

Abstract
Exchange proteins directly activated by cAMP (Epac) are a family of guanine nucleotide exchange factors that regulate a wide variety of intracellular processes in response to second messenger cAMP. To explore the structural determinants for Epac antagonist properties of high throughput screening (HTS) hit ESI-08, pyrimidine 1, a series of 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues have been synthesized and evaluated for their activities for Epac inhibition. Structure-activity relationship (SAR) analysis led to the identification of three more potent Epac antagonists (6b, 6g and 6h). These inhibitors may serve as valuable pharmacological probes for further elucidation of the physiological functions and mechanisms of Epac regulation. Our SAR results and molecular docking studies have also revealed that further optimization of the moieties at the C-6 position of pyrimidine scaffold may allow us to discover more potent Epac-specific antagonists.
Keywords: Pyrimidine analogues, Exchange Proteins Directly Activated by cAMP (Epac), Antagonists, Structure-activity relationship (SAR)
The cyclic adenosine monophosphate (cAMP) signaling pathway, widely viewed as the “prototypic” second messenger signaling pathway, regulates a myriad of biological processes, including cardiac and smooth contraction, insulin secretion, neurotransmitter release, learning and memory, inflammation, cell growth, cell differentiation and cell apoptosis.1–5 The effects of cAMP in mammalian cells are attributed to the activation of two intracellular cAMP targets: the classic cAMP-dependent protein kinase (PKA) and the more recently discovered exchange protein directly activated by cAMP (Epac, a.k.a. cAMP-GEF).6–8 By far the best-studied aspect of cAMP signaling involves cAMP-mediated activation of PKA. Since the discovery of the Epac proteins in 1998,9 significant progress has been made toward elucidating the molecular mechanism of Epac activation. These new findings have altered the prevailing viewpoints about cAMP signaling that previously had been ascribed mainly to PKA, and the contribution of Epac is now more and more appreciated.10,11 Epac proteins are guanine nucleotide exchange factors for Ras-like small GTPases, Rap1 and Rap2. There are two isoforms of Epac, Epac1 and Epac2, which have been implicated in a number of cellular processes.12–15 Epac proteins exert their diverse biological functions alone, in concert with PKA, and/or even in opposed to PKA.16 Therefore, it remains a great challenge to understand the complexity of Epac activation/inhibition within the cAMP signaling pathway and to provide insights into the molecular mechanisms of Epac in the regulation of multiple effectors and biological functions.
To distinguish the signaling pathway activated by Epac proteins, membrane-permeable cAMP analogues with high affinity and specificity for Epac proteins are needed. The 8-pCPT-2'-O-Me-cAMP (a.k.a. 007), a selective agonist for Epac proteins, is very useful in unraveling the role of Epac in a wide variety of biological processes, ranging from exocytosis of insulin to the regulation of calcium channels and permeability of the vascular endothelium.17–21 While 007 has become a widely used tool in Epac-related research, its application is greatly limited by its low membrane permeability because of the negatively charged phosphate group. To mitigate this issue, an AM (acetoxymethyl)-ester (007-AM) has been introduced to mask the negatively charged group.22 The ester appendage of 007-AM can be intracellularly removed by esterases to generate 007. Nevertheless, 007 can also bind and inhibit phosphodiesterase-1, -2, and -6, and thus indirectly increase intracellular cAMP or/and 3'-5'-cyclic guanosine monophosphate (cGMP) levels, and eventually activate PKA, PKG and/or cyclic nucleotide gated channels.23 Meanwhile, accumulating evidence demonstrated that Epac proteins may represent new mechanism-based therapeutic targets for the treatment of various human disorders such as diabetes, heart disease and cancer.24–28 Therefore, development of Epac antagonists as useful pharmacological probes to dissect the physiological functions that Epac proteins play in the overall cAMP-mediated signaling remains a major challenge in the research field.29
In an attempt to identify novel molecular antagonists that are capable of specifically inhibiting Epac, an automated, fluorescence-based high throughput screening (HTS) assay has been developed by our team using a fluorescent cyclic nucleotide analog, 8-NBD-cAMP (8-(2-[7-Nitro-4-benzofurazanyl]aminoethylthio)adenosine-3', 5'-cyclic monophosphate). This competitive assay which is performed in a “mix and measure” format, is based upon a dramatic dose-dependent change in fluorescence signal of the binding of 8-NBD-cAMP to Epac2.30 A Maybridge Hitfinder compound library of 14,400 chemically diversified small molecules has been screened. ESI-08 (1) has been identified as an Epac antagonist (Figure 1) which is capable of completely inhibiting both Epac1 and Epac2 activity at 25 μM in the presence of equal molar concentration of cAMP (unpublished data). Herein, we report the synthesis and the exploration of the structure-activity relationship (SAR) of the 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues as potent antagonists targeting Epac. Molecular modeling studies of these novel Epac inhibitors were also performed using AutoDock Vina docking approach.31
Figure 1.

