

Abstract
Ageing is a complex process that in muscle is usually associated with a decrease in mass, strength, and velocity of contraction. One of the most striking effects of ageing on muscle is known as sarcopenia. This inevitable biological process is characterized by a general decline in the physiological and biochemical functions of the major systems. At the cellular level, aging is caused by a progressive decline in mitochondrial function that results in the accumulation of reactive oxygen species (ROS) generated by the addition of a single electron to the oxygen molecule. The aging process is characterized by an imbalance between an increase in the production of reactive oxygen species in the organism and the antioxidant defences as a whole. The goal of this review is to examine the results of existing studies on oxidative stress in aging human skeletal muscles, taking into account different physiological factors (sex, fibre composition, muscle type, and function).
1. Human Aging Muscle: An Overview
Aging represents an inevitable and complex biological process that is characterized by a general time-dependent decline in the physiological and biochemical functions of the major systems [1]. Several changes can be observed during aging, which include a reduced capacity to use oxygen along with impaired cardiocirculatory capacity and respiratory adaptation, deterioration of nervous system (decrease in the form, width, and rate of conduction of evoked potential), and degeneration in muscle mass characterized by a reduction in muscle fiber diameters and by a qualitative and quantitative alteration in muscle fibres [2].
Also at the cellular level, morphological and biochemical changes are involved in this process. Skeletal muscle can be considered the largest organ in the body [3] and the age-associated loss of skeletal muscle mass and strength (i.e., sarcopenia) seems an unavoidable part of the aging process. After about the age of 50 years, there is a progressive decrease of muscle mass at the rate of 1-2% per year. Similarly but with different decline rate and timing, muscle strength also decreases by about 3% yearly after 60 years of age [4] while the cross-sectional area of skeletal muscle is reduced by 25–30% after age 70 [4–6]. Sarcopenia is, therefore, a multidimensional phenomenon of aging (someone indicates it as a syndrome) and represents a powerful risk factor for the development of negative health-related events in the elderly. In fact, the relationships of sarcopenia with impaired physical performance, frailty, loss of functional independence, and increased risk of falls are all well established in literature [7]. Moreover, decreased muscle strength is also highly predictive of incident disability, and all-cause mortality in older persons [8]. An important aspect regards the different functional decline associated with sarcopenia, which is more evident in men than in women [9]. Moreover, the extent of sarcopenia, and thus age-related atrophy, are higher in glycolytic muscles compared to oxidative muscles [10, 11]; Type I fibers are slow contracting, mainly oxidative, while type II fibres are fast contracting, mainly glycolytic with a lower number of mitochondria. In humans, the structural changes of responsible for age-related atrophy and decline in muscle strength are correlated to the progressive impairment of the cross-sectional fibre area [12] and to fibre denervation and fibre number loss, with type II fibres being the most affected by aging [13, 14]. The remaining type II fibres seems to maintain their efficiency probably by adjusting their capability to produce energy, as suggested by the absence of age-related changes in the enzymatic activities of the anaerobic machinery for energy production [15–17].
One of the most important endogenous causes of sarcopenia is likely correlated to the loss of a motor neuron input to the muscle [18]. This decline of muscle innervation may be one of the key events in the sarcopenic process since innervation is crucial to the maintenance of muscle mass, as well as strength. In the elderly, there is a decrease in the number of functional motor units associated with a concomitant enlargement of the cross-sectional area of the remaining units [19]. Together with neurological factors, a decline in anabolic hormones may also play a key role in the sarcopenic process. This reduction of anabolic hormones, namely, growth hormone (GH) and sexual steroid hormones, could be implicated in the aetiopathogenesys of the sarcopenic process. Many studies have demonstrated that GH levels begin to decline in the fourth decade and progressively continue to decline over ensuing years. Interestingly, it seems that sex hormones are an important factor in maintaining muscle mass and strength in men but not in women [20–23]. Regarding the multifactorial aetiology in recent years many assumptions were made about the causes of Sarcopenia that can be very schematically summarized as follows [24].
Mitochondrial deletion: a failure of replication of mitochondrial DNA (mtDNA) may be the cause of a significant deletion in the mitochondrial genome; the shorter genome is replicated more quickly by inducing the formation of malfunctioning or completely inactive mitochondria.
Alteration of protein synthesis.
Loss of the ability of reparative satellites cells (SC): the proliferation and fusion of the SC is regulated by specific growth factors protein (mainly IGF-1, mIGF-1, HGF) but is also influenced by hormones such as GH, testosterone, and estrogen. Moreover, satellite cells are also activated by mechanical stretch, and muscle or sarcolemmal damage. One of the factors that could play a key role in triggering sarcopenia is the accumulation of reactive oxygen species (ROS) that have produced throughout one's lifetime [2]. ROS, which are generated by the addition of a single electron to the oxygen molecule, are formed in all tissues including muscle fibers and, especially, in the mitochondrial respiratory chain.
2. Mitochondrial ROS Production, mtDNA Mainteinance, and Skeletal Muscle Aging
The generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is a normal process in the life of aerobic organism; ROS and RNS, in fact, are continually produced as a consequence of aerobic metabolism: up to 5% of oxygen reacting with the respiratory chain is incompletely reduced to ROS [15, 25, 26]. This production contributes to mitochondrial damage in a range of pathologies and is also important in redox signalling from the organelles to the rest of the cell. A schematic overview of the mitochondrial ROS production is shown in the Figure 1: ROS production by mitochondria can lead to oxidative damage to mitochondrial proteins, membranes, and DNA, impairing the ability of mitochondria to synthesize ATP and to carry out their wide range of metabolic functions, including the tricarboxylic acid cycle, fatty acid oxidation, the urea cycle, amino acid metabolism, haem synthesis, and FeS centre assembly that are central to the normal operation of most cells. Mitochondrial oxidative damage can also increase the tendency of mitochondria to release intermembrane space proteins such as cytochrome c (cyt c) to the cytosol by mitochondrial outer membrane permeabilization (MOMP) and consequently activate the cell's apoptotic machinery. It is well established that apoptosis is elevated during aging of cells and tissues and significantly contributes to cell loss and the pathogenesis of several age-related diseases [27]. In fact, mitochondrial ROS production also leads to induction of the mitochondrial permeability transition pore (PTP), which renders the inner membrane permeable to small molecules in situations such as ischaemia/reperfusion injury. Consequently, it is unsurprising that mitochondrial oxidative damage contributes to a wide range of pathologies. Moreover, mitochondrial ROS may act as a moduletable redox signal, reversibly affecting the activity of a range of functions in the mitochondria, cytosol, and nucleus [28].
Figure 1.
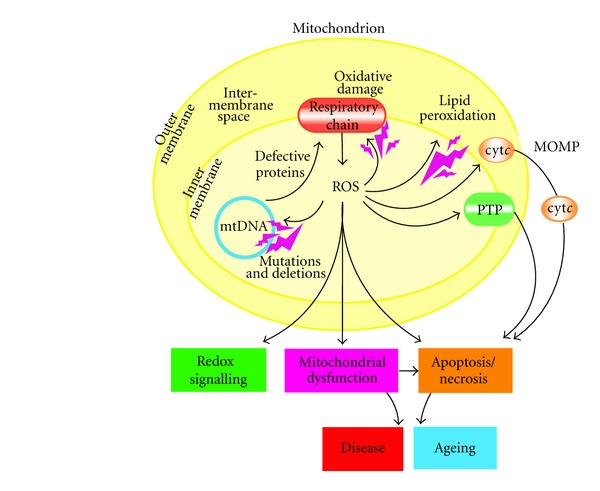
mitochondria ROS production by [28].
ROS include oxygen free radical, the superoxide radical anion (O2 ∙−, primary product of one-electron dioxygen reduction, precursor of most other reactive oxygen species and involved in the propagation of oxidative chain reactions, even though it is not a strong oxidant), the extremely aggressive hydroxyl radical (deriving from subsequent chemical reaction), and the strong nonradical oxidants, singlet oxygen and hydrogen peroxide [15]:
| (1) |
ROS-mediated damage is the result of an increase in electron flux and the corresponding leakage from the mitochondrial respiratory chain [15].
In muscle fibres, ROS, in particular the superoxide anion, can be produced in several cellular sites, including plasma membrane, mitochondria, sarcoplasmic reticulum (SR), T tubules, sarcolemma and cytosol, and they are generally released in the cytosol of the cell muscle [29].
