2, J = 7.2), 3.14-3.06 (m, 2), 1.60-1.42 (m, 4), 1.38-1.26 (m, 2).
To a solution of freshly prepared crude 8 (502 mg, from 400 mg (3.1 mmol) of 6) and NEt3 (0.83 mL, 6.0 mmol) in 10 mL of CH2Cl2 at 0 °C was added a solution of crude 10b (450 mg, from 350 mg (2.0 mmol) of 9b) in 4 mL of CH2Cl2. The reaction mixture was stirred at room temperature for 2 h and concentrated. To the residue was added 15 mL of ClCH2CH2Cl. The mixture was heated at 80 °C for 2 h and concentrated. To the residue was added 15 mL of saturated NaHCO3 solution. The mixture was extracted with CH2Cl2 (15 mL × 4). The combined organic layers were dried over Na2SO4 and concentrated. Flash chromatography of the residue on silica gel (6:1 CH2Cl2/EtOAc) gave 410 mg (63% from 9b, 40% from 6) of 11b as a beige solid: mp 58–59 °C; 1H NMR 8.16 (br, 1, NH) 7.63 (br d, 1, J = 8.0), 7.37 (br d, 1, J = 8.0), 7.22 (br dd, 1, J = 8.0, 8.0), 7.20 (br s, 1), 7.15 (br dd, 1, J = 8.0, 8.0), 4.29 (br, 1, NH), 4.22 (s, 2), 3.25 (t, 2, J = 6.8), 3.23 (dt, 2, J = 6.8, 6.8), 1.68-1.56 (m, 4), 1.49-1.37 (m, 2); 13C NMR 177.3, 168.6, 136.1, 126.7, 123.0, 122.5, 119.9, 118.7, 111.3, 108.1, 51.2, 43.1, 28.9, 28.5, 23.9, 23.4; IR (neat) 3306 (br), 2092, 1703, 1661, 1594, 1456, 1339, 1247, 740; HRMS (ESI) calcd for C16H20N7O (MH+) 326.1729, found 326.1727.
N-(5-Azidopentyl)-5-[(6-bromo-1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine (11a)
To a suspension of 9a (254 mg, 1.0 mmol) in 10 mL of CH2Cl2 at 0 °C was added DMF (2 drops) and oxalyl chloride (0.26 mL, 3.0 mmol, 3.0 equiv) successively. The mixture was stirred at 0 °C for 1.5 h and concentrated to give 320 mg of crude 10a, which was used for the synthesis of 11a without further purification.
To a solution of freshly prepared crude 8 (see preparation of 11b for procedure) (254 mg, from 200 mg (1.6 mmol) of 6) and NEt3 (0.42 mL, 3.0 mmol) in 5 mL of CH2Cl2 at 0 °C was added a solution of crude 10a (320 mg, from 254 mg (1.0 mmol) of 9a) in 2 mL of CH2Cl2. The reaction mixture was stirred at room temperature for 2 h and concentrated. To the residue was added 8 mL of ClCH2CH2Cl. The mixture was heated at 80 °C for 2 h and concentrated. To the residue was added 15 mL of saturated NaHCO3 solution. The mixture was extracted with CH2Cl2 (15 mL × 4). The combined organic layers were dried over Na2SO4 and concentrated. Flash chromatography of the residue on silica gel (6:1 CH2Cl2/EtOAc) gave 246 mg (61% from 9a, 39% from 6) of 11a as a beige solid: mp 86–87 °C; 1H NMR 8.21 (br, 1, NH) 7.51 (br s, 1), 7.47 (br d, 1, J = 8.4), 7.24 (br d, 1, J = 8.4), 7.16 (br s, 1), 4.29 (br, 1, NH), 4.18 (s, 2), 3.26 (t, 2, J = 6.8), 3.23 (dt, 2, J = 6.8, 6.8), 1.68-1.56 (m, 4), 1.49-1.37 (m, 2); 13C NMR 177.0, 168.6, 136.9, 125.6, 123.7, 123.2, 120.0, 116.1, 114.2, 108.3, 51.2, 43.1, 28.9, 28.5, 23.9, 23.3; IR (neat) 3296 (br), 2092, 1594, 1453, 1330, 1236, 893, 803, 735; HRMS (ESI) calcd for C16H19BrN7O (MH+) 404.0834, found 404.0833.
