

Abstract
Despite of established and effective therapy for epilepsy, 20–25% patients develop therapeutic failure; this encourages finding newer drugs. Novel approaches target receptors which remain unaffected by conventional therapy or inhibit epileptogenesis. AMPA receptor antagonists have shown faster and complete protection compared to diazepam. Protein kinase (PK) plays an important role in the development of epilepsy. PK inhibitors such as K252a, VID-82925, and Herbimycin A have been found effective in inhibition of spread of epileptiform activity and epileptogenesis. Metabotropic glutamate receptors (mGluRs) are G protein-coupled receptors classified into three groups. Group 1 mGluRs antagonist and Groups 2 and 3 mGluRs agonists inhibited pentylenetetrazole-induced kindled seizures. Combined use of these agents has also shown favorable results. Mammalian target of rapamycin (mTOR) plays a central role in multiple mechanisms of epileptogenesis. mTOR causes transcription, induction of proapoptotic proteins, and autophagy inhibition. Rapamycin was effective in suppression of recurrent seizures as well as in tuberous sclerosis and acute brain injury model. 5% CO2 showed potent effects on cortical epileptiform activity and convulsions in animal epilepsy models and in humans with drug-resistant partial epilepsy. It is found to be rapidly acting, safe and cheap, thus it can be a good option in emergency for suppression of seizure. Neurosteroids are considered as fourth generation neuromessengers, they act as positive allosteric modulators of γ-aminobutyric acid (GABAA) receptors. Clinical trial of ganaxolone, an allopregnanolone analogue, has shown a beneficial role in pharmacoresistant epilepsy. However, most of these drugs are tested in early phases of development and the possible use and safety in epilepsy has to be proven in clinical trials.
Keywords: AMPA receptor antagonists, mammalian target of rapamycin, metabotropic glutamate receptors, pharmacoresistant epilepsy, protein kinase inhibitors
INTRODUCTION
Epilepsy is the second most common neurological disorder with an incidence rate of 0.3–0.5% worldwide and prevalence of five to ten person per thousand population. Approximately 50 million people are suffering from epilepsy worldwide and among them 90% are in developing countries.[1] Management of epilepsy requires immediate attention and long-term treatment. About 75–80% patients achieve control with conventional anti-epileptic drugs like phenytoin, carbamazepine, and valproic acid. Most of these drugs have a narrow therapeutic margin and require therapeutic drug monitoring. Moreover, 20–25% patients develop therapeutic failure with these drugs.[2] This stimulates further research to find newer drugs and new management modalities. The present review discusses newer therapeutic targets [Table 1] which shown promising results and can be used in future for development of new antiepileptic drugs.
Table 1.
Novel therapeutic targets in epilepsy
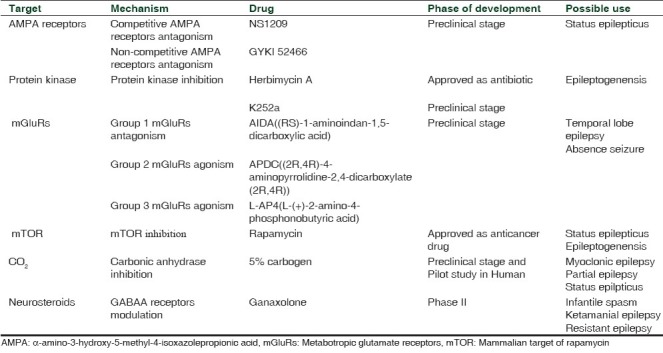
NEWER THERAPEUTIC TARGETS
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors
AMPA receptors play an important role in pathogenesis of refractory epilepsy. Transition from single self-limiting seizure to repeated prolong seizure is accompanied by externalization of AMPA and internalization of γ-aminobutyric acid (GABAA)receptors. This process of internalization leads to GABAA receptors agonist ineffective, but AMPA receptors antagonist might be useful due to externalization of AMPA receptors in refractory epilepsy.[3–5] GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive AMPA receptor antagonist has good penetration in the blood–brain barrier.[6] Recently, Fritsch et al. evaluated effects of GYKI 52466 on early and late kainic acid (KA)-induced status epilepticus.[7] GYKI 52466 administered in two doses 15 min before seizure induction have stopped seizure activity rapidly as well as its recurrence. All treated animals survived, even in the group that have received 45 mg/kg KA, while 33% mortality observed in untreated animals. When status epilepticus continues, it becomes refractory to conventional treatment. In this study, late administration of diazepam was slow to inhibit and unable to prevent recurrence. In contrast to diazepam, GYKI 52466 was able to terminate seizure and prevented recurrence when administered in the late phase. Pitkänen et al. demonstrated that competitive AMPA receptor antagonist NS1209 provided faster and more complete protection as compared to diazepam in status epileptics.[8] A major problem with the use of NS1209 is that it required continuous infusion whereas GYKI 52466 was effective in two bolus doses 15 min apart. Due to noncompetitive antagonism by GYKI 52466, it may have wider anticonvulsant activity as its blocking action cannot be overcome by even high level of glutamate.[9]