The skeletal muscle is the largest consumer of oxygen in the body, and in the literature, there are several debates about the relationship between the aging in this tissue and the changes of the cellular antioxidant defence mechanism with a progressive ROS increase [30]. Oxidative damage has been proposed as one of the major contributors of the skeletal muscle decline occurring with aging [31, 32]. In general, this lack of results homogeneity in literature could be related to the variety of mechanisms that can lead to free radical production and that can be modulated by aging, sex, fibre composition, muscle type, and the physiological state of the animals [15]. Summarizing the findings of the last years, if oxidative stress plays an important role in the aging of skeletal muscle, in particular of the satellite cells, that are usually recruited to replace damaged fibers and promote their regeneration, the susceptibility against oxidative stress seems to be less prominent in this kind of cells with respect to other cell systems, since oxidative stress is counteracted by an increase in the antioxidant defence system [20]. Cellular antioxidant defence, in fact, has been shown to play an interesting role in advanced age: in the liver, brain, heart, kidney, and rat skeletal muscle aging seems to be associated with reduced capacity of enzymatic and nonenzymatic antioxidant system to convert ROS into more inert species [33, 34]. The redox imbalance is this disparity between ROS generation and the counter-acting antioxidant forces, and it constitutes the major trigger of the imbalance between protein synthesis and degradation that in turn leads to muscle atrophy [35, 36]. How such an oxidative insult plays a role in the age-related decrease of muscle performance and mass has yet to be defined. In aged muscle, there is an increase in the presence of the products of oxidative damage to lipids, proteins, and DNA [37] together with a marked increase in the number and variety of mitochondrial DNA rearrangements [38]. On the other hand, a causal relationship between oxidative macromolecules modifications, mitochondrial DNA mutations, mitochondrial dysfunction and aging is now clearly demonstrable [39]. The increased ROS production in mitochondria, hence, can subsequently lead to a vicious cycle of exponentially increasing levels of mtDNA damage.
The lack of protective histones and close proximity to the ETC, whose complexes I and III are believed to be the predominant sites of ROS production inside the cell, make mtDNA extremely vulnerable to oxidative stress [38]. Although various genetic problems in mitochondria cause phenotypes that resemble premature aging [40], additional support for this theory was provided by studies showing a direct link between mtDNA mutations and mammalian aging. In particular, mice with a proofreading-deficient version of PolgA, the catalytic subunit of mitochondrial DNA polymerase γ (POLG), accumulate mtDNA mutations that are associated with impaired respiratory-chain function and increased levels of apoptosis. These mtDNA-mutator mice, with accelerated levels of mutations, had a shorter life span and displayed age-related phenotypes such as hair loss, kyphosis (curvature of the spine), osteoporosis, and sarcopenia at an early age [40]. Interestingly, these changes were not accompanied by increased levels of oxidative stress, a finding that has also been confirmed in humans [41]. This has resulted in much controversy regarding the idea that mtDNA mutations contribute to aging through increased ROS production and enhanced levels of oxidative stress in mitochondria [42]. However, it is possible that the accumulation of mtDNA mutations that occur with age leads to alterations in cell-signaling pathways that can induce cell dysfunction and initiate apoptosis, irrespective of increased ROS production and oxidative stress in mitochondria. Previous studies have provided strong, though correlative, experimental support for an association between mtDNA mutations and tissue dysfunction, particularly in long-lived postmitotic cells such as cardiomyocytes, skeletal muscle fibers, and neurons. Moreover, mtDNA mutations have been shown to accumulate with aging in several tissues, including skeletal muscle of various species. Therefore, it has been established that many of these mtDNA mutations start to occur after the mid-thirties and they accumulate with age in postmitotic tissues of the human body [43–45]. Beside this, it was observed that the extent of mtDNA mutation strongly correlates with the progressive decrease of cytochrome c oxidase activity in aging human muscle [46]. Actually, whether mtDNA mutations play a causal role in the aging process is still an ongoing debate; however, the fact that a functional decline in mitochondria occurs with age and that properly functioning mitochondria are crucial for longevity and minimizing age-related diseases cannot be refuted [42]. Another important aspect to be considered is the motility of mitochondria which continuously undergo fusion and fission events that actively alter their morphology [43]. Therefore, genetic defects in the proteins involved in mitochondrial fusion and fission lead to severely altered mitochondrial shape, loss of mtDNA integrity, increased oxidative stress, and apoptotic cell death; it has been shown that these alterations can subsequently cause developmental abnormality, neuromuscular degeneration, and metabolic disorders in humans [44]. On the other hand there are only few in-depth study confirming this relationship between the mitochondrial fusion and fission mechanism and aging, mostly because the molecular events that underlie the aging process have not yet been completely elucidated. But an important example that supports this concept is provided by recent studies of mitochondria in muscle [42]. The subcellular localization of mitochondria and the overall mitochondrial network in multinucleated muscle fibers are tightly controlled by mitochondrion-shaping proteins, and an imbalance in mitochondrial fusion and fission dynamics probably impairs their function and contributes to the age-related loss of muscle. Indeed, it has recently been shown that the expression of two genes codifying for proteins involved in mitochondrial fusion (Mfn1 and Mfn2) is reduced in the skeletal muscle of aging individuals [45]. Furthermore, the role of mitochondrion-shaping proteins has recently been investigated in mice with a muscle-specific deficiency in genes codifying for two key protein (Fis1 and Drp1) of the regulation of mitochondrial fission in mammalian cells: diminished mitochondrial fission was associated with reduced muscle atrophy and attenuated activation of atrophy-related genes during fasting [47]. Interestingly, the level of PGC-1α, a transcriptional coactivator that enhances the activity of specific transcription factors and that consequently stimulates the expression of Mfn2, is decreased in various models of muscle atrophy [48–50], whereas overexpression of PGC-1α protects skeletal muscle from atrophy, mainly by inhibiting the induction of genes that are crucial for the atrophy process [51, 52]. Many other studies indicate that alterations in mitochondrial dynamics are a key component of the mitochondrial adaptations that occur in response to mitochondrial biogenesis. These mitochondrial adaptations seem to be driven by regulatory pathways that can be activated by several physiological stimuli, including exercise and calorie restriction. However, these studies are not sufficient to reveal the functional relationship between mitochondrial dynamics and biogenesis, or to explain how fusion and fission events contribute to mitochondrial turnover [42]. Nevertheless, as the results of several studies on mammalian cells showed, there is a relationship between mitochondrial dynamics and genomic stability [50, 51]. Furthermore, following confirmation of the role of some genes like Mfn1 and Mfn2 in mtDNA maintenance, loss of mitochondrial fusion in skeletal muscle resulted in increased mtDNA point mutations and deletions as well as severe mtDNA depletion, which preceded the phenotypic changes observed in mice mutated for the genes above mentioned (Mfn1 and Mfn2) [53–55]. These studies support the hypothesis that mitochondrial fusion through intramitochondrial exchange increases the tolerance of a cell to mutant mtDNA and protects the integrity of the mitochondrial genome [42].
In general, several lines of evidence indirectly implicate mtDNA in longevity. The Framingham Longevity Study of Coronary Heart Disease has indicated that longevity is more strongly associated with age of maternal death than that of paternal death, suggesting that mtDNA inheritance might be involved [56]. On the other hand, longevity was shown to be associated with certain mtDNA polymorphisms, which also may play a role in aging process [57]. In the opinion of Alexeyef, the mitochondrial theory of aging can be considered as an extension and refinement of the free radical theory. Its major premise is that mtDNA mutations accumulate progressively during life and are directly responsible for a measurable deficiency in cellular oxidative phosphorylation activity, leading to an enhanced ROS production. In turn, increased ROS production results in an increased rate of mtDNA damage and mutagenesis, thus causing a “vicious cycle” of exponentially increasing oxidative damage and dysfunction, which ultimately culminates in death [58].
Regarding the genetic modifications induced by ROS and various other oxidants, it was also found that radicals induce MnSOD mRNA levels to a moderate extent in several cell types. This increase in SOD level is expected to result in increased generation of hydrogen peroxide which can in turn affect the expression of many redox-sensitive genes (such as MMP-1 and MMP-2 codifying for extracellular matrix-degrading metalloproteinases [59]) and consequently a displaced equilibrium in the steady-state levels of ROS. This process is associated with massive macrophage apoptosis and contributes thereby to the formation of the atherosclerotic lesions. The process may be further enhanced by cytokines and other factors such as TNF, interleukin-1β, angiotensin II, and interferon-γ [60–63].
