

Abstract
Background and Purpose
α-Tocotrienol (TCT) represents the most potent neuroprotective form of natural vitamin E that is Generally Recognized As Safe (GRAS) certified by US FDA. This work addresses a novel molecular mechanism by which α-TCT may be protective against stroke in vivo. Elevation of intracellular GSSG triggers neural cell death. Multidrug resistance-associated protein 1 (MRP1), a key mediator of GSSG efflux from neural cells, may therefore possess neuroprotective functions.
Methods
Stroke-dependent brain tissue damage was studied in MRP1 deficient mice and α-TCT supplemented mice.
Results
Elevated MRP1 expression was observed in glutamate challenged primary cortical neuronal cells and stroke-affected brain tissue. MRP1 deficient mice resulted in larger stroke-induced lesion recognizing a protective role of MRP1. In vitro, protection against glutamate-induced neurotoxicity by α-TCT was attenuated under conditions of MRP1 knockdown suggesting a role of MRP1 in α-TCT-dependent neuroprotection. In vivo studies demonstrated that oral supplementation of α-TCT protected against murine stroke. MRP1 expression was elevated in the stroke affected cortical tissue of α-TCT-supplemented mice. Efforts to elucidate the underlying mechanism identified MRP1 as a target of miR-199a-5p. In α-TCT supplemented mice, miR-199a-5p was downregulated in stroke affected brain tissue.
Conclusions
This work recognizes MRP1 as a protective factor against stroke. Furthermore, findings of this study adds a new dimension to the current understanding of the molecular bases of α-TCT neuroprotection by identifying MRP1 as a α-TCT-sensitive target and by unveiling the general prospect that oral α-TCT may regulate microRNA expression in stroke-affected brain tissue.
Keywords: Antioxidant, vitamin E, microRNA, glutathione
Introduction
Vitamin E is a generic term for tocopherols (TCPs) and tocotrienols (TCTs). Although TCPs have been widely studied for decades, the significance of α-TCT as the most potent neuroprotective forms of natural vitamin E has been uncovered recently. We have reported that nanomolar concentrations of α-TCT, not α-TCP, prevent stroke-associated neurodegeneration1–2. α-TCT protects neural cells by two major mechanisms including inhibition of inducible c-Src and 12-lipoxygenase pathways1,3. Recently, we have reported a key role of intracellular GSSG in causing neural cell death4. Under normal physiological conditions, in most tissues GSSG represents 1% of the total glutathione in the cell. However, under conditions of oxidative stress, reduced glutathione (GSH) is rapidly oxidized to GSSG. Once intracellular GSSG is formed, it may be recycled to GSH in the presence of reducing equivalents which are known to be depleted in the face of oxidant insult. Excessive cellular GSSG is toxic and therefore pumped out at the expense of ATP. We have demonstrated that inefficient clearance of intracellular GSSG may trigger neural cell death. These observations highlight the significance of cellular GSSG clearing systems in neuroprotection especially in the context of stroke where oxidative stress is overt4.
Multidrug resistance-associated protein 1 (MRP1) plays a key role in clearing intracellular GSSG5. MRP1 is an integral membrane glycophosphoprotein abundantly expressed in the brain. We sought to test the hypothesis that under conditions of oxidant insult, improved clearance of intracellular GSSG by augmented MRP1 function results in neuroprotective outcomes. Furthermore, this study tested orally supplemented natural vitamin E α-TCT for its ability to enhance MRP1 function in the context of stroke-induced brain injury.
Materials and Methods
For specific details, see online supplement.
Primary cortical neurons
Cells were isolated from the cerebral cortex of rat feti (Sprague-Dawley, day 17 of gestation; Harlan, Indianapolis, IN) (see online supplement).
siRNA delivery and analysis of genes
DharmaFECTTM1 transfection reagent was used to transfect cells with 100nmol/L siRNA pool (Dharmacon RNA Technologies, Lafayette, CO) (see online supplement).
Delivery of microRNA(miR) mimic and inhibitor
DharmaFECTTM 1 transfection reagent was used to transfect cells with miRIDIAN rno-miR-199a-5p mimic or inhibitor (Dharmacon RNA Technologies) (see online supplement).
pGL3-MRP1-3’fUTR luciferase reporter assay
(see online supplement)
Quantification of miR expression
miR fraction was isolated using miRVanaTM miRNA isolation kit (Ambion, Austin, TX). miR-199a-5p levels were quantified using Taqman Universal Master Mix (Applied Biosystems, Forster City, CA) (see online supplement).
