

Abstract
A cardinal feature of neurons in the cerebral cortex is stimulus selectivity, and experience-dependent shifts in selectivity are a common correlate of memory formation. We have used a theoretical “learning rule,” devised to account for experience-dependent shifts in neuronal selectivity, to guide experiments on the elementary mechanisms of synaptic plasticity in hippocampus and neocortex. These experiments reveal that many synapses in hippocampus and neocortex are bidirectionally modifiable, that the modifications persist long enough to contribute to long-term memory storage, and that key variables governing the sign of synaptic plasticity are the amount of NMDA receptor activation and the recent history of cortical activity.
Learning from Learning Rules
Among the insights attributed to Hebb (1) are the notions that memories of sensory experiences are stored by synaptic modifications, and that these changes occur in the same regions of the brain that are used to process sensory information. Thus, memories of visual experiences would be stored in visual cortex, auditory experiences would be stored in auditory cortex, and so on. Within each of these regions of cortex, memory of a sensory event would result from the permanent modification of the synapses between the cortical neurons that are activated by that event.
Research using neural network models confirms that memories can be stored by small but coherent modifications of synapses that may be widely distributed among many neurons (2). At the single cell level, the occurrence of such modifications would be manifest as a change in neuronal selectivity for particular input patterns (3). Consider as an example the simple model shown in Fig. 1. Three neurons (labeled 1, 2, and 3) receive excitatory synaptic inputs that convey information about three stimuli (labeled A, B, and C). Initially, before learning, each neuron responds similarly to each stimulus; there is no pattern of cellular output that uniquely represents each stimulus. After learning, however, the synapses have modified so that different stimuli yield different responses. Note that although each stimulus evokes a maximal response from a different neuron, the neural representation of a stimulus is distributed over all three cells. Stimulus A, for example, evokes a large response in cell 1, a moderate response in cell 2, and a weak response in cell 3. The representation of stimulus A is this unique combination of responses across the cells in the network. This is what is meant by distributed memory storage. Such representations are resistant to the loss of individual neurons. For example, loss of cell 1 would still leave an activity ratio in cells 2 and 3 that is unique to stimulus A. Also note that the memories are encoded by both increases and decreases in synaptic effectiveness. In principle, modifications of both signs can contribute equally to memory formation in neural networks.
Figure 1.
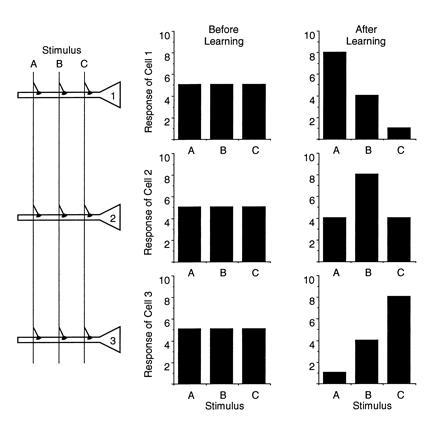
A model of distributed information storage. Three neurons (1, 2, and 3) receive inputs carrying information about three stimuli (A, B, and C). Before learning, all neurons respond equally to all stimuli. After learning, the neurons show stimulus selectivity, reflecting the modification of synapses in the network.
Models of distributed memory storage such as that in Fig. 1 suggest that a cellular correlate of memory is experience-dependent changes in neuronal stimulus selectivity. Indeed, neurophysiological studies of neurons in the hippocampus and neocortex have revealed precisely this type of change as animals learn to recognize and discriminate stimuli. For example, neurons in rat hippocampus show selectivity for positions in space, and this selectivity shifts as animals learn a new spatial environment (4, 5); neurons in primary auditory cortex show selectivity for tones of particular frequencies, and this selectivity is altered as animals learn to perform auditory discriminations (6, 7, 8); neurons in the primate inferior temporal cortex show selective responses to faces that are altered as the animal learns to recognize new faces (9), and so on (10, 11, 12, 13, 14). Thus, a question of extraordinary interest concerns how cortical synapses modify under the influence of experience such that stimulus selectivity is altered.