The structures of cAMP, Epac agonists: 2'-O-Me-cAMP analogs 007 and 007-AM, Epac antagonist: HTS hit 1 (ESI-08).
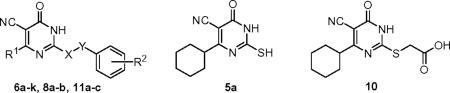
Using the HTS hit ESI-08 as a starting point, structural modifications of three major sites on the 5-cyano-6-oxo-1,6-dihydro-pyrimidine scaffold have been explored. The synthesis of various pyrimidine analogues is outlined in Scheme 1. The reaction of methyl cyanoacetate with the corresponding aldehydes 2a–f and thiourea in the presence of piperidine refluxing in EtOH afforded the pyrimidine thiones (or thiouracils) 5a–f.32 Selective S-alkylation of 5a–f with the appropriate benzyl halide in the presence of K2CO3 in acetone gave the desired products 6a–j.33 Removal of the Boc group from 6j with trifluoroacetic acid yielded the desired compound 6k. Condensation of 2a with methyl cyanoacetate 3 and appropriate phenylguanidines 7a–b provided analogues 8a–b. The S-alkylation reaction of 5a with bromoacetic acid 9 afforded the intermediate 10. Compound 10 was then condensed with the appropriate anilines in the presence of EDCI and DIPEA to form 11a–c. The structures and purity of all synthesized compounds were confirmed by 1H and 13C NMR, HR-MS and HPLC analysis.34
Scheme 1.

aReagents and conditions: (a) piperidine, EtOH, reflux; 5a R1 = cyclohexyl, 51%; 5b R1 = isopropyl, 55%; 5c R1 = cyclopropyl, 18%; 5d R1 = cyclopentyl, 62%; 5e R1 = 1-methyl-piperidin-4-yl, 69%; 5f R1 = 1-Boc-piperidin-4-yl, 59%; 8a R3 = H, 48%; 8b R3 = 4-Cl, 49%; (b) various benzyl halides, K2CO3, acetone, rt; 6a 93%, 6b 87%; 6c 91%; 6d 94%; 6e 95%; 6f 75%; 6g 80%; 6h 92%; 6i 65%; 6j 77%; (c) TFA, DCM, 0 °C, 92%; (d) EDCI, DIPEA, various anilines, DCM, rt; 11a 91%; 11b 80%; 11c 82%.
Synthesized 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues 5a, 6a–k, 8a–b, 10 and 11a–c were screened for their relative binding affinity of Epac2 by using dose-dependent titrations to compete the binding of 8-NBD-cAMP to Epac2.30 Apparent IC50 values of these analogues are summarized in Table 1. As illustrated in Table 1, the pharmacophore moieties at the 2-position of the pyrimidine scaffold have demonstrated very crucial to the activity, and an appropriate hydrophobic group such as S-benzyl moiety is required to achieve the activity of these analogues to target Epac. For example, compound 5a with a mercapto group at the 2-position that is not alkylated with a hydrophobic group displayed no activity (IC50 > 300 μM). Given that the hydrophobic group is important for the potency and the hit (1) itself has a 2,5-dimethyl substituent on the phenyl ring, analogues 6a–6e with substituents of proton, methyl, dimethyl, or trimethyl groups at various positions of the aromatic ring were initially explored and compared. Among compounds 6a–6e, 6c and 6e demonstrated a moderate potency against Epac2 with IC50 values of 15.7 μM and 11.6 μM, respectively. The 3,5-dimethyl analogue 6b, with an IC50 value of 7.0 μM, showed a slightly better activity than the hit ESI-08 (1), a corresponding 2,5-dimethyl analogue. Compounds 6a and 6d with the R2 of either 4-methyl or H displayed a much lower potency.
