

Abstract
Using a single-trajectory-based tempering method with a high-temperature dihedral bias, we repeatedly folded four helical proteins [α3D (PDB ID: 2A3D, 73 residues), α3W (1LQ7, 67 residues), Fap1-NRα (2KUB, 81 residues) and S-836 (2JUA, 102 residues)] and some of the mutants in explicit solvent within several microseconds. The lowest root-mean-square deviations of backbone atoms from the experimentally determined structures were 1.9, 1.4, 1.0, and 2.1 Å, respectively. Cluster analyses of folding trajectories showed the native conformation usually occupied the most populated cluster. The simulation protocol can be applied to large-scale simulations of other helical proteins on commonly accessible computing platforms.
Keywords: enhanced sampling, molecular dynamics, packing chirality, protein folding
Molecular dynamics (MD) allows one to study protein folding (1–5) in molecular details. It is, however, computationally demanding if we model a protein accurately using an all-atom representation and simulate it in explicitly solvent. Recent advances in simulation methods and improved computer hardware have made it possible to fold a protein up to about 30 to 80 residues (6–17).
In this study, we report folding simulations of four helical proteins of 67 to 102 amino acid residues, along with their mutants, via a dihedral-biased tempering method. The method couples accelerated dihedral dynamics (10, 18, 19) with a fast motion at a high temperature through a single-trajectory-based tempering method reported previously (12). As the dihedral bias was only added at higher temperatures, we were still able to recover unbiased room temperature properties. This method allowed us to study several interesting nonnative conformations in addition to the native one.
We simulated four proteins in Protein Data Bank (PDB): α3D (PDB ID: 2A3D, 73 residues) (20, 21), α3W (1LQ7, 67 residues) (22, 23), Fap1-NRα (2KUB, 81 residues) (24) and S-836 (2JUA, 102 residues) (25) in a few microseconds. The first three are parallel three-helix bundles, and the last is a parallel four-helix bundle. Among the four, only Fap1-NRα is a natural domain; the rest are artificially designed proteins.
Results
Overview of Folding of Helical Proteins.
Protein sequences, simulation conditions, and parameters for the dihedral bias are listed in Tables S1, S2, and S3, respectively. In Fig. 1, we show the trajectory frames with the lowest backbone root-mean-square deviations (RMSDs) 1.9 (α3D), 1.4 (α3W), 1.0 (Fap1-NRα), and 2.1 Å (S-836) from the respective experimental structures, which will be referred to as “native” structures even for the designed proteins. By comparison, control simulations using regular MD starting from the native conformations yielded higher average RMSDs (3.2, 2.2, 1.5, and 3.2 Å, respectively) at 300 K, suggesting that the native conformations were stably reached.
Fig. 1.

Best folded structures (blue) of four proteins: (A) α3D, (B) α3W, (C) Fap1-NRα, and (D) S-836 compared with the experimentally (NMR) determined structures (red).
Without the experimental structure, cluster analyses (see Methods and SI Text) showed that the native conformation occupied the largest cluster in most trajectories for α3D, α3W, and S-836 (see Table S3). For Fap1-NRα, the native conformation was found in the second largest cluster, possibly due to limited sampling and/or force field inaccuracy (see SI Text for a discussion).
For each protein, we show the time series of RMSD for a typical trajectory in Fig. 2. All simulations started from fully extended conformations (Fig. S1) with large RMSDs. Folding and unfolding events, as indicated by the rises and falls of RMSD, typically occurred on a time scale of 0.5 to 2 µs. We have accumulated around 10 folding events in each of α3D, α3W, and S-836, although longer and/or more simulations may be needed to further improve statistics.
Fig. 2.

The backbone root-mean-square deviation (RMSD) from the experimentally determined structure versus time in a typical simulation trajectory of (A) α3D, (B) α3W, (C) Fap1-NRα, (D) S-836.
Since the simulation method used a variable temperature, only a fraction of trajectory frames were pertinent to the room temperature. We only reported results after reweighted to 300 K using the multiple histogram method (26–28; see SI Text).
Geometry of a Parallel Helix Bundle.
Helical proteins are geometrically interesting, as they present different ways of packing helices. Here we discuss two features in packing helices by using the three proteins α3D, α3W, and S-836 as examples (we omitted Fap1-NRα as it shares the same overall geometry with α3W).