Serious theoretical analysis of the problem of experience-dependent changes in neuronal selectivity began in the 1970s (15, 16, 17, 18, 19). This work was inspired by experiments showing that neurons in the primary visual cortex exhibit selectivity for stimuli of particular orientations (20), and that elaboration of this property during postnatal development requires visual experience (21). A number of “learning rules” were proposed to account for experience-dependent development of orientation selectivity, each making slightly different assumptions about how synapses would modify during various combinations of presynaptic and postsynaptic activity. Although this work explicitly concerned visual cortical development, for which there was a wealth of experimental data, it was implicit in these studies that the same principles could apply to the synaptic changes that underlie adult learning and memory.
One such synaptic learning rule, from the work of Cooper and colleagues (19), is illustrated in Fig. 2. Cooper et al. (19) considered a single neuron receiving an array of excitatory synapses carrying information about the sensory environment. To account for the development and plasticity of neuronal stimulus selectivity, they proposed that active synapses are potentiated when the total postsynaptic response exceeds a critical value, called the “modification threshold” (θm), and that active synapses are depressed when the total postsynaptic response is greater than zero (assumed to be the average “spontaneous” level), but less than θm. It is readily apparent how this rule leads to stimulus selectivity. Consider the situation in Fig. 2 where postsynaptic responses to stimuli A–C are initially clustered around the value of θm. Because the response to C is greater than θm, the synapses that are active during presentation of C potentiate. Likewise, because the responses to A and B are less than θm, the synapses that are active during presentation of A and B depress, and the neuron becomes selectively responsive only to stimulus C.
Figure 2.
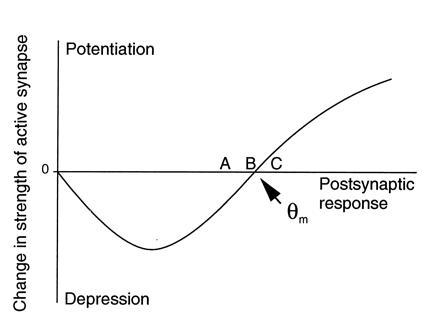
Function controlling synaptic plasticity at the Cooper synapse.
While “Cooper synapses” do yield stimulus selectivity under the appropriate conditions, a fixed θm can have unfortunate consequences. For example, if patterns A–C all initially yielded responses below θm, then all synapses would depress and the cell would cease responding to any stimulus. On the other hand, if patterns A–C all initially yielded responses greater than θm, then all synapses would potentiate to their saturation limit and the cell would lack selectivity. An elegant solution to this problem was introduced by Bienenstock, and colleagues (59) in what is known as the BCM algorithm. They showed that if the value of θm was allowed to vary as a nonlinear function of the average integrated postsynaptic activity, then the cell would evolve to a stable, selective state in a patterned input environment regardless of the initial condition.
Consideration of learning rules such as BCM is instructive because it allows one to connect the elementary mechanisms of synaptic plasticity with the systematics of learning and memory. However, showing that a learning rule yields desirable properties in artificial neural network models is no proof that it is actually implemented by synapses in the brain. Therefore, about 10 years ago I initiated a research program aimed at investigating whether the synaptic modifications assumed by the BCM theory have a biological basis in the cerebral cortex. In this paper, I will describe our progress to date.
Cooper Synapses in the Hippocampus
It has been known for many years that certain excitatory synapses in the hippocampus can be potentiated following high-frequency tetanic electrical stimulation (22). In the CA1 region of hippocampus, it has been shown that this long-term potentiation (LTP) of synapses is a specific consequence of the coincident release of the neurotransmitter glutamate from presynaptic axon terminals and the strong depolarization of the postsynaptic membrane (23, 24, 25, 26).
The initial steps in LTP induction have been well-characterized. Glutamate bound to N-methyl-d-aspartate (NMDA) receptors results in postsynaptic Ca2+ entry when the postsynaptic membrane is depolarized. Elevated [Ca2+] activates certain protein kinases, including Ca2+-calmodulin-dependent protein kinase II and protein kinase C. These kinases phosphorylate specific substrate proteins that ultimately lead to enhanced synaptic effectiveness, but precisely how and where synaptic effectiveness is increased remains controversial (27).