Western blot
Samples (40–50 μg of protein / lane) were separated on a 4–12% SDS-polyacrylamide and probed with anti-MRP1 (1:20 dilution, Enzo Life Sciences, Plymouth Meeting, PA) (see online supplement).
Cell viability assay
(see online supplement)
Immunocytochemistry
(see online supplement)
Calcein clearance assay
After MRP1 siRNA transfection, primary cortical neurons were treated with glutamate. Calcein-AM (5 μmol/L, Invitrogen, Carlsbad, CA) was loaded to the cells for 30min and loss of cellular fluorescence was analyzed (see online supplement).
Mouse stroke model
Transient focal cerebral ischemia was induced in 8 week old MRP1 deficient (n=20, male, Taconic, Hudson, NY) or background FVB mice (n=15, male, Taconic) by middle cerebral artery occlusion (MCAO) (see online supplement).
MRI
T2-weighted imaging was performed on stroke-affected mice. Imaging experiments were carried out using a 11.7T MR system comprised of a vertical bore magnet (Bruker Biospin, Ettlingen, Germany) (see online supplement).
α-Tocotrienol supplementation
C57BL/6 (5-week old, male, Harlan, Indianapolis) mice were randomly divided into two groups, control (n=18) and supplemented (n=23). The control group was orally gavaged with vitamin E stripped corn oil with volume matching the mean volume of α-TCT supplement group. The test group was orally gavaged with α-TCT (Carotech, Malaysia) in vitamin E-stripped corn oil at a dosage of 50 mg/kg body weight for 13 weeks as reported previously1. MCAO was performed at 20 to 24h after the last supplementation. After 48h of MCAO, T2-weighted images were recorded. Mice suffering from surgical complications (e.g. hemorrhage or death) during MCAO were excluded. Immediately after imaging, tissues from control and stroke-affected hemispheres of control (n=9) and α-TCT supplemented (n=10) mice were harvested. Mice were maintained under standard conditions at 22±2°C with 12:12 dark: light cycles. All animal protocols were approved by the Institutional Animal Care and Use Committee of the Ohio State University.
HPLC-electrochemical detection
(see online supplement)
mRNA expression assay from laser-captured microdissected somatosensory cortex of brain sample
Laser microdissection and pressure catapulting (LMPC) was performed on coronal slices of mouse brains using the microlaser system from PALM Microlaser Technologies AG. (see online supplement).
Histology
Coronal slices of cortical sections were stained with rabbit polyclonal antibody to MRP1 (1: 200, Abbiotec, San Diego, CA), 0.0001% Fluoro-JadeR C (Millipore, Billerica, MA) or 4-hydroxynonenal (4-HNE, 1:1000, Enzo Life Sciences, Plymouth Meeting, PA) (see online supplement)
Statistics
Data are reported as mean ± SD. Differences between means were tested using Student’s t-test or one-way ANOVA with Tukey’s test. P<0.05 was considered statistically significant.
Results
Neuroprotection of α-tocotrienol was compromised in MRP1 knockdown in primary cortical neurons
In neural cells, glutamate toxicity is associated with oxidant insult resulting in rapid elevation of intracellular GSSG. We were therefore led to test the hypothesis that under such conditions of GSSG loading, GSSG efflux mechanisms such as MRP1 would be augmented as a defense response in favor of neuronal survival. We observed that extracellular glutamate challenge may induce MRP1 expression in both primary cortical neurons (Figure 1A) as well as HT4 neural cells (Figure S1A).
Figure 1. MRP1 knockdown attenuated the neuroprotection of α-tocotrienol in primary cortical neurons.