N-[5-[[Bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]pentyl]-5-[(1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine (14b)
To a solution of 11b (410 mg, 1.26 mmol) in 8 mL of MeOH was added ammonium formate (397 mg, 6.30 mmol, 5.0 equiv) and activated zinc10 (328 mg, 5.04 mmol, 4.0 equiv) successively. The mixture was stirred at room temperature for 7 h. The reaction was quenched by addition of 1 M NaOH (15 mL). The mixture was extracted with CH2Cl2 (15 mL × 4). The combined organic layers were dried over Na2SO4 and concentrated to give 390 mg of crude 12b, which was used in the next step without purification: 1H NMR 8.14 (br, 1, NH), 7.63 (br d, 1, J = 8.0), 7.38 (br d, 1, J = 8.0), 7.22 (br dd, 1, J = 8.0, 8.0), 7.22 (br s, 1), 7.15 (br dd, 1, J = 8.0, 8.0), 4.26 (br, 1, NH), 4.22 (s, 2), 3.23 (dt, 2, J = 6.8, 6.8), 2.68 (t, 2, J = 6.8), 1.68-1.54 (m, 2), 1.52-1.32 (m, 4).
To a solution of crude 12b (390 mg) in 8 mL of DMF was added 1,3-bis(tert-butoxycarbonyl)-2-methylthiopseudourea (13) (561 mg, 1.89 mmol) and NEt3 (1.40 mL, 10.1 mmol) successively. To the resulting mixture at 0 °C was added AgNO3 (429 mg, 2.52 mmol) in small portions. The reaction was stirred at 0 °C for 2 h and at room temperature for 5 h. The reaction was quenched by addition of EtOAc (50 mL) and filtered through Celite. The filtrate was washed with saturated NaHCO3 (50 mL × 4), dried over Na2SO4, and concentrated. Flash chromatography of the residue on silica gel (2:3 EtOAc/hexanes) gave 417 mg (61% from 11b) of 14b as a pale yellow sticky oil: 1H NMR 11.49 (br s, 1, NH), 8.30 (br, 2, NH), 7.62 (br d, 1, J = 8.0), 7.37 (br d, 1, J = 8.0), 7.21 (br dd, 1, J = 8.0, 8.0), 7.20 (br s, 1), 7.14 (br dd, 1, J = 8.0, 8.0), 4.33 (br, 1, NH), 4.22 (s, 2), 3.37 (dt, 2, J = 6.8, 6.8), 3.20 (dt, 2, J = 6.8, 6.8), 1.64-1.54 (m, 4), 1.50 (s, 9), 1.49 (s, 9), 1.42-1.32 (m, 2); 13C NMR 177.2, 168.7, 163.6, 156.1, 153.3, 136.1, 126.7, 123.0, 122.3, 119.8, 118.7, 111.3, 108.0, 83.1, 79.3, 43.1, 40.7, 29.0, 28.6, 28.3 (3 C), 28.0 (3 C), 24.0, 23.4; IR (neat) 3324 (br), 1719, 1597, 1414, 1366, 1325, 1129, 1053, 912, 731; HRMS (ESI) calcd for C27H40N7O5 (MH+) 542.3091, found 542.3085.
N-[5-[[Bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]pentyl]-5-[(6-bromo-1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine (14a)
To a solution of 11a (246 mg, 0.61 mmol) in 5 mL of MeOH was added ammonium formate (192 mg, 3.05 mmol, 5.0 equiv) and activated zinc10 (159 mg, 2.44 mmol, 4.0 equiv) successively. The mixture was stirred at room temperature for 7 h. The reaction was quenched by addition of 1 M NaOH (15 mL). The mixture was extracted with CH2Cl2 (15 mL × 4). The combined organic layers were dried over Na2SO4 and concentrated to give 228 mg of crude 12a, which was used in the next step without purification: 1H NMR 8.15 (br, 1, NH) 7.53 (br s, 1), 7.48 (br d, 1, J = 8.4), 7.24 (br d, 1, J = 8.4), 7.19 (br s, 1), 4.29 (br, 1, NH), 4.18 (s, 2), 3.23 (dt, 2, J = 6.8, 6.8), 2.68 (t, 2, J = 6.8), 1.68-1.54 (m, 2), 1.52-1.32 (m, 4).
