

Abstract
Regions of low oxygen concentration (hypoxia) occur in both normal human physiology and under pathophysiological conditions. Fluorescent probes for the direct imaging of cellular hypoxia could be useful tools that complement radiochemical imaging and immunohistochemical staining methods. In this work, we set out to characterize the hypoxia-selective enzymatic conversion of a simple nitroaryl probe, 6-nitroquinoline (1). We envisioned that this compound might undergo hypoxia-selective, bioreductive conversion to the fluorescent product, 6-aminoquinoline (2, Scheme 2). The probe 1 was, indeed, converted to a fluorescent product selectively under hypoxic conditions by the one-electron reducing enzyme NADPH: cytochrome P450 reductase. However, inspection of the fluorescence spectrum and LC/MS analysis of the reaction mixture revealed that the expected product 2 was not formed. Rather, the 63-fold increase in fluorescence emission at 445 nm resulting from the hypoxic metabolism of 1 was due to formation of the azoxy-helicene product, pyrido[3,2-f]quinolino[6,5-c]cinnoline 3-oxide (4). The generation of 4 involves an unusual biaryl bond formation under reductive conditions. The mechanism of this process remains uncertain, but could proceed via combination of a nitroaryl radical anion with a neutral nitrosoaryl radical, followed by tautomerization and intramolecular condensation between the resulting hydroxylamine and nitroso functional groups. Bioreductive metabolism of nitroaryl compounds represents a promising strategy for the selective delivery of cytotoxic agents and fluorescent markers to hypoxic tissue, but the results described here provide a important glimpse of the chemical complexity that can be associated with the enzymatic one-electron reduction of nitroaryl compounds.
Introduction
The concentration of oxygen in normal human tissue is generally between 20–90 μM (14–65 mm Hg);1,2 however, regions of much lower oxygen concentration (hypoxia) occur in both normal human physiology and under pathophysiological conditions.1–7 For example, many solid tumors contain significant regions of hypoxia.1–3,5,6,8,9 In normal physiology, transient regions of hypoxia may play a role in regulating cell differentiation and organ development during embryogenesis.1,3,4,10 With growing interest in hypoxia, comes an increased need for new tools that researchers can use to detect hypoxia in biological systems. Fluorescent probes for the direct imaging of cellular hypoxia could provide a useful complement to radiochemical imaging11 and immunohistochemical staining methods.12
One strategy for the development of fluorescent probes of cellular hypoxia involves identification of nonfluorescent nitroaromatic compounds that can be metabolized to a fluorescent amine derivative.13–16 It is well established that nitroaromatic compounds can undergo intracellular enzymatic reduction selectively under hypoxic conditions.17–22 This process involves a series of enzymatic one-electron reductions that ultimately produce the corresponding arylamine (Scheme 1).17–25 It is generally assumed that the initially-generated nitro radical anion is the key oxygen-sensitive intermediate that confers hypoxia selectivity to these metabolic processes.20,23,24
Scheme 1.

Enzymatic Reduction of Nitroaryl Compounds.
Here we set out to characterize the hypoxic metabolism of a simple nitroaryl substrate, 6-nitroquinoline (1). We envisioned that this compound might undergo hypoxia-selective, bioreductive conversion to the fluorescent product, 6-aminoquinoline (2, Scheme 2). Compound 2 displays an impressive 205 nm Stokes shift26 and has been used as a fluorescent reporter in a number of applications.27–31 In the event, however, we found that the enzymatic reduction of 1 generated a fluorescent product of unexpected structure.
Scheme 2.

Anticipated Conversion of 6-Nitroquinoline to 6-Aminoquinoline.
Results and Discussion
Conversion of 1 to a Fluorescent Product by NADPH:Cytochrome P450 Reductase Under Hypoxic Conditions
We first showed that the nitro precursor 1 is non-fluorescent in aqueous solution (Figure 1A, column 1). We then employed the enzyme NADPH:cytochrome P450 reductase as a tool to carry out one-electron reductive metabolism of 1. We chose this enzyme because it plays an important role in the bioreductive metabolism of xenobiotics in humans.19,21,22,32–37 For reactions carried out under anaerobic conditions, molecular oxygen was removed from the solutions by three cycles of freeze-pump-thaw degassing and the assay mixtures assembled and incubated in an inert atmosphere glove bag. Assays were shielded from light to prevent possible photoreactions. Under hypoxic conditions, we observed that NADPH:cytochrome P450 reductase converted 1 to a fluorescent product, with a 63-fold increase in fluorescence emission at 445 nm (Figure 1A, column 5). In contrast, little or no fluorescence was observed in an aerobic control assay (Figure 1A, column 4) or in assays where a non-fluorescent electron acceptor was substituted for 1 (Figure 1A, columns 2 and 3). Both the enzyme and NADPH were required for the conversion of 1 to a fluorescent product. Curiously, the fluorescence spectrum of the mixture generated by hypoxic metabolism of 1, consisting of a pair of emission maxima at 440 and 460 nm, did not match that for the expected product 2, which displays broad emission peaks at 445 and 530 nm (Figure 1B). Therefore, we were motivated to characterize the products generated in the hypoxic metabolism of 1.