3. Biochemical Alterations and Oxidative Damage Induced by ROS
Therefore, currently, it is generally accepted that free radicals or ROS play a primary role in the aging process, especially in those tissues in which the generation of free radicals is more pronounced, such as skeletal muscle. This is a consequence of the high level of oxygen consumption, seen in skeletal muscle compared to other tissues, which results in higher concentrations of ROS [24], which are not only involved in muscle damage but are also able to modulate the contractile function of skeletal muscle [39]. Studies of reduced preparations have identified a number of regulatory sites that exhibit altered function when exposed to exogenous ROS donors ex vivo. There are insights have correlated oxidative damage to the mechanisms underlying EC (excitation-contraction) coupling. Since Ca2+ release channels are one of the major targets of oxidative damage [64, 65], the unbalance of Ca2+ transport that is present in the sarcopenic muscle could be due to the modified oxidative status of those structures involved in Ca2+ release and uptake [66]. Several studies have shown the presence of an enzyme, NAD(P)H-oxidase, that is associated with skeletal muscle SR (sarcoplasmic reticulum) [67]. In fact, the superoxide anion generated by this enzyme can stimulate the proper release of calcium from SR through oxidation of the Ryanodine receptor [67]. Ryanodine receptor 1 (RyR1) is the calcium release channel of the skeletal muscle SR required for muscle contraction. Related experiments showed as RyR1 from aged (24-month-old) rodents resulted oxidized and cysteine-nitrosylated compared to RyR1 from younger (3–6-month-old) adults. RyR1 channel activity is dramatically altered by redox modifications of critical thiols group contained in the protein (oxidation, S-nitrosylation, or alkylation); an oxidation of approximately 20% of these free thiols is enough to produce a significant effect on the channel activity that in some case can lead to an irreversible inactivation of the channel. These data indicate that leaky RyR1 contributes to age-related loss of muscle function [68, 69].
The involvement of other enzymes, such as Phospholipase A2 (PLA2) and xanthine oxidase, has been suggested. The activation of PLA2 may stimulate NAD(P)H oxidase, while it has been shown that increases in PLA2 activity stimulate ROS production in mitochondria and in cytosol, and subsequent release in extracellular space [70]. PLA2 enzyme is present in cells as two isoforms: calcium-dependent PLA2 (cPLA2) and non-calcium-dependent PLA2 (iPLA2). It has been suggested that iPLA2 is mostly responsible for ROS production at rest, while isoform cPLA2 seems to be activated during all processes that increase intracellular Ca-concentration, such as muscle contraction [70]. Xanthine dehydrogenase (XDH) is a cytoplasmic enzyme that is converted to Xanthine oxidase, which in turn is able to produce superoxide anion and, accordingly, hydrogen peroxide. Skeletal muscle generates superoxide and nitric oxide at rest, and this generation is increased by contractile activity. Proteins, such as superoxide dismutases (SODs), catalase (CAT), and heat shock proteins (HSPs), prevent oxidative damage to tissues.
The mechanisms underlying these adaptive responses to contraction include activation of redox-sensitive transcription factors, such as nuclear factor-kappaB (NF-κB), activator protein-1 (AP-1), and heat shock factor 1 (HSF1) [1]. Moreover the literature reports that postcontraction attenuation of the pathways involved in mitochondrial biogenesis in fast muscle fibers (type II) is far more severe in old rats than in those of young rats. ROS are known to play an important role in the activation of signalling cascades [71]. These latter suggest that ROS affect mitochondrial biogenesis via upregulation of transcriptional regulators, such as peroxisome proliferator-activated receptor-gamma coactivator-1 protein-alpha (PGC-1alpha) and that excessively high ROS generation subsequent to contractions may be responsible for diminished mitochondrial biogenesis in the muscles of old rats. The inability of skeletal muscle from old rats to activate redox-sensitive transcription factors such as NF-κB or AP-1 in response to stress is characterized by chronic activation of transcription factors at rest and by an inability to further activate these factors subsequent to muscle contraction. This lack of activation of redox-sensitive transcription factors with contractile activity is associated with an inability to increase the expression of various cytoprotective proteins and appears to influence the susceptibility of skeletal muscle from old animals to oxidative damage [72, 73].
3.1. Lipid Damage
One of the principal indicators of free radical damage during aging is lipid peroxide (LPO). Polyunsaturated fatty acids (PUFAs) of the biomembranes are attacked by free radicals in presence of the oxygen; malondialdeyde (MDA) and 4-hydroxy-2,3-trans-nonenal (4-HNE) are the main products of PUFA peroxidation because they bear numerous double bonds such as linoleic acid and arachidonic acid. The main effect on lipid peroxidation of biological membranes is the overall decrease in their fluidity, which makes it extremely easy for the two monolayers to exchange phospholipids, increases the permeability of the membrane to substances that are not normally able to cross other than by specific channels, and ultimately results in severe damage to membrane proteins. These latter tend to aggregate and to form high-molecular-weight complex [74].
In human skeletal muscle, LPO levels are significantly higher in aged than in young subjects [75]. There are works that have demonstrated that there is an age-dependent increase in MDA content in skeletal muscle [76, 77], and also data obtained from our research group in these years agree with those reported by previous authors. Furthermore, LPO levels in young women are significantly lower (by about 50%) than in young men [36, 75]. Possibly, the difference in lipid peroxidation between young and aged women could be caused by the reduction of estrogens' protective role after menopause. In any case, the level reached in elderly women is lower compared to men [31, 75]; in particular, MDA values in the VL (vastus lateralis) muscle in aged females (over 70) were comparable to those obtained from young males (under 40) [77].
In human skeletal muscle, the level of peroxidation also depends on muscle fiber composition and on muscle function. Fiber composition indicates that muscle with more than 40% type II fiber content shows statistically lower LPO levels than does muscle with less than 40% type II fiber content [75]. This suggests that increased lipid peroxidation prevails in type I fibers (which are mainly oxidative), where most ROS production probably takes place by mitochondrial alteration [1]. An increase in the LPO level during aging is evident in the VL [75, 77, 78] and in the external intercostal [75], but not in the RA (rectus abdominis) muscle [77]. Fiber I distribution is very similar in RA and VL, which suggests that functional differences in these muscles could be the cause of major lipid peroxidation. There are two possible explanations for the observation that oxidative lipid levels in RA differ from those in VL: the greater contractile capacity of VL muscle compared with RA muscle and/or the higher levels of peroxidable substrates present in VL compared with RA. On the base of these results, it seems that lipid peroxidation level are influenced by a variety of different factors: sex, fiber composition, and muscle specificity [1].
4. Protein Carbonylation
A great number of studies have shown an increase in oxidized proteins at the intracellular level during senescence. For the most part, oxidative-modified proteins are not repaired and must be removed by proteolytic degradation carried out by proteosome, which selectively degrades damaged proteins [15]. The molecular mechanism that causes the accumulation of oxidative-modified proteins is not completely clarified. Nevertheless, the oxidative damage to proteins has several physiological implications related to the loss of function in the affected proteins with deducible consequences for the organism. Protein oxidative damage involves both the loss of thiol groups, and modifications to amino acids that constitute the polypeptide chain, in particular, histidine. Protein carbonylation (PC) is used as a marker of oxidative damage, which acts, in particular, on the side chains of the amminoacid residues lysine, arginine, proline, and threonin; this may occur either directly or indirectly as a consequence of lipid peroxidation [79].
Since oxidized protein is harmful to the maintenance of cellular homeostasis, it requires rapid removal by proteolytic digestion. PC content increases drastically in the last third of the life span [80] and reaches a level such that on average one out of every three protein molecules carries modification. Both in men and in women, PC content tends to increase during aging, but in women these changes are not statistically significant [31]. Increasing amounts of PC content were observed and compared during aging in male skeletal muscles, but only in subjects younger than age 40 and older than age 70 [31]. Muscles with different functions in humans have been compared [77], and some authors reported an increase in PC level during aging in both the VL [81] and the external intercostal [78]; in other cases, the increase was not statistically significant in the VL [77, 78] and in the RA [77]. The difficulty in showing significant changes in PC in the chronic physiological process could be due to the dynamic features of the process: during aging, myofibrillar carbonyl content has been shown to increase significantly three hours after acute oxidative stress, only to return to its basic level within a few hours, in contrast with the normal condition in skeletal muscle as characterized by a relatively slow protein turnover [82]. Finally, protein carbonyl content tend to increase during aging and although data recently obtained from our research do not reach statistical significance, they are in line with those obtained in other studies on the brain, heart, and kidney [83] and skeletal muscle [84].
5. Mitochondrial DNA Damage
Mitochondrial DNA (mtDNA) is a double-stranded ring (about 16.5 kb) encoding 13 subunits of the ETC (electron transport chain) as well as 2rRNA and 22tRNA protein necessary for their translocation. Mitochondrial DNA lacks introns and is devoid of histones and other DNA-associated proteins; it is located in mitochondrial matrix, close to the major source of ROS [15]. For all these reasons, the probability of the oxidative modification of a coding region of mtDNA is very high. Moreover, the repair devices of mtDNA, even present, are largely insufficient to overcome extensive DNA damage which, furthermore, persist longer than that nuclear DNA, as demonstrated by in vitro experiments with cell cultures [85, 86].