To a solution of crude 12a (228 mg) in 4 mL of DMF was added 1,3-bis(tert-butoxycarbonyl)-2-methylthiopseudourea (13) (267 mg, 0.92 mmol) and NEt3 (0.70 mL, 5.06 mmol) successively. To the resulting mixture at 0 °C was added AgNO3 (207 mg, 1.22 mmol) in small portions. The reaction was stirred at 0 °C for 2 h and at room temperature for 5 h. The reaction was quenched by addition of EtOAc (30 mL) and filtered through Celite. The filtrate was washed with saturated NaHCO3 (30 mL × 4), dried over Na2SO4, and concentrated. Flash chromatography of the residue on silica gel (2:3 EtOAc/hexanes) gave 226 mg (60% from 11a) of 14a as a pale yellow sticky oil: 1H NMR 11.50 (br s, 1, NH), 8.70 (br, 1, NH), 8.31 (br, 1, NH), 7.52 (br s, 1), 7.47 (br d, 1, J = 8.0), 7.23 (br d, 1, J = 8.0), 7.16 (br s, 1), 4.47 (br, 1, NH), 4.18 (s, 2), 3.32 (dt, 2, J = 6.8, 6.8), 3.16 (dt, 2, J = 6.8, 6.8), 1.58-1.46 (m, 4), 1.49 (s, 18), 1.34-1.24 (m, 2); 13C NMR 176.9, 168.7, 163.5, 156.1, 153.3, 136.9, 125.7, 123.7, 123.1, 120.0, 115.9, 114.3, 108.2, 83.1, 79.4, 43.1, 40.7, 29.0, 28.6, 28.3 (3 C), 28.0 (3 C), 24.0, 23.3; IR (neat) 3322 (br), 1718, 1598, 1413, 1366, 1330, 1131, 1052, 910, 803, 730; HRMS (ESI) calcd for C27H39BrN7O5 (MH+) 620.2196, found 620.2187.
N-[5-[(Aminoiminomethyl)amino]pentyl]-5-[(1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine Trifluoroacetic Acid Salt (Phidianidine B, 1b)
14b (417 mg, 0.77 mmol) was taken up in 20 mL of 1:10 TFA/CH2Cl2, and the resulting solution was stirred at room temperature for 8 h. The mixture was diluted with 25 mL MeOH and concentrated to give 326 mg (93%) of >95% pure 1b as a pale yellow oil: 1H NMR (CD3OD) 7.52 (br d, 1, J = 8.0), 7.35 (br d, 1, J = 8.0), 7.21 (br s, 1), 7.11 (br dd, 1, J = 8.0, 8.0), 7.01 (br dd, 1, J = 8.0, 8.0), 4.22 (s, 2), 3.15 (t, 2, J = 6.8), 3.14 (t, 2, J = 6.8), 1.68-1.54 (m, 4), 1.46-1.36 (m, 2); (DMSO-d6) 11.04 (br s, NH), 7.55 (br t, 1, J = 5.6, NH), 7.51 (br d, 1, J = 8.0), 7.37 (br d, 1, J = 8.0), 7.31 (br d, 1, J = 2.2), 7.09 (br dd, 1, J = 8.0, 8.0), 6.99 (br dd, 1, J = 8.0, 8.0), 6.72 (br t, 1, J = 5.6, NH), 4.20 (s, 2), 3.06 (dt, 2, J = 5.6, 5.6), 3.01 (dt, 2, J = 5.6, 5.6), 1.56-1.40 (m, 4), 1.35-1.25 (m, 2); 13C NMR (CD3OD, referenced to the residual solvent peaks centered at δ 49.15) 179.5, 170.2, 158.8, 138.2, 128.3, 124.8, 122.9, 120.2, 119.4, 112.5, 108.3, 43.9, 42.5, 29.9, 29.7, 25.0, 24.2; (DMSO-d6, referenced to the residual solvent peaks centered at δ 39.51) 176.9, 168.5, 156.7, 136.2, 126.7, 124.2, 121.2, 118.7, 118.4, 111.5, 106.9, 42.3, 40.7, 28.1 (2 C), 23.4, 22.7; IR 3303 (br), 1671, 1599, 1201, 1137; HRMS (ESI) calcd for C17H24N7O (M+) 342.2042, found 342.2047. In both solvents, the indole carbons near the nitrogen are doubled in various ratios due to the presence of both NH and ND forms of the indole.15