Figure 1.

Conversion of 1 into a fluorescent product under hypoxic conditions. A. Fluorescence emission at 445 nm (λex 307 nm) for: (1) a control sample of compound 1 alone (0.8 mM), (2) a control reaction composed of NADPH:cytochrome P450 reductase (1.1 U/mL), NADPH (2.4 mM), and a non-fluorescent electron acceptor 1,2,4-benzotriazine 1,4-dioxide35 (6.4 mM) under aerobic conditions, (3) a control reaction composed of NADPH:cytochrome P450 reductase (1.1 U/mL), NADPH (2.4 mM), and a non-fluorescent electron acceptor 1,2,4-benzotriazine 1,4-dioxide35 (6.4 mM) under anaerobic conditions, (4) compound 1 (0.8 mM) + NADPH:cytochrome P450 reductase (1.1 U/mL) and NADPH (2.4 mM) under aerobic conditions, (5) compound 1 (0.8 mM) + cytochrome P450 reductase (1.1 U/mL) and NADPH (2.4 mM) under anaerobic conditions. Reactions were incubated for 18 h in sodium phosphate buffer at (12 mM, pH 7.4) at 24 °C, then diluted with aerobic sodium phosphate buffer (12 mM, pH 7.4), and the fluorescence measured (λex 307 nm, λem 445 nm). It is important to note that NADPH exhibits fluorescence with an emission maximum at 445 nm. However, control experiments showed that unconsumed NADPH in the assays was ultimately converted to the non-fluorescent NADP+ product by enzyme-driven redox cycling upon opening the reaction vessel to air and dilution with aerobic buffer prior to fluorescence measurements (Figure S2). B. Fluorescence spectrum of the reaction mixture generated in the anaerobic metabolism of 1 by NADPH:cytochrome P450 reductase as described for reaction 5 above (orange line, with emission maxima at 440 and 450 nm) and fluorescence spectrum of authentic 6-aminoquinoline (2, 50 μM, λex 340 nm, in sodium phosphate buffer, 10 mM, pH 7.4).
LC/MS Analysis of the Mixture Generated by the Enzymatic Reduction of 1 Under Hypoxic Conditions
LC/MS analysis of the reaction mixture resulting from anaerobic metabolism of 1 by NADPH:cytochrome P450 reductase revealed two major products in addition to the starting probe 1 (Figure 2A). The originally anticipated product, 6-aminoquinoline 2, was not observed in the LC/MS (this compound elutes at approximately 4.5 min under these chromatographic conditions, Figure S1). The peak eluting at 19.4 min in the chromatogram displayed an m/z of 301 (Figure 2B). This mass-to-charge ratio is consistent with the [M+H]+ ion of 6,6′-azoxyquinoline (3, Scheme 3). This compound can be envisioned to arise via a precedented type of condensation between the 6-nitrosoquinoline and 6-hydroxylaminoquinoline products expected from the reductive metabolism of 1.38,39 Indeed, the LC/MS spectra of the 19.4 min product matched that of an authentic sample of 3 prepared via reduction of 1 by hydrazine hydrate in the presence of Raney nickel (Figures 2E and F).40,41 Importantly, 3 displayed no fluorescence in aqueous buffer and, therefore, could not account for the fluorescence generated by the hypoxic metabolism of 1. Accordingly, we turned our attention to the product eluting near 15 min in the LC/MS. This compound displayed an m/z of 299 (Figure 2C). This mass-to-charge ratio is not consistent with either a simple 6-substituted quinoline reduction product or a nitrogen-linked dimer such as an azoxy or azo compound. Rather, this result was suggestive of a more complex dimeric structure such as pyrido[3,2-f]quinolino[6,5-c]cinnoline 3-oxide (4, Scheme 3) or dipyrido[3,2-a:3′,2′-h]phenazine 7-oxide (5). There was some indication that helicenes related in structure to 4 are fluorescent42,43 and we investigated the possibility that the helical molecule 4 might be the fluorescent product arising from the bioreductive metabolism of 1.
Figure 2.