Potentiation of synapses that are active at the same time as strong postsynaptic activation is an assumption of most “Hebbian” learning rules, including the Cooper synapse (28). Thus, we suggested that the modification threshold, θm, corresponds to the level of postsynaptic activation at which a critical level of Ca2+ passes through the glutamate-bound NMDA receptor channel (29, 30). Associating θm with a critical level of NMDA receptor activation and postsynaptic Ca2+ entry led to the additional hypothesis that input activity that fails to activate NMDA receptors beyond the level required to trigger LTP should cause long-term depression (LTD) of the active synapses. We initially turned to slices of hippocampus to test this hypothesis.
High-frequency stimulation is particularly effective in inducing LTP in CA1 because it produces a strong postsynaptic response due to the temporal summation of dendritic excitatory postsynaptic potentials. In an effort to provide a high level of presynaptic input activity without producing a postsynaptic response so large that it yielded LTP, we tried extended periods of low-frequency stimulation (LFS) in the range of 0.5–10 Hz. We discovered that stimulation of the Schaffer collaterals at frequencies between 0.5 and 3 Hz did indeed trigger a long-lasting, homosynaptic LTD in CA1 (31).
Considerable progress has been made identifying the mechanisms of LTD induction in hippocampus. Surprisingly, induction of LTD, like LTP, requires NMDA receptor activation and a rise in intracellular Ca2+ (31, 32). Available data suggest that the information encoded by the pattern or amount of NMDA receptor activation alone may be sufficient to trigger both forms of synaptic plasticity (33). Lisman (34) showed the feasibility of this type of regulation in a model in which modest elevations of postsynaptic Ca2+ caused LTD by selectively activating protein phosphatases, and large increases in Ca2+ caused LTP by activating protein kinases. Mulkey et al. (35, 36) provided data that support this model in CA1 by showing that LTD is completely blocked by application of several phosphatase inhibitors.
Although homosynaptic LTD has been widely replicated in slices of hippocampus from young animals (37), its relevance to memory has been questioned because of failures to observe it in the adult brain in vivo (38). We recently reexamined this important issue, and found that homosynaptic LTD with all the properties described in vitro can also be elicited in the adult hippocampus in vivo (ref. 39; see also ref. 40). LTD induced in vivo remained stable for many hours without any sign of decay back to baseline (Fig. 3). Thus, LTD, like LTP, can store information long enough to contribute to a hippocampal memory store.
Figure 3.
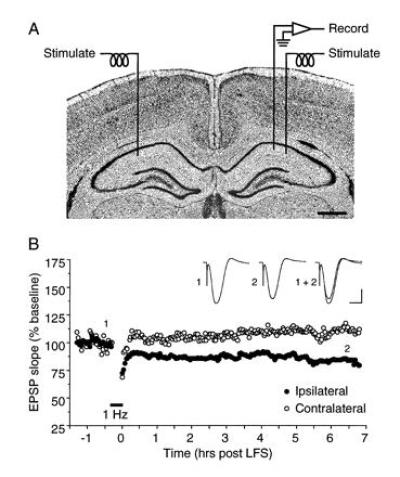
Homosynaptic LTD in adult hippocampus in vivo. (A) Schematic of the stimulation-recording configuration used in anesthetized rats. (B) LTD induced by a single episode of LFS (1 Hz, 900 pulses) is stable for as long as the preparation is viable. In this example, LFS of the ipsilateral Schaffer collaterals produced LTD that was stable for >6.5 hr. Displayed field potentials were evoked by stimulation of the ipsilateral Schaffer collaterals (averages of 10 consecutive sweeps) at times indicated by numerals. Scale bars: 2 mV, 10 ms. Figure modified from ref. 39.