N-[5-[(Aminoiminomethyl)amino]pentyl]-5-[(6-bromo-1H-indol-3-yl)methyl]-1,2,4-oxadiazol-3-amine Trifluoroacetic Acid Salt (Phidianidine A, 1a)
14a (100 mg, 0.16 mmol) was taken up in 5 mL of 1:10 TFA/CH2Cl2, and the resulting solution was stirred at room temperature for 8 h. The mixture was diluted with 5 mL MeOH and concentrated to give 79 mg (92%) of >95% pure 1a as a pale yellow oil: 1H NMR (CD3OD) 7.52 (br d, 1, J = 1.7), 7.45 (br d, 1, J = 8.0), 7.23 (br s, 1), 7.13 (br dd, 1, J = 8.0, 1.7), 4.20 (s, 2), 3.15 (br t, 4, J = 7.2), 1.66-1.54 (m, 6), 1.46-1.36 (m, 2); (DMSO-d6) 11.18 (br, s, NH), 7.56 (br d, 1, J = 1.7), 7.49 (br s, NH), 7.47 (br d, 1, J = 8.4), 7.35 (br d, 1, J = 2.5), 7.14 (br dd, 1, J = 8.4, 1.7), 6.73 (br t, 1, J = 6.0, NH), 4.20 (s, 2), 3.06 (dt, 2, J = 6.0, 6.0), 3.00 (dt, 2, J = 6.0, 6.0), 1.56-1.42 (m, 4), 1.34-1.22 (m, 2);13C NMR (CD3OD, referenced to the residual solvent peaks centered at δ 49.15) 179.1, 170.3, 158.8, 139.1, 127.3, 125.9, 123.4, 121.0, 116.3, 115.5, 108.8, 43.9, 42.5, 29.9, 29.7, 25.1, 24.1; (DMSO-d6, referenced to the residual solvent peaks centered at δ 39.51) 176.7, 168.5, 156.7, 137.0, 125.8, 125.3, 121.6, 120.2, 114.2, 114.0, 107.4, 42.3, 40.7, 28.1 (2 C), 23.3, 22.5; IR 3280 (br), 1670, 1601, 1184, 1137; HRMS (ESI) calcd for C17H23BrN7O (M+) 420.1147, found 420.1135. In both solvents, the indole carbons near the nitrogen are doubled in various ratios due to the presence of both NH and ND forms of the indole.15
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM-50151) for support of this work.
Footnotes
Supporting Information Available: Results from the NCI 60 cell screens of phidianidine A (1a), phidianidine B (1b), amine 12a, and amine 12b, tables of spectral data, and copies of 1H and 13C NMR spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Carbone M, Li Y, Irace C, Mollo E, Castelluccio F, Di Pascale A, Cimino G, Santamaria R, Guo Y-W, Gavagnin M. Org Lett. 2011;13:2516–2519. doi: 10.1021/ol200234r. [DOI] [PubMed] [Google Scholar]
- 2.For reviews of 1,2,4-oxadiazoles, see: Hemming K. J Chem Res, Synop. 2001:209–216.Kayukova LA. Pharm Chem J. 2005;39:539–547.Pace A, Pierro P. Org Biomol Chem. 2009;7:4337–4348. doi: 10.1039/b908937c.