The high turnover of mitochondria and the not sufficient mtDNA repair systems cannot counteract the oxidative damage that accumulates. This involves a progressive accumulation of mitochondrial functional impairment and, therefore, a lower availability of energy and consequent impairment of cell functions. Therefore, ROS can damage DNA either by an indirect mechanism, or by direct interaction with several molecules. Indirect mechanism involves the peroxides, which can oxidize-SH groups of Ca2+ channels in the endoplasmic reticulum, which contains ion cell, and thus determines its release [87]. Increases in intracellular calcium can stimulate Ca2+-dependent endonucleases and it can results in excessive and incorrect DNA fragmentations. The hydroxyl radical bears the greatest responsibility for the damage caused by ROS with a direct mechanism. HO∙ reacts indiscriminately with both sugars, ribose and deoxyribose, and with both nucleobases, purine and pyrimidine, by addition and H-abstraction. In this case, modified bases induce DNA strand breakage, during replication, and pairing errors with consequent mutations [88]. HO∙ reacts predominantly with Guanine in C8 and C4 of the ring, thus generating Guanine radical. In the presence of oxygen or other oxidizing agents, the Guanine radical is converted to stable product 8-hydroxy-2′-deoxyguanosine (8-OHdG) [89], which is considered a good biomarker of oxidative DNA damage [90].
Data obtained in male skeletal muscle indicate an increase in DNA damage during aging [32, 81] for pooled male and female data [37]. In fact, investigations of human skeletal muscle have established a correlation between age and the accumulation of mtDNA deletions an mutations [91]. It has also been suggested that mtDNA mutations start to occur after the fourth decade of life, and that they accumulate with age in postmitotic tissues [92]. There is strong evidence for continuous age-related increase in the levels of oxidative stress and oxidative damage on mitochondrial biomolecules, damage that becomes progressively more apparent with advancing age [93]. However, progressive loss of mitochondrial redundancy may not limit human skeletal muscle's capacity to supply the cellular energetic demands at basal metabolic conditions. This is important, because such limits might constrain the functioning of myofibers during situations with higher energy requirements. Moreover, an increase in MDA content is significantly correlated with an increase in 8-OHdG, a finding which suggests a direct correlation between lipid peroxidation and DNA damage in human skeletal muscles [37]. It has been demonstrated that deletion patterns of mtDNA over a lifetime are tissue specific and more pronounced in the skeletal muscle and heart [94].
It is also important to note that in human skeletal muscle oxidative damage is probably lower in aged women compared with aged men [32]. In particular, it seems that women are more protected against molecular oxidative stress during premenopausal life period suggesting a significative antioxidant role of estrogen, present in high levels in women. These hormones could contribute, therefore, to enhancing antioxidant defences in female muscles [95]. During the fertile age, these hormones probably induce a less accumulation of the oxidative damage with respect to males, determining a more favourable condition in the aging process [15].
5.1. The Antioxidant Defence in Aged Muscle
Antioxidants are substances able to inhibit the rate of oxidation [96, 97]. Biological antioxidant elements are present under diverse and numerous forms and they can be categorized as enzymatic (superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GR)) and nonenzymatic (ascorbate, tocopherol, beta-carotene, reduced coenzyme Q, or ubiquinol, reduced glutathione, uric acid, and minerals like selenium and zinc). These antioxidant systems act as primary intracellular defences against reactive oxygen species before their interaction with macromolecules protecting cells from free radical damage. Nevertheless, since free radicals are necessary for correct functioning of human organism, efficient mechanisms of enzymatic antioxidant defence had to be developed, especially in cells highly exposed to oxidation processes like those of skeletal muscle with the aim of protecting cellular constituents. The equilibrium between ROS production and the antioxidant defences at the level of the cellular structures determines whether a cell exposed to ROS increase will survive or will be seriously damaged or will die inducing apoptosis [98]. The enzymatic antioxidants serve mainly intracellularly, being absent or present in small amounts extracellularly, and they prevent free radical chain propagation by interrupting initiation. This is particularly important in the intravascular and connective-tissue spaces [99]. Nonenzymatic antioxidants can be divided into lipid- and water-soluble molecules. Among the nonenzymatic antioxidant molecules, the most important and studied are the vitamins C and E.
5.2. Non-Enzymatic Antioxidants
Endogenous non-enzymatic elements with antioxidant properties contribute to the maintenance of homeostasis by primarily acting as cofactors for the antioxidant enzymes. A major source of antioxidants is diet [100]. Among dietary antioxidants, the most important (and also largely available as supplements) are vitamin C, vitamin E, and carotenoids. Tocopherol (vitamin E) and beta-carotene, a precursor of vitamin A, are the main lipid-soluble nonenzymatic antioxidants. They are mainly localized to cell membranes and LDL. Alpha-tocopherol, the most active form of vitamin E, breaks free radical chain reactions. Given its location, tocopherol inhibits already initiated lipid peroxidation, in addition to having several other antioxidant effects. Beta-carotene interacts primarily with singlet oxygen by “de-exciting” it. Moreover it has been reported a significantly high correlation of α-tocopherol (the most diffuse form of circulating vitamin E) with physical performance, and of γ-tocopherol with skeletal muscle strength [101].
Ascorbate (AA) is the main water-soluble nonenzymatic antioxidant. AA interacts with a wide variety of free radicals intracellularly and forms a front-line extracellular defence against free radicals in plasma [102]. AA is moreover able to interact with superoxide and hydroxyl-free radicals, in addition to singlet oxygen. Ascorbate free radical, with its relatively low redox potential, does not propagate free radical chain reaction [103, 104]. One additional important antioxidant role ascorbate plays is in the regeneration of alpha-tocopherol (the most active form of vitamin E) from the tocopherol radical [105, 106]. Thus, AA does not only directly protect membranes and LDL from ROS generated in the aqueous phase but also indirectly protects them by reduction of vitamin E radical. Moreover, ascorbic acid concentrations are inversely associated with isoprostanes, a marker of lipid peroxidation [107] and it has a number of well-defined biological functions, including collagen, catecholamine, and carnitine biosynthesis [101–103].
Recent investigations are emerging that show that low serum/plasma carotenoids are independently associated with poor skeletal muscle strength and impaired physical performance. Among 669 women aged 70–79 years in the Women's Health and Aging Studies (WHAS) I and II, low serum carotenoid levels were associated with poor muscle strength [108]. Also, other studies showed carotenoids provide a balance to reactive oxygen species [109].
The diet is, moreover, a very important source of carotenoid. The six major dietary carotenoids, up-taken mostly from fruits and vegetables (α-carotene, β-carotene, β-cryptoxanthin, lutein, zeaxanthin, and lycopene) comprise an important component of the antioxidant defense system in humans protecting against oxidative stress by quenching singlet oxygen, scavenging free radicals, inhibiting lipid peroxidation, and modulating redox-sensitive transcription factors that are involved in the upregulation of proinflammatory cytokines [103, 107]. Carotenoids are hydrophobic molecules, and thus, interact with lipophilic elements of the cell, such as the lipid membrane bilayer. They are commonly located within cell membranes, and the location of specific carotenoids within the membrane structure depends upon the chemical structure of the carotenoid [109].
Other important and powerful nonenzymatic antioxidant is glutathione (GSH/GSSG). It is representing also the base material for several other important antioxidant enzymes including glutathione-peroxidase, glutathione-reductase, and glutathione-transferase. Glutathione is a compound classified as a peptide made of three amino acids: cysteine, glutamic acid, and glycine. Glutathione is also found in every part of the body, especially the lungs, intestinal tract, and liver. The body produces and stores the largest amounts of GSH in the liver, where it is used to detoxify harmful compounds so that they can be removed from the body through the bile. The liver also supplies GSH directly to red and white blood cells in the bloodstream; it helps keep red blood and white blood cells healthy to maximize the disease-fighting power of the immune system. Glutathione also appears to have an anti-aging affect on the body. Strong evidence that glutathione depletion causes cell death comes from cell culture studies by Li and colleague [110]. Also other works showed that direct depletion of mitochondrial and cytoplasmic GSH resulted in increased generation of ROS, disruption of the mitochondrial transmembrane potential, and rapid loss of mitochondrial function [111, 112].
The CoQ10 (Q10 coenzyme) is also an important endogenous compound in preserving mitochondrial function and it cooperates in maintaining a proper energy level, which serves to prevent the aging skin from switching to anaerobic energy production mechanisms. Furthermore, the antioxidant capacity of CoQ10 contributes to a positive effect against UV-mediated oxidative stress [103].
