

Abstract
To test the hypothesis that DNA polymerase ζ participates in immunoglobulin hypermutation, we generated two mouse models of Pol ζ function: a B-cell specific conditional knock-out and a knock-in strain with a Pol ζ mutagenesis-enhancing mutation. Pol ζ-deficient B-cells had a reduction in mutation frequency at immunoglobulin loci in the spleen and in Peyer’s Patches, while knock-in mice with a mutagenic Pol ζ, displayed a marked increase in mutation frequency in Peyer’s Patches revealing a pattern that was similar to mutations in yeast strains with a homologous mutation in the gene encoding the catalytic subunit of Pol ζ. Combined, these data are best explained by a direct role for DNA polymerase ζ in immunoglobulin hypermutation.
Introduction
The genes encoding the antigen-interacting portions of antibodies and immunoglobulin (Ig) receptors are subjected to a process of deliberate hypermutation during immune responses leading to enhanced affinity of antibodies to a specific antigen (1, 2). This mechanism, termed somatichypermutation (SHM), is triggered by the activation-induced deaminase (AID), a molecule expressed when B lymphocytes are activated by foreign antigen (3, 4). AID deaminates cytosines in the DNA encoding the Ig variable (V) regions (5). Mice and humans defective in AID lack SHM and class switch recombination (CSR), as AID is also required to generate switched antibodies such as IgG, IgA, etc. (3, 4). A subset of Hyper IgM syndrome patients are defective in AID and these patients lack CSR, SHM, and suffer from lymph node hyperplasia (6).
Despite the fact that AID is a cytosine deaminase, mice deficient in AID lack mutations at both G-C and A:T base pairs (3). This paradox is in part explained by the hypothesis that AID-mediated deamination of cytosines in Ig V regions triggers the recruitment of translesion synthesis DNA polymerases to Ig loci (7). These DNA polymerases have relaxed geometric requirements, and thus are prone to inserting incorrect bases during DNA synthesis (8). One such polymerase is DNA polymerase η, which has been shown to play a role in the misinsertion of bases at A:T sites during SHM (9, 10). Other DNA polymerases have also been implicated in SHM but, because they have important roles in other cellular functions such as cell division and DNA repair, discerning whether they play a direct or indirect role in SHM has been difficult (11–13).
The mutations made during SHM of Ig V genes are predominantly base substitutions. The pattern of hypermutation suggests that a putative error-prone DNA polymerase must not only insert the incorrect base but also extend from a mismatched terminus, a very difficult task for most DNA polymerases. Confounding this is the fact that some of the base substitutions in SHM occur in tandem, suggesting multiple misinsertion and mismatch extension events during a single DNA transaction (14). This lead to the hypothesis that DNA polymerase ζ plays a direct role in SHM, since it is a robust mismatch extender, alone and in conjunction with other translesion synthesis DNA polymerases, including Pol η (9, 14–16). However, demonstrating this has been difficult because mice deficient in DNA polymerase ζ are early embryonic lethals (17–19). A mouse expressing antisense RNA against Rev3 (encoding the catalytic subunit of Pol ζ) experienced decreased SHM frequency and severely impaired affinity maturation (20). However, because all cells, not just B cells, expressed the antisense transcript, it remained possible that the phenotype was due to indirect effects, such as diminished T cell function. In addition, SHM was reduced in human B cell lines in which the Rev3 gene encoding the catalytic domain of DNA polymerase ζ was inhibited by antisense oligonucleotides, suggesting a direct role for this polymerase in an in vitro model of hypermutation (21). A conditional knockout mouse model of Rev3 using the CD21 promoter also resulted in a reduced SHM frequency that was difficult to discern from a proliferation defect (22).
To circumvent the problem of embryonic lethality and non-specific effects from Pol ζ deficiency in non-B cells, we generated mice with B-cell specific deletion of Rev3. We also constructed mice with a knocked-in leucine (L) to phenylalanine (F) mutation at residue 2610 in Rev3 (Rev3L2610Fmice). The homologous change in Saccharomyces cerevisiae increases spontaneous and UV light-induced mutagenesis and is associated with a specific error signature (23, 24). We reasoned, that if Pol ζ plays a direct role in SHM, a more mutagenic variant would increase the frequency of mutation at Ig loci and its error signature would be accentuated. Indeed, we show here that Rev3 knock-out mice experienced a dramatic reduction in SHM frequency, while the Rev3L2610Fmice showed a significant increase in SHM frequency and an altered SHM specificity. The results indicate a direct role for DNA polymerase ζ in SHM.
Materials and Methods
Generation of B cell-specific Rev3 knock-outs and Rev3L2610Fknock-ins
A linearized targeting vector containing loxP sites flanking exon 26 of Rev3 was generated and electroporated into embryonic stem (ES) cells from C57BL6 mice (Supplemental Figure 1A and 1B). Exon 26 of Rev3 encodes the metal binding domain of DNA polymerase ζ and it is essential to its DNA synthesis function (25, 26). Recombinants were electroporated with Cre recombinase to eliminate the loxP site-flanked neo site. Clones still retaining the loxP sites flanking exon 26 of Rev3 (Rev3-floxed mice) were identified by Southern hybridization. Recombinant clones were confirmed by PCR analysis using primers N17delckF; 5’- GTTTGGGGCATTGGTTTACAGGTG - 3’ and N17del3R; 5’- CTCCTTACTGCTGGGGATACTCATGTG -3’. Several clones were selected for blastocyst injection resulting in male chimeras. Chimeras were breed with C57BL/6 Taconic mice and heterozygotes obtained. Rev3-floxed mice were crossed with the cre-recombinase transgenic line C.Cg-Cd19tm1(cre)Cgn (Balb/c) (27) which we will refer to as CD19+) and reporter mice B6.129X1-Gt(Rosa)26Sortm1(EYFP)Cos/J (C57BL/6) (which we will refer to as YFP+), strains purchased from The Jackson Laboratory(Bar Harbor, ME). CD19+ mice were crossed with YFP+ mice to give, CD19+/−, YFP+/− these mice were then backcrossed to the Rev3F/F background (10 generations) to give CD19+/−, YFP+/−, Rev3F/F mice in a C57BL6 background. The resulting mice had B cell specific deletion of Rev3 exon 26 and the recombinant B cells can be collected by FACS by virtue of their YFP expression (Supplemental Figure 1B). Alleles of the targeted floxed exon 26 Rev3 gene were examined by PCR with primers N1753F (5'-TTTCTATCAGCTTGTGCCCTTATCCTTACT-3') plus N17cKR (5'-ACAAGATTTCTTTGTGTAACAGCCCTGG-3') for the 5’ loxp site, give WT and mutant products of 474bp and 624bp, respectively. Genotyping of the whole exon 26 region using N1753F and N17del3R resulted in a WT product of 1330bp and mutant product of 1630bp (Supplemental Figure 1C). Upon cre-mediated deletion of the floxed region, the above primers resulted in a product of 632bp (Supplemental Figure 1C).
To detect the CD19 cre-recombinase transgene primers, CD19 5’ (5′-CTATCTGAAAAATATTTAACAGGTGCCAC-3′), CD19 3’ (5′-CACTATCCTCCACGTTCACTGTCCA-3′) and CD19 cre (5′-GGCAAATTTTGGTGTACGGTCAGTAA-3), resulting in a WT product of 950bp and a transgene product of 850bp were used ((Supplemental Figure 1D). YFP genotyping primers were oIMR0316 (5’-GGAGCGGGAGAAATGGATAT-3’), 0IMR0883 (5’-AAAGTCGCTCTGAGTTGTTAT-3’) and 0IMR4982 (5’AAGACCGCGAAGAGTTTGTC-3’) which amplify a WT fragment of 600bp and a mutant fragment of 320bp (Supplemental Figure 1E).