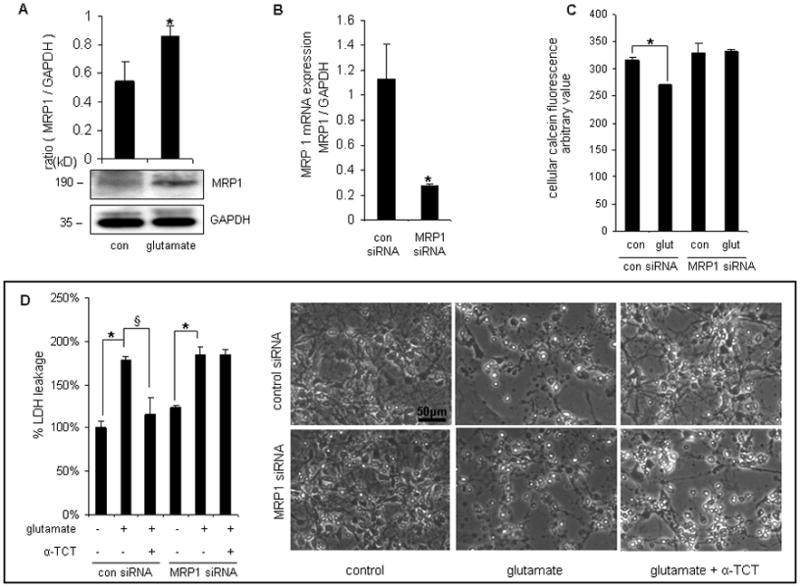
After 24h of seeding, primary cortical neurons were treated with glutamate (5mmol/L, 24h). Glutamate challenge induced the expression of MRP1 protein (A). After MRP1 siRNA transfection, MRP1 mRNA expression was significantly down-regulated (B). C, Glutamate-challenged primary neurons with MRP1 knockdown exhibited loss of functional MRP1 by retaining more calcein compared to corresponding control cells. D, α-TCT (1μmol/L) was added into cell culture medium 6h before glutamate treatment. After glutamate (5mmol/L, 24h) challenge, LDH leakage was measured. Neuroprotection of α-TCT was compromised under conditions of MRP1 knockdown. Bar=50μm, n=3, *P<0.05 compared with control; §P< 0.05 compared with control siRNA-transfected, glutamate-treated primary neurons.
Previously we have reported that α-TCT is potently neuroprotective against a number of insults including GSSG-induced cell death3–4,6. To test whether such neuroprotective property of α-TCT depends on MRP1 in neural cells, cellular GSSG clearance was studied utilizing a MRP1 knockdown approach (Figure 1B & S1 B-D). Higher MRP1 activity resulted in enhanced clearance of calcein from preloaded neurons. Glutamate challenge upregulated MRP1 function. Such improved clearance of calcein in glutamate challenged cells was blunted by MRP1 knockdown demonstrating specificity of the assay for MRP1 (Figure 1C). The ability of α-TCT to protect against glutamate challenge was compromised following MRP1 knockdown in primary cortical neurons (Figure 1D).
MCAO-induced brain injury was exacerbated in MRP1 deficient mice
Transient focal cerebral ischemia was induced by MCAO in MRP1 deficient mice or corresponding background FVB mice. Both FVB and MRP1 deficient mice showed comparably decreased levels of MCA-area blood flow during occlusion (not shown). Interestingly, MRP1 deficient mice showed a larger hemispherical infarct volume than that noted in FVB mice (Figure 2A). Increased abundance of MRP1 protein was observed in the infarct hemisphere of FVB mice (Figure 2B). MCAO-induced brain injury increased tissue GSSG levels in both MRP1 deficient as well as in FVB mice. Notably, MRP1 deficient mice contained 1.6-fold higher tissue levels of GSSG in the infarct hemisphere compared to that detected in background mice (Figure 2C). These data indicate that loss of MRP1 function impairs GSSG clearance during stroke resulting in increased brain injury. Histochemical studies depicted that increased MRP1 positive cells were selectively localized in the infarct hemisphere of FVB mice (Figure 3A). Both MRP1 deficient as well as FVB mice presented positive Fluoro-Jade immunofluorescence staining in the stroke-affected cortex supporting the incidence of neurodegeneration. Importantly, the infarct hemisphere of MRP1 deficient mice exhibited a higher level of neurodegenerative outcome (Figure 3B).
Figure 2. MCAO-induced brain injury was exacerbated in MRP1 deficient mice.