5.3. Enzymatic Antioxidants
Probably the main physiological mechanism by which cells regulate ROS activities (and thus protect against oxidative damage) consists, firstly, in modification of the expression and activities of the regulatory enzymes such as SOD, CAT, GPx, haem oxygenase-1 (HO-1) and, secondly, in increases in other cytoprotective proteins, such as HSPs [29]. These enzymes work to maintain a state of balance preventing the transformation of ROS and to convert them into more stable molecules (like water and molecular oxygen). There is a great controversy in the literature as to whether or not aging is associated with an increase or decrease of enzymatic antioxidant defence in the cell. Results reported in the literature need to be carefully interpreted taking into account sex, muscle fiber composition, and specificity [15].
5.4. Superoxide Dismutase (SOD)
SOD is one of the primary enzymatic antioxidant defences, and it readily converts superoxide radicals (O2−) in hydrogen peroxyde (H2O2). An increase in total SOD activity corresponds with enhanced resistance to oxidative stress [113]. SOD exists in two forms, cytosolic (Cu-ZnSOD) and mitochondrial (MnSOD). MnSOD activity in skeletal muscle, both in rats [114] and in men [2, 114, 115], is reported to increase significantly with aging and to respond to age-related oxidative stress on the mitochondria by upregulation [15]. In human, moreover, as in the rats, the age-dependent enhancement in MnSOD activity is particularly evident in the RA [77] and in external intercostal muscle [78] rather than in VL [75, 77, 82], suggesting a marked increase in ROS production mainly in the oxidative muscle respect to glycolytic muscle. Furthermore, MnSOD activity value is comparable between young men and women, doubling during aging in both sexes [31]. The enzymatic level seems to play an active role in both women and men during aging [15]. This regulatory mechanism, therefore, partially counteracts the increase of free radicals, in particular O2−, in the mitochondria in senescent tissues. This reinforces the hypothesis that mitochondrial stress, due to an increased leakage in ETC during aging, is associated with production of superoxide. Accordingly, cytoplasmic SOD did not show any change or decrease in activity during aging [2, 32, 75].
In human skeletal muscle, the relationship between SOD activity, aging, and sex was investigated [2], and the obtained results showed that enzymatic activity in this kind of muscle tissue in young women is about four-to-five times higher than in men of the same age, suggesting that total SOD activity could be a limiting factor in muscle defence against oxidative damage in humans [15, 116]. Moreover, total SOD activity appears to be significantly higher in enriched type II fiber muscle [31]. Strength training, which results in muscle hypertrophy and improved muscular strength and power [117, 118], could be an important factor in delaying the progressive loss of type II fibers in elderly subjects as well [118, 119].
5.5. Catalase (CAT) and Glutathione Peroxidase (GPx)
The decomposition of hydrogen peroxide to form water and oxygen is carried out in the cell by GPx and CAT. H2O2 has the ability to freely cross the membrane and has relatively long half life [120]. Like SOD, GPx and CAT are located in both mitochondria and cytosol, where they provide important cellular protection against free radical damage to membrane lipids, protein, and nucleic acids [15]. Most of the data on rats report an increase in CAT activity both in senescent oxidative muscles and glycolytic muscles [121]. This finding is consistent with the possibility that nonmitochondrial ROS production increases. In contrast, few and contradictory data are available on human skeletal muscle. Some authors report no change in CAT activity in men during aging [116]. No differences are observed in CAT activity with aging in all condition tested: between men and women, between muscles with different fiber compositions, and between oxidative and glycolytic muscles [75], while other studies describe a significant increase [76] or decrease in CAT activity in the VL [32]. Nevertheless, these contradictory data could be due to the dynamics of CAT activity during aging: in men, as in rats, a two-phase trend was described: an initial decrease in adult animal followed by a significant enhancement in aged animals [114, 122].
In human skeletal muscles, GPx does not change during aging, whether in glycolytic or oxidative muscle [123]. In young rats, skeletal muscle fibers with greater oxidative capacity (like the soleus muscle) have higher levels of GSH (reduced glutathione), the substrate for GPx to detoxify hydrogen peroxide, and total glutathione content than those with lower oxidative potential. In addition, one study reports that GPx activity was higher in oxidative muscles and thus caused less vulnerability and more resistance to an exercise-induced oxidative stress, while the modest levels of GSH and GPx in the deep or superficial VL muscle were associated with greater lipid peroxidation under exercise stress [15]. Probably, GSH and GSSG (oxidized glutathione) muscle levels, as well as the GSH/GSSG ratio, are closely correlated with differing muscle functions. Moreover, since some data showed that in type I fibers of both glycolytic and oxidative muscle in aging the GSH and GSSG levels remained unchanged [75], there is suggested that during aging there are no alterations of membrane glutathione transport and that there is fiber-specific adaptation of the GSH system in the skeletal muscle [123].
5.6. Role of Nutrition against Muscle Oxidative Damage
The beneficial effects of nutrition on the prevention of oxidative stress have been attributed to the presence of antioxidants contained mainly in vegetables: carotenoids, plant polyphenols, tocopherols, ascorbate, phytic acid, and selenium [1]. Recent epidemiological studies suggest that carotenoids or carotenoid-rich foods are protective against a decline in muscle strength and walking disability among older community-dwelling adults. As report above, the carotenoids protect against oxidative stress by quenching singlet oxygen, scavenging free radicals, and inhibiting lipid peroxidation [109]. The literature provides little evidence of the possible effects of antioxidant supplementation on skeletal muscle aging [124–126]. On an animal aged model, it has been showed that vitamin E and C supplementation as well as a blend of polyphenols and carotenoids for 10 months resulted in significantly increased activity of the GSH system in muscle cell [125]; another study indicated that mixed supplementation with rutin, vitamin E, vitamin A, Zinc, and selenium restores the ability to stimulate protein synthesis subsequent to leucine administration [124]; some authors showed that vitamin C and E supplementation attenuated the increase in markers of oxidative stress (H2O2, MDA, and 8-OHdG) in response to chronic repetitive muscle loading, with an improvement in the work output of aged muscles [125]. Altogether, the above studies suggest that a diet supplemented with a combination of antioxidants may possibly increase antioxidant defences, lower muscle oxidative damage, and improve muscle protein balance during senescence [1]. But if, overall, results consistently report significant improvements of antioxidant biomarkers after a period of specific supplementation, differently, the effect of antioxidant supplementation on muscle performance is still and largely controversial. In fact it is necessary to underline that there are currently no trials verifying the effects of antioxidant supplementation on sarcopenia (as identified by one of several the consensus definitions provided by international groups of experts). Interestingly, a recent statement from the society on sarcopenia, cachexia, and wasting disease does not even mention antioxidant supplementation as a possible tool to manage sarcopenia in older persons [127]. On the other hand, in order to explain these controversial findings, we must consider that the modification of a biomarker concentrations not automatically changes clinical parameters. It is more likely that subclinical effects are more sensible to changes than clinically evident manifestations.
6. Conclusions
During organism aging, production of reactive oxygen species is increased as a result of the functional deterioration of mitochondria. There is strong evidence that increased free radical generation may be the underlying reason for several age-related pathogeneses. However, few studies have actually measured aging-induced free radicals in human skeletal muscles. It has been suggested that age-related oxidative stress may be a function of a reduction in enzymatic antioxidant capacity, but this has not been demonstrated in human skeletal muscle.
The increased production of free radicals with aging plays a major role in targeting adaptive responses in human skeletal muscle. The adaptive mechanisms involve the antioxidant scavenger system and appear to be sex-, fiber-, and muscle-specific. Furthermore, the amount of introduced dietary antioxidants could play a specific and important role even if further trials in humans are required to clearly establish the hypothesized relationship between antioxidant and sarcopenia [15].