To generate Rev3L2610F (mutator knock-ins), a second vector was made with a mutation replacing the leucine at position 2610 to phenylalanine in exon 23 (Supplemental Figure 2A). The homologous mutation in yeast reveals a 2–3 fold increase in Rev3-mediated mutagenesis (23–24) Rev3L2610Fcells with the L to F mutation that had undergone homologous recombination were identified by Southern Hybridization and confirmed for the mutation by PCR using primers: N29B5PCRF: 5’- CTTTCATGTTCTCCATCAGCGTTTCC- 3’ and N29B5PCRR: 5’- GGTTAGCTGGGCTACATTCCAATTCATC -3’, followed by sequencing with primer: N29B5PCRR: 5’GGTTAGCTGGGCTACATTCCAATTCATC -3’. (Supplemental figure 2B–C).
For Rev3L2610Fmice, detection of the mutant allele was done by PCR with primers: N29BNeodelF 5’- GTTAAATCAGCTTCCGTTGCAGCACT- 3’ , and N29BNeodelR: 5’- GCTTCCACAAGTGTTTCCTATGAGAGTTG-3’. The presence of the mutation in the mRNA encoding Rev3 in thèse mice was confirmed using cDNA as template and using primers : F5’ATgAgAgCCCCACAgTgTgTT and R5’CAACCCTAgCACCCCATTTCT and the nested primers, 5’gTCTCgTTTCTATAgCAACTCTg and 5’ CTggCTTTgTgAACAATgCTATC. The nested primers were also used for sequencing (Supplemental Figure 2D). All mice were kept at the pathogen specific free animal facility at NIEHS and were maintained in microisolator cages on hardwood bedding, and provided with autoclaved food and reverse osmosis, deionized water.
Confirmation of Rev3 exon 26-deletion in YFP expressing B cells by PCR
For detection of WT transcripts, cDNA from Rev3-deleted B cells were amplified using Taq DNA polymerase (Invitrogen) and using Rev3 junct 25/26 F (5’-GCCGTGCATTGAGGTTGGTGATA-3’) and Rev3 exon 27 R (5’-CTTCAGTTTCACTGGCCTAGGATTAGTA-3’) resulting in a 229bp WT fragment. Mutant Rev3 transcripts were detected using Rev3 junct 25/27 F (5’-TGCCGTGCATTGAGTATGTTTGTACTACT-3’) and Rev3 exon 30 R (5’-CAGCGTTTCATACATGTAGCCCACAT-3’) resulting in a 191bp mutant Rev3 transcript (Supplemental Figure 1F). The RT-PCR of mouse GAPDH was performed with GAPDH F primer (5’-ACCACAGTCCATGCCATCAC-3’) and GAPDH R primer (5’-TCCACCACCCTGTTGCTGTA-3’).
Immunizations
8–12-week-old mice were immunized by intraperitoneal injection of 100µl (0.1ml of 1mg/ml) of 4-Hydroxy-3-nitrophenylacetyl hapten, or 2,4,6-Trinitrophenyl hapten, conjugated to chicken γ-globulin (NP-CGG, N-5055-5, and TNP-CGG, T5052-1; Biosearch Technologies, Novato, CA) prepared in an alum-precipitated suspension. Mice were euthanized and their spleens and serum recovered for analysis 14–15 days post-immunization.
Preparation of bone marrow B cells, and of splenic and Peyer’s Patches GC B cells
Spleen and bone marrow cell suspensions were created as describe in (28). Peyer’s patches were collected from mice at ages of 8, 12 and 26–52 weeks and cell suspensions generated as described previously (29).
Flow Cytometry analysis
Bone marrow progenitor B cells were enriched using allophycocyanin (APC)-labelled rat anti mouse B220/CD45R (clone RA3-6B2). Combined staining with either phycoerythrin (PE)-labelled rat anti mouse CD117/c-kit (clone ack45), CD25/IL-2Rα-chain (clone Pc61) or IgM (cloneR6-60.2) isolated pre-BI, pre-BII and immature B cells, respectively. Splenic B cells were isolated using PE-labelled rat anti mouse-CD19 (1D3) and IgM (cloneR6-60.2), plus APC-labelled rat anti mouse B220/CD45R (clone RA3-6B2) all from PharMingen (San Diego, CA). Activated splenic B cells from NP-immunized mice were stained for APC-labelled rat anti mouse B220/CD45R (clone RA3-6B2) (BD PharMingen, San Diego, CA) and biotinylated rat anti mouse Ig λ1, λ2 & λ3 light chain (clone R26-46) (BD PharMingen, San Diego, CA). Biotinylated antibodies were revealed using streptavidin-phycoerythrin (Southern Biotechnology Assoc, Birmingham, AL), strepavidin-allophycocyanin (BD PharMingen, San Diego, CA), or strepavidin-tri-colour (Caltag laboratories). Peyer’s Patches B cells were enriched by staining with PE-labelled rat anti mouse-B220/CD45R (clone RA3-6B2) and APC-labelled Anti-mouse Ly-77 (clone GL7) from eBioscience (San Diego, CA). Cell sorting was carried out using a Becton Dickinson FACSVantage SE Flow Cytometer or FACSDiVa (Franklin Lakes, NJ). Data was analysed using the FlowJo (Ashland, OR).
PCR cloning and sequencing of the intronic region downstream of rearranged V genes and of Vh186.2 regions
RNA from B220+ YFP+ λ+ cells sorted cells was prepared in TRIzol. One microgram of RNA was used as template for cDNA synthesis in the reverse transcriptase reaction by using a SuperScript II First-Strand Synthesis System (RT-PCR; Invitrogen). cDNA (2µl) was amplified using Phusion DNA polymerase (New England BioLab, Ipswich, MA) using the VH186.2 specific primers VH186.2 F (5’-AGCAGCCTGGGGGCTGAGCTT-3’) and IgH Cγ1 R (5’-CAGGGGCCAGTGGATAGACAGA-3’) and the following PCR conditions: 94°C for 2 mins; 35 cycles of 94°C for 1 minute, 60°C for 45 seconds and 72°C for 45 seconds; and 72°C for 5 mins, resulting in an ~400bp product. Sorting for λ+ cells and amplifying the VH186.2 region enriches for Ig sequences that have experienced SHM during the NP response (30). The reverse primer was anchored in the gamma constant domain to ensure amplification of the Ig HC from B cells likely to have participated in the GC reaction. The IgH intronic region downstream of rearranged V genes was amplified by PCR as previously described (28).
Spleen sectioning and immunohistochemistry
GC morphology was examined via biotin-labeled peanut agglutinin (PNA) (Vector Laboratories) as previously described (20). Briefly, spleens from 8–12 week-old mice were frozen in Tissue-Tek OCT (Sakura) and sectioned on a Leica CM 3050 S cryostat (6 µm) and affixed to slides with Rapid Fix solution (Shandon-Lipshaw). Protein blocking was carried out with an avidin-biotin blocking kit (Vector Laboratories). Incubation with PNA was done at a 1/1000 dilution and labeling with Biogenex streptavidin label. The stain was developed with diaminobenzidine chromogen (DakoCytomation) and the slides were counterstained with hematoxylin and visualized with a fluorescence microscope.