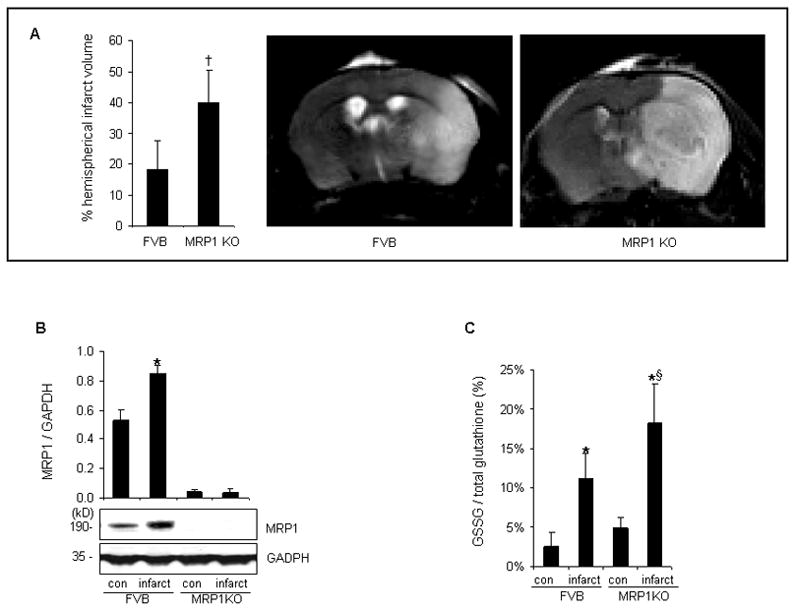
Transient focal cerebral ischemia was induced in MRP1 deficient (n=20) or FVB mice (n=15) by MCAO. A, MRP1 KO mice presented larger stroke-induced lesion compared to FVB mice after 48h reperfusion (FVB, n=8; MRP1 KO, n=12). B, Protein expression of MRP1 was increased in infarct hemisphere of FVB mice (n=3). C, MRP1 deficient mice contained significantly higher level of GSSG in stroke-affected hemisphere compared to that of FVB mice (n=4). *P<0.05 compared with corresponding contralateral hemisphere; †P<0.05 compared with FVB; §P<0.05 compared with infarct hemisphere of FVB.
Figure 3. MCAO-induced neurodegeneration was accentuated in MRP1 deficient mice.
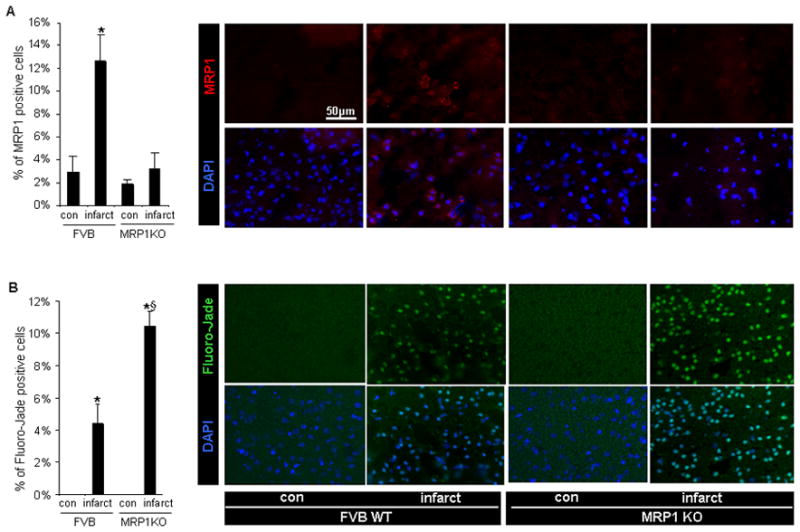
A, MRP1 expression was upregulated at the stroke affected cortex of FVB mice (red-MRP1 protein; blue-DAPI stained nuclei) B, The infarct hemisphere of MRP1 deficient mice presented abundant Fluoro-Jade signals in cortical region (green-Fluoro-Jade positive neurons; blue-DAPI stained nuclei). Bar=50μm, n=3, *P<0.05 compared with corresponding contralateral hemisphere; §P<0.05 compared with infarct hemisphere of FVB.