Table 1.
Apparent IC50 values of the 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues for competing with 8-NBD-cAMP in binding Epac2.
| Entry | R1 | X | Y | R2 | IC50 (μM) | Relative Potency (RA)* |
|---|---|---|---|---|---|---|
| cAMP | 48 | 1 | ||||
| 1 | cyclohexyl | S | CH2 | 2,5-dimethyl | 8.4 | 5.7 |
| 5a | >300 | <0.16 | ||||
| 6a | cyclohexyl | S | CH2 | 4-methyl | 38.7 | 1.2 |
| 6b | cyclohexyl | S | CH2 | 3,5-dimethyl | 7.0 | 6.9 |
| 6c | cyclohexyl | S | CH2 | 2,4-dimethyl | 15.7 | 3.1 |
| 6d | cyclohexyl | S | CH2 | H | 30.5 | 1.6 |
| 6e | cyclohexyl | S | CH2 | 2,4,6-trimethyl | 11.6 | 4.1 |
| 6f | isopropyl | S | CH2 | 2,5-dimethyl | 12.7 | 3.8 |
| 6g | cyclopropyl | S | CH2 | 2,5-dimethyl | 4.0 | 12 |
| 6h | cyclopentyl | S | CH2 | 2,5-dimethyl | 5.9 | 8.1 |
| 6i | 1-methyl-piperidin-4-yl | S | CH2 | 2,5-dimethyl | 127 | 0.4 |
| 6k | piperidin-4-yl | S | CH2 | 2,5-dimethyl | >300 | <0.16 |
| 8a | cyclohexyl | NH | - | H | >300 | <0.16 |
| 8b | cyclohexyl | NH | - | 4-Cl | >300 | <0.16 |
| 10 | >300 | <0.16 | ||||
| 11a | cyclohexyl | S | CH2C(O)NH | H | >300 | <0.16 |
| 11b | cyclohexyl | S | CH2C(O)NH | 2,4,6-trimethyl | >300 | <0.16 |
| 11c | cyclohexyl | S | CH2C(O)NH | 4-methyl | >300 | <0.16 |
RA = IC50, cAMP/IC50, compound
The effects of various alkyl substituents at the C-6 position of the pyrimidine ring have also been investigated. As shown in Table 1, compound 6g with the 6-cyclohexyl replaced by 6-cyclopropyl moiety displayed enhanced activity with an IC50 value of 4.0 μM when compared with the hit ESI-08 (1). 6-Cyclopentyl derivative 6h showed a similar potency with an IC50 value of 5.9 μM (Figure 2). In contrast, 6f and 6i with an isopropyl or a N-methyl piperidyl were found significantly less active, with IC50 values of 12.7 and 127 μM, respectively. Interestingly, compound 6k with 6-(piperidin-4-yl) group, which was initially designed with an attempt to improve the aqueous solubility, showed no activity (IC50 > 300 μM), indicating that a basic moiety at this position seems inappropriate. Taken together, these results suggest that a moderate hydrophobic group at the C-6 position of the pyrimidine scaffold is important to achieve the potent inhibition activity. In addition, the replacement of the S-benzyl group at the 2-position of the pyrimidine scaffold with either phenylamino (8a) or 4-chloro-phenylamino (8b) led to a dramatic loss of the inhibitory activity (IC50 > 300 μM). This is also true for analogues 10 and 11a–c, in which S-benzyl moiety was replaced with either 2-sulfanyl acetic acid or 2-sulfanyl acetamides, indicating that 2-benzylsulfanyl group is a crucial hydrophobic pharmacophore for this type of molecules to achieve the activity targeting Epac2.