Lymphocyte proliferation assay
For CFSE analysis, splenic B cells from 8–12 week-old C57BL/6J, CD19+(B6), Rev3L2610F and Rev3-deleted mice were CD43-depleted using CD43 MicroBeads and MACS cell separator magnetic columns following the manufacturer’s instructions (Miltenyi Biotech GmbH, Bergisch Gladbach, Germany). Cells were incubated with 5nM CFSE for 15 min at 37°C following the manufacturer’s instructions (Invitrogen, Molecular Probes Carlsbad, CA). Cells were washed twice with PBS and incubated in RPMI 1640 medium. Cells (106 cells/ml) were then stimulated over 4 days using LPS alone (20ug/ml, Sigma), or together with IL-4 (25ng/ml, R & D Systems. All samples were analyzed as duplicates. The absorbance (Ex. 492nm / Em. 517nm) was measured using BD Biosciences FACSVantage SE Flow Cytometer. Activated B cells were determined using flow cytometry using APC-Cy7-labelled rat anti mouse B220/CD45R (clone RA3-6B2) and PE-labelled IgM (cloneR6-60.2) and IgG1 (clone 15H6) all from BD PharMingen (San Diego, CA). There were 105 cells analysed for each sample. A total of 5 mice per group were used.
Statistics
The difference in the distribution of mutational classes (number of clones with a particular number of mutations) among groups was examined for significance using the Kolmogorv-Smirnov test. The differences in the mutation frequency among genotypes in the spleen and PP were examined using Mann Whitney statistic and Kolmogov-Smirnov . The difference in the proportion of insertions and deletions, of tandem multiples, or of mutations from deoxythymidine among all mutations was tested for significance among groups using Fisher’s exact test or Chi-Square statistic. All probability values were considered significant if less than 0.05.
Results
To test the hypothesis that DNA polymerase ζ directly induces mutations at Ig loci during SHM, we generated mice with B-cell specific deletion of Rev3 exon 26 which encodes the metal binding domain, and mice with a mutation in Rev3 that, in yeast, renders the polymerase more mutagenic (23, 24) (Supplemental Figures 1–3). Rev3 knock-out mice were immunized with the NP hapten and mutations at the Vh186.2 heavy chain (HC) were analyzed using cDNA from splenic B cells. To sort the cells, we used B220+, λ+, and YFP+B cells to make the cDNA and amplified rearranged Vh186.2 regions that had switched to IgG. This regimen enriches for NP-reactive, isotype switched B cells. YFP is a marker for expression of cre-recombinase activity and thus strongly correlates with Rev3 deletion, as YFP+ cells lacked the floxed exon (Supplemental Figures 1A–B, 1F). Vh186.2 is the Ig HC associated with enhanced affinity to NP, and Ig’s bearing Vh186.2 HCs and λ light chains experience a 10-fold increase in affinity when tryptophan at position 33 is mutated to leucine (30). Affinity maturation to a specific antigen such as NP can profoundly influence the mutational pattern and frequency at the relevant Ig loci. Therefore, we also examined the accumulation of mutations in the intronic region downstream of rearranged J558 Ig HC V genes in B cells from the Ileal Peyer’s Patches (PP) of Rev3-altered mice. Because the region analyzed is non-coding, mutations here are a more accurate representation of the intrinsic SHM machinery in the absence of selection.
B cell development in the bone marrow and the spleen appears normal in Rev3-altered B cells
Impaired Pol ζ function did not impact B cell development in the bone marrow or the spleen compared to CD19-driven cre-recombinase transgenics and C57BL6 controls (Figure 1A–B). Over 90% of B220+ B cells were also YFP+, the marker for Rev3 deletion (Figure 1C–D). The number of GC B cells (GL7+) in B-cell specific Rev3 knock-out mice was similar to C57BL6 controls. While over 90% of all splenic B cells were YFP+, the percentage decreased to 56% among GL7+ B cells (Figure 1B). This was accentuated dramatically in Peyer’s Patches, where only 3% of the GL7+ B cells were YFP+ (Figure 1B). Splenic or Peyer’s Patches B cells from Rev3L2610Fmice displayed no defects in B cell development or in the number of germinal center (GC) cells (Figure 1A; data not shown).
Figure 1.

Normal B cell development in Rev3 knock-out and Rev3L2610Fmice. (A) B cells from 8–12 week-old mice were sorted and examined by FACS. Development of Rev3 knock-out and Rev3L2610FB cells in bone marrow stage pre-BII (left (B220+ CD25+) and in the immature B cell stage (middle, B220+ IgM+) is comparable to CD19-promoter driven cre-recombinase transgenics (heterogyzotes) and C57BL6 controls (not shown). As the B cells mature and migrate to the spleen (right, B220+IgM+) normal numbers of mature/resting splenic B cell populations are observed in both the Rev3 knock-out and Rev3L2610F. (B) Unimmunized12 week-old mice were NP-CGG injected and their B cells examined 15 days later. The activated splenic GC B cell population in Rev3 knock-out mice undergoing somatic hypermutation (B220+GL7+) appears normal in numbers with 42–56% of this gated population being YFP+. However, while the percentage of GL7+ B cells in PP appears normal, only a tiny fraction of those (3%) were YFP+. (C) Enrichment for activated splenic B cells from conditional Rev3 knock-out, 15 days after immunization with 150µg NP-CGG in alum. The post-sort (B220+ YFP+ λ+) GC splenic B cell population is greatly enriched for the YFP (marker for Rev3 deletion). The λ light chain was selected because it is associated with the Vh186.2 HC for the high affinity response to the NP hapten. This pure Rev3-null population was used for subsequent RTPCR and mutation analysis (D) Over 90% of B220+ B cells in the spleen were YFP+ in Rev3 knock-out mice.
Rearranged Vh186.2 HCs from Rev3-deleted splenic B cells displayed a reduction in mutation frequency
Following immunization with NP-CGG in alum, YFP+, Rev3-deleted B cells from Rev3 conditional knock-outs that had been crossed to CD19 promoter-driven cre-recombinase transgenics were examined for mutations at the rearranged Vh186.2 locus. Over 40% of the clones from these mice had either zero or 1 mutation compared to less than 15% of the clones from either C57BL6 controls or CD19 cre-recombinase transgenics without the floxed Rev3 gene (Figure 2A –BSupplemental Figure 4). This distribution of mutational frequencies was significantly different from controls (Mann-Whitney and Kolmogorov-Smirnov test, p= 0.00041 and p = 0.001, respectively). In addition, among VH186.2 clones with at least one mutation, 46% had the affinity-enhancing mutation W to L at amino acid 33 (Figure 2B). Remarkably, in some clones this was the only mutation. GC’s from these mice were indistinguishable from those of control mice (Figure 1B) in terms of both morphology and abundance, 15 days following immunization. The fact that GC morphology was intact and that the same proportion of clones from these mice acquired the NP-affinity enhancing mutations as in controls suggests that the decrease in the mutation frequency is not due to a defect in proliferation in these cells (see below).
Figure 2.
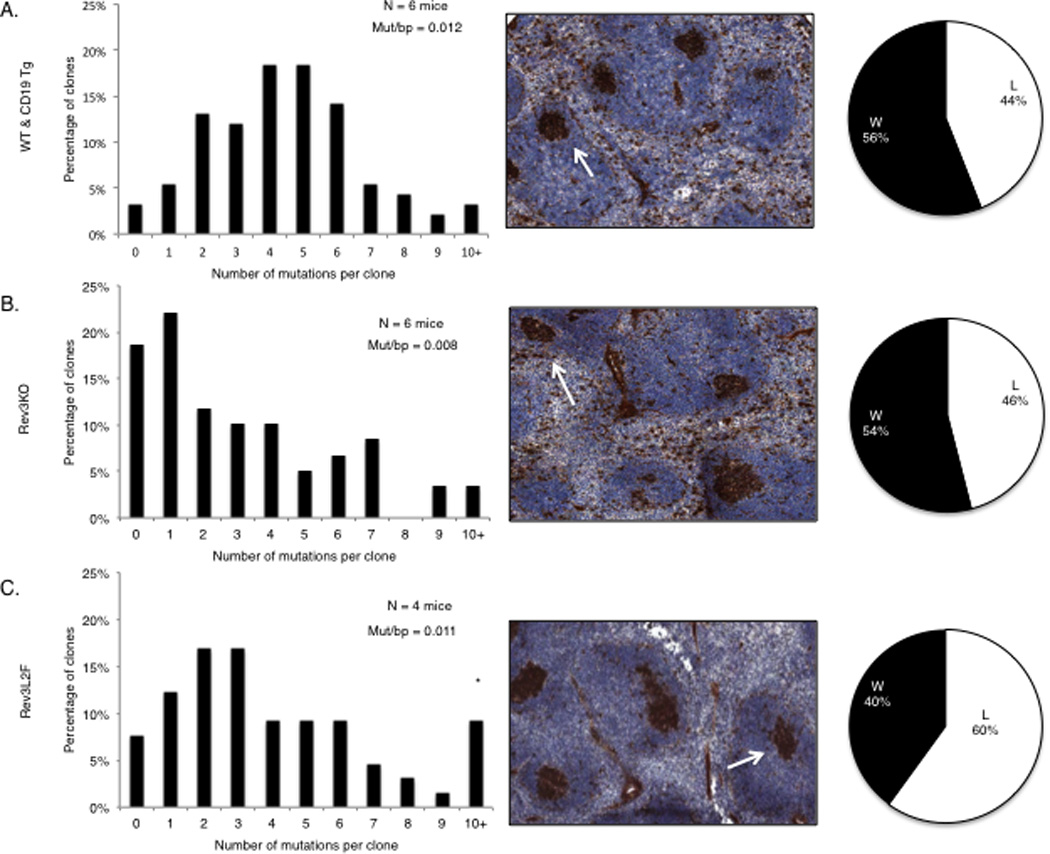
The immune response in mice with altered DNA polymerase ζ function. (A–B) Rev3 knock-out B cells experienced a decrease in the frequency of mutations at Ig V regions during the immune response to NP-CGG compared to WT mice and CD19 transgenics. Close to 40% of the clones in the Rev3 knock-out had 0–1 mutations, compared to less than 10% in the controls. Spleens from Rev3 knock-out mice displayed normal GC with 46% of the clones achieving the affinity-enhancing W to L mutation at position 33 of Vh186.2. (C) Rev3 knock-in mice (Rev3L2610F) had a similar overall distribution of mutational classes as controls but had a higher percentage of clones with more than 9 mutations. These mice displayed normal GC, but also displayed an increase in the fraction of Vh186.2 clones with the affinity enhancing W to L change (pie chart). The following mice were used for these experiments: C57BL/6 and CD19 promoter-driven cre recombinase transgenics in the C57BL/6 background, n = 6 mice and 75 unique clones; Rev3 knock-out, n = 6 mice and 59 unique clones; Rev3L2610Fclones, n = 4 mice and 50 unique clones. Unique clones were determined by the CDR3 amino acid sequence.
Altered immune responses to NP in Rev3L2610Fmice
Following immunization, most of the unique clones derived from Rev3L2610Fmice B cells contained slightly fewer mutations than C57BL6 controls (Figure 2C). However, there was an increase in the fraction of clones containing at least 10 mutations, with a few clones harboring more than 15 in the rearranged V region (Figure 2C). While it did not achieve significance at 0.05, there was a trend for an increase in the fraction of Vh186.2 clones with the characteristic W to L change at amino acid 33 that is associated with increased affinity to NP (> 60% of the clones, Figure 2C). These data may be explained by an increased rate of SHM due to an enhanced mutagenic potential or activity of DNA polymerase ζ. For example, B cells from these mice may acquire the affinity-enhancing mutations in VH186.2 more efficiently, and thus terminate SHM early in the GC reaction. The accumulation of mutations in conditions where the impact of selection is minimized (see below) helps clarify these data.
Proliferation among Rev3-altered B cells
Given that only 3% of GL7+ B cells in Peyer’s Patches and 56% of GL7+ B cells from the spleen were YFP+ (compared to over 90% of all B cells), and to rule out a defect in proliferation from Rev3 deficiency, we measured proliferation in Rev3-deleted B cells. Following activation with LPS, Rev3-deleted B cells consistently exhibited a significant defect in proliferation compared to CD19 transgenic WT controls (Figure 3A), as LPS-only treated Rev3-deleted B cells appeared to have an excess of cells in earlier stages of division. This defect was less obvious in LPS plus IL4 treated cells when compared to CD19 transgenics with normal Rev3 function (Figure 3B). Rev3L2610F mice activated with LPS and IL4 displayed normal proliferation (Figure 3B). As CSR is tied to proliferation, we examined switch to IgG1 following activation with LPS and IL4 and found that Rev3KO cells displayed decreased numbers of IgG1+ cells when compared to CD19 transgenic controls while Rev3L2610F had levels that were similar to C57Bl6 controls (Figure 3C).
Figure 3.
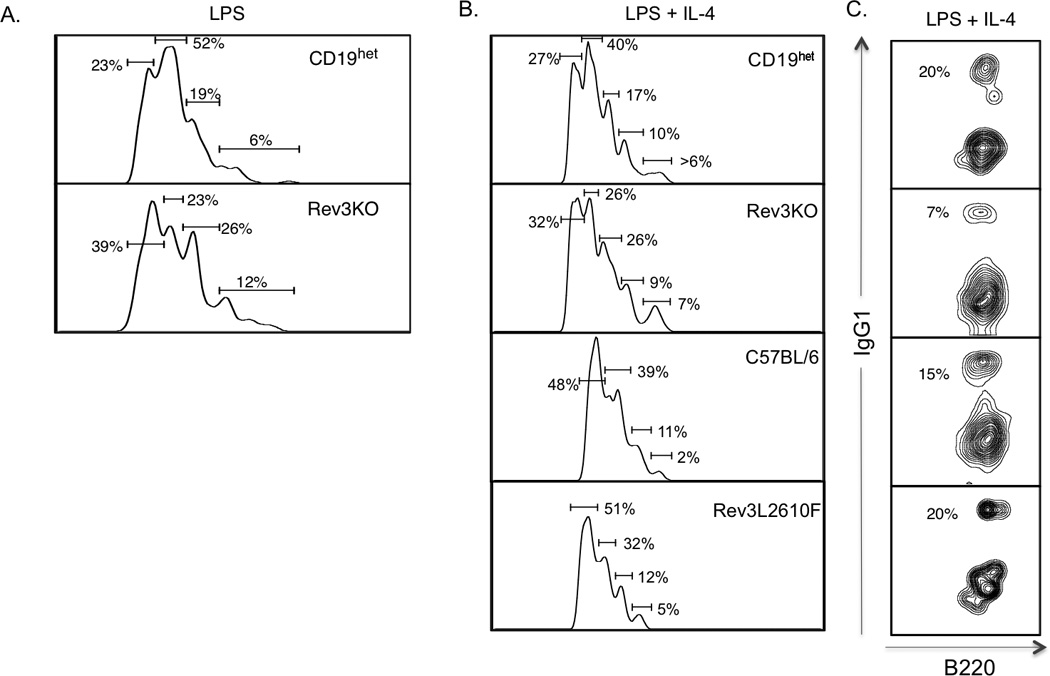
B cell proliferation in Rev3KO and Rev3L2610F mice. CD43-depleted splenic B cells from 8–12 week-old mice were isolated and incubated with 5nM CFSE in PBS to measure cell proliferation. B cells (106 cells/ml) were stimulated over 4 days using LPS alone (20ug/ml) (A), or together with IL-4 (25ng/ml) (B) to stimulate class switch recombination. In vitro stimulation of B cells was determined at day 0 and day 4 of stimulation. Flow cytometry was used to determine the B cell proliferation following in vitro stimulation as depicted by histograms. The numbered gated represent the percentage of B cells within sequential rounds of cell division. These proliferation peaks were based on 105 events, duplicate samples and a total of 5 mice per group. (C) Percentage of IgG1+ B cells following 4 days of activation with LPS and IL4.
Mutation accumulation at the intron downstream of rearranged V genes in PP B cells of Rev3-deleted B cells is dramatically reduced
Given the profound impact that antigen-driven selection has on the frequency and the pattern of SHM, we examined SHM in Rev3-altered mice in the intronic region downstream of rearranged VDJ segments from Peyer’s Patches B cells. This region has been characterized as a target of the SHM machinery with a reported peak mutation frequency of 0.01–0.15 mutations/base pairs (31, 32). Peyer’s Patches B cells are chronically stimulated and accumulate mutations over time, but by 5 months of age most mice reach their peak mutation frequency (33). All but one of the clones from mice with B-cell specific deletion of Rev3 were not mutated, even at 6 months of age (Figure 4A). In contrast, over 75% of the clones from control mice had at least one mutation by 6 months of age. Since only 3% of GL7+ B cells in Peyer’s Patches were YFP+ in the Rev3 knock-out mice, it is possible that the low mutation frequency of Rev3-deleted B cells puts them at a selective disadvantage in the GC environment of the Peyer’s Patches, specially given the pressure exerted by microbial flora in the small intestine. Alternatively, a proliferation defect may put these cells in a competitive disadvantage.
Figure 4.
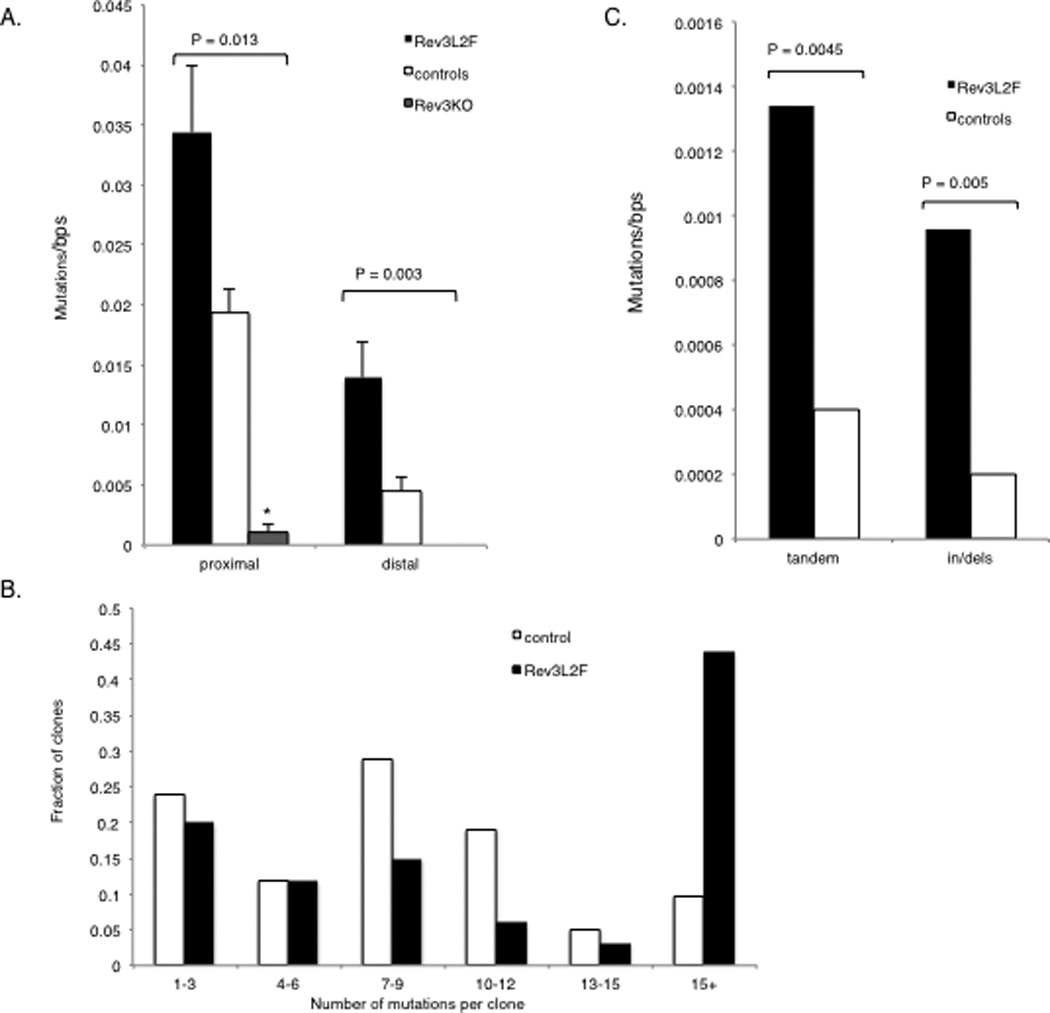
Mutations at the intronic region downstream of rearranged V genes in Peyer’s Patches B cells in mice with altered DNA polymerase ζ function. The IgH intronic region downstream of rearranged V genes of GC B cells was assessed for accumulation of mutations. Peyer’s Patches B220+ GL7+ (and YFP+ for Rev3 knock-out) B lymphocytes were isolated from mice aged 12 or 36 weeks: C57BL/6, n = 20 mice and 76 unique clones (with 53% of the mutated clones from 36 weeks old mice); Rev3L2610Fn = 8 mice and 84 unique clones (with 58% of the mutated clones from 36 weeks old mice); Rev3 knock-out, n = 4 mice and 18 unique clones (with 100% of the clones from 36 weeks old mice). Because the rearranged CDR3 was included in the PCR fragment, unique clones could be assessed and those differing by more than 5 mutations were considered as different clones, although shared mutations were only counted once. (A) Mutations per nucleotide among mutated clones were calculated for both the proximal (~ 430 bps, beginning immediately downstream of JH4) and distal ends of the IgH intronic region downstream of rearranged V genes (region was the last 590 bps of the 1.2kb PCR fragment). The regions were analyzed separately to examine mutation pattern and frequency in less saturated regions. In both the proximal and distal regions, the mutation rate in the Rev3L2610Fmice is at least twice that observed in controls and this difference was highly significant. Rev3 knock-out clones had an extremely low mutation frequency that was highly significantly different from controls. All except one of the unique Rev3 knock-out clones had no mutation; thus, all were included in the analysis. (B) The increase in mutation frequency among mutated clones in Rev3L2610Fmice was the result of clones with a large number of mutations: there was a dramatic increase in the fraction of Rev3L2610Fclones with >15 mutations, some having as many as 30 or more mutations in the proximal region alone (P = 0.037). 98% of the Rev3 knock-out clones were unmutated even in 6 month-old mice, compared to 33 and 57% for the control and the Rev3L2610Fclones, respectively. (C) Tandem doublets and triplets and insertions and deletions were significantly increased in Rev3L2610Fmice. The difference in mutation frequencies were tested for significance using the Mann-Whitney U test, the difference in the distribution of mutations among clones was tested for significance using the Kolmogorov-Smirnov test and the difference in the frequency of tandem and indels among all mutations was analyzed using the Fisher exact test (two-sided p values are given).
Mutation accumulation at the intronic region downstream of rearranged V genes in PP B cells of Rev3L2610Fmice is increased and the pattern is significantly altered
Rev3L2610Fmice were also examined for mutations at the intronic region downstream of rearranged V genes. There was a highly significant increase in the mutation frequency in GC B cells from Peyer’s Patches of these mice (Figure 4A). The number of mutations per base pair in Rev3 knock-in B cells was around 0.035, which represents a near 2-fold increase over the wild type (WT) controls. Because of the elevated mutation frequency, we divided the 1.2kb region for analysis into proximal and distal regions as defined by the distance from the rearranged V region, with the notion that the distal region may be less saturated and a more accurate assessment of the mutational frequency and pattern will be possible. This analysis revealed that the distal region from Rev3L2610Fmice had experienced a 3-fold increase in mutation frequency (0.014) over the same region in the WT controls (0.004) that was also statistically significant (Figure 4, Supplemental Figure 3D). Strikingly, 40% of the clones derived from Rev3L2610Fmice had accumulated more than 15 independent mutations, with many of those having 30–40 mutations in the 420bps of the proximal region alone (Figure 4B). There were also highly significant changes in the pattern of mutation in B cells derived from Rev3L2610Fmice. Tandem mutations, such as adjacent doublets and triplets, were increased 3–4 fold, and insertions or deletions (mostly +1 or −1 bp) were 5-fold higher than in controls (Figure 4C; Figure 5). Finally, when examining the non saturated sequences (distal region or mutations from 2 month old mice) mutations at thymidine bases were moderately increased in these cells (Figure 6), suggesting strand bias by Pol ζ in SHM at A:T base pairs.
Figure 5.
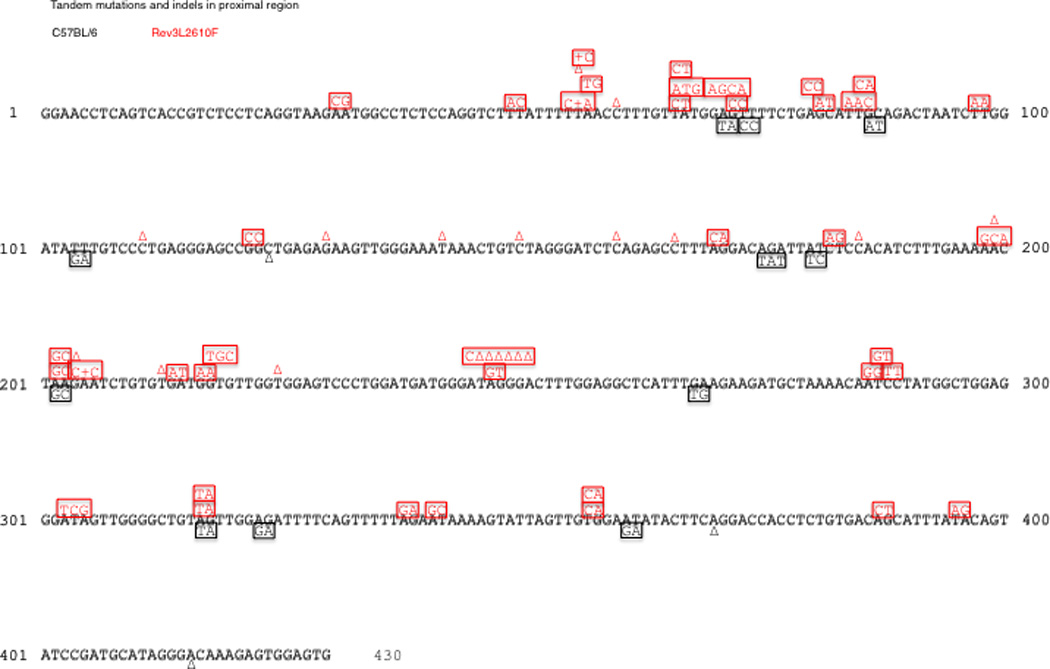
Tandem and indel mutations in the proximal HC intronic region. B cells from Rev3L2610F mice (33 unique clones) reveal an increase in tandem (squares) and indel mutations (+ or Δ) when compared to controls (41 unique clones). A similar pattern is seen in the distal region of the sequences (data not shown). Shared mutations from oligoclonality and single base substitutions are not shown.
Figure 6.
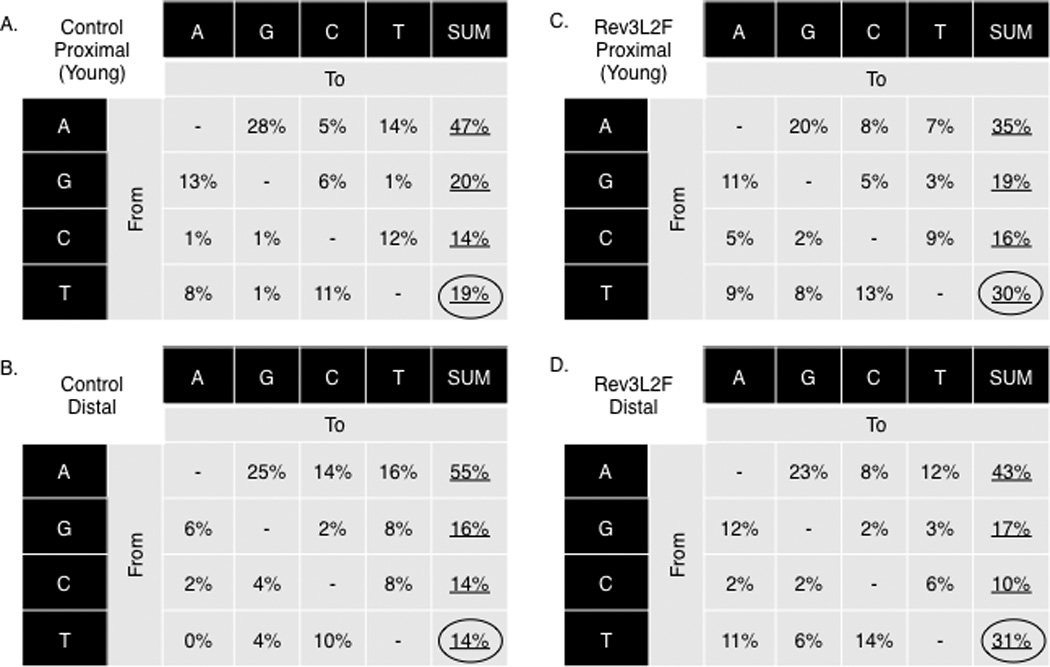
Mutational pattern of Rev3L2610Fmice reveals a moderate increase in mutations at T bases indicating Rev3L2610F-mediated strand bias. The mutation pattern at the IgH intronic proximal regions of B cells from young mice is depicted for controls (A) and Rev3L2610F(B) mice. The IgH intronic distal region of all controls (C) and all Rev3L2610F(D) mice B cells are also shown. The combined increase at thymidine was statistically significant (χ2 test, P = 0.04). The base composition in the proximal region is 57% A: T and 43% G:C, and for the distal region, it is 61% A:T and 39% G:C.
Discussion
In this study, we tested the hypothesis that DNA polymerase ζ plays a direct role in SHM using mice with altered Rev3 function, the gene encoding the catalytic subunit of DNA polymerase ζ. Mice with a B-cell specific deletion of Rev3 displayed a significant reduction in the accumulation of mutations at the VH186.2 locus during the immune response to the NP hapten, and PP B cells from these mice displayed a dramatic reduction in mutation frequency at the intronic region downstream of rearranged V genes. Indirect effects on SHM, a possibility plaguing previous studies, is minimized here as B-cell specific deletion of Rev3 eliminated the impact of Rev3 deletion in other cells such as in helper T cells or antigen presenting cells. Furthermore, in our model, Rev3-deleted B cells could be specifically recovered because cre-recombinase deletion of Rev3 (under control of the CD19 promoter) coincides with YFP expression (resulting from cre-recombinase-mediated deletion of a floxed stop codon upstream of the YFP gene). Thus, Rev3- deleted cells could be isolated based on YFP expression. This minimized the impact of incomplete Rev3 deletion, particularly in the highly competitive GC environment. Indeed, while over 90% of the splenic B cells were YFP+, only 56% of GL7+ B cells, a GC marker, were YFP+. PP B cells with Rev3 deletion displayed almost no mutation accumulation even at 6 months of age. B cells from Rev3 knock-out mice formed morphologically normal GC’s, yet displayed a proliferation defect following activation with LPS. Among those GC B cells that were mutated, a similar proportion had acquired the affinity-enhancing tryptophan to leucine change at amino acid 33 of VH186.2 following immunization with NP, suggesting that eventual acquisition of high affinity antibodies was not significantly altered in these cells, although it is possible that the affinity maturation rate may have been altered. These results are similar to a previous study using conditional Rev3 knock-out mice in terms of reduced mutation frequency but differ in that smaller germinal centers from Rev3 knock-out mice were reported in the previous study, while GC appeared normal in our study (22). This discrepancy may be due to the different cre-recombinase transgenic strains used in the studies (CD19 vs CD21). For instance, with earlier Rev3 deletion (as the CD19 promoter is active earlier than the CD21 promoter), it is possible Rev3-deleted cells may have partially compensated in terms of proliferative capacity from Rev3 deficiency, since this is not a polymerase that is essential for DNA replication. To circumvent the proliferation issue, and to determine if the SHM frequency reduction observed in both studies is the result of DNA polymerase ζ participating in the SHM process rather than entirely explained by a defect in proliferation, we examined mutation at Ig loci in Rev3 KI mice with a hypermutagenic polymerase ζ and an altered mutational spectra. An alteration in the pattern of mutations consistent with the observed error signature for the Rev3Ki polymerase, is strong evidence for a direct involvement and unlikely to be explained by a defect in proliferation.
In the Rev3Ki mice, the Rev3L2610F mice, a highly significant increase in mutation frequency at the intronic region downstream of rearranged V genes was seen in PP B cells. In yeast, the equivalent leucine to phenylalanine replacement in the Rev3 gene increases the error rate of DNA polymerase ζ about 2–3 fold without significantly altering its catalytic activity (23, 24). It is also associated with an increase in complex mutations (more than one mutation within a short patch presumed to occur in a single event), and with an increase in insertions and deletions. Given the high mutation frequency in the Rev3L2610F clones, it was difficult to ascertain which complex mutations originated from an accumulation of single mutational events or from a single DNA polymerase transaction that yielded complex mutational changes. However, limiting the analysis to tandem doublets or triplets revealed a 3- to 4- fold increase in these events. There was also a 5-fold increase in insertions and deletions, mostly adding or deleting a single nucleotide. These are similar to the mutational pattern observed in yeast (23, 24). Interestingly, an increase in mutations from thymine was also observed, suggesting Pol ζ strand bias during SHM. This is reminiscent of strand bias at A:T base pairs in Ig templates from DNA polymerase η deficient patients, and may imply complementary roles for Pol ζ and Pol η in SHM. Together, the results from both mouse models can best be explained by a direct role for DNA polymerase ζ in SHM.
While there were significant changes in both the frequency and pattern of SHM in mice with altered Rev3 function, there was no obvious difference between mutations at G:C vs A:T base pairs in either strain: all base pairs experienced either a decrease in Rev3-deleted B cells or an increase in Rev3L2610F B cells (with the exception of a modest increase in mutations from T’s but not from A’s in the Rev3L2610Fmice). The mutation frequency for Rev3 knock-out B cells and the lack of G:C bias is consistent with data from previously described models of Rev3-deletion, including mice expressing antisense transcripts to Rev3 (20, 22). These results are different from human B cells deficient in DNA polymerase η, where the loss of mutations was primarily at A:T base pairs (10), and suggests that DNA polymerase ζ participates in both the G:C and A:T phase of SHM.
On the surface, it appears difficult to reconcile the fact that Pol ζ is directly involved in introducing mutations at both G:C and A:T base pairs with the fact that AID triggers SHM by deaminating cytosines in both strands. One possibility, however, is that Pol ζ is involved in the mutagenic bypass of the abasic sites generated by the removal of AID-generated uracils in the DNA of Ig V regions. Continued synthesis pass the abasic site would also result in mutations at A:T base pairs, perhaps in complex with Pol η, and this would generate mutations at all base pairs. Pol ζ is uniquely efficient at extending synthesis past a variety of DNA lesions, including abasic sites, alone or in complex with REV1, a co-factor shown to contribute to mutations at AID-mediated abasic sites in a cell line (34, 35). Furthermore, it has been shown to extend mismatches generated by other TLS polymerases (36, 37). In Pol ζ deficient B cells, AID-generated abasic sites may be repaired in an error-free fashion reducing mutations at all base pairs, not just G:C base pairs. Inefficient recruitment of error-prone DNA polymerases to compensate for Pol ζ ablation may occur explaining why 15% of Rev3 deleted clones derived from splenic B cells had more than 6 mutations . Alternatively, Pol ζ may assemble a targeted mutasome complex to Ig loci which include AID. This would explain an impact on mutation at all base pairs, but it appears less likely since it would predict loss of all mutations, not just a reduction in the mutation frequency, with Rev3 deletion. Also, the results using Rev3L2610Fmice indicate a role for Pol ζ as a DNA polymerase, not just as a targeting complex.
It is interesting that Rev3-deleted B cells appear to be at a disadvantage in the PP environment compared to wild type B cells within the same animal. While close to 90% of B220+ B cells were YFP+ and therefore Rev3-deleted, only 3% of GC B cells in Peyer’s Patches expressed YFP. In the spleen, the reduction was less marked (56% of GL7+ B cells), and is more in agreement with other models of CD19 promoter-driven cre-recombinase deletion (38). This defect may be due to impaired proliferation as seen with splenic cells, or the result of intense competition in the GC environment for antigen binding and increased affinity through SHM. A reduced mutation frequency may impact the rate at which affinity-enhancing mutations are acquired, and this may place Rev3-deleted B cells in a competitive disadvantage in the small intestine Ileum, which is chronically stimulated by microbial flora. Interestingly, B cells from Rev3L2610Fmice in the spleen were more likely to have affinity-enhancing mutations to NP hapten (60% vs. 40% of controls). This raises the possibility that the converse is true: a higher rate of SHM with a mutagenic Pol ζ is associated with an enhanced affinity maturation rate for those cells. Having these novel strains that differ in the ability of their B cells to undergo SHM will be useful in examining the impact of SHM rate in the rate of affinity maturation to certain pathogens, specially those associated with specific antibodies bearing a large number of mutations, as appears to be the case for HIV (39) and influenza (40).
Supplementary Material
Acknowledgments
We are grateful to John W. Drake, and Dmitry Gordenin for critical readings of the manuscript. Natasha Clayton, Tiwanda Masinde for assistance with immunohistochemistry, and Carl Bortner, and Maria Sifre for assistance with flow cytometry. We are grateful to Yingbin Ouyang and Leanne Zhu from Xenogen and Manas Ray from NIEHS for chimera generation. We are also, grateful to Scott Jenkins for generation of the Rev3-deleted yeast strain.
Footnotes
References
- 1.Klinman NR. The mechanism of antigenic stimulation of primary and secondary clonal precursor cells. J Exp Med. 1972;136:241–260. doi: 10.1084/jem.136.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weigert MG, Cesari IM, Yonkovich SJ, Cohn M. Variability in the lambda light chain sequences of mouse antibody. Nature. 1970;228:1045–1047. doi: 10.1038/2281045a0. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 4.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 5.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 6.Durandy A. Hyper-IgM syndromes: a model for studying the regulation of class switch recombination and somatic hypermutation generation. Biochem Soc Trans. 2002;30:815–818. doi: 10.1042/bst0300815. [DOI] [PubMed] [Google Scholar]
- 7.Diaz M, Lawrence C. An update on the role of translesion synthesis DNA polymerases in Ig hypermutation. Trends Immunol. 2005;26:215–220. doi: 10.1016/j.it.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 9.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 10.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 11.Faili A, Aoufouchi S, Flatter E, Gueranger Q, Reynaud CA, Weill JC. Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymerase iota. Nature. 2002;419:944–947. doi: 10.1038/nature01117. [DOI] [PubMed] [Google Scholar]
- 12.Masuda K, Ouchida R, Takeuchi A, Saito T, Koseki H, Kawamura K, Tagawa M, Tokuhisa T, Azuma T, J OW. DNA polymerase theta contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc Natl Acad Sci U S A. 2005;102:13986–13991. doi: 10.1073/pnas.0505636102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zan H, Shima N, Xu Z, Al-Qahtani A, Evinger AJ, Iii, Zhong Y, Schimenti JC, Casali P. The translesion DNA polymerase theta plays a dominant role in immunoglobulin gene somatic hypermutation. Embo J. 2005;24:3757–3769. doi: 10.1038/sj.emboj.7600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz M, Flajnik MF. Evolution of somatic hypermutation and gene conversion in adaptive immunity. Immunol Rev. 1998;162:13–24. doi: 10.1111/j.1600-065x.1998.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 15.Diaz M, Flajnik MF, Klinman N. Evolution and the molecular basis of somatic hypermutation of antigen receptor genes. Philos Trans R Soc Lond B Biol Sci. 2001;356:67–72. doi: 10.1098/rstb.2000.0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz M, Velez J, Singh M, Cerny J, Flajnik MF. Mutational pattern of the nurse shark antigen receptor gene (NAR) is similar to that of mammalian Ig genes and to spontaneous mutations in evolution: the translesion synthesis model of somatic hypermutation. Int Immunol. 1999;11:825–833. doi: 10.1093/intimm/11.5.825. [DOI] [PubMed] [Google Scholar]
- 17.Bemark M, Khamlichi AA, Davies SL, Neuberger MS. Disruption of mouse polymerase zeta (Rev3) leads to embryonic lethality and impairs blastocyst development in vitro. Curr Biol. 2000;10:1213–1216. doi: 10.1016/s0960-9822(00)00724-7. [DOI] [PubMed] [Google Scholar]
- 18.Esposito G, Godindagger I, Klein U, Yaspo ML, Cumano A, Rajewsky K. Disruption of the Rev3l-encoded catalytic subunit of polymerase zeta in mice results in early embryonic lethality. Curr Biol. 2000;10:1221–1224. doi: 10.1016/s0960-9822(00)00726-0. [DOI] [PubMed] [Google Scholar]
- 19.Wittschieben J, Shivji MK, Lalani E, Jacobs MA, Marini F, Gearhart PJ, Rosewell I, Stamp G, Wood RD. Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol. 2000;10:1217–1220. doi: 10.1016/s0960-9822(00)00725-9. [DOI] [PubMed] [Google Scholar]
- 20.Diaz M, Verkoczy LK, Flajnik MF, Klinman NR. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase zeta. J Immunol. 2001;167:327–335. doi: 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- 21.Zan H, Komori A, Li Z, Cerutti A, Schaffer A, Flajnik MF, Diaz M, Casali P. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 2001;14:643–653. doi: 10.1016/s1074-7613(01)00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schenten D, Kracker S, Esposito G, Franco S, Klein U, Murphy M, Alt FW, Rajewsky K. Pol zeta ablation in B cells impairs the germinal center reaction, class switch recombination, DNA break repair, and genome stability. The Journal of experimental medicine. 2009;206:477–490. doi: 10.1084/jem.20080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakamoto AN, Stone JE, Kissling GE, McCulloch SD, Pavlov YI, Kunkel TA. Mutator alleles of yeast DNA polymerase zeta. DNA Repair (Amst) 2007;6:1829–1838. doi: 10.1016/j.dnarep.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone JE, Kissling GE, Lujan SA, Rogozin IB, Stith CM, Burgers PM, Kunkel TA. Low-fidelity DNA synthesis by the L979F mutator derivative of Saccharomyces cerevisiae DNA polymerase zeta. Nucleic Acids Res. 2009;37:3774–3787. doi: 10.1093/nar/gkp238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Sloun PP, Romeijn RJ, Eeken JC. Molecular cloning, expression and chromosomal localisation of the mouse Rev3l gene, encoding the catalytic subunit of polymerase zeta. Mutat Res. 1999;433:109–116. doi: 10.1016/s0921-8777(98)00067-6. [DOI] [PubMed] [Google Scholar]
- 26.Gibbs PE, McGregor WG, Maher VM, Nisson P, Lawrence CW. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc Natl Acad Sci U S A. 1998;95:6876–6880. doi: 10.1073/pnas.95.12.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic acids research. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, Diaz M. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cumano A, Rajewsky K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. Embo J. 1986;5:2459–2468. doi: 10.1002/j.1460-2075.1986.tb04522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krijger PH, Langerak P, van den Berk PC, Jacobs H. Dependence of nucleotide substitutions on Ung2, Msh2, and PCNA-Ub during somatic hypermutation. J Exp Med. 2009;206:2603–2611. doi: 10.1084/jem.20091707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabouri Z, Okazaki IM, Shinkura R, Begum N, Nagaoka H, Tsuchimoto D, Nakabeppu Y, Honjo T. Apex2 is required for efficient somatic hypermutation but not for class switch recombination of immunoglobulin genes. Int Immunol. 2009;21:947–955. doi: 10.1093/intimm/dxp061. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Fernandez A, Gilmore D, Milstein C. Age-related decrease in the proportion of germinal center B cells from mouse Peyer's patches is accompanied by an accumulation of somatic mutations in their immunoglobulin genes. Eur J Immunol. 1994;24:2918–2921. doi: 10.1002/eji.1830241151. [DOI] [PubMed] [Google Scholar]
- 34.Gibbs PE, McDonald J, Woodgate R, Lawrence CW. The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–582. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. Embo J. 2003;22:1654–1664. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 2002;16:1872–1883. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- 37.Zhu F, Zhang M. DNA polymerase zeta: new insight into eukaryotic mutagenesis and mammalian embryonic development. World J Gastroenterol. 2003;9:1165–1169. doi: 10.3748/wjg.v9.i6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 39.Verkoczy L, Kelsoe G, Moody MA, Haynes BF. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr Opin Immunol. 2011;23:383–390. doi: 10.1016/j.coi.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.