LC/MS analysis of the reaction mixture generated by anaerobic metabolism of 1 (0.8 mM) by NADPH:cytochrome P450 reductase (1.1 U/mL) and NADPH (6.4 mM). The enzymatic reduction of 1 was carried out as described in the Supporting Information and the Legend of Figure 1. The reaction was dried, products dissolved in methanol, and the mixture analyzed by LC/MS. The column was eluted with a gradient of 99% A (water containing 0.1% acetic acid) and 1% B (acetonitrile containing 0.1% acetic acid) followed by a linear increase to 90% B over 30 min. The elution was continued at 90% B for 3 min and then B decreased to 1% over next 8 min. A flow rate of 0.35 mL/min was used and the metabolites were detected by their absorbance at 254 nm. Mass spectra were obtained using electrospray ionization in the positive ion mode. Panel A: HPLC of the anaerobic reaction mixture monitoring absorbance at 254 nm. Panel B: LC/MS spectrum of the product eluting at 19.1 min. Panel C: LC/MS spectrum of the product eluting at 15.1 min. Panel D: LC/MS spectrum for of the product eluting at 17.1 min. Panel E: HPLC of authentic 3 monitoring absorbance at 254 nm. Panel F: LC/MS spectrum of authentic 3. Panel G: HPLC of authentic 4 monitoring absorbance at 254 nm. Panel H: LC/MS spectrum of authentic 4.
Scheme 3.

Products Obtained from Enzymatic Reduction of 6-Nitroarylquinoline Under Hypoxic Conditions.
Synthesis of the Azoxy Helicene 4 by Alkaline Glucose Reduction of 1
The suspected product 4 has an interesting history. This structure was first proposed in 1948 by Huisgen as a product resulting from the reaction of 1 with sodium methoxide.44,45 In 1965, this structural assignment was refuted by Farrar who suggested that the product of Huisgen’s reaction was actually the isomeric compound 5.46,47 However, as part of his studies, Farrar suggested that an alkaline glucose reduction of 1 carried out earlier by Galbraith et al. in 1951 and originally proposed to yield 6,6′-azoxyquinoline 3 (based upon nitrogen analysis) did, in fact, produce the helicene 4.46,48
Seeking a chemical synthesis of 4, we carried out the alkaline glucose reduction of 1 by the method of Galbraith48 and Farrar.46 A blue fluorescent compound generated in this reaction did indeed match the thin layer chromatographic (TLC) properties of the blue fluorescent product generated in the hypoxic metabolism of 1. Therefore, we isolated this compound from the alkaline glucose reduction by preparative TLC and carried out full spectroscopic characterization (Supporting Information). High-resolution mass spectrometric analysis gave a molecular formula for the compound that was consistent with either of the dimeric structures 4 or 5. NMR analysis readily confirmed that the product was not a simple 6-substituted quinoline; rather, the presence of 18 distinct resonances in the 13C-spectra was suggestive of an asymmetric quinoline dimer. The 1H-NMR displayed ten distinct resonances, consistent with the notion that two of the quinoline hydrogens had been replaced with an aryl-aryl bridge of some type. The proton at position 4 of the quinoline ring system in 5 was expected to be shifted substantially downfield to ≥ 9.3 ppm due to its proximity to the N-oxide oxygen (to facilitate comparison of the spectral data anticipated for 4 and 5 we employ a quinoline-based numbering system in this discussion).49 On the other hand, no such downfield proton resonance was expected for structure 4. Thus, it was significant that we did not observe any resonances at 9.3 ppm or above in the 1H-NMR spectrum of the product from the alkaline glucose reduction of 1. The TOCSY and COSY spectra allowed unambiguous assignment of the resonances for the 4 and 4′ protons in the spectra and strong support for the helicene structure 4 was provided by an NOE experiment showing that these protons are in close proximity. In the helicene structure 4, these protons are very close in space (2.5 Å, Figure 4), whereas, in the alternate structure 5, these protons are quite distant and therefore would not be expected to display a cross-peak in the NOE spectrum. Finally, the TOCSY, COSY, HMQC, and HMBC data were consistent with the azoxy helicene 4 (Table 1). The structure of 4 was ultimately confirmed by X-ray crystallographic analysis (Figure 4).50
Figure 4.
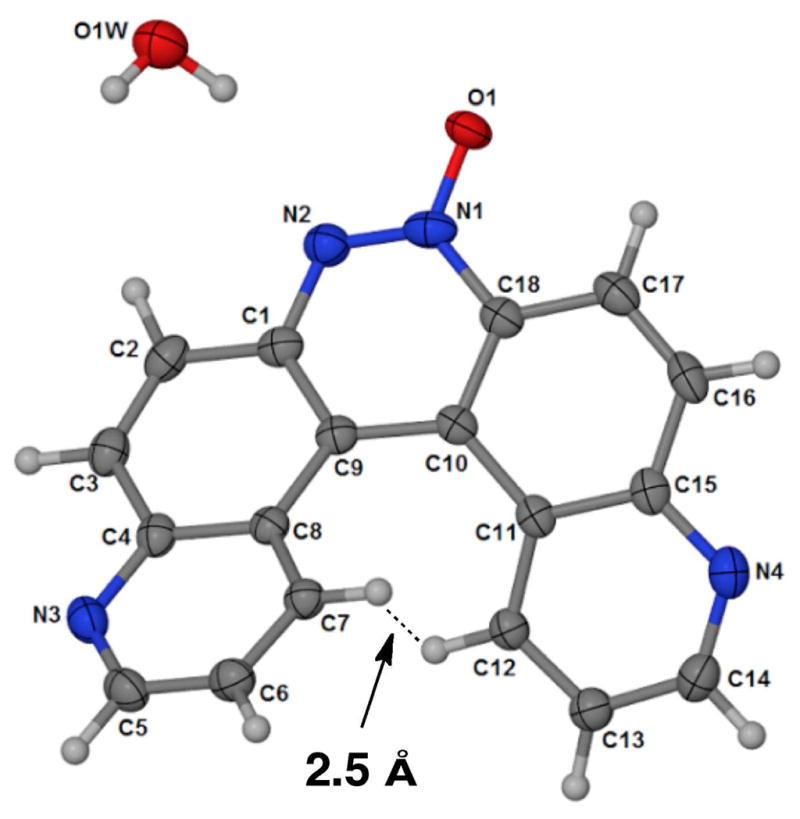
In the helicene structure 4, the 4- and 4′-hydrogens (see numbering system in Scheme 3) are close in space. X-ray structure from ref. 50
Table 1.