References
- 1.Buonocore D, Rucci S, Vandoni M, Negro M, Marzatico F. Oxidative system in aged skeletal muscle. Muscles, Ligaments and Tendons Journal. 2011;1(3):85–90. [PMC free article] [PubMed] [Google Scholar]
- 2.Pansarasa O, Bertorelli L, Vecchiett J, Felzani G, Marzatico F. Age-dependent changes in oxidative damage to DNA, lipids and protein in human skeletal muscle. Free Radical Biology and Medicine. 1999;27:617–622. doi: 10.1016/s0891-5849(99)00108-2. [DOI] [PubMed] [Google Scholar]
- 3.Preedy VR, Afachi J, Ueno Y. Alcoholic skeletal muscle myopathy: definition, features, cotribution of neuropathy, impact and diagnosis. European Journal of Neurology. 2001;8:677–687. doi: 10.1046/j.1468-1331.2001.00303.x. [DOI] [PubMed] [Google Scholar]
- 4.von Haehling S, Morley JE, Anker SD. An overview of sarcopenia: facts and numbers on prevalence and clinical impact. Journal of Cachexia, Sarcopenia and Muscle. 2010;1(2):129–133. doi: 10.1007/s13539-010-0014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu CJ, Latham NK. Progressive resistance strength training for improving physical function in older adults. Cochrane Database of Systematic Reviews. 2009;(3) doi: 10.1002/14651858.CD002759.pub2. Article ID CD002759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenk K, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. Journal of Cachexia, Sarcopenia and Muscle. 2010;1:9–21. doi: 10.1007/s13539-010-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roubenoff R. Sarcopenia and its implications for the elderly. European Journal of Clinical Nutrition. 2000;54(3):S40–S47. doi: 10.1038/sj.ejcn.1601024. [DOI] [PubMed] [Google Scholar]
- 8.Metter EJ, Talbot LA, Schranger M, Conwit R. Skeletal muscle strength as a pedictor of all-cause mortality in healthy men. The Journals of Gerontology A. 2002;57:359–365. doi: 10.1093/gerona/57.10.b359. [DOI] [PubMed] [Google Scholar]
- 9.Neder JA, Nery LE, Silva AC, Andreoni S, Whipp BJ. Maximal aerobic power and leg muscle mass and strength related to age in non-athletic males and females. European Journal of Applied Physiology and Occupational Physiology. 1999;79(6):522–530. doi: 10.1007/s004210050547. [DOI] [PubMed] [Google Scholar]
- 10.Baumgatner RN, Koehler KM, Gallagher D, et al. Epidemiodology of sarcopenia among the elderly in New Mexico. American Journal of Epidemiology. 1998;147:755–763. doi: 10.1093/oxfordjournals.aje.a009520. [DOI] [PubMed] [Google Scholar]
- 11.Cartee GD. Aging skeletal muscle: response to exercise. Exercise and Sport Sciences Reviews. 1994;22:91–120. [PubMed] [Google Scholar]
- 12.Porter MM, Vandervoort AA, Lexell J. Aging of human muscle: structure, function and adaptability. Scandinavian Journal of Medicine & Science in Sports. 1995;5(3):129–142. doi: 10.1111/j.1600-0838.1995.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 13.Orlander J, Kiessling KH, Larsson L, Karlsson J, Aniansson A. Skeletal muscle metabolism and ultrastructure in relation to age in sedentary men. Acta Physiologica Scandinavica. 1978;104:249–261. doi: 10.1111/j.1748-1716.1978.tb06277.x. [DOI] [PubMed] [Google Scholar]
- 14.Faulkner JA, Brooks SV. Muscle fatigue in old animals: unique aspects of fatigue in elderly humans. Advances in Experimental Medicine and Biology. 1995;384:471–480. [PubMed] [Google Scholar]
- 15.Rossi P, Marzani B, Giardina S, Negro M, Marzatico F. Human skeletal muscle aging and the oxidative system: cellular events. Current aging science. 2008;1(3):182–191. doi: 10.2174/1874609810801030182. [DOI] [PubMed] [Google Scholar]
- 16.Lexell J. Human aging, muscle mass, and fiber type composition. Journals of Gerontology A. 1995;50:11–16. doi: 10.1093/gerona/50a.special_issue.11. [DOI] [PubMed] [Google Scholar]
- 17.Larsson L, Karlsson J. Isometric and dynamic endurance as a function of age and skeletal muscle characteristics. Acta Physiologica Scandinavica. 1978;104(2):129–136. doi: 10.1111/j.1748-1716.1978.tb06259.x. [DOI] [PubMed] [Google Scholar]
- 18.Brown WF. A method for estimating the number of motor units in thenar muscles and the changes in motor unit count with ageing. Journal of Neurology Neurosurgery and Psychiatry. 1972;35(6):845–852. doi: 10.1136/jnnp.35.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gundersen K. Determination of muscle contractile properties: thimportance of the nerve. Acta Physiologica Scandinavica. 1998;162:333–341. doi: 10.1046/j.1365-201X.1998.0336e.x. [DOI] [PubMed] [Google Scholar]
- 20.Fulle S, Di Donna S, Puglielli C, et al. Age-dependent imbalance of the antioxidative system in human satellite cells. Experimental Gerontology. 2005;40(3):189–197. doi: 10.1016/j.exger.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Korenman SG, Morley JE, Mooradian AD, et al. Secondary hypogonadism in older men: its relation to impotence. Journal of Clinical Endocrinology and Metabolism. 1990;71(4):963–969. doi: 10.1210/jcem-71-4-963. [DOI] [PubMed] [Google Scholar]
- 22.Morley JE, Kaiser FE, Perry HM, et al. Longitudinal changes in testosterone, luteinizing hormone, and follicle- stimulating hormone in healthy older men. Metabolism. 1997;46(4):410–413. doi: 10.1016/s0026-0495(97)90057-3. [DOI] [PubMed] [Google Scholar]
- 23.Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ. Predictors of skeletal muscle mass in elderly men and women. Mechanisms of Ageing and Development. 1999;107(2):123–136. doi: 10.1016/s0047-6374(98)00130-4. [DOI] [PubMed] [Google Scholar]
- 24.Fulle S, Protasi F, Di Tano G, et al. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Experimental Gerontology. 2004;39(1):17–24. doi: 10.1016/j.exger.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 26.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(17):6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marzetti E, Lawler JM, Hiona A, Manini T, Seo AY, Leeuwenburgh C. Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radical Biology and Medicine. 2008;44(2):160–168. doi: 10.1016/j.freeradbiomed.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 28.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiological Reviews. 2008;88(4):1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McArdle A, Pattwell D, Vasilaki A, Griffiths RD, Jackson MJ. Contractile activity-induced oxidative stress: cellular origin and adaptive responses. American Journal of Physiology. 2001;280(3):C621–C627. doi: 10.1152/ajpcell.2001.280.3.C621. [DOI] [PubMed] [Google Scholar]
- 31.Pansarasa O, Castagna L, Colombi B, Vecchiet J, Felzani G, Marzatico F. Age and sex differences in human skeletal muscle: role of reactive oxygen species. Free Radical Research. 2000;33(3):287–293. doi: 10.1080/10715760000301451. [DOI] [PubMed] [Google Scholar]
- 32.Fanò G, Mecocci P, Vecchiet J, et al. Age and sex influence on oxidative damage and functional status in human skeletal muscle. Journal of Muscle Research and Cell Motility. 2001;22(4):345–351. doi: 10.1023/a:1013122805060. [DOI] [PubMed] [Google Scholar]
- 33.Ji LL. Antioxidant enzyme response to exercise and aging. Medicine and Science in Sports and Exercise. 1993;25(2):225–231. [PubMed] [Google Scholar]
- 34.Matsuo M. Age-related alteration in antioxidant defense . In: Yu BP, editor. Free Radical in Aging. Boca Raton, Fla, USA: CRC Press; 1993. pp. 143–181. [Google Scholar]
- 35.Powers SK, Duarte J, Kavazis AN, Talbert EE. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Experimental Physiology. 2010;95(1):1–9. doi: 10.1113/expphysiol.2009.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle and Nerve. 2007;35(4):411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- 37.Mecocci P, Fanó G, Fulle S, et al. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radical Biology and Medicine. 1999;26(3-4):303–308. doi: 10.1016/s0891-5849(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 38.Melov S, Shoffner JM, Kaufman A, Wallace DC. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Research. 1995;23(20):4122–4126. doi: 10.1093/nar/23.20.4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC, Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. Journal of Physiology. 2011;589(9):2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kujoth CC, Hiona A, Pugh TD, et al. Medicine: mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 41.Hütter E, Skovbro M, Lener B, et al. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007;6(2):245–256. doi: 10.1111/j.1474-9726.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 42.Reid MB. Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Medicine and Science in Sports and Exercise. 2001;33(3):371–376. doi: 10.1097/00005768-200103000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB Journal. 2001;15(2):322–332. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- 44.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutation Research. 1992;275(3-6):169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 45.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature Genetics. 1992;2(4):324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 46.Muller-Hocker J, Seibel P, Schneiderbanger K, Kadenbach B. Different in situ hybridization patterns of mitochondrial DNA in cytochrome c oxidase-deficient extraocular muscle fibres in the elderly. Virchows Archiv. 1993;422(1):7–15. doi: 10.1007/BF01605127. [DOI] [PubMed] [Google Scholar]