By varying the stimulation frequency, but holding the stimulation intensity and total number of pulses constant, it is possible to derive a function relating synaptic plasticity to stimulation frequency (31, 39, 41). Little or no plasticity is observed at frequencies less than 0.1 Hz, robust LTD is observed using 1 Hz stimulation, and LTP is observed using stimulation frequencies greater than 10 Hz (Fig. 4). Interestingly, 10 Hz stimulation produces, on average, neither LTD nor LTP. Because the amount of presynaptic activity is held constant (900 pulses) in these experiments, the frequency-response function of Fig. 4D is equivalent to the Cooper synapse modification function in Fig. 2, if it is assumed that the measure of postsynaptic response relevant to synaptic plasticity (possibly Ca2+ current) is proportional to the stimulation frequency (see 1). Indeed, it is now well-established that the critical variables are postsynaptic depolarization and Ca2+ entry, and not stimulation frequency per se. For example, while 1 Hz stimulation normally produces LTD, postsynaptic hyperpolarization during conditioning prevents any change, and depolarization leads to induction of LTP (42). Likewise, while high-frequency stimulation normally produces LTP, it produces LTD instead if delivered in the presence of subsaturating concentrations of an NMDA receptor antagonist (33).
Figure 4.
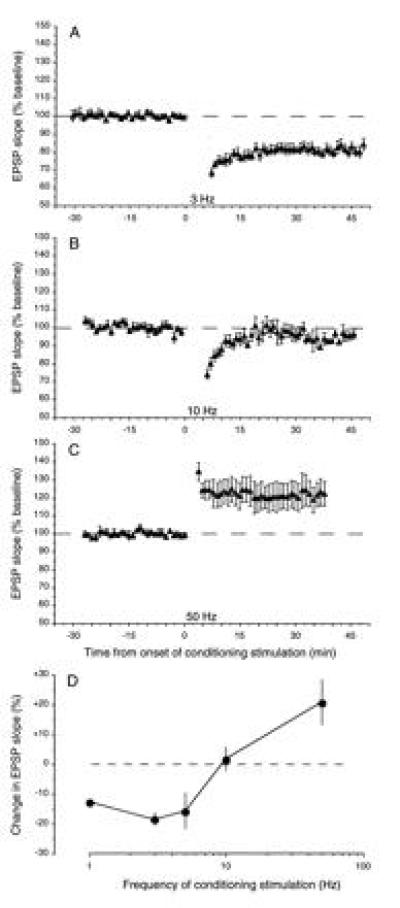
Effects of conditioning stimulation delivered to the Schaffer collaterals at different frequencies. (A–C) Normalized averages (±SEM) of experiments in which 900 pulses were delivered at different frequencies: A, 3 Hz, n = 5; B, 10 Hz, n = 6; C, 50 Hz, n = 5. (D) The mean (±SEM) effect of 900 pulses of conditioning stimulation delivered at various frequencies on the response measured 30 min postconditioning. For each point, n ≥ 5. Figure modified from ref. 31.
Additional experiments, performed both in vitro and in vivo, have shown that the same CA1 synapses support both LTD and LTP; that is, synapses are bidirectionally modifiable (35, 39, 43). Together, these data show that Cooper synapses exist in the CA1 region of hippocampus, and suggest that the mechanisms of LTP and LTD are equal partners in the storage of information in hippocampal neural networks.
Cooper Synapses in the Neocortex
It has been appreciated for some time that the neocortex is the major site of storage of nonspatial declarative memory. However, for many years it appeared that NMDA-receptor-dependent LTP might be a phenomenon expressed primarily in the hippocampus because of difficulties in reliably eliciting it in neocortex. Fortunately, the procedural difficulties have been overcome, and recent studies have shown that neocortical synapses also support both robust LTP and LTD (44, 45).
In rat visual cortex, Artola et al. (46) presented evidence suggesting that active synapses (i) are not modified if the level of postsynaptic activation during a high-frequency tetanus is low, (ii) are depressed if the level of postsynaptic activation is moderate, and (iii) are potentiated if the level of postsynaptic activation is high. We used a different approach but reached a similar conclusion. We found that stimulation of neocortical layer IV reliably induces synaptic LTP and LTD in layer III with precisely the same types of stimulation protocols that are effective in the CA1 region of hippocampus (47). Specifically, low frequency stimulation produces LTD and high frequency stimulation produces LTP (Fig. 5). As for hippocampus, neocortical LTP and LTD are specific to the conditioned pathway and are dependent upon activation of NMDA receptors (47, 48). Furthermore, homosynaptic LTD in visual cortex, like in hippocampus, is blocked by phosphatase inhibitors (48).