NMR Data (CDCl3) for compound 4.
| position | δC | δH (J in Hz) | COSY | TOCSY | HMBCa | NOE |
|---|---|---|---|---|---|---|
| 2′ | 153.4 | 9.15 1H, d (4.5) | 3′ | 3′, 4′ | 4′, 8a′ | 3′ |
| 2 | 151.5 | 9.05 1H, m | 3 | 3, 4 | 4, 8a | 3 |
| 3′ | 120.5 | 7.46 1H, dd (9.0, 4.5) | 2′, 4′ | 2′, 4′ | 4a′ | 2′, 4′ |
| 3 | 120.7 | 7.41 1H, dd (9.0, 4.5) | 2, 4 | 2, 4 | 4a | 2, 4 |
| 4′ | 136.0 | 8.87 1H, d (9.0) | 3′ | 3′, 2′ | 2′, 8a′ | 4 |
| 4 | 134.8 | 8.67 1H, d (9.0) | 3 | 3, 2 | 2, 8a | 4′ |
| 5′ | 128.0 | |||||
| 5 | 114.2 | |||||
| 4′a | 123.4 | |||||
| 4a | 123.5 | |||||
| 7′ | 121.8 | 9.05 1H, m | 8′ | 8′ | 8a′, 5′ | 8′ |
| 7 | 127.4 | 8.23 1H, d (9.0) | 8 | 8 | 8a, 5 | 8 |
| 8′ | 133.4 | 8.43 1H, d (9.0) | 7′ | 7′ | 6′, 4a′ | 7′ |
| 8 | 134.3 | 8.41 1H, d (9.0) | 7 | 7 | 6, 4a | 7 |
| 6′ | 137.1 | |||||
| 6 | 144.1 | |||||
| 8′a | 149.7 | |||||
| 8a | 148.5 |
HMBC correlations are from the proton to the stated carbon(s).
Evidence That the Fluorescent Product Generated in the Hypoxic Metabolism of 1 by NADPH:Cytochrome P450 Reductase is the Azoxy Helicene 4
First, the results described above confirmed Farrar’s 1965 supposition that 4 was produced in the alkaline glucose reduction of 1. Second, and more important to the current study, our characterization of the product arising from the alkaline glucose reduction of 1 provided us with an authentic standard of the helicene 4 that could be compared to the fluorescent product generated in the anaerobic, enzymatic metabolism of 1. Along these lines, LC/MS analysis showed that the retention time and mass spectrum of the product generated in the enzymatic metabolism of 1 eluting near 15 min matches that of the authentic helicene (Figure 2). Furthermore, the proton NMR spectrum of the enzymatically-generated product mirrors that of the authentic helicene 4. Finally, an NOE experiment confirmed the proximity of the 4 and 4′ protons in the enzymatically-generated product (Figures S12 and S13).
The fluorescence spectrum of the authentic helicene (Figure 3) closely resembles that produced by the hypoxic metabolism of 1 (Figure 1B), with emission maxima at 440 and 460 nm. Calibration curves generated using authentic 4, allowed us to estimate that the enzymatic reduction of 1 generates the helicene 4 in about 10% yield. Although NADPH:cytochrome P450 reductase has shown the ability to reduce an azoxy functional group to the corresponding azo group in other molecules,51,52 we find that the azoxy helicene 4 is refractory to reduction by this enzyme system under our reaction conditions.
Figure 3.

Fluorescence spectrum of authentic 4 (50 μM, λex 307 nm) in sodium phosphate buffer (12 mM, pH 7.4).