- 47.Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiological Reviews. 2009;89(3):799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 48.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Human Molecular Genetics. 2009;18(2):R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. Journals of Gerontology A. 2010;65(2):119–128. doi: 10.1093/gerona/glp179. [DOI] [PubMed] [Google Scholar]
- 50.Romanello V, Guadagnin E, Gomes L, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO Journal. 2010;29(10):1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adhihetty PJ, O’Leary MFN, Chabi B, Wicks KL, Hood DA. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. Journal of Applied Physiology. 2007;102(3):1143–1151. doi: 10.1152/japplphysiol.00768.2006. [DOI] [PubMed] [Google Scholar]
- 52.Sandri M, Lin J, Handschin C, et al. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes. 2006;55(6):1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- 54.Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato A, Nakada K, Hayashi JI. Mitochondrial dynamics and aging: mitochondrial interaction preventing individuals from expression of respiratory deficiency caused by mutant mtDNA. Biochimica et Biophysica Acta. 2006;1763(5-6):473–481. doi: 10.1016/j.bbamcr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Brand FN, Kiely DK, Kannel WB, Myers RH. Family patterns of coronary heart disease mortality: the Framingham Longevity Study. Journal of Clinical Epidemiology. 1992;45(2):169–174. doi: 10.1016/0895-4356(92)90009-c. [DOI] [PubMed] [Google Scholar]
- 57.Ross OA, McCormack R, Curran MD, et al. Mitochondrial DNA polymorphism: its role in longevity of the Irish population. Experimental Gerontology. 2001;36(7):1161–1178. doi: 10.1016/s0531-5565(01)00094-8. [DOI] [PubMed] [Google Scholar]
- 58.Alexeyev MF, LeDoux SP, Wilson GL. Mitochondrial DNA and aging. Clinical Science. 2004;107(4):355–364. doi: 10.1042/CS20040148. [DOI] [PubMed] [Google Scholar]
- 59.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radical Biology and Medicine. 2004;37(6):768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 60.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 61.Kinscherf R, Wagner M, Kamencic H, et al. Characterization of apoptotic macrophages in atheromatous tissue of humans and heritable hyperlipidemic rabbits. Atherosclerosis. 1999;144(1):33–39. doi: 10.1016/s0021-9150(99)00037-4. [DOI] [PubMed] [Google Scholar]
- 62.Reid VC, Mitchinson MJ, Skepper JN. Cytotoxicity of oxidized low-density lipoprotein to mouse peritoneal macrophages: an ultrastructural study. Journal of Pathology. 1993;171(4):321–328. doi: 10.1002/path.1711710413. [DOI] [PubMed] [Google Scholar]
- 63.Shull S, Heintz NH, Periasamy M, et al. Differential regulation of antioxidant enzymes in response to oxidants. Journal of Biological Chemistry. 1991;266(36):24398–24403. [PubMed] [Google Scholar]
- 64.Belia S, Pietrangelo T, Fulle S, et al. Sodium nitroprusside, a NO donor, modifies Ca2+ transport and mechanical properties in frog skeletal muscle. Journal of Muscle Research and Cell Motility. 1998;19(8):865–876. doi: 10.1023/a:1005499606155. [DOI] [PubMed] [Google Scholar]
- 65.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102(4):499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 66.Dargelos E, Poussard S, Brulé C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90(2):359–368. doi: 10.1016/j.biochi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 67.Hidalgo C, Sánchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. Journal of Biological Chemistry. 2006;281(36):26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- 68.Andersson DC, Betzenhauser MJ, Reiken S, et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metabolism. 2011;14(2):196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radical Biology and Medicine. 2008;44(2):193–201. doi: 10.1016/j.freeradbiomed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 70.Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. Journal of Applied Physiology. 2006;100(2):399–405. doi: 10.1152/japplphysiol.00873.2005. [DOI] [PubMed] [Google Scholar]
- 71.Ljubicic V, Hood DA. Kinase-specific responsiveness to incremental contractile activity in skeletal muscle with low and high mitochondrial content. American Journal of Physiology. 2008;295:E195–E204. doi: 10.1152/ajpendo.90276.2008. [DOI] [PubMed] [Google Scholar]
- 72.Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. American Journal of Physiology. 2009;296(1):C116–C123. doi: 10.1152/ajpcell.00267.2007. [DOI] [PubMed] [Google Scholar]
- 73.Vasilaki A, Mansouri A, Van Remmen H, et al. Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell. 2006;5(2):109–117. doi: 10.1111/j.1474-9726.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- 74.Richter C. Biophysical consequences of lipid peroxidation in membranes. Chemistry and Physics of Lipids. 1987;44(2-4):175–189. doi: 10.1016/0009-3084(87)90049-1. [DOI] [PubMed] [Google Scholar]
- 75.Marzani B, Pansarasa O, Marzatico F. ‘Oxidative stress’ and muscle aging: influence of age, sex, fiber composition and function. Basic And Applied Myology. 2004;14:37–44. [Google Scholar]
- 76.Yagi K. Lipid peroxides and human diseases. Chemistry and Physics of Lipids. 1987;45(2-4):337–351. doi: 10.1016/0009-3084(87)90071-5. [DOI] [PubMed] [Google Scholar]
- 77.Marzani B, Felzani G, Bellomo RG, Vecchiet J, Marzatico F. Human muscle aging: ROS-mediated alterations in rectus abdominis and vastus lateralis muscles. Experimental Gerontology. 2005;40(12):959–965. doi: 10.1016/j.exger.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 78.Barreiro E, Coronell C, Laviña B, Ramírez-Sarmiento A, Orozco-Levi M, Gea J. Aging, sex differences, and oxidative stress in human respiratory and limb muscles. Free Radical Biology and Medicine. 2006;41(5):797–809. doi: 10.1016/j.freeradbiomed.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 79.Grune T, Shringarpure R, Sitte N, Davies KJA. Age-related changes in protein oxidation and proteolysis in mammalian cells. Journals of Gerontology A. 2001;56(11):B459–B467. doi: 10.1093/gerona/56.11.b459. [DOI] [PubMed] [Google Scholar]
- 80.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radical Biology and Medicine. 2002;32(9):790–796. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 81.Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Experimental Gerontology. 2004;39(9):1391–1400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 82.Nagasawa T, Hatayama T, Watanabe Y, Tanaka M, Niisato Y, Kitts DD. Free radical-mediated effects on skeletal muscle protein in rats treated with Fe-nitrilotriacetate. Biochemical and Biophysical Research Communications. 1997;231(1):37–41. doi: 10.1006/bbrc.1996.6034. [DOI] [PubMed] [Google Scholar]
- 83.Youngman LD, Park JYK, Ames BN. Protein oxidation associated with aging is reduced by dietary restriction of protein or calories. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(19):9112–9116. doi: 10.1073/pnas.89.19.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pryor WA. The free radical theory of aging revisited: a critique and a suggested disease-specific theory. In: Warner HR, Butler RN, Richard L, editors. Modern Biological Theories of Aging. New York, NY, USA: Raven Press; 1987. pp. 89–112. [Google Scholar]
- 85.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(2):514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barja G. Free radicals and aging. Trends in Neurosciences. 2004;27(10):595–600. doi: 10.1016/j.tins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 87.Nicotera P, Orrenius S. Molecular mechanism of toxic cell death: an overview. Toxicology Methods. 1994;1B:p. 23. [Google Scholar]
- 88.Van Remmen H, Hamilton ML, Richardson A. Oxidative damage to DNA and aging. Exercise and Sport Sciences Reviews. 2003;31(3):149–153. doi: 10.1097/00003677-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 89.Candeias LP, Steenken S. Reaction of HO• with guanine derivatives in aqueous solution: formation of two different redox-active OH-adduct radicals and their unimolecular transformation reactions. Properties of G(-H)• . Chemistry. 2000;6(3):475–484. doi: 10.1002/(sici)1521-3765(20000204)6:3<475::aid-chem475>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 90.Shigenaga MK, Ames BN. Assays for 8-hydroxy-2’-deoxyguanosine: a biomarker of in vivo oxidative DNA damage. Free Radical Biology and Medicine. 1991;10(3-4):211–216. doi: 10.1016/0891-5849(91)90078-h. [DOI] [PubMed] [Google Scholar]