Figure 5.
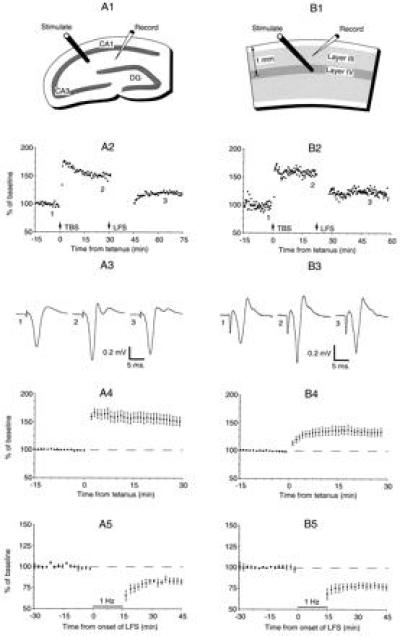
Common forms of synaptic plasticity in slices of adult rat hippocampus (A) and adult rat visual cortex (B). The top row shows the stimulation-recording configurations. DG, dentate gyrus. The second row shows changes in the extracellular field potential induced by theta-burst stimulation (TBS) and by LFS (900 pulses delivered at 1 Hz in A2 and at 3 Hz in B2). Response magnitude was measured as the change in the initial slope of the negative field potential in A2 and as the peak negativity in B2. The third row shows averages of four consecutive field potentials taken in each preparation before conditioning stimulation, after TBS, and after LFS for the experiments in row 2. The fourth row shows the average change in response magnitude after TBS (n = 4 for A4; n = 19 for B4). The fifth row shows the average change in response after LFS (900 pulses at 1 Hz), starting from an unpotentiated state (n = 5 for A5; n = 5 for B5). Figure modified from ref. 47.
We have obtained similar results using slices of mouse, rat, and cat visual cortex, prepared from neonatal and adult animals. LTP and LTD of synaptic responses in layer III following high-frequency stimulation and LFS of layer IV has also been observed in slices of rat somatosensory cortex (49). In guinea pig visual cortex, it has been shown that low-frequency electrical stimulation of white matter paired with postsynaptic depolarizing or hyperpolarizing current injections can produce transient synaptic enhancements and decrements, respectively (50). Very recently, by recording from synaptically coupled layer V neurons, Markram and Sakmann (51) beautifully confirmed that neocortical synapses are bidirectionally modifiable, with a critical variable being the pattern or amount of NMDA receptor activation. Finally, in slices of human inferotemporal cortex, a region believed to be a repository of visual memories (10), Lee et al. (52) have demonstrated LTD and LTP using the same protocols we introduced to study rat visual cortex. Together, the data support the idea that very similar principles guide synaptic plasticity in widely different regions of the cerebral cortex. Excitatory synapses in the CA1 region of hippocampus and in the superficial layers of sensory neocortex are bidirectionally modifiable, and these modifications follow the Cooper synapse learning rule.
Are the receptive fields of cortical neurons actually modified by these rules and mechanisms? It is possible to manipulate receptive field properties in visual cortex by pairing visual stimulation with various imposed patterns of postsynaptic activity (53, 54). The modifications of receptive fields observed in these studies conform well with theoretical predictions. It will be interesting to see if these changes employ the same mechanisms as LTP and LTD.
Evidence for a Sliding Modification Threshold in Visual Cortex
With the addition of a variable modification threshold proposed by Bienenstock et al. (59), Cooper synapses can account for many aspects of naturally occurring receptive field plasticity. Thus, we have searched for evidence that the value of θm varies according to the history of postsynaptic cortical activity. Postsynaptic activity can be manipulated in slices simply by stimulating synaptic inputs, and this has been shown in hippocampus to alter the thresholds for LTP and LTD (55). So far, however, these alterations in plasticity have been restricted to the stimulated synapses. Theoretical requirements for the BCM modification threshold, θm, are that its value (i) is a function of the history of the entire cell’s (or relevant computational unit’s) postsynaptic activity, and (ii) is the same at all modifiable synapses on this cell. It is possible that the failure to observe the heterosynaptic “sliding threshold” envisaged by BCM is because detectable alterations require more hours of stimulation than are available in a slice experiment, or that the mechanism is greatly reduced or absent in vitro.