The first feature is the packing “chirality.” If a parallel three-helix bundle protein structure is viewed as three connected rods, after the first two helices are aligned, the third one can be packed against either side of the plane extended by the former two. We call the bundle right-handed if the first helix, when viewed along the middle helix from N to C terminus, can be rotated clockwise to overlap with the third one by an angle less than 180°; otherwise, we call the bundle left-handed. A three-helix bundle of either chirality exists; α3D is right-handed, while α3W and Fap1-NRα are left-handed.
Another packing feature is the helix orientation. Although two successive helices are usually aligned antiparallel, a parallel alignment via a long loop is also possible.
In our trajectories, we found several compact but less stable nonnative conformations, which differed from the native one by the packing chirality or helix orientation. We wished to see what determines the natural geometry.
α3D.
We started with the first three-helix bundle, α3D (20, 21). A typical trajectory is shown in Fig. S2A. The three helices are labeled as A, B, and C from N to C terminus. In Fig. 3A, we show the distributions of the chiral distance D (defined in SI Text, positive for a right-handed bundle, negative a left-handed one) from three independent folding trajectories, as well as a 300 K regular MD trajectory starting from the native structure. The distributions were dominated by a right-handed (native) peak around D = 9 ∼ 10 Å, coinciding with that at D = 9.7 Å from the native simulation. The widths of the peak still differed among the trajectories, however.
Fig. 3.

The packing-chirality D distributions at 300 K of (A) α3D and a mutant (B) α3DL. In each panel, the shaded peak was obtained from a 300 K regular MD simulation of the respective native conformation. For each protein, interaction patterns of charged residues are also shown schematically in top and side views, in which the right and left sides represent the right-handed and left-handed conformations, respectively. For α3D (A), only the right-handed (native) conformation exists; the left-handed conformation is hypothetical. But the opposite is true for the mutant α3DL (B). Inset of A: helix heptad. In the top views and heptad illustration, crosses and dots in circles mean pointing into and out of the paper, respectively. The sequences of the two proteins are compared in the middle of the figure.
The dominance of the right-handed conformation can be explained by a favorable interhelical charge interaction (20). In the protein, residues in all three helices follow the heptad repeat a - b - c - d - e - f - g (see Fig. 3A, Left, Inset), in which the a and d residues are hydrophobic and the e and g ones charged or polar. The charge interactions among the e and g residues of different helices help secure the skeleton.
For α3D, the e and g residues on helix A are all positively charged, while those on helix B are negatively charged; on helix C, the e residues are positively charged but the g ones are negatively charged. In the center diagram of Fig. 3A, we show schematically the distributions of these residues in the two opposite chiral conformations (Right, native; Left, hypothetical) in both top and side views. In the right-handed native conformation (the right panel), the positively charged e residues (Arg57 and Arg64) on helix C contact with the negatively charged g residues (Glu39 and Glu32) on helix B, and the negatively charged g residues (Glu59 and Glu66) on helix C with the positively charged e residues (Lys8 and Lys15) on helix A; both are favorable. By contrast, in the left-handed conformation, the e and g residues on helix C have to contact unfavorably with their counterparts on helices A and B with the same charges.
We further show that the packing chirality can be reversed by a mutation, in which we flipped the charges on the e and g residues of helix C through two local swaps: R57E-E59R and R64E-E66R (R57E means mutating Arg57 to Glu57). We also made three local swaps (A5E-E6A, E54A-A54E, and R71N-N71R) to help stabilize the left-handed conformation. The mutant is referred to as α3DL. A sequence comparison with α3D is shown in the middle of Fig. 3.
We simulated the mutant α3DL in three trajectories (Figs. S2B and S3). As shown in Fig. 3B, the left-handed conformation became dominant with a signature peak at D = -9.7 Å, while the right-handed one almost vanished. Cluster analyses confirmed that the left-handed conformation occupied the largest cluster in 300 K.
We further investigated the local stability of the mutant α3DL in comparison with that of the original α3D. The cluster center from trajectory 1 was extracted and energy minimized as the putative native conformation of α3DL, as illustrated in Fig. S4A. We then ran a separate 300 K regular MD simulation starting from this point for local properties (the local chiral distribution was shown as the shaded peak in Fig. 3B). The average RMSD among all trajectory frames from the native simulation was 2.0 Å, which was comparable to that of α3D, 2.1 Å, suggesting a similar conformational stability. Further, as shown in Table S4, the hydrophobic contact number, interhelical distances, and angles were similar to the native conformation of α3D. Finally, the side chain χ1 (C-Cα-Cβ-Cγ) dihedrals of α3D and α3DL had similar rotameric distributions, as shown in Fig. S5 A and B, as well as similar rotameric entropy of the hydrophobic core, as shown in Table S5. Thus, the mutations might not affect the stability of the hydrophobic core significantly, and the charge interaction should be responsible for the chirality change.
The comparison of α3D and α3DL also suggested that the stability of the native right-handed conformation of α3D was limited by the energy of forming a few favorable pairs of charged residues. Other energy contributions, such as hydrophobic interactions, were unlikely to inhibit the chirality flip.
α3W. We now turn to the second protein α3W (PDB ID: 1LQ7) (22, 23), which was previously simulated in implicit solvent (29–31). However, our trajectories revealed in addition to the native conformation two compact, geometrically different (albeit less populated) conformations.
Like α3D, α3W is a parallel bundle of three helices, again labeled as A, B, and C from N to C terminus. Its strategy to secure the chirality is similar to that of α3D (22): the heptad pattern is also strictly followed in all three helices; the e and g residues on helix B are positively charged; those on helix C are negatively charged; and the e and g residues on helix A are negatively and positively charged, respectively. As shown in Fig. 4A, Upper, Inset, the arrangement now favors the left-handed conformation.
Fig. 4.

The packing-chirality D distributions at 300 K of (A) α3W and (B) a mutant LQLQ with a symmetric sequence. The native and two nonnative conformations are also shown in either case. The colors of the three helices are blue, yellow, and red, sequentially, from N to C terminus. The shade peaks (right vertical scales) in both panels were obtained from 300 K regular MD simulations starting from the representative structures.A, Upper Inset for charge interactions is similar to that in Fig. 3A or B.
The chiral distribution computed from the folding trajectories indeed showed a prominent left-handed (D < 0) peak as well as a suppressed right-handed (D > 0) one, as seen in Fig. 4A. In fact, the right-handed peak covered two different conformations, both of which had corresponding clusters under cluster analyses (300 K, cluster centers also shown in Fig. 4A). We ran respective 300 K MD simulations starting from the two nonnative conformations as well as the native one. The chiral distributions of the three conformations are shown as shaded peaks in Fig. 4A.
The first nonnative conformation (conf. 2) had the opposite chirality to the native one. Its metastability was achieved by sliding two helices along the parallel axis, which somewhat relieved unfavorable charge interactions (see Fig. S6). The conformation had similar interhelical distances and angles as the native one, but a lowered contact number [from 49 (native) to 40] among hydrophobic residues, as shown in Table S4.
The second nonnative conformation (conf. 3) differed from both the native one and conf. 2 by a reversely oriented helix C (see Fig. 4A). It was stabilized through a few interhelical hydrophobic and charged interactions, as shown in Fig. S7A.
We further demonstrate that if all charged residues on the helices were turned to polar or neutral ones, the protein could indeed switch among conformations of different geometries. We simulated a highly symmetric three-helix bundle, named LQLQ, in which all a and d residues were Leus and all b, c, e, f, and g residues were Glns in two independent trajectories. Cluster analyses revealed three clusters, which we refer to as conf. 1, 2, and 3, as shown in Fig. 4B.
Due to the symmetric sequence, conf. 1 and 2 (left- and right-handed, respectively) were similar in all geometric measures except the opposite chirality, as shown in Table S4. They represented the two most populated clusters with similar populations. Conf. 3 (a smaller cluster) was similar to, but more compact than, the conf. 3 in α3W, with the middle helix B being almost parallel, instead of orthogonal, to the other two helices. Note helix B was closer to helix C, not A, although the two choices appeared to be symmetric; we propose a possible explanation in Fig. S7B.
Additionally, we simulated another mutant LALQ in which the b and c residues were Alas instead Glns with similar results (Fig. S8), suggesting some generality of LQLQ in modeling a symmetric three-helix bundle. However if the e, f, and g residues were also replaced Alas, helices in the resulting mutant LALA tended to be bent (see Fig. S9), suggesting that the polar and/or charged residues with long side chains might help maintaining a straight skeleton.
S-836.
The last protein is the four-helix bundle S-836 (PDB ID: 2JUA) (25). The four helices are labeled as A, B, C, and D from N to C terminus. In five trajectories (the total simulation time was 17 μs), the first four reached the native conformation, while the last sampled two different nonnative ones.
To describe the geometry of the protein, we first extended the packing chirality for a three-helix bundle to a four-helix one. We view the four helices as two successive three-helix groups, A-B-C and B-C-D, each of which is thus equivalent to a three-helix bundle and has its own chirality. We partition the conformation space to four quadrants according to the signs of the two chiral distances DABC and DBCD, denoted as LR (DABC < 0, DBCD > 0), RR (DABC > 0, DBCD > 0), RL (DABC > 0, DBCD < 0) and LL (DABC < 0, DBCD < 0). A quadrant can also be represented by either the native (LR) conformation or some hypothetic one that differs from the native only by chirality; e.g., the representative RR conformation differs from the native only by the chirality of the A-B-C helix group.
We computed the joint distribution of DABC and DBCD after combining data from all trajectories. As shown in Fig. 5, the distribution revealed four major clusters. The four corresponding conformations were the native one (LR), conf. 2 (RR) and 3 (RL), and conf. 4, which differed from conf. 3 by a reversely oriented helix A. Although the competing conformations exist, the native one still occupied the most populated cluster in the first four folding trajectories, and the population ratios to the first runner-up clusters were 2.1, 3.8, 1.1, and 1.9, respectively.
Fig. 5.

The logarithm of the packing-chirality distribution 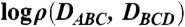 of S-386 at 300 K with representative conformations at the four most populated clusters. The colors of the four helices are blue, green, orange, and red, sequentially, from N to C terminus.
of S-386 at 300 K with representative conformations at the four most populated clusters. The colors of the four helices are blue, green, orange, and red, sequentially, from N to C terminus.
Except conf. 4 (which is not a simple chiral image of the native), the chiral conformations could be sorted as LR (native) > RR and RL (conf. 2 and 3) > LL (missing) by their populations. We attempt to explain the order by the pairing pattern of charged residues, as shown in Fig. S10. The missing LL conformation was possibly due to inescapable repulsion between His83, His86, Lys90, His94 on helix D, and Lys32, His36 on helix B (all His are positively charged). Helices A and D in the RL conformation encountered a similar but less severe problem, causing a kink in helix D to avoid direct repulsion between His86 and Lys17. Unfavorable interactions between helices B and D in the RR conformation could bend or dislocate helix D. The LR (native) conformation appeared to have the best pairing pattern, although unfavorable contacts exist; e.g., Glu91 and Asp11.
In nature, four-helix bundles adopting the RR, RL, and LL conformations do exist. In fact, the majority [e.g., cytochrome ć (PDB ID: 3ZTM) (32) and cytochrome b562 (PDB ID: 1QPU) (33)] adopt the RL conformation, the opposite of the native LR conformation of S-836. Two examples of the RR and LL conformations are the C-terminal domain of KaiA (PDB ID: 1Q6A) (34) and domain I of the transcription factor TFIIS (PDB ID: 1EO0) (35), respectively.
Additional results of the simulated proteins are discussed in the SI Text and Figs. S11–S17.
Conclusions
We have shown the usefulness of the single-trajectory-based tempering with a dihedral bias in studying helical proteins in explicit solvent. We were able to fold, within a few microseconds, four helical proteins (with the native conformation usually found in the largest cluster in simulation trajectories) as well as their mutants.
Our trajectories showed that a parallel helix bundle, especially one with a symmetric sequence, often has competing conformations that differ from the native one only by packing chirality and/or helix orientation. Similarly, the existence of competing conformations was previously reported for the Rop-dimer and its mutants (36–38). While the ability to identify the native geometry is essential in studying helical proteins, the competing conformations can help to design new proteins, either as targets or as pitfalls to avoid (39).
Despite continuous improvement, the current force field might have yet reached perfection. As suggested by Best and Hummer (40), the current AMBER force field still underestimates both the enthalpy of hydrogen bonds (rendering helical conformations less energetically favorable) and the entropy of unfolded states. Our sampling strategy, which used a helical bias and a high-temperature motion, could hopefully alleviate both problems.
Methods
We describe the principles of computational methods below; details are supplied in the SI Text. The source code can be downloaded from http://sigler.bioch.bcm.tmc.edu/MaLab/.