- 91.Murdock DG, Christacos NC, Wallace DC. The age-related accumulation of a mitochondrial DNA control region mutation in muscle, but not brain, detected by a sensitive PNA-directed PCR clamping based method. Nucleic Acids Research. 2000;28(21):4350–4355. doi: 10.1093/nar/28.21.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei YH, Ma YS, Lee HC, Lee CF, Lu CY. Mitochondrial theory of aging matures—roles of mtDNA mutation and oxidative stress in human aging. Chinese Medical Journal. 2001;64(5):259–270. [PubMed] [Google Scholar]
- 93.Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(15):5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meissner C, Bruse P, Oehmichen M. Tissue-specific deletion patterns of the mitochondrial genome with advancing age. Experimental Gerontology. 2006;41(5):518–524. doi: 10.1016/j.exger.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 95.Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. International Journal of Developmental Neuroscience. 2000;18(4-5):347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 96.Maxwell SRJ. Prospects for the use of antioxidant therapies. Drugs. 1995;49(3):345–361. doi: 10.2165/00003495-199549030-00003. [DOI] [PubMed] [Google Scholar]
- 97.Clarkson PM, Thompson HS. Antioxidants: what role do they play in physical activity and health? American Journal of Clinical Nutrition. 2000;72(2):37S–46S. doi: 10.1093/ajcn/72.2.637S. [DOI] [PubMed] [Google Scholar]
- 98.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. American Journal of Physiology. 2007;292(1):R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 99.Block G, Dietrich M, Norkus EP, et al. Factors associated with oxidative stress in human populations. American Journal of Epidemiology. 2002;156(3):274–285. doi: 10.1093/aje/kwf029. [DOI] [PubMed] [Google Scholar]
- 100.Fusco D, Colloca G, Lo Monaco MR, Cesari M. Effects of antioxidant supplementation on the aging process. Clinical Interventions in Aging. 2007;2(3):377–387. [PMC free article] [PubMed] [Google Scholar]
- 101.Cesari M, Pahor M, Bartali B, et al. Antioxidants and physical performance in elderly persons: the Invecchiare in Chianti (InCHIANTI) study. American Journal of Clinical Nutrition. 2004;79(2):289–294. doi: 10.1093/ajcn/79.2.289. [DOI] [PubMed] [Google Scholar]
- 102.Frei B, England L, Ames BN. Ascorbate is an outstanding antioxidant in human blood plasma. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(16):6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Niki E, Yamamoto Y, Takahashi M, et al. Free radical-mediated damage of blood and its inhibition by antioxidants. Journal of Nutritional Science and Vitaminology. 1988;34:507–512. doi: 10.3177/jnsv.34.507. [DOI] [PubMed] [Google Scholar]
- 104.Rose RC, Bode AM. Biology of free radical scavengers: an evaluation of ascorbate. FASEB Journal. 1993;7(12):1135–1142. [PubMed] [Google Scholar]
- 105.Beyer RE. The role of ascorbate in antioxidant protection of biomembranes: interaction with vitamin E and coenzyme Q. Journal of Bioenergetics and Biomembranes. 1994;26:349–358. doi: 10.1007/BF00762775. [DOI] [PubMed] [Google Scholar]
- 106.Chan AC. Partners in vitamin E and vitamin C. Canadian Journal of Physiology and Pharmacology. 1993;71:725–731. doi: 10.1139/y93-109. [DOI] [PubMed] [Google Scholar]
- 107.Hu P, Reuben DB, Crimmins EM, Harris TB, Huang MH, Seeman TE. The effects of serum beta-carotene concentration and burden of inflammation on all-cause mortality risk in high-functioning older persons: MacArthur Studies of Successful Aging. Journals of Gerontology A. 2004;59(8):849–854. doi: 10.1093/gerona/59.8.m849. [DOI] [PubMed] [Google Scholar]
- 108.Walston J, Xue Q, Semba RD, et al. Serum antioxidants, inflammation, and total mortality in older women. American Journal of Epidemiology. 2006;163(1):18–26. doi: 10.1093/aje/kwj007. [DOI] [PubMed] [Google Scholar]
- 109.Young AJ, Phillip DM, Lowe GM. Carotenoid antioxidant activity. In: Krinsky NI, Mayne ST, Sies H, editors. Carotenoids in Health and Disease. New York, NY, USA: Marcel Dekker; 2004. pp. 105–126. [Google Scholar]
- 110.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19(2):453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 111.Èllner W, Seyfried U, Groscurth J, et al. Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Research. 1999;826:53–62. doi: 10.1016/s0006-8993(99)01228-7. [DOI] [PubMed] [Google Scholar]
- 112.Seyfried J, Soldner F, Schulz JB, Klockgether T, Kovar KA, Wüllner U. Differential effects of L-buthionine sulfoximine and ethacrynic acid on glutathione levels and mitochondrial function in PC12 cells. Neuroscience Letters. 1999;264(1-3):1–4. doi: 10.1016/s0304-3940(99)00107-x. [DOI] [PubMed] [Google Scholar]
- 113.Fielding RA, Meydani M. Exercise, free radical generation, and aging. Aging. 1997;9(1-2):12–18. doi: 10.1007/BF03340124. [DOI] [PubMed] [Google Scholar]
- 114.Ji LL, Dillon D, Wu E. Alteration of antioxidant enzymes with aging in rat skeletal muscle and liver. American Journal of Physiology. 1990;258(4):R918–R923. doi: 10.1152/ajpregu.1990.258.4.R918. [DOI] [PubMed] [Google Scholar]
- 115.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and cortosterone exacerbates excitotoxicity, oxidative injury, and amyloid B-Peptide toxicity in hippocampal neurons. Journal of Neurochemistry. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 116.Pansarasa O, Felzani G, Vecchiet J, Marzatico F. Antioxidant pathways in human aged skeletal muscle: relationship with the distribution of type II fibers. Experimental Gerontology. 2002;37(8-9):1069–1075. doi: 10.1016/s0531-5565(02)00085-2. [DOI] [PubMed] [Google Scholar]
- 117.Campbell AJ, Robertson MC, Gardner MM, Norton RN, Tilyard MW, Buchner DM. Randomised controlled trial of a general practice programme of home based exercise to prevent falls in elderly women. British Medical Journal. 1997;315(7115):1065–1069. doi: 10.1136/bmj.315.7115.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fiatarone MA, Marks EC, Ryan ND. High-intensity strength training in nonagenarians. Effects on skeletal muscle. Journal of the American Medical Association. 1999;263:3029–3034. [PubMed] [Google Scholar]
- 119.Bassey EJ, Fiatarone MA, O’Neill EF, Kelly M, Evans WJ, Lipsitz LA. Leg extensor power and functional performance in very old men and women. Clinical Science. 1992;82(3):321–327. doi: 10.1042/cs0820321. [DOI] [PubMed] [Google Scholar]
- 120.Matsuo M, Kaneko T. The chemistry of reactive oxygen species and related free radicals. In: Radack Z, editor. Free Radicals in Exercise and Aging. Champaign, Ill, USA: Human Kinetics; 2000. [Google Scholar]
- 121.Capel F, Buffière C, Mirand PP, Mosoni L. Differential variation of mitochondrial H2O2 release during aging in oxidative and glycolytic muscles in rats. Mechanisms of Ageing and Development. 2004;125(5):367–373. doi: 10.1016/j.mad.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 122.Paradies G, Petrosillo G, Ruggiero FM. Cardiolipin-dependent decrease of cytochrome c oxidase activity in heart mitochondria from hypothyroid rats. Biochimica et Biophysica Acta. 1997;1319:5–8. doi: 10.1016/s0005-2728(97)00012-1. [DOI] [PubMed] [Google Scholar]
- 123.Leeuwenburgh C, Fiebig R, Chandwaney R, Ji LL. Aging and exercise training in skeletal muscle: responses of glutathione and antioxidant enzyme systems. The American Journal of Physiology. 1994;267(2):R439–R445. doi: 10.1152/ajpregu.1994.267.2.R439. [DOI] [PubMed] [Google Scholar]
- 124.Marzani B, Balage M, Vénien A, et al. Antioxidant supplementation restores defective leucine stimulation of protein synthesis in skeletal muscle from old rats. Journal of Nutrition. 2008;138(11):2205–2211. doi: 10.3945/jn.108.094029. [DOI] [PubMed] [Google Scholar]
- 125.Ryan MJ, Dudash HJ, Docherty M, et al. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Experimental Gerontology. 2010;45(11):882–895. doi: 10.1016/j.exger.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rebrin I, Zicker S, Wedekind KJ, Paetau-Robinson I, Packer L, Sohal RS. Effect of antioxidant-enriched diets on glutathione redox status in tissue homogenates and mitochondria of the senescence-accelerated mouse. Free Radical Biology and Medicine. 2005;39(4):549–557. doi: 10.1016/j.freeradbiomed.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morley JE, Abbatecola AM, Argiles JM, et al. Sarcopenia with limited mobility: an international consensus. Journal of the American Medical Directors Association. 2011;12(6):403–409. doi: 10.1016/j.jamda.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]