Standard treatment of status epileptics with diazepam is associated with a decrease in mean arterial pressure (MAP). When MAP falls below 70 mmHg, cerebral blood flow regulation falls and irreversible brain damage occurs. GYKI 52466 at therapeutic dose caused fall in the pulse rate, but no affect observed on MAP. Mice treated with GYKI 52466 appeared to be sedated but responded on stimulation showing moderate sedation. Animals treated with GYKI 52466 gained weight normally after they had recovered from the status epileptics episode showing good overall tolerability.[10]
AMPA receptor antagonists may be useful as single drug therapy in status epileptics with less cardiovascular complications compared to standard treatment with diazepam or phenytoin. AMPA receptor antagonists are promising targets in status epileptics.
Protein kinase
Protein kinase (PK) is an enzyme protein which primarily phosphorylates receptors, transport molecules, ion channels and affects excitability and functions of neurons and glias.[11] Experimental models have shown the involvement of protein tyrosine phosphorylation via PK in the development of status epilepticus. Phosphorylation of receptors could occur at least through three different intracellular pathways: (1) Src non-receptor tyrosine kinase (2) Eph receptors for the ephrins cell-to-cell adhesion molecules, and (3) Trk receptors for neurotrophins. These three kinases have been implicated in the epileptogenesis.[12] Queiroz and Mello showed the preventive role of tyrosin PK inhibitors K252a and herbimycin A in KA induced epileptiform activity and mossy fiber sprouting (MFS).[13] KA intracerebral injection leads to selective destruction of pyramidal CA3 neurons, which is associated with epileptiform discharges. It also leads to hippocampal neuronal loss, reactive gliosis, MFS, and spontaneous recurrent seizures. Herbimycin A and K252a modified the electrographic epileptiform activity induced by intrahippocampal KA administration but did not alter the cell loss pattern. Only K252a treatment reduced supragranular MFS. Herbimycin A inhibited the spread of the epileptiform activity to the contra lateral hippocampus; this might be due to its strong inhibitory activity on the family of the Src kinases. Src kinases have been described as “a hub for NMDA receptor regulation” and have been involved in NMDA receptor phosphorylation during status epilepticus.[14] K252a reduced intermittent burst activity by inhibition of BDNF-induced inhibition of GABAA receptor-mediated inhibitory postsynaptic currents in CA1 slices. Both the PK inhibitors failed to prevent limbic cell death. This may be due to animal models with frequent seizures have such widespread cell damage that this cannot be counter-affected by local (intra ventricular) administration of protein tyrosine kinase inhibitors. In future PK inhibitors can be evaluated with the use of more restricted damage animal model to show its effect on cellular death. A study by Zita Gajda et al. demonstrated the antiepileptogenic effect of VID-82925, a kinase inhibitor. VID-82925 was found to inhibit the epileptiform activity not only during developmental phase, but also during the long period of stable phase of focus. While levitiracetam pretreatment failed to exert antiepileptogenic effect, the effect of VID-82925 was comparable in intensity with carbenoxolone, a broad-spectrum gap junction channel blocker. It suggests VID-82925 also exerts antiepileptogenic effect by blocking of gap junctional communication.[15]
Tyrosine kinase inhibitors may cause some serious side effects like cardiotoxicity, anemia, and thrombocytopenia.[16] This drawback can be overcome by the development of PK inhibitors that specifically affect epileptogenesis without affecting normal cell growth. PK may be an effective target for the development of a drug which not only antiepileptic but also prevents the course of epileptogenesis.