miR-199a-5p silences MRP1
To test whether MRP1 expression is subject to post-transcriptional gene silencing by microRNA (miR), we first performed in silico studies using databases such as Target Scan (www.targetscan.org), Miranda (www.microrna.org) and PicTar (www.pictar.org). miR-199a-5p emerged as a major candidate miR which is likely to target MRP1 exhibiting a single conserved binding site to the 3’ untranslated region (3’UTR) of MRP1 transcript. Next, we turned towards biological validation of MRP1 as a target of miR-199a-5p. In cultured cells, miR-199a-5p levels were modulated by delivery of miR-199a-5p mimic or miR-199a-5p inhibitor (Figure 4A-B & Figure S2A). miR-199a-5p silenced MRP1 in both primary (Figure 4C-D) as well as HT4 cells (Figure S2B-C). To determine whether MRP1 is a direct target of miR-199a-5p in HT4 neural cells, a fragment of the 3’UTR of MRP1 mRNA containing the putative miR-199a-5p binding sequence cloned into a firefly luciferase reporter construct was used. This construct was co-transfected with a control renilla luciferase reporter construct into cells along with miR-199a-5p mimic or inhibitor. miR-199a-5p mimic significantly decreased luciferase activity while miR-199a-5p inhibitor significantly increased the activity of the miR-199a-5p firefly luciferase reporter recognizing MRP1 as a direct target of miR-199a-5p (Figure S2D).
Figure 4. miR-199a-5p silences MRP1 in primary cortical neurons.
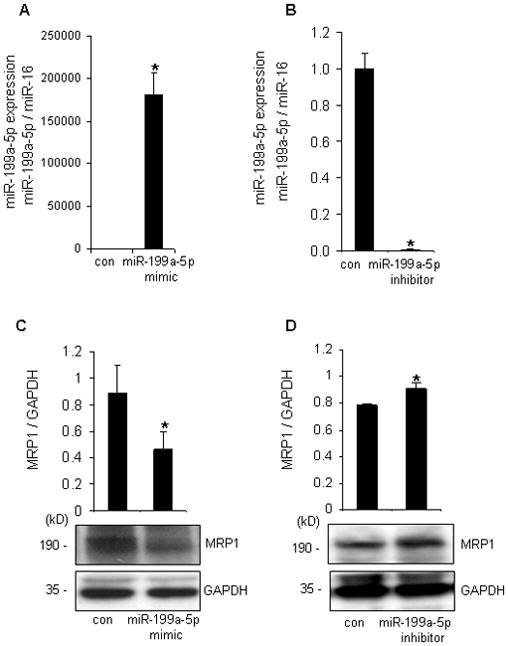
After 72h of miR-199a-5p mimic delivery, miR-199a-5p expression was significantly upregulated (A) while miR-199a-5p hairpin inhibitor significantly downregulated miR-199a-5p (B). miR-199a-5p mimic/inhibitor negatively regulated protein (C-D) of MRP1 in primary cortical neurons. n=3, *P<0.05 compared with control.
Oral α-tocotrienol protected against stroke via MRP1 upregulation
To test the neuroprotective effects of α-TCT against stroke in vivo, randomly divided mice were gavaged with either vitamin E stripped corn oil or α-TCT (50mg/ kg body weight) for 13 weeks. MCAO was performed one day after the last supplementation. MCAO affected brains were harvested 48h after reperfusion (Figure S3A). α-TCT supplementation significantly increased brain α-TCT level (Figure S3B) without changing α-TCP levels (Figure S3C). MCAO limited blood flow in α-TCT supplemented as well as control groups comparably (Figure S3D). However, MCAO-induced hemispherical infarct volume was significantly attenuated in α-TCT supplemented group (Figure 5A).
Figure 5. Orally supplemented α-tocotrienol protected against stroke via MRP1 upregulation.
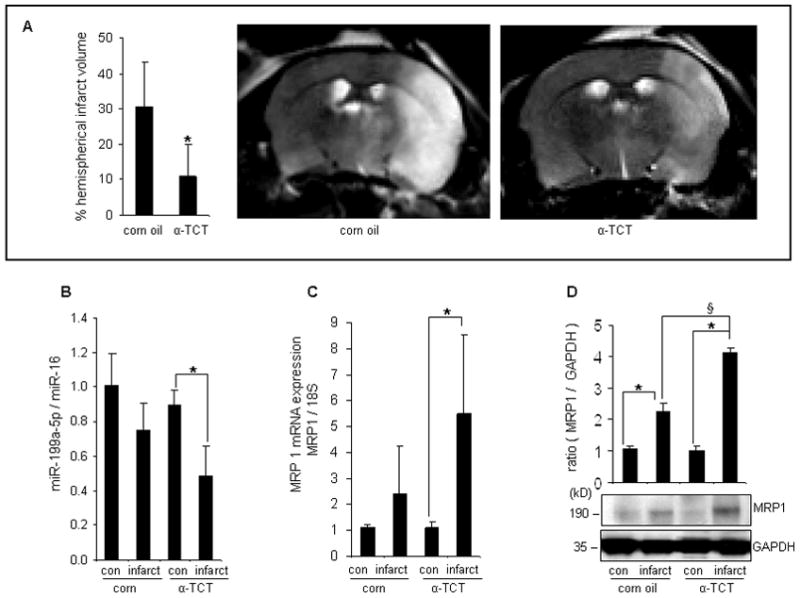
C57BL/6 mice were orally gavaged with vitamin E-stripped corn oil (n=18) or 50mg α-TCT per kg body weight (n=23) for 13 weeks then MCAO was performed. A, α-TCT supplementation significantly reduced MCAO-induced brain injury (corn oil supplemented mice, n=9; α-TCT supplemented mice, n=10). miR-199a-5p expression was significantly down-regulated (B) while MRP1 mRNA (C) and protein (D) levels were significantly elevated in the stroke affected tissue of α-TCT fed mice (n=3). *P<0.05 compared with corresponding control; §P<0.05 compared with infarct hemisphere of corn oil fed mice.
To test whether the neuroprotective properties of α-TCT against stroke in vivo was dependent on the ability of α-TCT to upregulate MRP1, cortical tissue elements were microdissected from infarct as well as contralateral non-infarct sites using a LMPC system (Figure S4). Notably, α-TCT supplementation induced MRP1 but lowered miR199a-5p level in the infarct hemisphere (Figure 5B-D). Immunohistochemical studies demonstrated higher abundance of MRP1 positive cells at the infarct hemisphere of α-TCT-supplemented mice (Figure 6A). α-TCT supplementation attenuated stroke-induced neurodegeneration (Figure 6B). Stroke induced lipid peroxidation as evident by elevated levels of 4-HNE-positive cells in the infarct tissue. Consistent with other observations indicating a beneficial influence of α-TCT supplementation, 4-HNE-positive cells were fewer in the infarct hemisphere of α-TCT supplemented mice (Figure 6C).
Figure 6. Orally supplemented α-tocotrienol attenuated MCAO-induced neurodegeneration and lipid peroxidation.
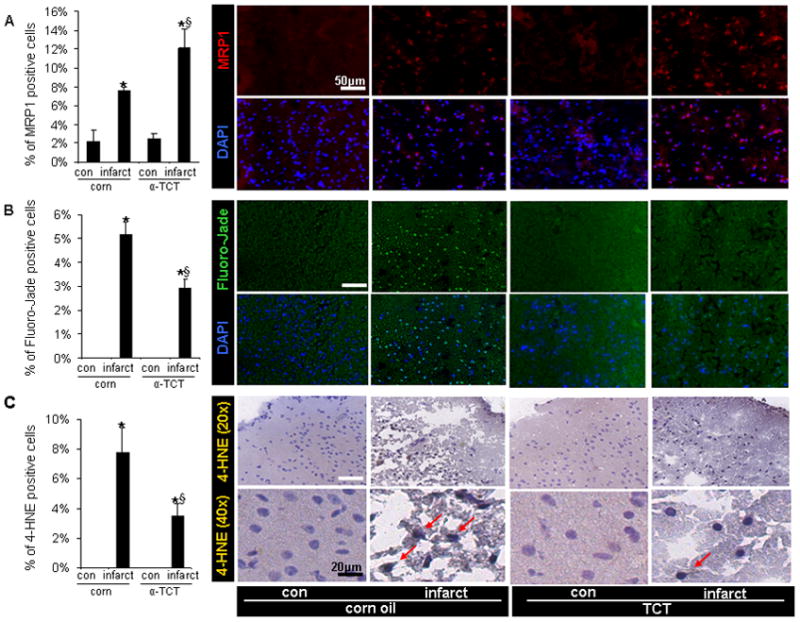
A, The abundance of MRP1 positive cells were significantly upregulated in the stroke affected cortex of α-TCT supplemented mice (red-MRP1 protein; blue-DAPI stained nuclei). B, Fluoro-Jade positive neurons were fewer in the infarct hemisphere of α-TCT fed group compared to those at the infarct site of corn oil fed group (green-Fluoro-Jade; blue-DAPI stained nuclei). C, The abundance of 4-HNE-positive cells was lower in infarct hemisphere of α-TCT supplemented mice. Bar(white)=50μm, Bar(black)=20μm. n=3, *P<0.05 compared with corresponding contralateral hemisphere; §P<0.05 compared with infarct hemisphere of corn oil fed mice.
Discussion
Following a number of failed clinical trials testing the conventional form of vitamin E, α-TCP7–8, interest in naturally occurring forms of vitamin E such as γ-TCP as well as the TCT family is sharply rising. Meta-analyses of clinical trials testing the efficacy of vitamin E in human health suffer from a blind spot because they fail to recognize that α-TCP, the only form of vitamin E tested in such trials, represent only a fraction of the natural vitamin E family7–8. Because TCPs and TCTs have unique functional properties it is important to limit title claims to the specific form of vitamin E studied. Neuroprotection by α-TCT at nanomolar concentration represents the most potent functional property of the entire vitamin E family6. Mechanisms explaining such property include inhibition of inducible c-Src as well as 12-lipoxygenase activity in stroke-related neurodegenerative settings1,9.
Stroke associated ischemic insult is known to compromise ATP production as well as depletes the reducing equivalent pool of the brain. This, in turn, compromises the function of ATP-dependent ion pumps as well as the ability of the brain to manage oxidant insult. As a result, on one hand K+ efflux and Ca2+ influx are increased in response to membrane depolarization. On the other hand, excessive GSSG builds up within the cell. Both elevated intracellular Ca2+ as well as GSSG are directly implicated in cell death signaling10–11. Our previous work highlights the critical significance of elevated and trapped cellular GSSG in executing neural cell death4.
MRP1 represents a major GSSG clearing system in neural cells12. The current work is the first to recognize MRP1 as a potential therapeutic target for stroke. During stroke-related oxidative insult, GSSG is rapidly formed but its redox cycling to GSH is severely arrested. GSSG reductase activity is impaired following stroke13. In addition, ischemia-reperfusion depletes NADPH impairing all reductase functions dependent on this reducing equivalent14. Therefore, a sharp rise in brain tissue GSSG/GSH ratio occurs following stroke4. In the current work we note that such elevation of intracellular GSSG is associated with the induction of MRP1 perhaps as an adaptive survival response. This contention is consistent with previous reports demonstrating upregulation of MRP1 in response to oxidant insult as an adaptive response defending cell survival12. Mechanisms of multidrug resistance and clinical outcomes in response to manipulation of MRP1 expression have been extensively studied in the context of various types of cancers. However, the significance of MRP1 in brain-related pathologies other than cancer is poorly developed. Currently, the limited information available consistently demonstrates that under pathological conditions known to be associated with oxidant insult, i.e Alzheimer’s and epilepsy, MRP1 expression is elevated in the brain15–16. Our observation that stroke related injury to the brain is exacerbated in MRP1 deficient mice establishes a key significance of MRP1 in determining stroke outcomes.
The current study identified MRP1 as a biologically validated target of miR-199a-5p.This constitutes first evidence that MRP1 is subject to post-transcriptional gene silencing by miRs. This finding provided a great segue to address the significance of miRs in stroke17. Antagomir miR-497 treatment has been recognized for its potential to attenuate injury related to acute ischemic stroke18. The miR-200 family and miR-182 has been found to be upregulated in ischemic pre-conditioning19. Although the significance of miR-199a-5p in stroke has not been studied, miR-199a-5p is recognized as a hypoxia-sensitive miR20. Thus, the hypoxia component of stroke-related ischemia may be responsible for downregulating miR-199a-5p following stroke21.
Conclusions
Findings of this study adds a new dimension to the current understanding of the molecular bases of α-TCT neuroprotection by identifying MRP1 as a α-TCT-sensitive target and by unveiling the general prospect that oral α-TCT may regulate microRNA expression in stroke-affected brain tissue. Neuroprotective as well as hypocholesterolemic properties of α-TCT make it a good candidate for nutrition-based intervention in people at high risk for stroke. Transient ischemic attack (TIA), or mini-stroke, serves as a sentinel warning sign for high-risk stroke patients22. Prophylactic stroke therapy therefore provides an opportunity for intervention in TIA patients prior to a major stroke event. Outcomes of the current study warrant clinical assessment of α-TCT in TIA patients. Furthermore, α-TCT is a nutrient that is certified by the US FDA to be Generally Recognized As Safe (GRAS; GRN307) and not a drug with potential side effects. Thus, α-TCT may be considered as a preventive nutritional countermeasure for people at high risk for stroke.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported by NIH grant NS42617.
Footnotes
Disclosures
None.
References
- 1.Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, et al. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005;3610:2258–2264. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem. 2000;27517:13049–13055. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- 3.Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK. Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem. 2006;985:1474–1486. doi: 10.1111/j.1471-4159.2006.04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park HA, Khanna S, Rink C, Gnyawali S, Roy S, Sen CK. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. 2009;168:1167–1179. doi: 10.1038/cdd.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minich T, Riemer J, Schulz JB, Wielinga P, Wijnholds J, Dringen R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J Neurochem. 2006;972:373–384. doi: 10.1111/j.1471-4159.2006.03737.x. [DOI] [PubMed] [Google Scholar]
- 6.Sen CK. Cellular thiols and redox-regulated signal transduction. Curr Top Cell Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- 7.Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;1421:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 8.Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta- analysis of randomised controlled trials. BMJ. 2010;341:c5702. doi: 10.1136/bmj.c5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khanna S, Roy S, Park HA, Sen CK. Regulation of c-Src activity in glutamate-induced neurodegeneration. J Biol Chem. 2007;28232:23482–23490. doi: 10.1074/jbc.M611269200. [DOI] [PubMed] [Google Scholar]
- 10.Schanne FA, Kane AB, Young EE, Farber JL. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;2064419:700–702. doi: 10.1126/science.386513. [DOI] [PubMed] [Google Scholar]
- 11.Leslie SW, Brown LM, Trent RD, Lee YH, Morris JL, Jones TW, et al. Stimulation of N-methyl-D-aspartate receptor-mediated calcium entry into dissociated neurons by reduced and oxidized glutathione. Mol Pharmacol. 1992;412:308–314. [PubMed] [Google Scholar]
- 12.Hirrlinger J, Konig J, Keppler D, Lindenau J, Schulz JB, Dringen R. The multidrug resistance protein MRP1 mediates the release of glutathione disulfide from rat astrocytes during oxidative stress. J Neurochem. 2001;762:627–636. doi: 10.1046/j.1471-4159.2001.00101.x. [DOI] [PubMed] [Google Scholar]
- 13.Adibhatla RM, Hatcher JF, Dempsey RJ. Effects of citicoline on phospholipid and glutathione levels in transient cerebral ischemia. Stroke. 2001;3210:2376–2381. doi: 10.1161/hs1001.096010. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Kim KY, Jang HS, Yoshida T, Tsuchiya K, Nitta K, et al. Role of cytosolic NADP+-dependent isocitrate dehydrogenase in ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2009;2963:F622–633. doi: 10.1152/ajprenal.90566.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. 2002;125(Pt 1):22–31. doi: 10.1093/brain/awf002. [DOI] [PubMed] [Google Scholar]
- 16.Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer's disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res. 2004;2912:2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- 17.Rink C, Khanna S. MicroRNA in Ischemic Stroke Etiology and Pathology. Physiol Genomics. 2010 doi: 10.1152/physiolgenomics.00158.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, et al. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 2010;381:17–26. doi: 10.1016/j.nbd.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee ST, Chu K, Jung KH, Yoon HJ, Jeon D, Kang KM, et al. MicroRNAs induced during ischemic preconditioning. Stroke. 2010;418:1646–1651. doi: 10.1161/STROKEAHA.110.579649. [DOI] [PubMed] [Google Scholar]
- 20.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;1047:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khanna S, Roy S, Maurer M, Ratan RR, Sen CK. Oxygen-sensitive reset of hypoxia-inducible factor transactivation response: prolyl hydroxylases tune the biological normoxic set point. Free Radic Biol Med. 2006;4012:2147–2154. doi: 10.1016/j.freeradbiomed.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;1217:948–954. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.