We recently approached the issue of a sliding modification threshold by comparing the frequency-response function in visual cortex of animals reared in complete darkness with that in normally reared animals (56). In accordance with theoretical predictions, we found in visual cortex of light-deprived rats that LTP is enhanced, and LTD is diminished, over a range of stimulation frequencies (Fig. 6). Control experiments suggested that the alteration in synaptic plasticity was restricted to visual cortex, as similar changes were not observed in hippocampus. These findings support the concept that θm is set according to the activation history of the cortex.
Figure 6.
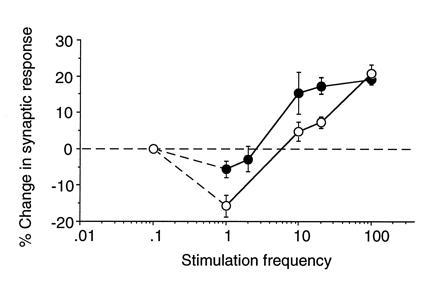
Evidence for a sliding modification threshold. Frequency-response functions derived from visual cortex of light-deprived (solid symbols) and normal (open symbols) rats. Data points for stimulation frequencies ≥ 10 Hz represent the average change (±SEM) 20 min after the delivery of 120 pulses of conditioning stimulation. Data points for 1 and 2 Hz stimulation represent the average change (±SEM) 30 min after delivery of 900 pulses of conditioning stimulation. The data point for 0.07 Hz is inferred from the fact that baseline stimulation once every 15 sec does not appear to induce synaptic modification in light-deprived or normal cortex. Figure modified from ref. 56.
As a test of the hypothesis that the value of θm actually adjusts to a change in cortical activity, visually deprived rats were exposed to light for various times, and the effects of LFS were investigated. We found that the magnitude of LTD in light-deprived visual cortex returned nearly to control levels after only 2 days of light exposure. These data are consistent with the hypothesis that θm “slides” as average cortical activity increases. It is of interest to note that the time course of the observed change in visual cortical LTD closely corresponds to that predicted for θm in modeling studies of visual cortical plasticity (57).
There are many questions remaining. Are changes caused by dark rearing, which is a profound form of sensory deprivation, also produced by brief deprivation of pattern vision in light-reared animals? Do manipulations of other types of sensory experience produce similar changes in other areas of cortex and at all ages? Does θm move in both directions at the same rate? Is the critical determinant of the value of θm actually the average postsynaptic response and, if so, what is the relevant measure of the postsynaptic response? And of course the big question remains: What is the mechanism whereby θm adjusts? Work is in progress to answer these questions.
In the meantime, however, it is interesting to consider the consequences of what apparently is a slow rate of change for θm. First, it suggests that under natural conditions the magnitude of a synaptic change occurring over a period of seconds (the “step size”) is quite small. Large, rapid synaptic modifications would require a faster moving θm to avoid stability problems; for example, to prevent runaway potentiation and saturation of synaptic weights. Thus, the commonly used experimental model of LTP, while very useful for understanding the principles and mechanisms of synaptic plasticity, may be a gross exaggeration of the changes that occur naturally. This may explain why changes equivalent to the magnitude of experimental LTP have not been observed in behaving animals during learning (58). The slow time course of θm adjustment, and the fact that it has the same value at all synapses on the same neuron, also places constraints on the possible mechanisms. Activity-dependent regulation of gene expression in the postsynaptic neuron seems ideally suited to account for this form of cellular memory.