Metabotropic glutamate receptors
Glutamate receptors are classified into two types: ionotropic (iGluRs) and metabotropic glutamate receptors (mGluRs). Ionotropic glutamate receptors are divided into three subtypes: kainate, AMPA, and NMDA receptors.[17] iGluRs are involved in fast synaptic transmission at glutamatergic synapses.[18] mGluRs were discovered in mid-1990s. mGluRs are G protein coupled receptors of the glutamate receptor family. It plays an important role in glutaminergic transmission.[19–21] Based on signal transduction mechanisms, the pharmacological profile and receptor protein mGluRs are classified into three groups: group 1 (mGluR1 and 5), 2 (mGluR2 and 3), and 3 (mGluR4, 6, 7, and 8).[18,22] Postsynaptically mGluRs have a role of modulating membrane properties by the second messenger while presynaptic mGluRs are involved in control synaptic release. These properties can be used to control glutamatergic signaling in the central nervous system (CNS), without interfering functions of iGluRs.[23] Group 1 mGluRs through Gq protein are involved in intracellular mobilization, upregulation of these receptors is seen in patients with epilepsy.[24] Groups 2 and 3 receptors are located presynaptically where they decrease glutamate release; reduction in their expression has been found in patient with temporal lobe epilepsy.[25] Recent studies have shown that Group 2 mGluRs inhibit cortical excitation of thalamic neurons, cortical layer 5, and intracortically projecting layer 2/3.[26] In pentylenetetrazole-induced kindled seizures (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA), a selective mGluR1 antagonist and (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (2R,4R)-APDC), a selective mGluR2/3 agonist have been shown dose-dependent inhibition of both seizure stage and EEG pattern. l-(+)-2-Amino-4-phosphonobutyric acid (l-AP4), a selective mGluR4/8 agonist, at a dose of 10 and 20 nmol/site injected at the lateral ventricle has also shown dose-dependent inhibition of pentylenetetrazole-induced kindled seizures. The combined use of AIDA, APDC, and l-AP4 has shown significant inhibition of seizure stages and EEG pattern with decreased individual dose of these drugs. There was no significant effect when AIDA was used by an intraperitoneal route, possibly due to prevention of its entry by blood–brain barrier. l-AP4 at low dose is found to have antiepileptic effect but when used in high doses, it induces convulsion. It could be due to depolarization of NMDA receptors at higher dose. Combined use of these agents also shown favorable results.[27]
Group 1 mGluRs are involved in cerebellar motor function and blockage of these receptors may lead to motor incordination and ataxia. This could be major hindrance in the development of group 1 mGluRs antagonist. Group 2 mGluRs may be a promising target due to their selectivity and inhibitory action on cortical and thalamic neuron.[26] Group 3 mGluRs agonists have shown mixed effects; some studies have demonstrated it as pro-convulsive while in other studies found it to have a protective role in absence seizure and proconvulsant in generalized seizure. This shows involvement of multiple pathways in Group 3 mGluRs action. Further studies required before labeling it as an important target.