- 3.(a) Byrde RJW, Clifford DR, Woodcock D. Ann Appl Biol. 1962;50:291–298. [Google Scholar]; (b) Srivastava SK, Smith TA. Phytochemistry. 1981;21:997–1008. [Google Scholar]; (c) Cabral JPS. Antimicrob Agents Chemother. 1991;35:341–344. doi: 10.1128/aac.35.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Yu M, Pochapsky SS, Snider BB. J Org Chem. 2008;73:9065–9074. doi: 10.1021/jo801956w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu M, Snider BB. Org Lett. 2009;11:1031–1032. doi: 10.1021/ol802981h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yu M, Pochapsky SS, Snider BB. Org Lett. 2010;12:828–831. doi: 10.1021/ol902895e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Barykina OV, Snider BB. Org Lett. 2010;12:2664–2667. doi: 10.1021/ol100896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Saunders J, MacLeod AM, Merchant K, Showell GA, Snow RJ, Street LJ, Baker R. J Chem Soc, Chem Commun. 1988:1618–1619. [Google Scholar]; (b) Saunders J, Cassidy M, Freedman SB, Harley EA, Iversen LL, Kneen C, MacLeod AM, Merchant KJ, Snow RJ, Baker R. J Med Chem. 1990;33:1128–1138. doi: 10.1021/jm00166a008. [DOI] [PubMed] [Google Scholar]; (c) Street LJ, Baker R, Book T, Kneen CO, MacLeod AM, Merchant KJ, Showell GA, Saunders J, Herbert RH, Freedman SB, Harley EA. J Med Chem. 1990;33:2690–2697. doi: 10.1021/jm00172a003. [DOI] [PubMed] [Google Scholar]; (d) Showell GA, Gibbons TL, Kneen CO, MacLeod AM, Merchant K, Saunders J, Freedman SB, Patel S, Baker R. J Med Chem. 1991;34:1086–1094. doi: 10.1021/jm00107a032. [DOI] [PubMed] [Google Scholar]; (e) Chen C-y, Senanayake CH, Bill TJ, Larsen RD, Verhoeven TR, Erider PJ. J Org Chem. 1994;59:3738–3741. [Google Scholar]; (f) Ahmad S, Ngu K, Combs DW, Wu SC, Weinstein DS, Liu W, Chen B-C, Chandrasena G, Dorso CR, Kirby M, Atwal KS. Bioorg Med Chem Lett. 2004;14:177–180. doi: 10.1016/j.bmcl.2003.09.066. [DOI] [PubMed] [Google Scholar]; (g) Vieira E, Huwyler J, Jolidon S, Knoflach F, Mutel V, Wichmann J. Bioorg Med Chem Lett. 2005;15:4628–4631. doi: 10.1016/j.bmcl.2005.05.135. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lee JW, Jun SI, Kim K. Tetrahedron Lett. 2001;42:2709–2711. [Google Scholar]; (b) Srinivasan R, Tan LP, Wu H, Yang PY, Kalesh KA, Yao SQ. Org Biomol Chem. 2009;7:1821–1828. doi: 10.1039/b902338k. [DOI] [PubMed] [Google Scholar]
- 7.(a) Snider BB, O’Hare SM. Tetrahedron Lett. 2001;42:2455–2458. [Google Scholar]; (b) Kumar V, Kaushik MP, Mazumdar A. Eur J Org Chem. 2008:1910–1916. [Google Scholar]; (c) Sathe M, Karade HN, Kaushik MP. Synth Commun. 2008;38:1375–1380. [Google Scholar]
- 8.(a) Pufahl RA, Nanjappan PG, Woodard RW, Marletta MA. Biochemistry. 1992;31:6822–6828. doi: 10.1021/bi00144a024. [DOI] [PubMed] [Google Scholar]; (b) Xian M, Fujiawara N, Wen Z, Cai T, Kazuma S, Janczuk AJ, Tang X, Telyatnikov VV, Zhang Y, Chen X, Miyamoto Y, Taniguchi N, Wang PG. Bioorg Med Lett. 2002;10:3049–3055. doi: 10.1016/s0968-0896(02)00155-4. [DOI] [PubMed] [Google Scholar]; (c) Cho JY, Dutton A, Miller T, Houk KN, Fukuto JM. Arch Biochem Biophys. 2003;417:65–76. doi: 10.1016/s0003-9861(03)00335-7. [DOI] [PubMed] [Google Scholar]; (d) Schade D, Kotthaus J, Klein N, Kotthaus J, Clement B. Org Biomol Chem. 2011;9:5249–5259. doi: 10.1039/c0ob01117g. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cai SX, Zhang H-z, Kuemmerle JD, Zhang H, Kemnitzer WE. WO 2004058253 A1 PCT Int Appl. 2004; Chem Abstr. 2004;141:123632. [Google Scholar]; (b) Fox BM, Iio K, Inaba T, Kayser F, Li K, Sagawa S, Tanaka M, Yoshida A. WO 2005013907 A2 PCT Int Appl. 2005; Chem Abstr. 2005;142:240441. [Google Scholar]; (c) Kubota H, Nakamura Y, Higashijima T, Yamamoto Y, Oka K, Igarashi S. US 20070032485 A1 US Pat Appl Publ. 2007; Chem Abstr. 2007;146:229322. [Google Scholar]; (d) Kubota H, Sugahara M, Furukawa M, Takano M, Motomura D. US 20070082896 A1 US Pat Appl Publ. 2007; Chem Abstr. 2007;146:421853. [Google Scholar]; (e) Lachance N, Li CS, Leclerc J-P, Ramtohul YK. WO 2008064474 A1 PCT Int Appl. 2008; Chem Abstr. 2008;149:32315. [Google Scholar]; (f) Bradbury RH, Hales NJ, Rabow AA. WO 2009081197 A1 PCT Int Appl. 2009; Chem Abstr. 2009;151:124013. [Google Scholar]; (g) Li X, Liu X, Loren J, Molteni V, Nabakka J, Yeh V, Chianelli D. WO 2009105712 A1 PCT Int Appl. 2009; Chem Abstr. 2009;151:313557. [Google Scholar]; (h) Bertram LS, Fyfe MCT, Gattrell W, Jeevaratnam RP, Keily J, Procter M. WO 2010004348 A1 J PCT Int Appl. 2010; Chem Abstr. 2010;152:144685. [Google Scholar]; (i) Fox BM, Iio K, Li K, Choi R, Inaba T, Jackson S, Sagawa S, Shan B, Tanaka M, Yoshida A, Kayser F. Bioorg Med Chem Lett. 2010;20:6030–6033. doi: 10.1016/j.bmcl.2010.08.066. [DOI] [PubMed] [Google Scholar]
- 10.Hannick SM, Kishi Y. J Org Chem. 1983;48:3833–3835. [Google Scholar]
- 11.(a) Srinivasa GR, Nalina L, Abiraj K, Gowda DC. J Chem Res, Synop. 2003:630–631. [Google Scholar]; (b) Boruah A, Baruah M, Prajapati D, Sandhu JS. Synlett. 1997:1253–1253. [Google Scholar]; (c) Lin W, Zhang X, He Z, Jin Y, Gong L, Mi A. Synth Commun. 2002;32:3279–3284. [Google Scholar]; (d) Amantini D, Fringuelli F, Pizzo F, Vaccaro L. Org Prep Proced Int. 2002;34:109–147. [Google Scholar]
- 12.(a) Ma D, Xia C, Jiang J, Zhang J. Org Lett. 2001;3:2189–2191. doi: 10.1021/ol016043h. [DOI] [PubMed] [Google Scholar]; (b) Han S, Moore RA, Viola RE. Bioorg Chem. 2002;30:81–94. doi: 10.1006/bioo.2001.1228. [DOI] [PubMed] [Google Scholar]; (c) DeMong DE, Williams RM. J Am Chem Soc. 2003;125:8561–8565. doi: 10.1021/ja0351241. [DOI] [PubMed] [Google Scholar]
- 13.(a) Rasmussen T, Jensen J, Anthoni U, Christophersen C, Nielsen PH. J Nat Prod. 1993;56:1553–1558. [Google Scholar]; (b) Baran PS, Shenvi RA. J Am Chem Soc. 2006;128:14028–14029. doi: 10.1021/ja0659673. [DOI] [PubMed] [Google Scholar]
- 14.Thomas JR, Liu X, Hergenrother PJ. J Am Chem Soc. 2005;127:12434–12435. doi: 10.1021/ja051685b. [DOI] [PubMed] [Google Scholar]
- 15.(a) Morales-Ríos MS, Del Río RE, Joseph-Nathan P. Magn Reson Chem. 1988;26:552–558. [Google Scholar]; (b) Joseph-Nathan P, Del Río RE, Morales-Ríos MS. Heterocycles. 1988;27:377–383. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.