Conclusion
In this paper, I have attempted to show how consideration of a theoretical “learning rule” has helped guide experiments into the elementary mechanisms of synaptic plasticity in the cerebral cortex. The data clearly indicate that the key assumptions of the BCM theory have a biological basis in hippocampus and neocortex. Active synapses are bidirectionally modifiable, the key variable determining the sign of the modification is the level of postsynaptic response, and the depression-potentiation crossover point θm varies depending on the history of cortical activity. In addition to serving as a guide, theoretical analysis can also be used as a bridge to connect these mechanisms with their consequences. A natural consequence is the experience-dependent modification of stimulus-selective receptive fields, reflecting the distributed storage of information in the cortical network.
Acknowledgments
I thank Drs. Serena Dudek, Arnold Heynen, Alfredo Kirkwood, and Marc Rioult for performing much of the work described here. I also thank Dr. Michael Paradiso for comments on the manuscript, and Dr. Leon Cooper for many valuable discussions. The work has been supported by the National Eye Institute, the Office of Naval Research, the National Science Foundation, the Charles A. Dana Foundation, and the Howard Hughes Medical Institute.
Relating Theoretical and Experimental Modification Functions
According to BCM,
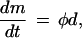 |
1 |
where m is the weight of a synaptic junction, d is the presynaptic axon activity of that synaptic junction, and φ is a function of the integrated response of the postsynaptic neuron (c). The theoretical modification function φ (c) is shown in Fig. 7A.
Figure 7.
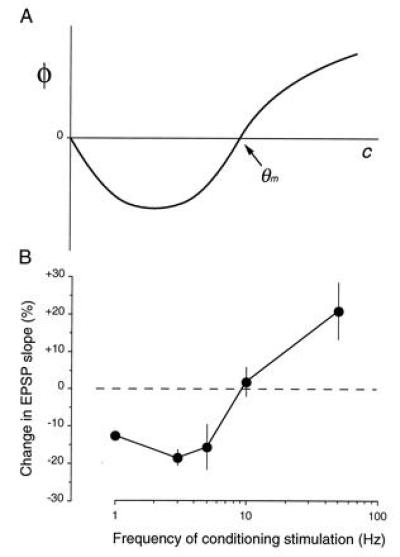
Comparison of theory and experiment.
From Eq. 1, it follows that the total change in synaptic weight (Δm) across n time steps is
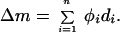 |
2 |
Because frequency (f) is constant during conditioning stimulation, it is assumed that the integrated postsynaptic response (c) is constant across the n iterations. If we also assume that the value of θm is unchanged during this short period, then the value of φ is constant during a bout of conditioning stimulation. Therefore,
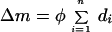 |
3 |
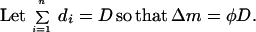 |
Because the same number of stimulation pulses are delivered, D is the same for each frequency. The measured Δm therefore is proportional to the value of φ. Thus, a plot of Δm (change in synaptic effectiveness) against log f (Fig. 7B) is equivalent to plotting φ against c, if we assume that c ∝ f. Plotting f on a logarithmic scale is for convenience.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: LTP, long-term potentiation; NMDA, N-methyl-d-aspartate; LTD, long-term depression; LFS, low-frequency stimulation.