Dihedral-biased Tempering.
Tempering is a technique (12, 41–48) that accelerates sampling by occasionally raising and lowering the temperature. In this study, we adopted a single-trajectory-based tempering method reported previously (12) for computing efficiency. We then added a dihedral bias on top of it to facilitate simulations of helical proteins.
In the tempering method, temperature T is a continuous variable in (TL,TH) covering the room temperature T0. Here, TL is slightly below T0, while TH(TH > T0) accelerates the system motion. Tempering preserves the Boltzmann distribution at any temperature cross-section such that the corresponding generalized ensemble is simply a superposition of canonical ensembles of the temperature range.
We now introduce a bias potential V to the generalized ensemble through a temperature-dependent Hamiltonian; i.e., the energy function, H(T) = H0 + (T/T0 - 1)V, where H0 is the unbiased Hamiltonian. Since V has the maximal magnitude at TH, but is zero at the room temperature T0, the new Hamiltonian is reduced to the original one at T = T0, and unbiased room temperature T0 properties can thus be recovered through a modification of the multiple histogram method (26–28).
We let the bias potential V target the two backbone dihedral angles [i.e., φ (C-N-Cα-C) and ψ(N-Cα-C-N)], as they critically influence protein dynamics (10, 18, 19). It is parameterized in terms of three intrinsic dihedral modes (see Fig. S18 and Table S6), one of which corresponds to the helical conformation. The magnitudes of the modes are adjustable in each trajectory. The bias potential V applies uniformly to all except glycine and proline residues.
The method was coded to GROMACS 4.0.7 (49). We used a modified version (40) of AMBER03 (50, 51) as the force field (the modification slightly reduced helical content), TIP3P (52) as the water model, the particle-meshed Ewald (PME) method (53) for long-range electrostatic interaction, and the velocity-rescaling method for thermostat (54); SETTLE (55) and the parallel LINCS (56) were used as constraint algorithms for waters and proteins, respectively.
The verifications of the methods are shown in SI Text; see also Figs. S19 and S20.
Although the method is designed for helical proteins, extensions to other types of proteins might be achieved via a random trial of various bias potentials, as discussed in the SI Text. To confirm a folding, we can compare several independent trajectories to see if a common structure is reached.
Clustering Method.
We used a deterministic clustering method. The method finds a balance between the intracluster distance (the distance of two protein structures is defined as their backbone RMSD after alignment; it is then averaged over all pairs of frames within the cluster) and the total number of clusters. It also allows each frame to carry the weight from the multiple histogram method (26–28) to filter out frames irrelevant to 300 K.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Fengyun Ni and Dr. Qinghua Wang for helpful discussions. J.M. acknowledges support from National Institutes of Health (Grant R01-GM067801), National Science Foundation (NSF) (Grant MCB-0818353), The Welch Foundation (Grant Q-1512), the Welch Chemistry and Biology Collaborative Grant and the Faculty Initiatives Fund from Rice University. This work was supported in part by the Shared University Grid at Rice funded by NSF under Grant EIA-0216467, a 2010 IBM Shared University Research (SUR) Award on IBM’s Power7 high performance cluster (BlueBioU) to Rice University, the Cyberinfrastructure for Computational Research funded by NSF under Grant CNS-0821727, and the NSF through TeraGrid resources provided by Texas Advanced Computing Center under Grant TG-MCB100013. Use of Visual Molecular Dynamics (57) is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112143109/-/DCSupplemental.