Figure 2. Relative potency of Epac2 antagonists.

Dose-dependent competition of Epac2 antagonists with 8-NBD-cAMP in binding to Epac2: closed circles, 6g (HJC0198); closed triangles up, 6h (HJC0197); closed diamonds, 1 (ESI-08); open triangles down, 6a (HJC0167); closed squares, cAMP.
The in vitro SAR results allowed us to select compounds exhibiting a high Epac2 inhibitory activity for further investigation. Compounds 6g (HJC0198) and 6h (HJC0197) have therefore been selected for further evaluation in suppressing cAMP-mediated Epac1 and Epac2 GEF activities to determine their specificity using purified recombinant full-length Epac1 and Epac2 proteins (Figure 3).30, 35, 36 These two analogues were shown to be able to inhibit Epac2 GEF activity to basal levels at 25 μM concentration in the presence of equal concentration of cAMP. Compound 6h was found to also inhibit Epac1-mediated Rap1-GDP exchange activity at 25 μM in the presence of equal concentration of cAMP, while compound 6g is more Epac2-specific (Figure 3). From these results it appears that smaller alkyl substituent on the C-6 position of the pyrimidine ring may be more beneficial for the specificity of Epac2. These findings suggest that further optimization on the moieties at the C-6 position of pyrimidine scaffold may provide a great potential to develop new and more Epac2-specific inhibitors.
Figure 3. Specificity of Epac antagonists 6g and 6h.

(A) cAMP-mediated Epac1 GEF activity measured in the presence or absence of Epac antagonists: open squares, Epac1 alone; closed squares: Epac1 in the presence of 25 μM cAMP; open circles, Epac1 with 25 μM cAMP and 25 μM 6g; closed circles, Epac1 with 25 μM cAMP and 25 ȝM 6h. (B) cAMP-mediated Epac2 GEF activity measured in the presence or absence of Epac antagonists: open squares, Epac2 alone; closed squares: Epac2 in the presence of 25 μM cAMP; open circles, Epac2 with 25 μM cAMP and 25 μM 6g; closed circles, Epac2 with 25 μM cAMP and 25 μM 6h. Similar results were obtained from two independent experiments.
To further characterize the relative potency of these Epac antagonists, we have performed counter-screening assays that measure type I and II PKA holoenzyme activities, respectively.30 As shown in Figure 4, compounds ESI-08, 6g (HJC0198) and 6h (HJC0197) at 25 μM have been found not to alter cAMP-induced type I and II PKA holoenzymes activation while H89, a selective PKA inhibitor, blocked the type I or II PKA activities completely. These results suggest that compounds ESI-08, 6g (HJC0198) and 6h (HJC0197) are Epac-specific inhibitors that selectively block cAMP-induced Epac activation, but do not inhibit cAMP-mediated PKA activation.
Figure 4. Effects of Epac antagonists ESI-08, 6g (HJC0198) and 6h (HJC0197) on type I and II PKA activities.

Relative Type I (filled bars) and II (open bars) PKA holoenzyme activities in the presence of 100 μM cAMP plus vehicle control, 25 μM H89 or 25 μM ESI-08 or 25 μM 6g (HJC0198) or 25 μM 6h (HJC0197). Data are presented in the format of means and standard deviations (n = 3).
Epac proteins are also known to activate the Akt/PKB signaling pathways.16 To determine if our newly synthesized Epac specific inhibitors are capable of blocking Epac1- or Epac2-mediated Akt activation, the phosphorylation statuses of T308 and S473 of Akt in HEK293 cells ectopically expressing Epac1 or Epac2 were monitored using anti-phospho-Akt antibodies. As shown in Figure 5, pretreatment of HEK293/Epac1 and HEK293/Epac2 cells with 10 μM of HJC0197 (6h) and HJC0198 (6g) for 5 min before the administration of 007-am, a membrane permeable Epac selective agonist, completely blocked Epac1 and Epac2-mediated Akt phosphorylation. These results demonstrate that in addition to inhibiting Epac1 and Epac2 biochemically, HJC0197 (6h) and HJC0198 (6g) can also suppress Epac1 and Epac2 function in cells.