Conclusions
We did not observe the expected hydroxylamino or amino products in the one-electron enzymatic reduction of 1 by NADPH:cytochrome P450 reductase under hypoxic conditions. Rather, we obtained a helicene product 4 arising from an unusual biaryl bond formation under reductive conditions. The mechanism underlying formation of the helicene 4 remains uncertain, but one plausible pathway involves combination of a nitroaryl radical anion with a neutral nitrosoaryl radical, followed by tautomerization and intramolecular condensation of the resulting hydroxylamine and nitroso functional groups as shown in Scheme 4. The initial reaction could be driven by formation of hydrogen bonded and stacked aggregates similar to those proposed in the radical polymerization of aniline derivatives.53
Scheme 4.

Possible Mechanism for the Formation of Azoxyhelicine 4.
Bioreductive metabolism of nitroaryl compounds represents a promising strategy for the selective delivery of cytotoxic agents and fluorescent markers to hypoxic tissue.13–16,54,55 However, in many studies, even when hypoxia-selective agents have been developed, the products resulting from enzymatic reduction of the nitroaryl precursors have not been chemically characterized.15,16,54,56 The results described here provide a useful indication of the chemical complexity that can be associated with the bioreduction of nitroaryl compounds.
Experimental Section
Materials and methods
The compound 1,2,4-benzotriazine-1,4-di-N-oxide was synthesized according to literature methods.57
Hypoxic metabolism of 1 by NADPH:cytochrome P450 reductase and fluorescence analysis of the resulting mixture
For anaerobic reactions, all reagents except NADPH and cytochrome P450 reductase were degassed in Pyrex tubes by three cycles of freeze-pump-thaw. The tubes were torch-sealed, transferred to an inert gas glove bag, scored, broken open, and bubbled with argon (5 min). Solutions of NADPH were prepared in the glove bag using degassed water. Assay mixtures were assembled in the glove bag, wrapped in foil to protect them from light, and incubated in the glove bag. In a typical reaction, 1 (4 μL of a 50 mM solution in DMF, final concentration 800 μM) was mixed with NADPH (20–160 μL of a 10 mM solution in water, final concentration 0.8 mM–6.4 mM), NADPH:cytochrome P450 reductase (2 μL of a 140 U/mL stock solution, final concentration 1.1 U/mL), sodium phosphate buffer (6 μL of a 500 mM, pH 7.4 solution, final concentration 12 mM) and water to obtain a final volume of 0.25 mL at room temperature (24 °C). After 18 h, the reaction was brought to a final volume of 1 mL with aerobic sodium phosphate buffer (50 mM, pH 7.4). After incubation at room temperature for 1 h, the fluorescence was analyzed. The fluorescence spectra were obtained on a Varian Cary Eclipse Fluorescence Spectrophotometer equipped with a xenon flash lamp using 10 nm slit widths and a 10 mm path length cuvette.
LC/MS analysis of products generated in the hypoxic metabolism of 1 by NADPH:cytochrome P450 reductase
In vitro enzymatic metabolism of 1 was carried out as described above. The resulting products were extracted into ethyl acetate, the organic layer washed with brine, and the ethyl acetate removed by rotary evaporation. The resulting solid was re-dissolved in methanol and analyzed by LC/MS in the positive ion mode. Separation was carried out using a C18 reverse phase Phenomenex Luna column (5 μm particle size, 100 Å pore size, 150 mm length, 2.00 mm i.d.) on a ThermoSeparations liquid chromatograph (TSP4000) and the metabolites were detected by their UV-absorbance at 254 nm. The elution began with a 99:1 mixture of A (water containing 0.1% acetic acid) and B (acetonitrile containing 0.1% acetic acid), followed by a linear increase to 90% B over the course of 30 min. The elution was continued at 90% B for 3 min and then returned to 1% B over the next 8 min at a flow rate of 0.35 mL/min. The LC/ESI-MS analyses were carried out in the positive ion mode on a Finnigan TSQ 7000 triple quadrupole instrument using a 250 kV needle voltage and a capillary temperature of 250 °C.