References
- 1.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 2.Daggett V, Fersht A. The present view of the mechanism of protein folding. Nat Rev Mol Cell Biol. 2003;4:497–502. doi: 10.1038/nrm1126. [DOI] [PubMed] [Google Scholar]
- 3.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Scheraga HA, Khalili M, Liwo A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu Rev Phys Chem. 2007;58:57–83. doi: 10.1146/annurev.physchem.58.032806.104614. [DOI] [PubMed] [Google Scholar]
- 5.Dill KA, Ozkan SB, Weikl TR, Chodera JD, Voelz VA. The protein folding problem: When will it be solved? Curr Opin Struct Biol. 2007;17:342–346. doi: 10.1016/j.sbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 7.Seibert MM, Patriksson A, Hess B, van der Spoel D. Reproducible polypeptide folding and structure prediction using molecular dynamics simulations. J Mol Biol. 2005;354:173–183. doi: 10.1016/j.jmb.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 8.Ensign DL, Kasson PM, Pande VS. Heterogeneity even at the speed limit of folding: Large-scale molecular dynamics study of a fast-folding variant of the villin headpiece. J Mol Biol. 2007;374:806–816. doi: 10.1016/j.jmb.2007.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juraszek J, Bolhuis PG. Rate constant and reaction coordinate of Trp-cage folding in explicit water. Biophys J. 2008;95:4246–4257. doi: 10.1529/biophysj.108.136267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kannan S, Zacharias M. Folding simulations of Trp-cage mini protein in explicit solvent using biasing potential replica-exchange molecular dynamics simulations. Proteins. 2009;76:448–460. doi: 10.1002/prot.22359. [DOI] [PubMed] [Google Scholar]
- 11.Freddolino PL, Schulten K. Common structural transitions in explicit-solvent simulations of villin headpiece folding. Biophys J. 2009;97:2338–2347. doi: 10.1016/j.bpj.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C, Ma J. Enhanced sampling and applications in protein folding in explicit solvent. J Chem Phys. 2010;132:244101. doi: 10.1063/1.3435332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 14.Best RB, Mittal J. Balance between alpha and beta structures in ab initio protein folding. J Phys Chem B. 2010;114:8790–8798. doi: 10.1021/jp102575b. [DOI] [PubMed] [Google Scholar]
- 15.Day R, Paschek D, Garcia AE. Microsecond simulations of the folding/unfolding thermodynamics of the Trp-cage miniprotein. Proteins. 2010;78:1889–1899. doi: 10.1002/prot.22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikebe J, Standley DM, Nakamura H, Higo J. Ab initio simulation of a 57-residue protein in explicit solvent reproduces the native conformation in the lowest free-energy cluster. Protein Sci. 2011;20:187–196. doi: 10.1002/pro.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y, Karplus M. Folding of a model three-helix bundle protein: A thermodynamic and kinetic analysis. J Mol Biol. 1999;293:917–951. doi: 10.1006/jmbi.1999.2936. [DOI] [PubMed] [Google Scholar]
- 19.Jang S, Shin S, Pak Y. Replica-exchange method using the generalized effective potential. Phys Rev Lett. 2003;91:058305. doi: 10.1103/PhysRevLett.91.058305. [DOI] [PubMed] [Google Scholar]
- 20.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. From coiled coils to small globular proteins: Design of a native-like three-helix bundle. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh ST, Cheng H, Bryson JW, Roder H, DeGrado WF. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc Natl Acad Sci USA. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson JS, Gibney BR, Skalicky JJ, Wand AJ, Dutton PL. A native-like three-alpha-helix bundle protein from structure-based redesign: A novel maquette scaffold. J Am Chem Soc. 1998;120:3881–3886. [Google Scholar]
- 23.Dai QH, et al. Structure of a de novo designed protein model of radical enzymes. J Am Chem Soc. 2002;124:10952–10953. doi: 10.1021/ja0264201. [DOI] [PubMed] [Google Scholar]
- 24.Ramboarina S, et al. Structural insights into serine-rich fimbriae from Gram-positive bacteria. J Biol Chem. 2010;285:32446–32457. doi: 10.1074/jbc.M110.128165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Go A, Kim S, Baum J, Hecht MH. Structure and dynamics of de novo proteins from a designed superfamily of 4-helix bundles. Protein Sci. 2008;17:821–832. doi: 10.1110/ps.073377908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrenberg AM, Swendsen RH. New Monte-Carlo technique for studying phase-transitions. Phys Rev Lett. 1988;61:2635–2638. doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- 27.Ferrenberg AM, Swendsen RH. Optimized Monte-Carlo data-analysis. Phys Rev Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 28.Chodera JD, Swope WC, Pitera JW, Seok C, Dill KA. Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. J Chem Theory Comput. 2007;3:26–41. doi: 10.1021/ct0502864. [DOI] [PubMed] [Google Scholar]
- 29.Yang JS, Chen WW, Skolnick J, Shakhnovich EI. All-atom ab initio folding of a diverse set of proteins. Structure. 2007;15:53–63. doi: 10.1016/j.str.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 30.Meinke JH, Hansmann UH. Free-energy-driven folding and thermodynamics of the 67-residue protein GS-alpha3W—a large-scale Monte Carlo study. J Comput Chem. 2009;30:1642–1648. doi: 10.1002/jcc.21321. [DOI] [PubMed] [Google Scholar]
- 31.Czaplewski C, Kalinowski S, Liwo A, Scheraga HA. Application of multiplexed replica exchange molecular dynamics to the UNRES force field: Tests with alpha and alpha+beta proteins. J Chem Theory Comput. 2009;5:627–640. doi: 10.1021/ct800397z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonyuk SV, et al. Carbon monoxide poisoning is prevented by the energy costs of conformational changes in gas-binding haemproteins. Proc Natl Acad Sci USA. 2011;108:15780–15785. doi: 10.1073/pnas.1109051108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnesano F, et al. The solution structure of oxidized Escherichia coli cytochrome b562. Biochemistry. 1999;38:8657–8670. doi: 10.1021/bi982785f. [DOI] [PubMed] [Google Scholar]
- 34.Vakonakis I, Langenhan T, Promel S, Russ A, Campbell ID. Solution structure and sugar-binding mechanism of mouse latrophilin-1 RBL: A 7TM receptor-attached lectin-like domain. Structure. 2008;16:944–953. doi: 10.1016/j.str.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Booth V, Koth CM, Edwards AM, Arrowsmith CH. Structure of a conserved domain common to the transcription factors TFIIS, elongin A, and CRSP70. J Biol Chem. 2000;275:31266–31268. doi: 10.1074/jbc.M002595200. [DOI] [PubMed] [Google Scholar]
- 36.Levy Y, Cho SS, Shen T, Onuchic JN, Wolynes PG. Symmetry and frustration in protein energy landscapes: A near degeneracy resolves the Rop dimer-folding mystery. Proc Natl Acad Sci USA. 2005;102:2373–2378. doi: 10.1073/pnas.0409572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schug A, Whitford PC, Levy Y, Onuchic JN. Mutations as trapdoors to two competing native conformations of the Rop-dimer. Proc Natl Acad Sci USA. 2007;104:17674–17679. doi: 10.1073/pnas.0706077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gambin Y, et al. Direct single-molecule observation of a protein living in two opposed native structures. Proc Natl Acad Sci USA. 2009;106:10153–10158. doi: 10.1073/pnas.0904461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baltzer L, Nilsson H, Nilsson J. De novo design of proteins—what are the rules? Chem Rev. 2001;101:3153–3163. doi: 10.1021/cr0000473. [DOI] [PubMed] [Google Scholar]
- 40.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyubartsev AP, Martsinovski AA, Shevkunov SV, Vorontsovvelyaminov PN. New approach to Monte-Carlo calculation of the free-energy—method of expanded ensembles. J Chem Phys. 1992;96:1776–1783. [Google Scholar]
- 42.Marinari E, Parisi G. Simulated tempering—a new Monte-Carlo scheme. Europhys Lett. 1992;19:451–458. [Google Scholar]
- 43.Zhang C, Ma J. Simulation via direct computation of partition functions. Phys Rev E. 2007;76:036708. doi: 10.1103/PhysRevE.76.036708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Fajer M, Yang W. Simulated scaling method for localized enhanced sampling and simultaneous “alchemical” free energy simulations: A general method for molecular mechanical, quantum mechanical, and quantum mechanical/molecular mechanical simulations. J Chem Phys. 2007;126:024106. doi: 10.1063/1.2424700. [DOI] [PubMed] [Google Scholar]
- 45.Geyer CJ. Proceedings of the 23rd Symposium on the Interface; New York: Am Statistical Association; 1991. [Google Scholar]
- 46.Hansmann UHE. Parallel tempering algorithm for conformational studies of biological molecules. Chem Phys Lett. 1997;281:140–150. [Google Scholar]
- 47.Hukushima K, Nemoto K. Exchange Monte Carlo method and application to spin glass simulations. J Physical Soc Japan. 1996;65:1604–1608. [Google Scholar]
- 48.Swendsen RH, Wang JS. Replica Monte-Carlo simulation of spin-glasses. Phys Rev Lett. 1986;57:2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 49.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 50.Duan Y, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 51.Sorin EJ, Pande VS. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys J. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 53.Essmann U, et al. A smooth particle messh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 54.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 55.Miyamoto S, Kollman PA. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J Comput Chem. 1992;13:952–962. [Google Scholar]
- 56.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2007;4:116–142. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 57.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.