Figure 5. Effects of Epac antagonists on Epac-mediated Akt/PKB phosphorylation in HEK293/Epac cells.

HEK293/Epac1 and HEK293/Epac2 cells with or without pretreatment of 10 μM Epac antagonists (HJC0197 and HJC0198, respectively) were stimulated with 10 μM 007-AM. Cell lysates were subjected to Western blot analyses using anti-phospho-Ser473-specific (PKB-P473) and anti-phospho-Thr308-specific (PKB-P308) PKB antibodies.
Molecular docking studies were performed to investigate the conformation and the required spatial relationship between the pyrimidine scaffold and Epac2 protein.37 Since our robust HTS assay is particularly sensitive for searching compounds that directly compete with 8-NBD-cAMP in binding to Epac2, we predicted that our compounds may bind to the cAMP binding domain (CBD) of Epac2. AutoDock Vina docking data indeed revealed that our compounds could fit nicely into the functional cAMP binding packet of Epac2.38 To further characterize the binding pose, we selected analogue 6h as a case study for our theoretical investigation. As depicted in Figures 6A & B, the molecular docking results showed that the position and size of substituted groups played a key role in the binding of the compound with Epac2. The hydrophobic S-benzyl moiety interacts with the hydrophobic residues of Val447, Phe403, Lys405 and Leu406. Meanwhile, the cyclopentyl moiety at the C-6 position of the pyrimidine ring was also found to form hydrophobic interactions with Phe367, Ala407, Ala412, Pro413, Ala415, Ala416 and Ile418. These results are in full agreement with our established SAR data that hydrophobic groups at the C-6 position and 2-position of the pyrimidine scaffold are crucial to target Epac. These docking findings may also help us understand why analogues with the moderate hydrophobic substituent at the C-6 position have better potency while analogue 6k with 6-(piperidin-4-yl) group displays no activity. Moreover, this binding mode is further stabilized by the occurrence of one hydrogen bond between the oxygen of carbonyl group in the pyrimidine ring with the residue Lys450. Taken together, these studies suggest that optimization of the moieties at the C-6 position and 5-cyano of the pyrimidine scaffold may provide further opportunities to improve the binding affinity and specificity of the pyrimidine derivatives.
Figure 6. Predicted binding mode and molecular docking of compound 6h into the cAMP binding domain (CBD) of Epac2 protein.

(A) Nitrogen, oxygen, sulfur and phosphorus are shown in blue, red, orange and pale red, respectively. 6h is shown in big sticks and in green color. cAMP is shown in small sticks and in yellow color. Protein residues likely to be involved in polar and hydrophobic interactions are shown in sticks. Hydrogen bonds are indicated by dashed lines. (B) 6h is shown in yellow color while cAMP is shown in pink color.
In summary, a series of 5-cyano-6-oxo-1,6-dihydro-pyrimidine derivatives have been synthesized and biologically evaluated as a novel class of Epac antagonists. We have identified important structural features of the small molecule inhibitors targeting Epac. The SAR results and docking studies indicate that further modification at the C-6 position of the pyrimidine scaffold can tune the original hit 1 to achieve a more specific Epac2 antagonist. The pharmacological probes identified such as 6g (HJC0198) and 6h (HJC0197) will facilitate our efforts in elucidating the physiological functions of Epac and mechanisms of cAMP-mediated cell signaling. These probes will also have potential for the development of novel therapeutics against human diseases associated with dysfunction of specific cAMP-signaling components. Further studies of a systematic lead optimization on the C-6 position and 5-cyano of the pyrimidine scaffold by appropriate modifications of length, size, and chemical nature of the substituents are undergoing and will be reported in due course.