Mammalian target of rapamycin
Mammalian target of rapamycin (mTOR) is a conserved serine/threonine PK. mTOR activity is upregulated or downregulated in the tuberous sclerosis complex, cortical dysplasia, brain tumors, neurodegenerative disorders, and traumatic brain injury.[28] mTOR plays the central role in multiple mechanisms of epileptogenesis[29] [Figure 1]. A variety of extracellular and intracellular signals like nutrients, energy status, growth factors, and stress regulate the upstream pathway which in turn regulates mTOR activity.[30,31] mTOR responds to this pathway by modulating downstream pathways by increasing or decreasing growth, proliferation, metabolism, and survival. These effects are produced by direct translation of proteins.[32] Therefore, in anabolic states it increases cellular growth and catabolic states decreases it. It has been demonstrated that the mTOR pathway upregulated during 1–24 hours (acute phase) after seizure onset and 3 days–5 weeks (second phase) after status epilepticus (SE). Acute phase of mTOR activation can be induced directly by initial SE-evoked synaptic activity; while the second activation may be a consequence of ensuing spontaneous action potential discharges due to enhancement of synaptic efficacy after the initial episode of SE. mTOR causes transcription, induction of proapoptotic proteins such as BAX and leads to activation of intrinsic cell pathway.[33] Another mechanism proposed is the role of mTOR in autophagy inhibition.[34] Autophagic stress-induced neuronal death in in vivo models of neuron death and neurodegeneration, including the kainate-induced seizure model has been found.[35] Zeng et al. demonstrated the role of rapamycin in neurogenesis and MFS, which are major mediators in epileptogenesis. Rapamycin was effective in the suppression of recurrent seizure when given before and after seizure induction by kainate.[36] Wong demonstrated the antiepileptogenic role of rapamycin in tuberous sclerosis and acute brain injury model.[29]
Figure 1.
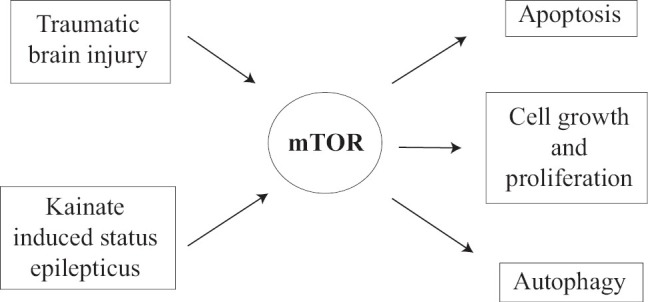
Role of a mammalian target of the rapamycin pathway in epileptogenesis
Safety is a major concern in usefulness of mTOR inhibitors. Rapamycin approved by U.S. Food and Drug Administration (FDA) as an immunosuppressant drug, and there are known risks associated with it. Rapamycin increases risk of infection, adversely affects growth and development on the long term use in children.[29] The question needs to be answered in the use of rapamycin is therapeutic window or up to how much time after brain injury rapamycin treatment can be initiated to exert antiepileptic effect. Other factor needs to be considered is the mTOR pathway that may also be associated with recovery of brain after injury and inhibition of these may adversely affect recovery process. The future use of mTOR inhibitors depends on careful titration of dose which inhibits pathological process but allows the normal physiological process involving the mTOR pathway.
However, it is a good signal of shifting epilepsy treatment from seizure suppression to prevention of epilepsy.
Role of CO2 in epilepsy
Suppression of the electroencephalography (EEG) spike-wave pattern in a patient of petit mal epilepsy by carbogen (10% CO2) was demonstrated in early 1990s. Number of studies performed on various concentrations (10–30%) of CO2 have demonstrated the antiepileptic effect. Recently, the effect of standard medical carbogen with low level of CO2 (5%) has been investigated for antiepileptic activity in the rat model of myoclonic epilepsy, non-human primate epilepsy model and in humans with partial epilepsy. In this study 5% CO2 showed potent effects on cortical epileptiform activity and convulsions in animal epilepsy models and in humans with drug-resistant partial epilepsy.[37] Epilepsy terminated within 1 min after administration of CO2 and prolonged administration was not needed. The elevated level of pCO2 causes acidosis which has a direct suppressant effect on brain excitability. Acetazolamide, an carbonic anhydrase inhibitor, has shown antiepileptic activity in previous studies.[38,39] Changes in brain pH through systemic carbonic anhydrase inhibition and retention of CO2 may account for its anticonvulsant properties. It is also proposed that direct effects on the interneuron carbonic anhydrase inhibitor may be an alternative mechanism. pH also plays an important role in the antiepileptic effect of CO2. In vivo and In vitro studies demonstrated that excessive neuronal activity during seizure leads to acidosis, which helps in termination of seizure.[40,41] A slight change in ambient pressure of CO2 precipitates seizures shows that seizure ishighly sensitive to pCO2.[42] Safety is major deciding factor in treatment with CO2; however, little discomfort was reported in patients breathing 5% CO2 for a couple of minutes. High percentage of CO2[43] for the prolonged period[44] may lead to symptoms such as anxiety. It has a very small effect on non-epileptic cortical activity as compared to antiepileptic activity.
On the basis of these findings CO2 due to its rapid action, safety and low cost may be a good option to treat prolonged seizures in the setting of the emergency room, intensive care unit, or ambulance especially in developing countries.
Neurosteroids
Neurosteroids are considered as fourth generation neuromessengers which are synthesized within the neurons. Neurosteroids are synthesized directly by brain from cholesterol. Pregnenolone is synthesized from cholesterol, which is then converted to allopregnanolone and allotetrahydrodeoxycorticosterone. Gonadal sex steroids and dehydroepiandrosterone (DHEA) are not considered as neurosteroids, as they are not synthesized in the brain. Only PREG(S) sulfur ester of DHEA, pregnanolone, allopregnanolone, and allotetrahydrocorticosterone are considered as ‘true’ neurosteroids. Neurosteroids have ability to modulate neurotransmission and act as positive allosteric modulators of GABAA receptors.[45] Studies have shown that during development of seizure, the level of neurosteroids fluctuates (expression of GABAA receptors increases when neurosteroids administered chronically and withdrawn). Enzymes 5-a-reductase (5aR) and 3-a-hydroxysteroid dehydrogenase (3aHSD) are found in brain that may be involved in their synthesis and metabolism. Epilepsy-like disorder (EL) mice is a genetic model for idiopathic complex partial seizures in humans.[46] During the development of seizure in EL mice, interictal period levels of neurosteroids, enzymes and GABAA receptor alpha4, gamma2, and delta subunits change. In third week of EL mice life, the level of neurosteroids increases which later at 5th to 10th week decreases and there is a sharp withdrawal of neurosteroids just before mice exhibit frequent seizures. Along with the decrease in the level of neurosteroids, there are upregulation of receptor alpha4, gamma2, and delta subunits.[47]
A recent clinical trial of ganaxolone, an allopregnanolone analogue, has shown the beneficial role in pharmacoresistant epilepsy.[48] No serious adverse reactions have been noted with ganaxolone treatment in this trial. In future neurosteroids which have shown to act on GABAA receptors may be a target for new drug development.
Future prospects
In summary, various therapeutic targets described in this review may have possible advantages to the available antiepileptic drugs (Some of these targets have shown to be effective in resistant cases, which may be an important option in therapeutic failure cases). Apart from the development of drug suppressing seizure, a new concept of prevention of epileptogenesis has arrived. Drugs acting through mTOR and PK pathways may be new rays in this direction. However, most of these drugs are tested in animals only and the possible use and safety in epilepsy have to be proven in clinical trials.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.de Boer HM, Mula M, Sander JW. The global burden and stigma of epilepsy. Epilep Behav. 2008;12:540–6. doi: 10.1016/j.yebeh.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 2.Rout SK, Kar DM. A review on antiepileptic agents, current research and future prospectus on conventional and traditional drugs. Int J Pharma Sci Rev Res. 2010:3. [Google Scholar]
- 3.Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–33. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–20. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JW, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand Suppl. 2007;186:7–15. [PubMed] [Google Scholar]
- 6.Vizzi ES, Mike A, Tarnawa I. 2,3-benzodiazepines (GYKI 52466 and analogs): Negative allosteric modulators of AMPA receptors. CNS Drug Rev. 1996;2:91–126. [Google Scholar]
- 7.Fritsch B, Stott JJ, Donofrio J Joelle, Rogawski MA. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia. 2010;51:108–17. doi: 10.1111/j.1528-1167.2009.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitkänen A, Mathiesen C, Rønn LC, Møller A, Nissinen J. Effect of novel AMPA antagonist, NS1209, on status epilepticus: An experimental study in rat. Epilepsy Res. 2007;74:45–54. doi: 10.1016/j.eplepsyres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi S, Donevan SD, Rogawski MA. Anticonvulsant activity of AMPA/kainate antagonists: Comparison of GYKI 52466 and NBOX in maximal electroshock and chemoconvulsant seizure models. Epilepsy Res. 1993;15:179–84. doi: 10.1016/0920-1211(93)90054-b. [DOI] [PubMed] [Google Scholar]
- 10.Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handb Exp Pharmacol. 2008;182:335–60. doi: 10.1007/978-3-540-74806-9_16. [DOI] [PubMed] [Google Scholar]
- 11.Engel J, Pedley TA, Aicardi J. Epilepsy: A Comprehensive Textbook. 1-3. Philadelphia: Lippincott-Raven Publishers; 1997. [Google Scholar]
- 12.Purcell AL, Carew TJ. Tyrosine kinases, synaptic plasticity and memory: Insights from vertebrates and invertebrates. Trends Neurosci. 2003;26:625–30. doi: 10.1016/j.tins.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Queiroz CM, Mello LE. Protein tyrosine kinase inhibitors modify kainic acid-induced epileptiform activity and mossy fiber sprouting but do not protect against limbic cell death. Braz J Med Biol Res. 2008;41:403–10. doi: 10.1590/s0100-879x2008000500009. [DOI] [PubMed] [Google Scholar]
- 14.Salter MW, Kalia LV. Src kinases: A hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–28. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 15.Gajda Z, Török R, Horváth Z, Szántai-Kis C, Orfi L, Kéri G, et al. Protein kinase inhibitor as a potential candidate for epilepsy treatment. Epilepsia. 2011;52:579–88. doi: 10.1111/j.1528-1167.2011.02979.x. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors: A review on pharmacology, metabolism and side effects. Curr Drug Metab. 2009;10:470–81. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- 17.Pellicciari R, Costantino G, Macchiarulo A. Metabotropic glutamate receptors: A structural view point. Pharm Acta Helv. 2000;74:231–7. doi: 10.1016/s0031-6865(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 18.Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. 2009;6:94–107. doi: 10.1016/j.nurt.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sladeczek F, Pin JP, Récasens M, Bockaert J, Weiss S. Glutamate stimulates inositol phosphate formation in striatal neurones. Nature. 1985;317:717–9. doi: 10.1038/317717a0. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama H, Ito I, Hirono C. A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature. 1987;325:531–3. doi: 10.1038/325531a0. [DOI] [PubMed] [Google Scholar]
- 21.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–79. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 22.Miller S, Kesslak JP, Romano C, Cotman CW. Roles of metabotropic glutamate receptors in brain plasticity and pathology. Ann N Y Acad Sci. 1995;757:460–74. doi: 10.1111/j.1749-6632.1995.tb17506.x. [DOI] [PubMed] [Google Scholar]
- 23.Alexander GM, Godwin DW. Metabotropic glutamate receptors as a strategic target for the treatment of epilepsy. Epilepsy Res. 2006;71:1–22. doi: 10.1016/j.eplepsyres.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Blümcke I, Becker AJ, Klein C, Scheiwe C, Lie AA, Beck H, et al. Temporal lobe epilepsy associated up-regulation of metabotropic glutamate receptors: Correlated changes in mGluR1 mRNA and protein expression in experimental animals and human patients. J Neuropathol Exp Neurol. 2000;59:1–10. doi: 10.1093/jnen/59.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Durand D, Pampillo M, Caruso C, Lasaga M. Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology. 2008;55:577–83. doi: 10.1016/j.neuropharm.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Alexander GM, Godwin DW. Metabotropic glutamate receptors as a strategic target for the treatment of epilepsy. Epilepsy Res. 2006;71:1–22. doi: 10.1016/j.eplepsyres.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe Y, Kaida Y, Fukuhara S, Takechi K, Uehara T, Kamei C. Participation of metabotropic glutamate receptors in pentetrazol-induced kindled seizure. Epilepsia. 2011;52:140–50. doi: 10.1111/j.1528-1167.2010.02764.x. [DOI] [PubMed] [Google Scholar]
- 28.Cao R, Li A, Cho HY. mTOR signaling in epileptogenesis: Too much of a good thing? J Neurosci. 2009;29:12372–3. doi: 10.1523/JNEUROSCI.3486-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 31.Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, et al. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 32.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–87. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 34.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 35.Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tolner EA, Hochman DW, Hassinen P, Otáhal J, Gaily E, Haglund MM, et al. Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia. 2011;52:104–14. doi: 10.1111/j.1528-1167.2010.02731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson RE, Howard RA, Woodbury DM. Correlation between effects of acute acetazolamide administration to mice on electroshock seizure threshold and maximal electroshock seizure pattern, and on carbonic anhydrase activity in subcellular fractions of brain. Epilepsia. 1986;27:504–9. doi: 10.1111/j.1528-1157.1986.tb03575.x. [DOI] [PubMed] [Google Scholar]
- 39.Rollins DE, Withrow CD, Woodbury DM. Tissue acid-base balance in acetazolamide-treated rats. J Pharmacol Exp Ther. 1970;174:535–40. [PubMed] [Google Scholar]
- 40.Xiong ZQ, Stringer JL. Extracellular pH responses in CA1 and the dentate gyrus during electrical stimulation, seizure discharges, and spreading depression. J Neurophysiol. 2000;83:3519–24. doi: 10.1152/jn.2000.83.6.3519. [DOI] [PubMed] [Google Scholar]
- 41.Wang RI, Sonnenschein RR. PH of cerebral cortex during induced convulsions. J Neurophysiol. 1955;18:130–7. doi: 10.1152/jn.1955.18.2.130. [DOI] [PubMed] [Google Scholar]
- 42.Doherty MJ, Youn C, Gwinn RP, Haltiner AM. Atmospheric pressure and seizure frequency in the epilepsy unit: Preliminary observations. Epilepsia. 2007;48:1764–7. doi: 10.1111/j.1528-1167.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- 43.Ley R. Ventilatory control of heart rate during inhalation of 5% CO2 and types of panic attacks. J Behav Ther Exp Psychiatry. 1991;22:193–201. doi: 10.1016/0005-7916(91)90016-x. [DOI] [PubMed] [Google Scholar]
- 44.Griez EJ, Colasanti A, van Diest R, Salamon E, Schruers K. Carbon dioxide inhalation induces dose-dependent and age-related negative affectivity. PLoS One. 2007;2:e987. doi: 10.1371/journal.pone.0000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawato S, Yamada M, Kimoto T. Brain neurosteroids are 4th generation neuromessengers in the brain: cell biophysical analysis of steroid signal transduction. Adv Biophys. 2003;37:1–48. doi: 10.1016/s0065-227x(03)80002-3. [DOI] [PubMed] [Google Scholar]
- 46.Seyfried TN, Todorova MT, Poderycki MJ. Experimental models of multifactorial epilepsies: The EL mouse and mice susceptible to audiogenic seizures. Adv Neurol. 1999;79:279–90. [PubMed] [Google Scholar]
- 47.Murashima YL, Yoshii M. New therapeutic approaches for epilepsies, focusing on reorganization of the GABAA receptor subunits by neurosteroids. Epilepsia. 2010;51:131–4. doi: 10.1111/j.1528-1167.2010.02627.x. [DOI] [PubMed] [Google Scholar]
- 48.Biagini G, Panuccio G, Avoli M. Neurosteroids and epilepsy. Curr Opin Neurol. 2010;23:170–6. doi: 10.1097/WCO.0b013e32833735cf. [DOI] [PMC free article] [PubMed] [Google Scholar]