Synthesis of 1,2-di(quinolin-6-yl)diazene oxide (3)
We employed a variation of the general method of Böge et al. for the reduction of an aromatic nitro group to a hydroxylamino group.58 Earlier work indicated that azoxy compounds were generated in the reduction of nitroaryl residues by Raney nickel.40,41 To a solution of compound 1 (0.5 g, 2.87 mmol) in a mixture of EtOH:CH2Cl2 (1:1, 20 mL) at 0 °C in an ice/salt bath was added a Raney nickel slurry (0.5 mL, active catalyst in water, Sigma-Aldrich catalog number 221678). To this mixture, five portions of hydrazine hydrate (approximately 0.3 mL each) were added at approximately 15 min intervals until 1 was almost completely consumed at judged by TLC (1.5 mL, 30 mmol total amount of hydrazine hydrate added) and the resulting mixture then stirred overnight. The solid was removed by filtration and the resulting solution washed with brine and then dried over sodium sulfate. Column chromatography on silica gel eluted with ethyl acetate gave impure 3 and a second silica gel column eluted with methanol:CH2Cl2 (99:1) gave 3 as a pure yellow solid (100 mg, Rf value = 0.25 in 4:96 methanol:CH2Cl2) m.p. 207–209 °C: 1H NMR (CDCl3, 300 MHz): δ ppm 9.14 (d, J = 1.5 Hz, 1H), δ 9.05 (dd, J = 4.5 Hz, J = 1.5 Hz, 1H), δ 8.93 (dd, J = 4.0 Hz, J = 1.5 Hz, 1H), δ 8.82 (d, J = 2.5 Hz, 1H), δ 8.67 (dd, J = 9.0 Hz, J = 2.5 Hz, 1H), δ 8.31 (dd, J = 8.5 Hz, J = 1.0 Hz, 1H), δ 8.21 (m, 4H), δ 7.49 (dd, J = 8.5 Hz, J = 4.5 Hz, 1H), 7.42 (dd, J = 8.5 Hz, J = 4.5 Hz, 1H). 13C NMR (CDCl3, 75 MHz): δ 152.9, 152.0, 149.6, 148.7, 146.0, 141.9, 137.9, 137.8, 130.9, 130.3, 129.3, 128.5, 127.7, 123.8, 123.4, 122.7, 122.6, 122.1; HRMS (ESI, [M+H]+) m/z calcd for C18H13N4O 301.1089, found 301.1080.
Preparation of pyrido[3,2-f]quinolino[6,5-c]cinnoline 3-oxide (4) via alkaline glucose reduction of 1.48
A solution of 1 (1.0 g, 5.74 mmol) in aqueous NaOH (20% solution, 10 mL) was heated to 90 °C with stirring. To this solution, D-glucose (1.3 g, 7.21 mmol) was added over 30 min and the reaction then stirred for an addition 1 h. The mixture was extracted with ethyl acetate (20 mL), the organic extract washed with brine, and then dried over magnesium sulfate. Column chromatography on silica gel eluted with ethyl acetate and methanol (99:1) gave the helicene 4 as a yellow solid (300 mg, 18% yield) m.p. 264–266 °C: 1H NMR (CDCl3, 500 MHz,): δ 9.15 (d, J = 4.5 Hz 1H), 9.05 (m, 2H), 8.87 (d, J = 9.0 Hz, 1H), 8.67 (d, J = 9.0 Hz, 1H), 8.44 (d J = 9.0 Hz, 1H), 8.40 (d J = 9.0 Hz, 1H), 8.23 (d, J = 9.0 Hz, 1H), 7.46 (dd, J = 9.0 Hz, J = 4.5 Hz, 1H), 7.41 (dd, J = 9.0 Hz, J = 4.5 Hz, 1H). 13C-NMR (CDCl3, 125 MHz,): δ 153.3, 151.5, 149.7, 148.5, 144.1, 137.1, 136.0, 134.8, 134.3, 133.4, 128.0, 127.5, 123.5, 123.3, 121.8, 120.6, 120.5, 114.2; HRMS (ESI, [M+H]+) m/z calcd C18H11N4O calculated mass 299.0933; actual mass 299.0934. Crystals for X-ray analysis were obtained by dissolving the pure compound in a minimum amount of warm methanol, followed by slow evaporation over the course of 3 d in a 2 mL vial.
Enzymatic generation of pyrido[3,2-f]quinolino[6,5-c]cinnoline 3-oxide (4)
Compound 1 (0.2 mL, of a 50 mM solution in DMF, final concentration 0.8 mM) was mixed with sodium phosphate buffer (0.3 mL, of a 500 mM solution, pH 7.4) and water (12.0 mL) in an argon-purged glove bag and the solution bubbled with argon (20 min). To this mixture was added NADPH (25 mg of the tetrasodium salt, 0.033 mmol, final concentration 2.6 mM) and NADPH:cytochrome P450 reductase (0.03 mL of a 140 U/mL solution, 0.34 U/mL final concentration) and the reaction stirred inside the glove bag for 18 h. The reaction was extracted with ethyl acetate (20 mL), the organic extract washed with brine, and then dried over magnesium sulfate. Column chromatography on silica gel eluted with ethyl acetate and methanol (99:1), followed by preparative TLC eluted with ethyl acetate and methanol (99:1), gave 4 (five reactions were combined to yield 0.3 mg, 2% yield). The 1H-NMR and LC/MS properties of this material matched that of authentic 4 prepared as described above.
Supplementary Material
Acknowledgments
We thank the NIH for partial support of this work (KSG CA 83925 and 119131). We are also grateful to Dr. Fabio Gallazzi, Dr. Nathan Leigh, and Dr. Wei G. Wycoff for experimental assistance and Professor Rainer Glaser for helpful discussions.