Acknowledgements
This work was supported by grants P30DA028821 (JZ), R01GM066170 and R21NS066510 (XC) from the National Institute of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, and John Sealy Memorial Endowment Fund. We thank Drs. Lawrence C. Sowers, Carol L. Nilsson, Jacob A. Theruvathu and Huiling Liu for mass spectrometry assistance and Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Beavo JA, Brunton LL. Nat. Rev. Mol. Cell Biol. 2002;3:710. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 2.Houslay MD, Kolch W. Mol. Pharmacol. 2000;58:659. [PubMed] [Google Scholar]
- 3.Zaccolo M, Movsesian MA. Circ. Res. 2007;100:1569. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 4.Bourne HR, Lichtenstein LM, Melmon KL, Henney CS, Weinstein Y, Shearer GM. Science. 1974;184:19. doi: 10.1126/science.184.4132.19. [DOI] [PubMed] [Google Scholar]
- 5.Follin-Arbelet V, Hofgaard PO, Hauglin H, Naderi S, Sundan A, Blomhoff R, Bogen B, Blomhoff HK. BMC Cancer. 2011;11:301. doi: 10.1186/1471-2407-11-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith FD, Scott JD. Eur. J. Cell Biol. 2006;85:585. doi: 10.1016/j.ejcb.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X, Ji Z, Tsalkova T, Mei FC. Acta. Biochim. Biophys. Sin. (Shanghai) 2008;40:651. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. Science. 1998;282:2275. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 9.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Nature. 1998;396:474. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 10.Gloerich M, Bos JL. Annu. Rev. Pharmacol. Toxicol. 2010;50:355. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 11.Grandoch M, Roscioni SS, Schmidt M. Br. J. Pharmacol. 2010;159:265. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc'h F. Cell. Signal. 2011;23:1257. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Sehrawat S, Cullere X, Patel S, Italiano J, Mayadas TN., Jr Mol. Biol. Cell. 2008;19:1261. doi: 10.1091/mbc.E06-10-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honegger KJ, Capuano P, Winter C, Bacic D, Stange G, Wagner CA, Biber J, Murer H, Hernando N. Proc. Natl. Acad. Sci. U.S.A. 2006;103:803. doi: 10.1073/pnas.0503562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zieba BJ, Artamonov MV, Jin L, Momotani K, Ho R, Franke AS, Neppl RL, Stevenson AS, Khromov AS, Chrzanowska-Wodnicka M, Somlyo AV. J. Biol. Chem. 2011;286:16681. doi: 10.1074/jbc.M110.205062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. J. Biol. Chem. 2002;277:11497. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- 17.Holz GG, Chepurny OG, Schwede F. Cell. Signal. 2008;20:10. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL. J. Cell Biol. 2003;160:487. doi: 10.1083/jcb.200209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. Nat. Cell Biol. 2002;4:901. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 20.Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. J. Biol. Chem. 2003;278:8279. doi: 10.1074/jbc.M211682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FE. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H1188. doi: 10.1152/ajpheart.00937.2007. [DOI] [PubMed] [Google Scholar]
- 22.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. ChemBioChem. 2008;9:2052. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 23.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Nat. Methods. 2008;5:277. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 24.Holz GG. Diabetes. 2004;53:5. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. Science. 2009;325:607. doi: 10.1126/science.1172256. [DOI] [PubMed] [Google Scholar]