References
- 1.Hebb D O. Organization of Behavior. New York: Wiley; 1949. [Google Scholar]
- 2.Anderson J A, Rosenfeld E, editors. Neurocomputing: Foundations of Research. Cambridge, MA: MIT Press; 1988. [Google Scholar]
- 3.Anderson J A, Mozer M C. In: Parallel Models of Associative Memory. Hinton G E, Anderson J A, editors. Hillsdale, NJ: Erlbaum; 1981. [Google Scholar]
- 4.Breese C, Hampson R, Deadwyler S. J Neurosci. 1989;9:1097–1111. doi: 10.1523/JNEUROSCI.09-04-01097.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson M, McNaughton B. Science. 1993;261:1055–1058. doi: 10.1126/science.8351520. [DOI] [PubMed] [Google Scholar]
- 6.Weinberger N, Diamond D. Prog Neurobiol. 1987;29:1–55. doi: 10.1016/0301-0082(87)90014-1. [DOI] [PubMed] [Google Scholar]
- 7.Edeline J-M, Pham P, Weinberger N M. Behav Neurosci. 1993;107:539–551. doi: 10.1037//0735-7044.107.4.539. [DOI] [PubMed] [Google Scholar]
- 8.Recanzone G H, Schreiner C E, Merzenich M M. J Neurosci. 1993;13:87–103. doi: 10.1523/JNEUROSCI.13-01-00087.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rolls E, Baylis G, Hasselmo M, Nalwa V. Exp Brain Res. 1989;76:153–164. doi: 10.1007/BF00253632. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita Y. Annu Rev Neurosci. 1993;16:245–263. doi: 10.1146/annurev.ne.16.030193.001333. [DOI] [PubMed] [Google Scholar]
- 11.Sakai K, Miyashita Y. Curr Opin Neurobiol. 1993;3:166–170. doi: 10.1016/0959-4388(93)90205-d. [DOI] [PubMed] [Google Scholar]
- 12.Fahy F L, Riches I P, Brown M W. Exp Brain Res. 1993;96:457–472. doi: 10.1007/BF00234113. [DOI] [PubMed] [Google Scholar]
- 13.Lin L I, Miller E K, Desimone R. J Neurophysiol. 1993;69:1918–1929. doi: 10.1152/jn.1993.69.6.1918. [DOI] [PubMed] [Google Scholar]
- 14.Rolls E T. Behav Brain Res. 1995;66:177–185. doi: 10.1016/0166-4328(94)00138-6. [DOI] [PubMed] [Google Scholar]
- 15.Cooper L N. In: Proceedings of the Nobel Symposium on Collective Properties of Physical Systems. Lindquist B, Lindquist S, editors. New York: Academic; 1973. pp. 252–264. [Google Scholar]
- 16.von der Malsburg C. Kybernetik. 1973;14:85–100. doi: 10.1007/BF00288907. [DOI] [PubMed] [Google Scholar]
- 17.Nass M M, Cooper L N. Biol Cybernet. 1975;19:1–18. doi: 10.1007/BF00319777. [DOI] [PubMed] [Google Scholar]
- 18.Grossberg S. Biol Cybernet. 1976;23:121–134. doi: 10.1007/BF00344744. [DOI] [PubMed] [Google Scholar]
- 19.Cooper L N, Liberman F, Oja E. Biol Cybernet. 1979;33:9–28. doi: 10.1007/BF00337414. [DOI] [PubMed] [Google Scholar]
- 20.Hubel D H, Wiesel T N. J Physiol (London) 1962;160:106–154. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fregnac Y, Imbert M. J Physiol (London) 1978;278:27–44. doi: 10.1113/jphysiol.1978.sp012290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 23.Kelso S R, Ganong A H, Brown T. Proc Natl Acad Sci USA. 1986;83:5326–5330. doi: 10.1073/pnas.83.14.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malinow R, Miller J P. Nature (London) 1986;320:529–530. doi: 10.1038/320529a0. [DOI] [PubMed] [Google Scholar]
- 25.Sastry R B, Goh J W, Auyeung A. Science. 1986;232:988. doi: 10.1126/science.3010459. [DOI] [PubMed] [Google Scholar]
- 26.Wigstrom H, Gustafsson B, Huang Y-Y, Abraham W C. Acta Physiol Scand. 1986;126:317–319. doi: 10.1111/j.1748-1716.1986.tb07822.x. [DOI] [PubMed] [Google Scholar]
- 27.Bear M F, Malenka R C. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 28.Brown T H, Kairiss E W, Keenan C L. Annu Rev Neurosci. 1990;13:475–511. doi: 10.1146/annurev.ne.13.030190.002355. [DOI] [PubMed] [Google Scholar]
- 29.Bear M F, Cooper L N, Ebner F F. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 30.Bear M F, Cooper L N. In: Neuroscience and Connectionist Theory. Gluck M A, Rumelhart D E, editors. Hillsdale, NJ: Erlbaum; 1990. pp. 65–93. [Google Scholar]
- 31.Dudek S M, Bear M F. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulkey R M, Malenka R C. Soc Neurosci Abstr. 1992;18:1498. [Google Scholar]
- 33.Cummings J A, Mulkey R M, Nicoll R A, Malenka R C. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 34.Lisman J. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulkey R M, