

Abstract
Kallikrein 6 (Klk6) is a secreted serine protease that is elevated in active multiple sclerosis lesions and patient sera. To further evaluate the involvement of Klk6 in chronic progressive demyelinating disease, we determined its expression in the brain and spinal cord of SJL mice infected with Theiler's murine encephalomyelitis virus (TMEV) and assessed the effects of Klk6‐neutralizing antibodies on disease progression. Klk6 RNA expression was elevated in the brain and spinal cord by 7 days postinfection (dpi). Thereafter, Klk6 expression persisted primarily in the spinal cord reaching a peak of fivefold over controls at mid‐chronic stages (60 dpi–120 dpi). Significant elevations in Klk6 RNA were also induced in splenocytes stimulated with viral capsid proteins in vitro and in activated human acute monocytic leukemia cells. Klk6‐neutralizing antibodies reduced TMEV‐driven brain and spinal cord pathology and delayed‐type hypersensitivity (DTH) responses when examined at early chronic time points (40 dpi). Reductions in spinal cord pathology included a decrease in activated monocytes/microglia and reductions in the loss of myelin basic protein (MBP). By 180 dpi, pathology scores no longer differed between groups. These findings point to regulatory activities for Klk6 in the development and progression of central nervous system (CNS) inflammation and demyelination that can be effectively targeted through the early chronic stages with neutralizing antibody.
Keywords: poliovirus, serine protease, spinal cord, TMEV
INTRODUCTION
An emerging concept in the pathogenesis of central nervous system (CNS) demyelinating disorders is that of the “degradome,” which collectively refers to all proteases within or secreted by cells positioned to drive discrete facets of disease 42, 48. Proteolysis in multiple sclerosis (MS) has been the subject of considerable prior research in part because of the therapeutic potential of targeting key proteolytic cascades. Proteolytic events play a central role in many discrete steps culminating in inflammatory demyelination including immune cell activation, extravasation, proinflammatory cytokine and complement activation, demyelination as well as oligodendroglial and axon injury. Of the known protease families (including serine, metallo‐ and cysteine), there is documented involvement of each at one or more of these steps 3, 9, 11, 17, 22, 25, 27, 35, 41, 57. Of all protease families studied with regard to MS, the matrix metalloprotease (MMP) family has historically received most attention. While growing evidence suggests involvement of several MMPs 1, 5, 21, 60, not all of which are necessarily pathogenic 56, 65, most is known regarding MMP‐9. MMP‐9 is secreted by T cells, degrades blood‐brain barrier (BBB) components, activates tumor necrosis factor alpha, is detectable in MS lesions 22, 25, 46 and is elevated in MS patient cerebrospinal fluid (CSF) and serum 13, 15, 23, 28, 37, 47, 64. Minocycline, which, among other activities, inhibits MMP‐9, reduces enhancing magnetic resonance imaging lesions (32).
Emerging studies highlight likely key roles for several serine protease cascades in MS including the fibrinolytic and blood coagulation systems. For example, fibrin depletion delays disease onset and demyelination in experimental autoimmune encephalomyelitis (EAE) 2, 66. A recent proteomic analysis of MS lesions pointed to a role for tissue factor and protein C in chronic active MS lesions. Indeed, activated protein C was shown to play a key role in ameliorating EAE (19). T cell‐derived granzyme B was also recently shown to trigger neurotoxicity (18). In aggregate, these studies underscore pivotal roles for several protease families at numerous steps in inflammatory demyelinating disease. Identifying the full complement of proteolytic enzymes that participate in MS, that is the “MS degradome”(48), appears critical to understanding demyelinating disease pathophysiology and, thus, to the development of new therapeutic strategies.
We cloned a novel serine protease, Kallikrein 6 (KLK6, upper case denoting human form), and determined this to be a trypsin‐like protease with abundant expression in the CNS and therein to be regulated by injury (49). Emerging studies link altered tissue, sera or CSF KLK6 levels to several debilitating neurological disorders. Levels of KLK6 are up‐regulated in active MS lesions (50), in human spinal cord injury (52) and in animal models of stroke (59) but down‐regulated in brain lesions 36, 70 and sera (34) of Alzheimer's patients. Of note, KLK6 is elevated in the serum (53) and CSF (20) of patients with progressive MS. Particularly relevant to the current study of viral‐mediated CNS disease, KLK6 also appears to be elevated in CSF of patients with post‐polio syndrome (16).
Further implicating KLK6 in key facets that drive demyelinating disease is its elevated expression in activated T cells as well as those stimulated by glucocorticoids, androgens or progesterone 10, 50, 51. In excess, KLK6 contributes to oligodendrogliopathy (50) as well as neuron and axon injury (53). Interestingly, KLK6 rapidly hydrolyzes myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein (MOG) 6, 7, 8, 50 as well as laminin, fibronectin and heat‐denatured collagen, key components of the BBB. Among 50 substrates tested in one comprehensive study, KLK6 showed highest hydrolytic activity toward MBP (4). More recently, KLK6 was shown to activate protease‐activated receptor 1 (PAR1) and PAR2 to mediate intracellular signaling in neurons, astrocytes and immune cells including T and B cells 54, 62. Importantly, with regard to inhibiting KLK6 enzymatic activity to attenuate neuroinflammation and neurodegeneration in MS patients, blocking Klk6 (lower case to indicate rodent form) enzymatic activity using a function‐blocking antibody approach diminished clinical and histological disease in mice with proteolipid protein (PLP) peptide or MOG peptide‐induced EAE as well as Th1 inflammatory responses (7). Taken together, these data support the hypothesis that KLK6 is a key player in multiple aspects of the “MS degradome”(48) and may therefore serve as a useful therapeutic target. Herein, we further evaluate this hypothesis by determining the effects of Klk6‐neutralizing antibodies on the development and progression of CNS inflammatory demyelinating disease induced by a virus. Theiler's murine encephalomyelitis virus (TMEV) is a picornavirus that induces acute polioencephalomyelitis and chronic progressive spinal cord inflammation and demyelination and is considered to be the best currently available model of progressive MS 26, 33. Results of this study indicate that Klk6 is likely to play roles in both the adaptive and innate immune system and that targeting Klk6 enzymatic activity is capable of delaying the progression of inflammation and demyelination through early chronic stages of TMEV‐induced demyelinating disease (TMEV‐IDD).
MATERIALS AND METHODS
TMEV model
Eight‐week‐old female SJL/J (H‐2S) mice (Jackson Laboratories, Bar Harbor, ME, USA) were injected intracerebrally with 2 × 105 plaque‐forming units (PFU) of the Daniel's strain (DA) of TMEV in a 10‐µL volume. Analyses of Klk6 and viral expression levels were performed at 7, 21, 30, 60, 90, 120 and 180 days postinfection (dpi). All studies were performed according to the guidelines of the Institutional Animal Care and Use Committee at the Mayo Clinic. Unless otherwise indicated, all reagents were obtained from Sigma (St. Louis, MO, USA).
Quantification of Klk6 and virus RNA
Quantitative real‐time polymerase chain reaction (RT‐PCR) was used to evaluate the expression of Klk6 and TMEV RNA in the brain and spinal cord at acute (7 days) through late chronic time points (180 days) post‐TMEV infection. The brain and spinal cord were obtained from at least five mice at acute (7 days), subacute (21 days) and chronic (30, 60, 90, 120 and 180 days) time points, and snap frozen in liquid nitrogen prior to RNA isolation using RNA STAT‐60 (Tel‐Test, Inc. Friendswood, TX, USA). Also, 0.5 µg of total RNA was subject to RT‐PCR using the Light Cycler‐RNA Amplification Kit SYBR Green I in a Roche Light Cycler apparatus following the manufacturer's instructions (Roche Diagnostics, Mannheim Germany). Primers specific for Mus musculus Klk6 were, forward, 5′CCTACCCTGGCAAGATCAC3′ and, reverse, 5′GGATCCATCTGATATGAGTGC3′(10). To gauge viral replication over the course of disease, RNA coding for the DA VP2 capsid protein were amplified using 5′‐TGGTCGACTCTGTGGTTACG‐3′ as the forward primer and 5′‐GGCATGGACTGTGGTCAGA‐3′ as the reverse primer (45). To control for loading, the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was amplified in the same RNA samples using 5′‐ACCACCATGGAGAAGGC‐3′ as the forward and 5′‐GGCATGGACTGTGGTCATGA‐3′ as the reverse primers. To examine KLK6 in human acute monocytic leukemia cells (THP‐1), primers specific for Homo sapiens KLK6 (forward, 5′TGCCAGGGTGATTCTGGG3′ and, reverse, 5′TGCAGACGTTGGTGTAGACT3′) were utilized (51). Expression levels were quantified relative to M. musculus Klk6, H. sapiens KLK6, GAPDH and DA VP2 nucleic acid templates. Serial dilutions of each gene‐specific clone containing known copy number were used to create standard curves. Relative gene expression levels were calculated during the logarithmic amplification phase and correlated to the copy number of each standard. Changes in Klk6 gene expression were reported as percent change relative to uninfected control mice or untreated cell culture samples. VP2 RNA levels were expressed as copy number on a logarithmic scale.
Analysis of Klk6 protein
To determine whether transcriptional changes observed in Klk6 in the spinal cord of TMEV‐infected mice were reflected at a protein level, we examined spinal cord Klk6 by Western blot in uninfected mice and in the spinal cord of infected mice at 21, 60, 90 and 180 dpi. We collectively homogenized three freshly isolated spinal cords at each time point in radio‐immunopercipitation assay buffer. Also, 50 µg aliquots of each protein lysate were separated on sodium dodecyl sulfate‐polyacrylamide gels prior to transfer onto nitrocellulose membranes. Blots were probed with a rabbit polyclonal Klk6‐specific antibody (Rb008) as previously described (7). In each case, membranes were stripped and re‐probed for GAPDH to control for loading, and all proteins of interest were detected on film using chemiluminescence Supersignal (Pierce, Rockford, IL, USA). For quantification, films were scanned and images were quantified using Image Lab 2.0 software (Bio‐Rad, Hercules, CA, USA). Relative changes in Klk6 protein in TMEV‐infected relative to control spinal cords were determined by normalizing optical density measurements for Klk6 to those of GAPDH detected on the same membrane. All Western blots were repeated at least three times providing similar results.
In vitro analysis of T cell and monocyte function
To determine whether the expression of Klk6 RNA is regulated in T cells in a virus‐dependent manner, whole splenocyte cultures were prepared from TMEV‐infected mice and stimulated with a combination of VP1 and VP2 viral capsid proteins, each at 5 µg/mL for 72 h. The constructs encoding TMEV VP1 and VP2 capsid proteins were expressed in Escherichia coli and purified as previously described 24, 38. Briefly, the proteins were purified over a HisTag column and dialyzed into phosphate buffered saline (PBS) before use. The VP1 construct encodes for 274 amino acids of VP1 and the VP2 construct for 276 amino acids of VP2. To determine whether the expression of Klk6 RNA shows regulated expression in activated monocytes, the level of KLK6 RNA was quantified in resting THP‐1 monocyte cultures or in parallel cultures stimulated by phorbol 12‐myristate‐13‐acetate (PMA), or a combination of PMA (10 ng/mL, Promega, Madison, WI, USA) and lipopolysaccharide (LPS) (10 µg/mL). THP‐1 cells are a human monocytic leukemia cell line obtained from the American Type Culture Collection. Cultured cells were grown in Roswell Park Memorial Institute 1640 media with 10% heat‐inactivated fetal calf serum, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid buffer and 10 µM 2‐mecaptoethanol.
Recombinant Klk6 and generation of Klk6‐neutralizing antibodies
Klk6‐neutralizing antibodies were generated in an autonomous fashion by immunizing naïve SJL mice with recombinant Rattus norvegicus Klk6 as described in prior studies (8). R. norvegicus Klk6 shares 91.4% sequence homology with M. musculus Klk6 and is expected to exert comparable enzyme activity and specificity, as has been established for R. norvegicus Klk6 and H. sapiens KLK6, which share 66.8% sequence homology (6). We have previously shown that this immunization approach results in the generation of high titers of Klk6‐specific antibodies that effectively block the activity of Klk6 in vitro and abrogate disease in murine EAE models of MS (8). R. norvegicus Klk6 was expressed in the baculovirus system, purified and activated as described in detail in prior studies (7).
Klk6 antibody production was initiated in SJL mice 5 weeks prior to TMEV infection by immunization with 100 µg of recombinant Klk6 in 200 µL of complete Freund's Adjuvant (CFA, Difco, Detroit, MI, USA), containing 200 µg of heat‐killed Mycobacterium tuberculosis. Control animals were immunized similarly with the non‐self antigen ovalbumin (OVA), with CFA alone or did not receive any prior immunization. One week prior to TMEV infection, sera were obtained by tail bleed to confirm Klk6 or OVA antibody titers by enzyme‐linked immunosorbent assay (ELISA) and the mice received an additional 20 µg of antigen subcutaneously to boost antibody production. Sera were also obtained at the termination of each experiment to verify high antibody titers.
The ability of antibodies generated by Klk6 immunization to block Klk6 hydrolytic activity was evaluated in vitro by determining their ability to inhibit Klk6‐mediated hydrolysis of an arginine‐specific chromogenic substrate, Ac‐Ala‐Thr‐Arg‐pNA (Bachem, King of Prussia, PA, USA). Immunoglobulin was isolated from pooled sera of mice immunized with Klk6 or CFA alone using protein G sepharose (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Purified IgG (4 µg) was preincubated with 20 ng of recombinant Klk6 (35:1 molar ratio) at RT for 15 minutes in 50 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA) and pH 8.5. The ability of Klk6 to subsequently hydrolyze 400 µM AcATRpNA was then examined by incubation at 37°C with absorbance read at 405 nm at 15‐minute intervals over a period of 2 h on a Beckman Coulter DU640 spectrophotometer (Beckman Coulter, Fullerton, CA, USA) interfaced with temperature controller.
Spinal cord morphometry
At 40 days or 180 days post‐TMEV infection, animals were euthanized (150 mg/kg sodium pentobarbital) and the brain and spinal cord were perfusion fixed with 4% paraformaldehyde (pH 7.4) for histological analysis. Spinal cords were sectioned transversely into 1‐mm blocks and every third block embedded in glycol methacrylate and 1‐µm thin sections stained with eriochrome and cresyl violet (39) to assess meningeal inflammation and parenchymal pathology (demyelination and inflammation within the substance of the spinal cord). This methodology reproducibly allows the visualization of inflammatory cells and the extent of demyelination within the entire spinal cord. An additional seven segments of the spinal cord spanning the cervical, thoracic, lumbar and sacral segments were embedded in paraffin to examine the appearance of MBP and activated monocytes and microglia using immunohistochemical techniques. MBP was detected using a rat monoclonal antibody (MAB386, Chemicon, Temecula, CA, USA) and microglia using biotinylated Bandeiraea simplicifolia (BS‐1, Sigma). Both antigens were visualized using standard avidin‐biotin immunoperoxidase techniques (Vector Laboratories, Burlingame, CA, USA) 50, 52.
Quantitative morphologic analysis was performed on 10 to15 plastic‐embedded sections, corresponding to 10 to 15 different spinal cord segments per mouse. We used two methods to determine the extent of white matter pathology. First, we calculated the total white matter area and the total lesion area (in mm2) using a Zeiss photomicroscope with attached camera lucida and the Zeiss interactive digital analysis system (ZIDAS). Total lesion area was expressed as a percent of total white matter area (63). Second, we assigned a pathologic score reflecting the frequency of pathology to each animal based on meningeal inflammation or demyelination. This score is expressed as a percentage of the total number of spinal cord quadrants positive for each pathologic measurement, divided by the total number of spinal cord quadrants examined 8, 12, 38. The quadrant method was also used to assess the appearance of BS‐1‐positive monocytes and the loss of immunoreactivity for MBP within spinal cord white matter. A maximum score of 100 reflects the presence of pathology in all four quadrants of every spinal cord segment examined from an individual spinal cord. All pathological scores in the spinal cord and brain were assigned without knowledge of experimental groups.
Brain pathology
The brain of each mouse was blocked into three pieces using two coronal cuts, one at the level of the optic chiasm and infundibulum, the other at the pontomedullary junction, and embedded together in a single paraffin block. The resulting slides were stained with hematoxylin and eosin. Brain pathology was assessed morphologically by grading the extent of pathology in six specific brain regions: cerebellum, brain stem, striatum, cortex, hippocampus and corpus callosum as well as in the meninges. Each area of the brain was graded on a scale of 0 to 4 with the score reflecting the maximum amount of damage in each area 12, 38. A score of 0 reflects the absence of pathology; a 1 reflects no tissue destruction but minimal inflammation; a 2 reflects early tissue destruction (loss of architecture) and moderate inflammation; 3 reflects definite tissue destruction (demyelination, parencymal damage, cell death, neurophagia, neuronal vacuoloation); and a 4 represents necrosis (complete loss of all tissue elements with associated cellular debris). Meningeal inflammation was also graded on a 1–4 scale: 0, no inflammation; 1, one cell layer of inflammation; 2, two cell layers of inflammation; 3, three cell layers of inflammation; 4, four or more cell layers of inflammation.
Delayed type 2 hypersensitivity response
Delayed‐type hypersensitivity (DTH) responses were elicited by the intradermal injection of ultraviolet (UV)‐inactivated, cesium chloride‐purified TMEV (5 µg in 10 µL PBS) into the ventral surface of the right ear using a 100 µL Hamilton syringe (Hamilton Co., Reno, NV, USA). PLP139‐151 (HSLGKWLGHPDKF) was injected into the opposite ear to examine the specificity of the response. At 24 h after challenge, increases in ear thickness over prechallenge measurements were determined using a dial gauge micrometer. Results were expressed in units of 10−4 inches ± standard error of the mean. PLP peptide was synthesized by the protein core facility at the Mayo Clinic, with amino acid composition and purity (>98%) verified by mass spectroscopy.
Virus‐specific antibody ELISA
Total serum immunoglobulins directed against purified TMEV were assessed by ELISA as previously described (32). Ninety‐six well Immulon II plates coated with TMEV were blocked with 1.0% bovine serum albumin and incubated with serial dilutions of serum obtained at the end point of each experiment. TMEV‐specific antibodies contained in serum were detected using biotinylated secondary antibodies and streptavidin‐labeled alkaline phosphatase with p‐nitrophenyl phosphate as the substrate.
Statistical analysis
All measurements were made without knowledge of treatment groups. Comparisons between two groups were made using the Student's t‐test, except when data were not normally distributed, when the Mann–Whitney U rank sum test was applied. Comparisons between multiple groups were made using one‐way analysis of variance (ANOVA) with the Student–Newman–Keul's (SNK) post hoc test or Kruskal–Wallis ANOVA on ranks for data that were not normally distributed with Dunn's test for pairwise multiple comparisons. The level for significance was set as P < 0.05 for all tests.
RESULTS
Spatial and temporal characteristics of CNS Klk6 in virus‐IDD
The expression of Klk6 RNA was differentially regulated in the brain and spinal cord across the acute (7 days), subacute (21) and more chronic phases (30–180 days) of TMEV‐IDD examined (P < 0.05, Figure 1A,B). In the brain, Klk6 was elevated by 2.5‐fold at 7 days post‐TMEV infection, paralleling acute viral infection of the brain and the acute inflammatory response known to ensue (58). By 21 days, Klk6 RNA expression was no longer elevated relative to uninfected control brain. A second peak in Klk6 RNA expression (twofold) was seen in the brain at 120 days, but levels had returned to baseline when examined 180 dpi (Student's t‐test P < 0.05).
Figure 1.
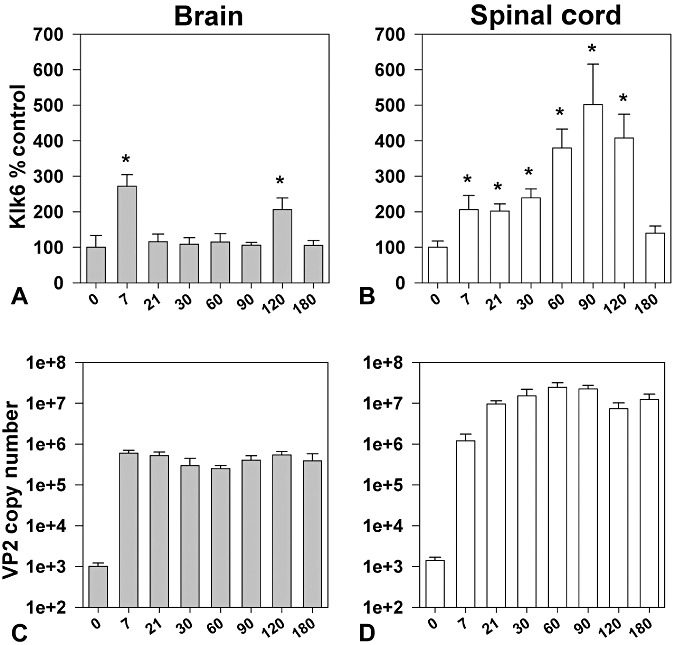
Expression of Klk6 RNA is differentially regulated in the brain and spinal cord during the acute and chronic phases of TMEV‐induced demyelinating disease. Histograms show real‐time PCR for Klk6 RNA isolated from the brain (A) or spinal cord (B) of SJL mice at acute through chronic time points after infection with the Daniel's strain of TMEV (n = 5–6 per group). Klk6 RNA copy number was determined by real‐time PCR, normalized to GAPDH copy number in the same sample and expressed as a percent control. In brain, Klk6 was significantly elevated acutely (7 days) after infection and again at 120 days. In spinal cord, Klk6 RNA was elevated at acute, subacute and chronic time points with levels progressively increasing out to 120 days, but decreasing thereafter (asterisks indicate significant difference from baseline levels, *P < 0.05, Student's t‐test). C and D show quantification of viral VP2 RNA expression in the brain and spinal cord over the course of disease. Raw copy number values illustrate that TMEV RNA transcripts reach highest levels in the spinal cord, which can be a log higher than that seen in the CNS from subacute through chronic time points examined. The levels of GAPDH were consistent across the time points examined (Brain, Log10 4.9 ± 0.1, spinal cord Log10 6.2 ± 0.7).
In the TMEV‐infected spinal cord, Klk6 RNA expression was already elevated by twofold at acute time points (7 days). Elevations in Klk6 RNA persisted at this level throughout the subacute period examined. Klk6 RNA expression progressively increased thereafter reaching more than fourfold elevations over control levels when examined at 60, 90 and 120 dpi (Student's t‐test P < 0.05). At 180 dpi, Klk6 RNA levels in the spinal cord were no longer elevated relative to controls.
Transcriptional analysis of viral RNA levels showed elevated VP2 RNA in the brain at 7 days, which persisted at a relatively stable level out to the 180‐day time point examined. These findings are consistent with the natural history of TMEV infection, which includes persistence of virus RNA within the brain. In the spinal cord, VP2 levels were already elevated at 7 dpi and increased thereafter to reach a peak at 60 to 90 days and which was at least one log higher than that detected in the brain. VP2 RNA in the spinal cord persisted at a high level out to 180 dpi.
Klk6 protein expression
To determine the extent to which Klk6 RNA transcriptional changes in the spinal cord were reflected in changes in protein, we also examined Klk6 levels by Western blot (Figure 2). Paralleling the peak of Klk6 RNA seen in the spinal cord at 90 days post‐TMEV infection, maximal levels of Klk6 protein were also detected in the spinal cord at this time point. Levels reached more than 10‐fold that seen in controls (Student's t‐test, P < 0.003). Some increases in Klk6 at a protein level were also seen at 21, 60 and 180 days post‐TMEV infection, but these changes did not reach the level of statistical significance.
Figure 2.
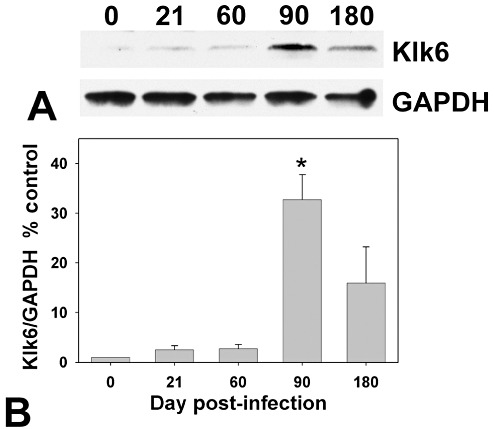
Klk6 protein levels are up‐regulated in the TMEV‐infected spinal cord. A. Western blot shows Klk6 protein in the spinal cord of uninfected mice at 21, 60, 90 or 180 days post‐TMEV infection. B. Protein levels were quantified by assessment of relative optical density and normalized to GAPDH levels to control for loading and expressed as a percent control. Relative to uninfected spinal cord, Klk6 protein levels reached a peak at 90 days postinfection (*Student's t‐test, P < 0.003 relative to control, n = 3 per time point).
Regulated expression of Klk6 in immune cells
Klk6 RNA was up‐regulated by more than twofold in splenocytes derived from TMEV‐infected mice when stimulated with VP1 and VP2 TMEV viral capsid proteins in vitro (Figure 3A, Student's t‐test, P < 0.05). Parallel treatment of splenocytes derived from uninfected control mice did not alter Klk6 RNA transcription. In addition, the expression of KLK6 RNA by THP‐1 monocytes was also significantly elevated by known monocyte activators, PMA or PMA in addition to LPS, which resulted in twofold to 2.5‐fold increases, respectively (Figure 3B, Student's t‐test, P < 0.05).
Figure 3.
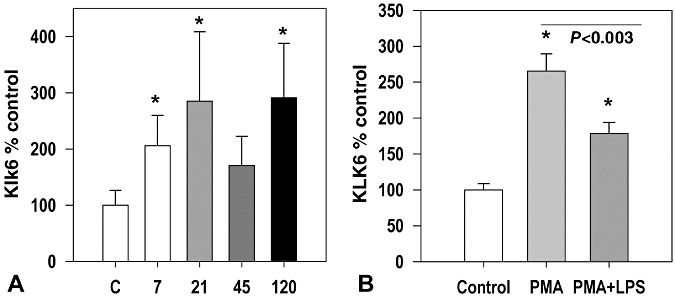
Klk6 RNA is up‐regulated in splenocytes in a viral capsid protein dependent fashion and in THP‐1 monocytes activated by PMA and LPS. A. Histogram shows changes in Klk6 RNA expression seen in splenocyte cultures derived from uninfected control mice or mice at acute through chronic time points post‐TMEV infection treated with a combination of VP1 and VP2 capsid proteins (each at 5 µg/mL) for 72 h. Viral antigen‐elicited recall responses promoted significant elevations in Klk6 RNA transcription at 7, 21 and 120 days postinfection. B. KLK6 RNA detected by real‐time PCR was significantly elevated in RNA isolated from THP‐1 monocytes activated by PMA or a combination of PMA and LPS (*P < 0.05, Student's t‐test).
Klk6‐neutralizing antibodies diminish spinal cord inflammation and demyelination to early chronic phases of disease
Immunization of SJL mice with recombinant Klk6 5 weeks prior to infection with TMEV resulted in the establishment of high levels of circulating Klk6 antibodies which persisted out to the 180‐day time point examined (Figure 4A–C). IgG isolated from the sera of Klk6‐immunized mice effectively neutralized Klk6‐mediated substrate hydrolysis in vitro (Figure 4B).
Figure 4.
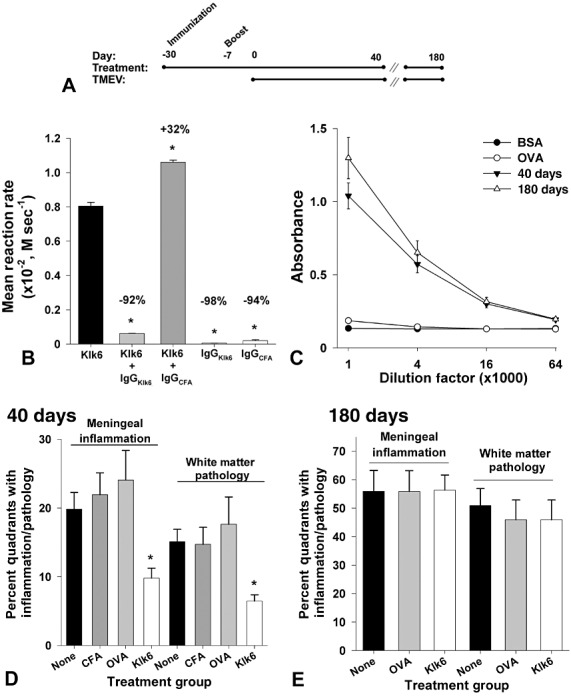
Klk6‐neutralizing antibodies attenuate white matter pathology and meningeal inflammation at early, but not chronic, stages of TMEV‐induced demyelinating disease. Time line (A) shows strategy used to generate SJL mice with high levels of circulating Klk6‐neutralizing antibodies prior to intracranial TMEV infection. Histogram (B) shows that IgG isolated from the serum of Klk6‐immunized mice significantly reduced the rate of Klk6‐mediated hydrolysis of AcATRpNA substrate in vitro. By contrast, IgG isolated from CFA‐immunized mice did not reduce but rather slightly enhanced Klk6‐mediated substrate hydrolysis. IgG isolated from Klk6‐ or CFA‐immunized mice did not promote significant substrate hydrolysis alone. Data are expressed as mean reaction rate of replicate experiments (*P < 0.05, Student's t‐test relative to Klk6 alone). (C) Serum Klk6 antibody titers were high and similar when examined at the 40‐ and 180‐day end points of each experiment. Histograms in (D) and (E) show quantification of the percent of spinal cord quadrants with meningeal inflammation or white matter pathology observed at 40 or 180 days post‐TMEV infection in Klk6‐immunized mice and in mice immunized with CFA alone, with OVA or without any immunization prior to injection with infectious virus (None). Significantly less meningeal inflammation and white matter pathology were observed in mice that had been immunized with Klk6 prior to TMEV infection relative to the immunization controls, when spinal cord pathology was assessed at 40 days after infection (ANOVA on ranks, P = 0.002; *Dunn's, P < 0.05). However, when spinal cord pathology was examined at chronic time points (180 days, E), no significant differences were observed betweenthe immunization groups. See also Table 1.
Mice with Klk6‐neutralizing antibodies showed significant attenuation of spinal cord and brain pathology when examined at 40 days post‐TMEV infection (4, 5, 6, 1, 2). At this early chronic time point, the number of spinal cord quadrants with white matter pathology was reduced by approximately half in Klk6‐immunized mice (ANOVA on ranks, P = 0.002; Dunn's P < 0.05) (Figure 4D, Table 1). Area measurements of white matter pathology confirmed these results (ANOVA, P = 0.009; SNK P < 0.05) (Figure 4D, Table 2). Mice with Klk6‐neutralizing antibodies also exhibited significantly reduced meningeal inflammation at the 40‐day time point (ANOVA, P = 0.001; SNK, P < 0.05) (Figure 4D, Table 1). In addition, evaluation of the number of spinal cord quadrants exhibiting a loss of immunoreactivity for MBP was also significantly reduced in Klk6‐immunized mice when examined 40 days post‐TMEV infection (ANOVA on ranks, P = 0.031; Dunn's, P < 0.05) (Figure 5H, Table 2). Decreased spinal cord white matter pathology in Klk6‐immunized mice was paralleled by significant reductions in the number of spinal cord quadrants containing BS‐1‐positive macrophages and activated microglia (ANOVA, P = 0.003; SNK, P < 0.05) (Figure 5I, Table 2).
Figure 5.
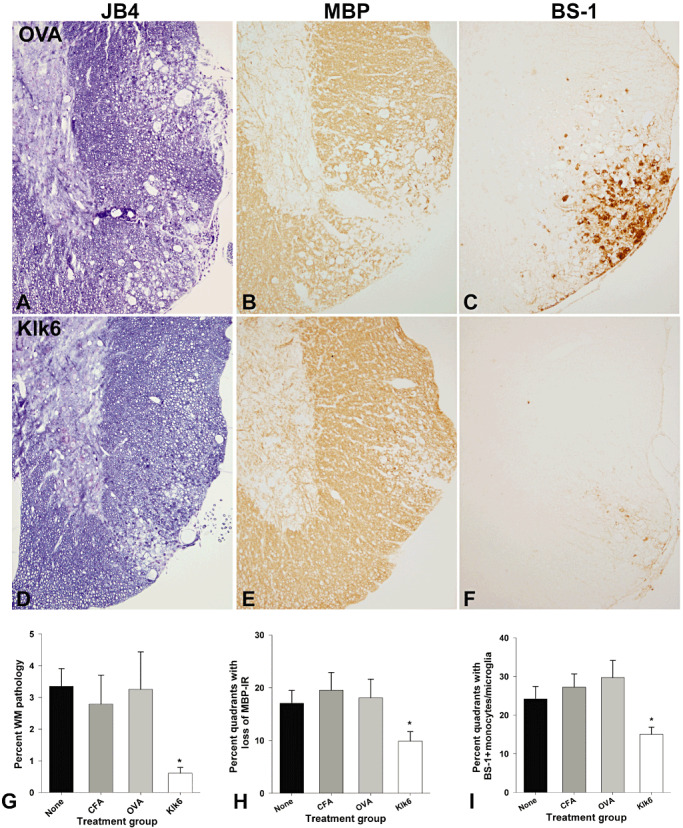
Decreased white matter pathology in the spinal cord of mice immunized with KLK6 in comparison to controls at 40 days post‐TMEV infection. A,D. Glycol methacrylate plastic‐embedded sections were stained with modified eriochrome/cresyl violet stain and the area of white matter pathology determined and expressed as a percent of total white matter (G). Parallel 1‐mm segments of spinal cord from the same mice were embedded in paraffin and 5‐µm sections stained for MBP (B,E) or for monocytes and activated microglia using a biotinylated BS‐1 antibody (C,F). The spinal cord of mice immunized with Klk6 5 weeks prior to TMEV infection showed significant reductions in the percent of white matter pathology (G, ANOVA, P = 0.009; *SNK P < 0.05) and in the percent of spinal cord quadrants exhibiting a loss of MBP staining (H, ANOVA on ranks, P = 0.03; *Dunn's P < 0.5) compared with immunization controls. Mice immunized with Klk6 prior to TMEV infection also showed fewer spinal cord quadrants associated with BS‐1‐positive monocytes and activated microglia (I, ANOVA, P = 0.003; *SNK, P < 0.05). See also Table 2.
Figure 6.
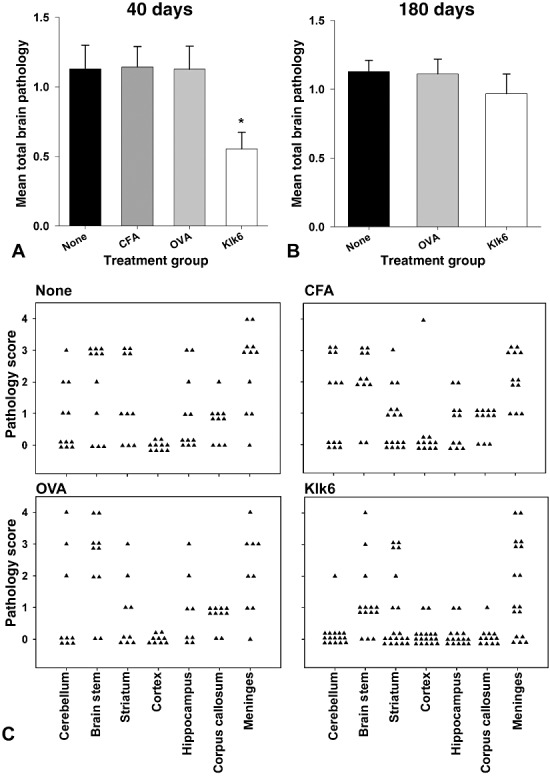
Klk6‐neutralizing antibodies attenuate brain pathology at early but not chronic stages of TMEV‐induced demyelinating disease. Analysis of total brain pathology based on the average of all pathology assessed in the cerebellum, brain stem, striatum, cortex, hippocampus, corpus callosum and meninges at 40 (A, C) and 180 days (B) post‐TMEV infection. (A) Histogram depicting total brain pathology scores shows significantly less pathology in mice with high levels of circulating Klk6‐neutralizing antibodies prior to TMEV infection when assessed at the 40‐day time point (ANOVA, P = 0.006; *SNK, P < 0.05). At 180 days post‐TMEV‐infection (B), there were no statistically significant differences between the groups examined. (C) shows the distribution of pathology severity in each region of the brain examined at 40 days after TMEV infection.
Table 1.
Spinal cord and brain pathology in TMEV‐infected mice at 40 and 180 dpi
| Group | No. of mice | Meningeal inflammation | White matter pathology | Total brain pathology |
|---|---|---|---|---|
| 40 days | ||||
| None | 10 | 19.8 ± 2.4 | 15.1 ± 1.8 | 1.13 ± 0.2 |
| CFA | 10 | 21.9 ± 3.1 | 14.7 ± 2.5 | 1.14 ± 0.1 |
| OVA | 9 | 24.1 ± 4.3 | 17.6 ± 3.9 | 1.13 ± 0.2 |
| Klk6 | 16 | 9.8 ± 1.4* | 6.5 ± 0.9** | 0.55 ± 0.1* |
| 180 days | ||||
| None | 13 | 55.9 ± 10.3 | 50.9 ± 8.1 | 1.13 ± 0.1 |
| OVA | 12 | 55.9 ± 7.3 | 45.9 ± 6.9 | 1.10 ± 0.1 |
| Klk6 | 12 | 56.3 ± 4.1 | 46.9 ± 4.1 | 0.97 ± 0.1 |
Data are expressed as the percentage of spinal cord quadrants showing the pathological abnormality (mean ±standard error). Significantly lower pathology scores in mice with Klk6‐neutralizing antibodies were observed relative to immunization control groups at 40 days but not at 180 days post‐TMEV infection (*ANOVA P ≤ 0.006, SNK P < 0.05; **ANOVA on ranks P = 0.002, Dunn's P < 0.05).
Table 2.
White matter pathology in TMEV‐infected mice at 40 dpi
| Group | No. of mice | Area of white matter pathology | Loss of MBP‐IR | BS1+monocytes |
|---|---|---|---|---|
| 40 days | ||||
| None | 10 | 3.4 ± 0.6 | 17.1 ± 2.4 | 24.2 ± 3.6 |
| CFA | 10 | 2.8 ± 0.9 | 19.5 ± 3.4 | 27.3 ± 3.4 |
| OVA | 9 | 3.3 ± 1.2 | 18.1 ± 3.5 | 29.7 ± 4.5 |
| Klk6 | 16 | 0.6 ± 0.2* | 9.9 ± 1.8** | 15.1 ± 1.8* |
The area of spinal cord white matter pathology was measured and expressed as a percent of total spinal cord white matter in each mouse. The percentage of spinal cord quadrants with a loss of MBP immunoreactivity (IR) or containing BS‐1+ monocytes/activated microglia is also provided. (*ANOVA P < 0.009, SNK P < 0.05; **ANOVA on ranks P = 0.03, Dunn's P < 0.05).
While a significant reduction in histopathological signs of disease was observed in mice with Klk6‐neutralizing antibodies at 40 dpi, no significant decreases in pathological outcome measures were observed when mice were allowed to progress to 180 dpi (Figure 4E). Meningeal inflammation and white matter pathology in the spinal cord were each observed to increase by two‐ to threefold between the early (40 days) and late chronic (180 days) time points examined (Figure 4D,E, Table 1).
Klk6‐neutralizing antibodies reduce overall brain pathology through early chronic disease
The effects of Klk6‐neutralizing antibodies on pathology in the brain paralleled results seen in spinal cord white matter with reductions in overall pathology seen at early (40 days) but not at the late chronic stage (180 days) of disease examined (Figure 6). Comparison of total pathology scores based on the average of all seven distinct regions examined using a semi‐quantitative four‐point scale demonstrated significant reductions in the average pathology score in Klk6‐immunized mice relative to the immunization controls examined (ANOVA, P = 0.006; SNK, P < 0.05). At 180 days, total mean pathology scores in TMEV‐infected SJL mice with no immunization, or those immunized with OVA, did not progress significantly from scores seen at 40 days. However, there was an increase in brain pathology seen in Klk6‐immunized mice at 180 days relative to that seen at 40 days, such that the significant attenuation of pathology seen at earlier time points was no longer apparent (Table 1).
Klk6‐immunized mice showed reduced DTH responses
To gain insight into the potential mechanisms by which Klk6 immunization ameliorates TMEV‐induced disease, we examined its effects on the development of DTH responses upon challenge with UV‐inactivated TMEV (Figure 7). TMEV‐induced ear swelling (DTH) initiated on Day 37 postinfection and examined 24 h later was suppressed twofold in Klk6‐immunized mice relative to those immunized with CFA alone (ANOVA, P = 0.03; SNK P < 0.05). Challenge with a myelin peptide (PLP139‐151) elicited no significant differences in DTH between groups.
Figure 7.
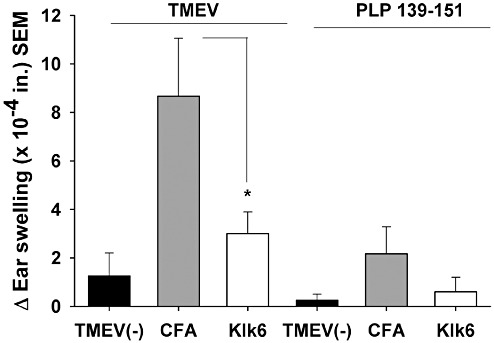
Antigen‐specific delayed‐type hypersensitivity (DTH) responses are decreased in mice immunized with Klk6. DTH responses were determined in Klk6‐ or CFA‐immunized mice at Day 37 post‐TMEV infection and in noninfected (TMEV(–)) control mice. DTH responses (ear thickness inches ± SE) to challenge with 10 µg of UV‐inactivated TMEV or PLP139‐151. Significantly less swelling was observed in Klk6‐immunized mice relative to CFA‐immunized controls at 24 h post‐challenge (ANOVA, P = 0.03; *SNK, P < 0.05).
Changes in virus RNA or antibody responses to virus antigen do not explain differences in pathology early in the disease course
To determine whether effects on virus could explain differences in pathological outcomes in mice with Klk6‐neutralizing antibodies, we assessed levels of TMEV RNA in the brain and spinal cord and determined serum IgG and IgM antibody responses directed toward TMEV antigen across the experimental groups examined (Figure 8). RNA encoding the VP2 capsid protein of TMEV in the brain and spinal cord of Klk6‐immunized mice did not differ significantly from controls (Figure 8A,B). Virus‐specific humoral responses were also similar across the experimental groups examined (Figure 8C).
Figure 8.
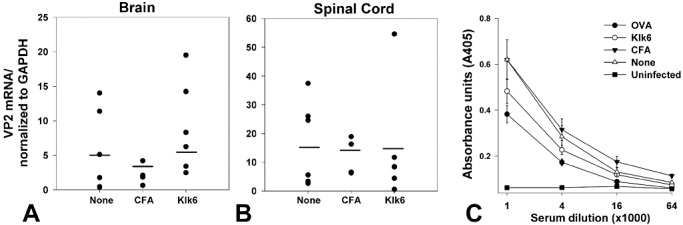
TMEV RNA in brain and spinal cord and TMEV antibody titers in serum do not differ between control and Klk6‐immunized mice. RNA encoding the VP2 capsid protein of TMEV was assessed by real‐time PCR in RNA isolated from the brain and spinal cord at the 40‐day end point of each experiment. A,B. Histograms show that VP2 RNA copy number, normalized to GAPDH to control for loading, did not differ between mice immunized with Klk6, CFA alone, or in those receiving no immunization prior to TMEV infection. C. Virus‐specific humoral immune responses were assessed at the 4‐day end point of each experiment and did not differ significantly between the groups of TMEV‐infected mice. Shown are the results of an ELISA for serum IgG antibodies directed against purified TMEV antigens across the different experimental groups examined. Negative controls are serum samples from mice not infected with TMEV.
DISCUSSION
Accumulating evidence places KLK6 among the serine proteases implicated in MS, as it is localized to active MS lesions (50) and is elevated in the serum (53) and CSF (20) of MS patients with progressive disease. To gain further insight into the activity of KLK6 in progressive CNS inflammatory disease, we examined its expression in the brain and spinal cord of mice at acute‐through‐chronic stages of TMEV‐IDD and determined the effects of Klk6‐neutralizing antibodies on disease progression. Theiler's virus induces a chronic progressive inflammatory demyelinating disease in the spinal cord of susceptible mice and is an important model of progressive MS. The dynamically regulated patterns of Klk6 expression, which are observed in the TMEV‐infected brain and spinal cord and within activated immune cells in the present studies, point to Klk6 as an important participant in the development of TMEV‐induced CNS polioencephalomyelitis and in the development and progression of inflammatory demyelinating disease that ensues in the spinal cord of SJL mice. Moreover, we demonstrate that Klk6‐neutralizing antibodies delay the progression of brain and spinal cord immunopathology, including demyelination within the spinal cord white matter and Th1‐driven DTH responses, at least through early chronic time points postinfection. These findings support the hypothesis that KLK6 is an important component of the “MS degradome.” Certainly, KLK6 warrants additional investigation as a potential therapeutic to be targeted alone or adjunctively to delay disease progression in patients with MS and other encephalitogenic demyelinating disorders.
Klk6 expression in the brain and spinal cord of TMEV‐infected mice showed dynamic changes that largely correspond to the sequence of inflammatory pathogenic events that occur during the acute and chronic time points of disease in susceptible strains of mice 30, 31. In the brain, Klk6 showed transiently elevated expression levels 1 week postinfection, corresponding to the acute phase of TMEV‐induced polioencephalomyelitis characterized by infection and neuron apoptosis. During more chronic stages (1 month or more after infection), inflammation in the gray matter of the brain subsides (44). Mirroring this sequence of events, Klk6 expression in the brain returned to baseline by 3 weeks post‐TMEV infection. There was a late second elevation in Klk6 RNA expression in the brain at 4 months postinfection that may relate to late reactivation of virus (12).
Paralleling early transcriptional changes seen in the brain, elevations in Klk6 RNA in the spinal cord were also seen by 1 week postinfection corresponding to the acute infectious and inflammatory events that characterize early viral poliomyelitis. Sharply contrasting the relatively transient nature of Klk6 transcriptional changes seen in the brain, elevations in Klk6 in the spinal cord progressively increased through mid‐chronic stages, mirroring the persistence of viral infection and second wave of chronic progressive inflammatory demyelination known to manifest in cords of TMEV‐infected SJL mice (12). Peak levels of Klk6 in the spinal cord were observed approximately 3 months postinfection for both RNA and protein. As TMEV‐induced spinal cord demyelination is known to reach a plateau approximately 100 dpi, it is tempting to speculate on the involvement of coordinately elevated levels of Klk6. Supporting this, Klk6 cleaves myelin proteins (MBP and MOG) and promotes direct oligodendrogliopathy in vitro 6, 7, 8, 50. After 90 days, spinal cord atrophy progresses, in part, through the loss of medium‐ and large‐diameter axons (31). By late chronic time points (6 months postinfection), when active demyelination is largely complete and remyelination can be observed (63), Klk6 RNA expression levels in the spinal cord had returned to near baseline. Taken together, these findings indicate key roles for Klk6 in the inflammatory and demyelinating phases of TMEV‐IDD that occur at acute through early to mid‐chronic stages in the brain and spinal cord of susceptible mice.
To directly address the involvement of Klk6 in TMEV‐IDD in vivo, we used a direct immunization approach with recombinant Klk6 to generate groups of mice with high levels of circulating Klk6‐neutralizing antibodies. In prior studies, this approach abrogated clinical and histological signs of disease in the spinal cord of mice with active PLP139‐159‐induced EAE (8). TMEV is a particularly relevant model of MS. It produces lesions resembling those seen in MS, including significant inflammation, demyelination and axon injury, and it has a viral etiology like the one proposed for MS 29, 40. Compared to several immunization controls examined in parallel, mice with Klk6‐neutralizing antibodies showed significantly reduced meningeal inflammation and white matter pathology when assessed for pathological outcomes at an early chronic (40 days) time point postinfection. Decreases in histopathology included reductions in the appearance of activated monocytes/microglia within spinal cord white matter and in the magnitude of MBP loss.
Reduced pathology in the spinal cord of mice with high levels of circulating Klk6‐neutralizing antibodies was paralleled by reductions in total brain pathology when evaluated at 40 dpi. However, when TMEV‐IDD was allowed to progress to a late chronic (180 days) time point, brain and spinal cord pathology in Klk6‐immunized mice ceased to differ substantially from controls, even though antibody titers remained high. These results suggest that Klk6‐neutralizing antibodies can reduce TMEV‐induced disease from acute through early chronic stages, but by late chronic time points, any beneficial effects are lost. The fact that neuroprotection was observed only until early chronic time points in the spinal cord may reflect the steadily rising levels of Klk6 observed as TMEV‐IDD progresses. By 90 dpi, Klk6 RNA and protein reached a peak of five‐ to 10‐fold control levels. The neutralizing antibody approach may not be effective at these high Klk6 levels, and the development of more potent Klk6 targeting strategies will be important. In this regard, emerging data appear to place Klk6 within a larger proteolytic cascade that is functioning within the CNS including activation relationships with other kallikreins such as Klk1, Klk5 and Klk9 68, 69. Even 1%–2% of residual Klk6 activity would permit such a cascade to slowly proceed. Targeting multiple members of this presumptive cascade, therefore, would be an additional therapeutic approach. Another key consideration is that numerous other pathophysiological processes are at play in TMEV‐IDD, particularly at chronic stages, as extensive virally induced inflammation and tissue destruction, likely not dependent on Klk6, accrue. Therefore, combining therapies that target multiple pathogenic players, including Klk6, will be an important line of future investigation to determine potential additive effects.
In previous reports, we documented elevations in Klk6 at sites of CNS inflammation that are related in part to the high levels expressed by infiltrating CD4 and CD8 T cells in both TMEV and EAE models of MS 8, 50. We have also shown that lymphocyte Klk6 transcriptional activity can be induced in vitro using pan T‐cell activators such as CD3 T‐cell receptor cross‐linking antibodies, interleukin 2, concanavalin A, or by steroid hormones such as glucocorticoids, androgens and progesterone 8, 10, 51. The participation of Klk6 in the T‐cell activation program is further supported by results herein that show prominent elevations in Klk6 RNA expression as a recall response to VP1 and VP2 viral capsid proteins, which are known to exacerbate disease in susceptible SJL mice (67) and, when transgenically, expressed in resistant strains (12). These results indicate that T cells encountering TMEV capsid proteins in vivo may up‐regulate Klk6 as part of the adaptive immune response. This is of particular interest in light of recent findings demonstrating that Klk6 promotes the survival of T and B cells by cleaving PAR1 to trigger intracellular signaling cascades that increase the prosurvival protein Bcl‐XL and decrease the proapoptotic protein Bim (54). Thus, at sites of viral infection, viral capsid‐induced elevations in T‐cell Klk6 may act in an autocrine or paracrine fashion to enhance lymphocyte survival and prolong the chronic inflammatory response. Reduced inflammation seen in mice with Klk6‐neutralizing antibodies, therefore, may in part reflect a loss of this prosurvival effect.
To further evaluate the impact of Klk6 function‐blocking antibodies on the adaptive immune response, we examined DTH responses to UV‐inactivated TMEV. Mice with Klk6‐neutralizing antibodies showed reduced TMEV‐driven DTH responses, pointing to reductions in CD4 T‐cell Th1 activity, despite the fact that viral RNA levels and viral antibody responses were largely unchanged. As Th1 skewed CD4 T cells play an important role in demyelination in TMEV‐IDD (14), reduced CD4 T‐cell Th1 responses may partially account for the reductions in immunopathology seen in mice with Klk6‐neutralizing antibodies. We did not directly examine the effects of Klk6 in CD8 T cell‐driven pathology, but given the expression of Klk6 by these cells (8) and their prominent role in the development of axon damage 43, 61, this will be an important line of future investigation. Notably in this regard, in vitro studies indicate that excess recombinant Klk6 promotes a dying back of neurites as well as neuron cell death (53).
As the host response to TMEV involves activation of the innate in addition to the adaptive arms of the immune system, we examined the regulation of KLK6 in activated monocytes in vitro as well as the effects of Klk6‐neutralizing antibodies on monocytes/microglia in vivo. Like activated T cells, activated monocytes responded with transcriptional elevations in KLK6. Elevated levels of Klk6 have been previously observed in macrophages and/or microglia at sites of CNS inflammation in both TMEV 7, 50 and EAE (8) models of MS, in human MS lesions (50), and in traumatic SCI (52). Activated astrocytes are also a rich source of de novo Klk6 52, 55. Importantly, in mice with high levels of circulating Klk6‐function‐blocking antibodies, detection of BS‐1‐positive monocytes/microglia was significantly reduced. These findings indicate key roles for Klk6 in monocyte/microglial driven processes governing innate immune responses in TMEV‐IDD.
In summary, the data presented support the conclusion that Klk6 is an important participant in the immunopathogenic events that drive TMEV‐IDD at acute through early chronic time points. The decreases observed in Th1‐driven DTH responses, taken with reductions in the extent of meningeal inflammation and monocyte/microglial activation, point to effects of Klk6‐neutralizing antibodies on the development of TMEV‐driven inflammatory responses and, therefore, TMEV‐mediated immunopathology (immune‐mediated tissue injury). Importantly, however, neutralizing Klk6 did not significantly alter viral levels detected in the spinal cord or the development of TMEV‐specific antibody responses, such that pan‐immunosuppressive effects were likely not invoked. Instead, it is likely that Klk6‐neutralizing antibodies also served to directly reduce Klk6‐mediated oligo‐ (50) and neurotoxic events (53) and its ability to directly degrade myelin proteins 6, 7, 50, thereby reducing the loss of MBP in the spinal cord through early chronic time points. Taken together, these findings support the idea of KLK6 as a key component of degradome‐regulating inflammatory demyelinating disease. Further efforts to determine how KLK6 can be therapeutically targeted to delay the development of disease in MS patients and promote neuroprotection are warranted.
ACKNOWLEDGMENTS
These studies were supported by R01NS052741, The Christopher and Dana Reeve Paralysis Foundation, RG3367 and a Collaborative MS Research Award CA1060A11‐02 from the National Multiple Sclerosis Society. The technical expertise of Louisa Papke, Mabel Pierce, Laurie Zoecklein and Jason Kerkvliet and the editorial assistance of Lea Dacy are also gratefully acknowledged.
REFERENCES
- 1. Abraham M, Shapiro S, Karni A, Weiner HL, Miller A (2005) Gelatinases (MMP‐2 and MMP‐9) are preferentially expressed by Th1 vs Th2 cells. J Neuroimmunol 163:157–164. [DOI] [PubMed] [Google Scholar]
- 2. Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H et al (2004) Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A 101:6698–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alvord EC, Hruby S, Sires LR (1979) Degradation of myelin basic protein by cerebrospinal fluid: preservation of antigenic determinants under physiological conditions. Ann Neurol 6:474–482. [DOI] [PubMed] [Google Scholar]
- 4. Angelo PF, Lima AR, Alves FM, Blaber SI, Scarisbrick IA, Blaber M et al (2006) Substrate specificity of human kallikrein 6: salt and glycosaminoglycan activation effects. J Biol Chem 281:3116–3126. [DOI] [PubMed] [Google Scholar]
- 5. Bar‐Or A, Nuttall RK, Duddy M, Alter A, Kim HJ, Ifergan I et al (2003) Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 126:2738–2749. [DOI] [PubMed] [Google Scholar]
- 6. Bernett MJ, Blaber SI, Scarisbrick IA, Dhanarajan P, Thompson SM, Blaber M (2002) Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin‐like kallikrein is expressed in the central nervous system. J Biol Chem 277:24562–24570. [DOI] [PubMed] [Google Scholar]
- 7. Blaber SI, Scarisbrick IA, Bernett MJ, Dhanarajan P, Seavy MA, Jin Y et al (2002) Enzymatic properties of rat myelencephalon‐specific protease. Biochemistry 41:1165–1173. [DOI] [PubMed] [Google Scholar]
- 8. Blaber SI, Ciric B, Christophi GP, Bernett MJ, Blaber M, Rodriguez M, Scarisbrick IA (2004) Targeting kallikrein 6‐proteolysis attenuates CNS inflammatory disease. FASEB J 19:920–922. [DOI] [PubMed] [Google Scholar]
- 9. Cammer W, Brosnan CF, Basile C, Bloom BR, Norton WT (1986) Complement potentiates the degradation of myelin proteins by plasmin: implications for a mechanism of inflammatory demyelination. Brain Res 364:91–101. [DOI] [PubMed] [Google Scholar]
- 10. Christophi GP, Isackson PJ, Blaber SI, Blaber M, Rodriguez M, Scarisbrick IA (2004) Distinct promoters regulate tissue‐specific and differential expression of kallikrein 6 in CNS demyelinating disease. J Neurochem 91:1439–1449. [DOI] [PubMed] [Google Scholar]
- 11. Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J (1996) The expression of tissue‐type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol 55:1194–1204. [DOI] [PubMed] [Google Scholar]
- 12. Denic A, Zoecklein L, Kerkvliet J, Papke L, Edukulla R, Warrington A et al (2011) Transgenic expression of viral capsid proteins predisposes to axonal injury in a murine model of multiple sclerosis. Brain Pathol 21:501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fainardi E, Castellazzi M, Bellini T, Manfrinato MC, Baldi E, Casetta I et al (2006) Cerebrospinal fluid and serum levels and intrathecal production of active matrix metalloproteinase‐9 (MMP‐9) as markers of disease activity in patients with multiple sclerosis. Mult Scler 12:294–301. [DOI] [PubMed] [Google Scholar]
- 14. Gerety SJ, Rundell MK, Dal Canto MC, Miller SD (1994) Class II‐restricted T cell responses in Theiler's murine encephalomyelitis virus‐induced demyelinating disease. VI. Potentiation of demyelination with and characterization of an immunopathologic CD4+ T cell line specific for an immunodominant VP2 epitope. J Immunol 152:919–929. [PubMed] [Google Scholar]
- 15. Gijbels K, Masure S, Carton H, Opdenakker G (1992) Gelatinase in cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol 41:29–34. [DOI] [PubMed] [Google Scholar]
- 16. Gonzalez H, Ottervald J, Nilsson KC, Sjogren N, Miliotis T, Von Bahr H et al (2009) Identification of novel candidate protein biomarkers for the post‐polio syndrome—implications for diagnosis, neurodegeneration and neuroinflammation. J Proteomics 71:670–681. [DOI] [PubMed] [Google Scholar]
- 17. Gveric D, Hanemaaijer R, Newcombe J, van Lent NA, Sier CF, Cuzner ML (2001) Plasminogen activators in multiple sclerosis lesions: implications for the inflammatory response and axonal damage. Brain 124:1978–1988. [DOI] [PubMed] [Google Scholar]
- 18. Haile Y, Simmen KC, Pasichnyk D, Touret N, Simmen T, Lu JQ et al (2011) Granule‐derived granzyme B mediates the vulnerability of human neurons to T cell‐induced neurotoxicity. J Immunol 187:4861–4872. [DOI] [PubMed] [Google Scholar]
- 19. Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS et al (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451:1076–1081. [DOI] [PubMed] [Google Scholar]
- 20. Hebb AL, Bhan V, Wishart AD, Moore CS, Robertson GS (2011) Human kallikrein 6 cerebrospinal levels are elevated in multiple sclerosis. Curr Drug Discov Technol 7:137–140. [DOI] [PubMed] [Google Scholar]
- 21. Kumnok J, Ulrich R, Wewetzer K, Rohn K, Hansmann F, Baumgartner W, Alldinger S (2008) Differential transcription of matrix‐metalloproteinase genes in primary mouse astrocytes and microglia infected with Theiler's murine encephalomyelitis virus. J Neurovirol 14:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leppert D, Waubant E, Burk MR, Oksenberg JR, Hauser SL (1996) Interferon beta‐1b inhibits gelatinase secretion and in vitro migration of human T cells: a possible mechanism for treatment efficacy in MS. Ann Neurol 40:846–852. [DOI] [PubMed] [Google Scholar]
- 23. Leppert D, Ford J, Stabler G, Grygar C, Lienert C, Huber S et al (1998) Matrix metalloproteinase‐9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 121:2327–2334. [DOI] [PubMed] [Google Scholar]
- 24. Lin X, Njenga MK, Johnson AJ, Pavelko KD, David CS, Pease LR, Rodriguez M (2002) Transgenic expression of Theiler's murine encephalomyelitis virus genes in H‐2(b) mice inhibits resistance to virus‐induced demyelination. J Virol 76:7799–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindberg RLP, De Groot CJA, Montagne L, Freitag P, van der Valk P, Kappos L, Leppert D (2001) The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain 124:1743–1753. [DOI] [PubMed] [Google Scholar]
- 26. Lipton HL (1975) Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun 11:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maeda A, Sobel RA (1996) Matrix metalloproteinases in the normal human central nervous system, microglia nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol 55:300–309. [DOI] [PubMed] [Google Scholar]
- 28. Mandler RN, Dencoff JD, Midani F, Ford CC, Ahmed W, Rosenberg GA (2001) Matrix metalloproteinases and tissue inhibitors of metalloproteinases in cerebrospinal fluid differ in multiple sclerosis and Devic's neuromyelitis optica. Brain 124:493–498. [DOI] [PubMed] [Google Scholar]
- 29. McFarlin DE, McFarland HF (1982) Multiple sclerosis (first of two parts). N Engl J Med 307:1183–1188. [DOI] [PubMed] [Google Scholar]
- 30. McGavern DB, Zoecklein L, Drescher KM, Rodriguez M (1999) Quantitative assessment of neurologic deficits in a chronic progressive murine model of CNS demyelination. Exp Neurol 158:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McGavern DB, Zoecklein L, Sathornsumetee S, Rodriguez M (2000) Assessment of hindlimb gait as a powerful indicator of axonal loss in a murine model of progressive CNS demyelination. Brain Res 877:396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Metz LM, Ahang Y, Yeung M, Party DG, Bell RB, Stoian CA et al (2004) Minocycline reduces gadolinium‐enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann Neurol 55:756–758. [DOI] [PubMed] [Google Scholar]
- 33. Miller SD, Gerety SJ (1990) Immunologic aspects of Theiler's murine encephalomyelitis virus (TMEV)‐induced demyelinating disease. Semin Virol 1:263–272. [Google Scholar]
- 34. Mitsui S, Okui A, Uemura H, Mizuno T, Yamada T, Yamamura Y, Yamaguchi N (2002) Decreased cerebrospinal fluid levels of neurosin (KLK6), an aging‐related protease, as a possible new risk factor for Alzheimer's disease. Ann N Y Acad Sci 977:216–223. [DOI] [PubMed] [Google Scholar]
- 35. Norga K, Paemen L, Masure S, Dillen C, Heremans H, Billiau A et al (1995) Prevention of acute autoimmune encephalomyelitis and abrogation of relapses in murine models of multiple sclerosis by the protease inhibitor d‐penicillamine. Inflamm Res 44:529–534. [DOI] [PubMed] [Google Scholar]
- 36. Ogawa K, Yamada T, Tsujioka Y, Taguchi J, Takahashi M, Tsuboi Y et al (2000) Localization of a novel type trypsin‐like serine protease, neurosin, in brain tissues of Alzheimer's disease and Parkinson's disease. Psychiatry Clin Neurosci 54:419–426. [DOI] [PubMed] [Google Scholar]
- 37. Paemen L, Olsson T, Soderstron M, Damme JV, Opdenakker G (1994) Evaluation of gelatinases and IL‐6 in the cerebrospinal fluid of patients with optic neuritis, multiple sclerosis and other inflammatory neurological diseases. Eur J Neurosci 1:55–63. [DOI] [PubMed] [Google Scholar]
- 38. Pavelko KD, Pease LR, David CS, Rodriguez M (2007) Genetic deletion of a single immunodominant T‐cell response confers susceptibility to virus‐induced demyelination. Brain Pathol 17:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pierce ML, Rodriguez M (1989) Erichrome stain for myelin on osmicated tissue embedded in glycol methacrylate plastic. J Histotechnol 12:35–36. [Google Scholar]
- 40. Poser CM (1986) Pathogenesis of multiple sclerosis. A critical reappraisal. Acta Neuropathol (Berl) 71:1–10. [DOI] [PubMed] [Google Scholar]
- 41. Proost P, Van Damme J, Opdenakker G (1993) Leukocyte gelatinase B cleavage releases encephalitogens from human myelin basic protein. Biochem Biophys Res Commun 192:1175–1781. [DOI] [PubMed] [Google Scholar]
- 42. Puente SS, Sanchez LM, Overall CM, Lopez‐Otin C (2003) Human and mouse proteases: a comparative genomic approach. Nat Rev Genet 4:544–558. [DOI] [PubMed] [Google Scholar]
- 43. Rivera‐Quinones C, McGavern D, Schmelzer JD, Hunter SF, Low PA, Rodriguez M (1998) Absence of neurological deficits following extensive demyelination in a class I‐deficient murine model of multiple sclerosis. Nat Med 4:187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriguez M, Oleszak E, Leibowitz J (1987) Theiler's murine encephalomyelitis: a model of demyelination and persistence of virus. Crit Rev Immunol 7:325–365. [PubMed] [Google Scholar]
- 45. Rodriguez M, Zoecklein L, Kerkvliet JG, Pavelko KD, Papke L, Howe CL et al (2008) Human HLA‐DR transgenes protect mice from fatal virus‐induced encephalomyelitis and chronic demyelination. J Virol 82:3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rosenberg GA, Estrada EY, Dencoff JE, Stetler‐Stevenson WG (1995) TNF‐alpha‐induced gelatinase beta causes delayed opening of the blood‐brain barrier: an expanded therapeutic window. Brain Res 703:151–155. [DOI] [PubMed] [Google Scholar]
- 47. Rosenberg GA, Dencoff JE, Correa NJ, Reiners M, Ford CC (1996) Effect of steroids on CSF matrix metalloproteinases in multiple sclerosis: relation to blood‐brain barrier injury. Neurology 46:1626–1632. [DOI] [PubMed] [Google Scholar]
- 48. Scarisbrick IA (2008) The multiple sclerosis degradome: enzymatic cascades in development and progression of central nervous system inflammatory disease. Curr Top Microbiol Immunol 318:133–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scarisbrick IA, Towner MD, Isackson PJ (1997) Nervous system specific expression of a novel serine protease: regulation in the adult rat spinal cord by excitotoxic injury. J Neurosci 17:8156–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scarisbrick IA, Blaber SI, Lucchinetti CF, Genain CP, Blaber M, Rodriguez M (2002) Activity of a newly identified serine protease in CNS demyelination. Brain 125:1283–1296. [DOI] [PubMed] [Google Scholar]
- 51. Scarisbrick IA, Blaber SI, Tingling JT, Rodriguez M, Blaber M, Christophi GP (2006) Potential scope of action of tissue kallikreins in CNS immune‐mediated disease. J Neuroimmunol 178:167–176. [DOI] [PubMed] [Google Scholar]
- 52. Scarisbrick IA, Sabharwal P, Cruz H, Larsen N, Vandell A, Blaber SI et al (2006) Dynamic role of kallikrein 6 in traumatic spinal cord injury. Eur J Neurosci 24:1457–1469. [DOI] [PubMed] [Google Scholar]
- 53. Scarisbrick IA, Linbo R, Vandell AG, Keegan M, Blaber SI, Blaber M et al (2008) Kallikreins are associated with secondary progressive multiple sclerosis and promote neurodegeneration. Biol Chem 389:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scarisbrick IA, Epstein B, Cloud BA, Yoon H, Wu J, Renner DN et al (2011) Functional role of kallikrein 6 in regulating immune cell survival. PLoS ONE 6:e18376. 1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scarisbrick IA, Radulovic M, Larson N, Burda J, Blaber I, Giannini C et al (2012) Kallikrein 6 is a novel molecular trigger of reactive astrogliosis. Biol Chem (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Skuljec J, Gudi V, Ulrich R, Frichert K, Yildiz O, Pul R et al (2011) Matrix metalloproteinases and their tissue inhibitors in cuprizone‐induced demyelination and remyelination of brain white and gray matter. J Neuropathol Exp Neurol 70:758–769. [DOI] [PubMed] [Google Scholar]
- 57. Stuve O, Dooley NP, Uhm JH, Antel JP, Francis GS, Williams G, Yong VW (1996) Interferon beta‐1b decreases the migration of T lymphocytes in vitro: effects on matrix metalloproteinase‐9. Ann Neurol 40:853–863. [DOI] [PubMed] [Google Scholar]
- 58. Tsunoda I, Fujinami RS (2010) Neuropathogenesis of Theiler's murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol 5:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uchida A, Oka Y, Aoyama M, Suzuki S, Yokoi T, Katano H et al (2004) Expression of myelencephalon‐specific protease in transient middle cerebral artery occlusion model of rat brain. Brain Res 126:129–136. [DOI] [PubMed] [Google Scholar]
- 60. Ulrich R, Baumgartner W, Gerhauser I, Seeliger F, Haist V, Deschl U, Alldinger S (2006) MMP‐12, MMP‐3, and TIMP‐1 are markedly upregulated in chronic demyelinating theiler murine encephalomyelitis. J Neuropathol Exp Neurol 65:783–793. [DOI] [PubMed] [Google Scholar]
- 61. Ure DR, Rodriguez M (2002) Preservation of neurologic function during inflammatory demyelination correlates with axon sparing in a mouse model of multiple sclerosis. Neuroscience 111:399–411. [DOI] [PubMed] [Google Scholar]
- 62. Vandell AG, Larson N, Laxmikanthan G, Panos M, Blaber SI, Blaber M, Scarisbrick IA (2008) Protease activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J Neurochem 107:855–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV et al (2000) Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U S A 97:6820–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Waubant E, Goodkin DE, Gee L, Bacchetti P, Sloan R, Stewart T et al (1999) Serum MMP‐9 and TIMP‐1 levels are related to MRI activity in relapsing multiple sclerosis. Am Acad Neurol 53:1397–1401. [DOI] [PubMed] [Google Scholar]
- 65. Weaver A, Goncalves da Silva A, Nuttall RK, Edwards DR, Shapiro SD, Rivest S, Yong VW (2005) An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB J 19:1668–1670. [DOI] [PubMed] [Google Scholar]
- 66. Yang Y, Tian SJ, Wu L, Huang DH, Wu WP (2011) Fibrinogen depleting agent batroxobin has a beneficial effect on experimental autoimmune encephalomyelitis. Cell Mol Neurobiol 31:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yauch RL, Palma JP, Yahikozawa H, Koh CS, Kim BS (1998) Role of individual T‐cell epitopes of Theiler's virus in the pathogenesis of demyelination correlates with the ability to induce a Th1 response. J Virol 72:6169–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoon H, Laxmikanthan G, Lee J, Blaber SI, Rodriguez A, Kogot JM et al (2007) Activation profiles and regulatory cascades of the human kallikrein‐related peptidases. J Biol Chem 282:31852–31864. [DOI] [PubMed] [Google Scholar]
- 69. Yoon H, Blaber SI, Debela M, Goettig P, Scarisbrick IA, Blaber M (2009) A completed KLK activome profile: investigation of activation profiles of KLK9, 10, and 15. Biol Chem 390:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zarghooni M, Soosaipillai A, Grass L, Scorilas A, Mirazimi N, Diamandis EP (2002) Decreased concentration of human kallikrein 6 in brain extracts of Alzheimer's disease patients. Clin Biochem 35:225–231. [DOI] [PubMed] [Google Scholar]