- 26.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F. Circ. Res. 2005;97:1296. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 27.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc'h F. Circ. Res. 2008;102:959. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 28.Lissitzky JC, Parriaux D, Ristorcelli E, Verine A, Lombardo D, Verrando P. Cancer Res. 2009;69:802. doi: 10.1158/0008-5472.CAN-08-2391. [DOI] [PubMed] [Google Scholar]
- 29.(a) Rehmann H, Schwede F, Døskeland SO, Wittinghofer A, Bos JL. J. Biol. Chem. 2003;278:38548. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]; (b) Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Døskeland SO. J. Biol. Chem. 2006;281:21500. doi: 10.1074/jbc.M603116200. [DOI] [PubMed] [Google Scholar]; (c) Das R, Chowdhury S, Mazhab-Jafari MT, Sildas S, Selvaratnam R, Melacini G. J. Biol. Chem. 2009;284:23682. doi: 10.1074/jbc.M109.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6133. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsalkova T, Mei FC, Cheng X. PLos One. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trott O, Olson AJ. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdou IM, Strekowski L. Tetrahedron. 2000;56:8631. [Google Scholar]
- 33.Ram VJ, Goel A, Nath M, Srivastava P. Bioorg. Med. Chem. Lett. 1994;4:2653. [Google Scholar]
- 34.Spectra data of the representative compounds: 4-Cyclopropyl-2-(2,5-dimethylbenzylsulfanyl)-6-oxo-1,6-dihydro-pyrimidine-5-carbonitrile (6g): White solid, mp 235–236 °C. 1H NMR (600 MHz, CDCl3/CD3OD 1:2) δ 6.89 (s, 1H), 6.86 (d, 1H, J = 7.8 Hz), 6.80 (d, 1H, J = 7.8 Hz), 4.15 (s, 2H), 2.12–2.08 (m, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 1.14–1.12 (m, 2H), 1.05–1.02 (m, 2H). 13C NMR (150 MHz, CDCl3/CD3OD 1:3) δ 176.4, 165.6, 160.3, 135.6, 133.5, 132.0, 130.3, 130.3, 128.8, 114.7, 94.1, 33.3, 20.3, 18.3, 16.6, 11.4 (2C); ESI-MS (m/z): 645 (2M + Na)+; HRMS-ESI Calcd for C17H18N3OS: 312.1171 (M + H)+; found: 312.1158. HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent; tR = 13.15 min; purity (by peak area) 97.3%. 4-Cyclopentyl-2-(2,5-dimethyl-benzylsulfanyl)-6-oxo-1,6-dihydro-pyrimidine-5-carbonitrile (6h): White solid, mp 217–219 °C. 1H NMR (600 MHz, CDCl3) δ 13.00 (bs, 1H), 7.15 (s, 1H), 7.08 (d, 1H, J = 7.8 Hz), 7.30 (d, 1H, J = 7.2 Hz), 4.47 (s, 2H), 3.48–3.44 (m, 1H), 2.34 (s, 3H), 2.25 (s, 3H), 2.08–2.05 (m, 2H), 1.92–1.87 (m, 4H), 1.76–1.74 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 180.0, 165.9, 162.6, 136.0, 133.9, 132.4, 130.8, 130.8, 129.3, 114.2, 95.4, 46.0, 33.8, 32.7 (2C), 26.8 (2C), 20.9, 19.0; ESI-MS (m/z): 362 (M + Na)+; HRMS-ESI Calcd for C19H22N3OS: 340.1484 (M + H)+; found: 340.1472. HPLC analysis conditions are same as described above; tR = 13.75 min; purity (by peak area) 98.7%.
- 35.Tsalkova T, Blumenthal DK, Mei FC, White MA, Cheng X. J. Biol. Chem. 2009;284:23644. doi: 10.1074/jbc.M109.024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D, Woods VL, Jr., Cheng X. J. Biol. Chem. 2011;286:17889. doi: 10.1074/jbc.M111.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Nature. 2008;455:124. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 38.Molecular docking protocol: The pyrimidine analogues were docked with Epac2 using the X-ray structure of Epac2 (PDB code: 3CF6) and AutoDock Vina 1.1.2. Water molecules and the ligand (Sp-cAMPS) within the crystal structure were removed and polar hydrogens were added using AutoDockTools. In the structures of the analogues, all bonds were treated as rotatable except for the aromatic, amide, cyano and double bonds, whereas the protein was treated as rigid. Docking runs were carried out using the standard parameters of the program for interactive growing and subsequent scoring, except for the parameters for setting grid box dimensions and center. For all of the docking studies, a grid box size of 40 Å × 60 Å × 40 Å, centered at coordinates 11.0 (x), −20.0 (y), and −70.0 (z) of the PDB structure. The binding affinities of the 6h output structures ranged from −9.6 to −8.4 kcal/mol.