Footnotes
Supporting Information Available. LC/MS of 2 and spectral data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Simon MC, Keith B. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown JM. Cancer Res. 1999;59:5863–5870. [PubMed] [Google Scholar]
- 3.Cejudo-Martin P, Johnson RS. Cell. 2005;9:575–576. doi: 10.1016/j.devcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Parmar K, Mauch P, Vergillo J, Sackstein R, Down JD. Proc Nat Acad Sci USA. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris AL. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 6.Shi CQX, Sinusas AJ, Dione DP, Singer MJ, Young LH, Heller EN, Rinker BD, Wackers FJT, Zaret BL. J Nucl Med. 1995;36:1078–1086. [PubMed] [Google Scholar]
- 7.Blikslager AT. Gastroenterology. 2008;134:346–348. doi: 10.1053/j.gastro.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 8.Wilson WR, Hay MP. Nat Rev Cancer. 2011;11:393–409. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 9.Chang Q, Jurisica I, Do T, Hedley DW. Cancer Res. 2011;71:3110–3120. doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 10.Chen EY, Fujinaga M, Giaccia AJ. Teratology. 1999;60:215–225. doi: 10.1002/(SICI)1096-9926(199910)60:4<215::AID-TERA6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Evans BJ, Doi JT, Musker WK. J Org Chem. 1990;55:2337–2344. [Google Scholar]
- 12.Koch CJ. Methods Enzymol. 2002;352:3–31. doi: 10.1016/s0076-6879(02)52003-6. [DOI] [PubMed] [Google Scholar]
- 13.Wardman P, Clarke ED, Hodgkiss RJ, Middleton RW, Parrick J, Stratford MRL. Int J Radiat Oncol Biol Phys. 1984;10:1347–1351. doi: 10.1016/0360-3016(84)90346-8. [DOI] [PubMed] [Google Scholar]
- 14.Stratford MRL, Clarke ED, Hodgkiss RJ, Middleton RW, Wardman P. Int J Radiat Oncol Biol Phys. 1984;10:1353–1356. doi: 10.1016/0360-3016(84)90347-x. [DOI] [PubMed] [Google Scholar]
- 15.Dai M, Zhu W, Xu Y, Qian X, Liu Y, Xiao Y, You Y. J Fluoresc. 2008;18:591–597. doi: 10.1007/s10895-007-0303-0. [DOI] [PubMed] [Google Scholar]
- 16.Zhu W, Dai M, Xu Y, Qian X. Bioorg Med Chem Lett. 2008;16:3255–3260. doi: 10.1016/j.bmc.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Wardman P, Dennis MF, Everett SA, Patel KB, Stratford MRL, Tracy M. Biochem Soc Trans. 1995;61:171–194. doi: 10.1042/bss0610171. [DOI] [PubMed] [Google Scholar]
- 18.Fitzsimmons SA, Workman PA, Grever M, Paull K, Camalier R, Lewis AD. J Natl Cancer Inst. 1996;88:259–269. doi: 10.1093/jnci/88.5.259. [DOI] [PubMed] [Google Scholar]
- 19.Rooseboom M, Commandeur JNM, Vermeulen NPE. Pharm Rev. 2004;56:53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 20.Wilson WR, Anderson RF, Denny WA. J Med Chem. 1989;32:23–30. doi: 10.1021/jm00121a006. [DOI] [PubMed] [Google Scholar]
- 21.Walton MI, Wolf CR, Workman P. Int J Radiat Oncol Biol Phys. 1989;16:983–986. doi: 10.1016/0360-3016(89)90900-0. [DOI] [PubMed] [Google Scholar]
- 22.Wen B, Coe KJ, Rademacher P, Fitch WL, Monshouwer M, Nelson SD. Chem Res Toxicol. 2008;21:2393–2406. doi: 10.1021/tx800281h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Hu L. Med Res Rev. 2009;29:29–64. doi: 10.1002/med.20137. [DOI] [PubMed] [Google Scholar]
- 24.Denny WA, Wilson WR. J Med Chem. 1986;29:879–887. doi: 10.1021/jm00156a001. [DOI] [PubMed] [Google Scholar]
- 25.James AL, Perry JD, Jay C, Monget D, Rasburn JW, Gould FK. Lett Appl Microbiol. 2001;33:403–408. doi: 10.1046/j.1472-765x.2001.01021.x. [DOI] [PubMed] [Google Scholar]
- 26.Molecular-Probes-Website. 2009 http://www.invitrogen.com/site/us/en/home/References/Molecular-Probes-The-Handbook.html.
- 27.Danieli E, Shabat D. Bioorg Med Chem. 2007;15:7318–7324. doi: 10.1016/j.bmc.2007.08.046. [DOI] [PubMed] [Google Scholar]
- 28.Azimi NT, Lytle FE, Huber DM, Whitaker JE, Haugland RP. Applied Spectros. 1990;44:400–403. [Google Scholar]
- 29.Tanaka F, Thayumanavan R, Barbas CF. J Am Chem Soc. 2003;125:8523–8528. doi: 10.1021/ja034069t. [DOI] [PubMed] [Google Scholar]
- 30.Brynes PJ, Bevilacqua P, Green A. Anal Biochem. 1981;116:408–413. doi: 10.1016/0003-2697(81)90381-x. [DOI] [PubMed] [Google Scholar]
- 31.Huang W, Hicks SN, Sondek J, Zhang Q. ACS Chem Biol. 2011;6:223–228. doi: 10.1021/cb100308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith GCM, Tew DG, Wolf CR. Proc Nat Acad Sci USA. 1994;91:8710–8714. doi: 10.1073/pnas.91.18.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solano B, Junnotula V, Marin A, Villar R, Burguete A, Vicente E, Perez-Silanes S, Monge A, Dutta S, Sarkar U, Gates KS. J Med Chem. 2007;50:5485–5492. doi: 10.1021/jm0703993. [DOI] [PubMed] [Google Scholar]
- 34.Junnotula V, Rajapakse A, Abrillaga L, Lopez de Cerain A, Solano B, Villar R, Monge A, Gates KS. Bioorg Med Chem. 2010;18:3125–3132. doi: 10.1016/j.bmc.2010.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Junnotula V, Sarkar U, Sinha S, Gates KS. J Am Chem Soc. 2009;131:1015–1024. doi: 10.1021/ja8049645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chowdhury G, Junnutula V, Daniels JS, Greenberg MM, Gates KS. J Am Chem Soc. 2007;129:12870–12877. doi: 10.1021/ja074432m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chowdhury G, Kotandeniya D, Barnes CL, Gates KS. Chem Res Toxicol. 2004;17:1399–1405. doi: 10.1021/tx049836w. [DOI] [PubMed] [Google Scholar]
- 38.Pizzolatti MG, Yunes RA. J Chem Soc Perkin. 1990;2:759–764. [Google Scholar]
- 39.Agrawal A, Tratnyek PG. Env Sci Technol. 1996;30:153–160. [Google Scholar]
- 40.Fletcher TL, Namkung MJ. J Org Chem. 1958;23:680–683. [Google Scholar]
- 41.Furst A, Moore RE. J Am Chem Soc. 1957;79:5492–5493. [Google Scholar]
- 42.Holt PF, Went CW. J Chem Soc. 1963:4099–4102. [Google Scholar]
- 43.Morrison DJ, Trefz TK, Piers WE, McDonald R, Parvez M. J Org Chem. 2005;70:5309–5312. doi: 10.1021/jo0506231. [DOI] [PubMed] [Google Scholar]
- 44.Knueppel CA. Liebigs Ann. 1899;310:75–88. [Google Scholar]
- 45.Huisgen R. Liebigs Ann. 1948;559:101–152. [Google Scholar]
- 46.Farrar WV. J Chem Soc. 1965:799–800. [Google Scholar]
- 47.Rummens FHA, Bellaart AC. Tetrahedron. 1967;23:2735–2738. [Google Scholar]
- 48.Galbraith HW, Degering EF, Hitch EF. J Am Chem Soc. 1951;73:1323–1324. [Google Scholar]
- 49.Silva RSF, de Amorim MB, Pinto MdCFR, Emery FS, Goulart MOF, Pinto AV. J Braz Chem Soc. 2007;18:759–764. [Google Scholar]
- 50.Rajapakse A, Barnes CL, Gates KS. J Chem Crystallog. 2011 doi: 10.1007/s10870-011-0162-z. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuchs T, Chowdhary G, Barnes CL, Gates KS. J Org Chem. 2001;66:107–114. doi: 10.1021/jo001232j. [DOI] [PubMed] [Google Scholar]
- 52.Fitzsimmons SA, Lewis AD, Riley RJ, Workman P. Carcinogenesis. 1994;15:1503–1510. doi: 10.1093/carcin/15.8.1503. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Kolla HS, Wang X, Raja K, Manohar SK. Adv Mater. 2006;16:1145–1152. [Google Scholar]
- 54.Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L, Lorente G, Banica M, Jung D, Wang J, Ma H, Li X, Yang Z, Hoffman RM, Ammons WS, Hart CP, Matteucci M. J Med Chem. 2008;51:2412–2420. doi: 10.1021/jm701028q. [DOI] [PubMed] [Google Scholar]
- 55.Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel KB, Pullen SM, Hicks KO, Syddall SP, Atwell GJ, Yang S, Denny WA, Wilson WR. Clin Cancer Res. 2007;13:3922–3932. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 56.Mulcahy RT, Gipp JJ, Schmidt JP, Joswig C, Borch RF. J Med Chem. 1994;37:1610–1615. doi: 10.1021/jm00037a011. [DOI] [PubMed] [Google Scholar]
- 57.Kelson AB, McNamara JP, Pandey A, Ryan KJ, Dorie MJ, McAfee PA, Menke DR, Brown JM, Tracy M. Anti-Cancer Drug Design. 1998;13:575–592. [PubMed] [Google Scholar]
- 58.Böge N, Kruger S, Schroder M, Meier C. Synthesis. 2007;24:3907–3914. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.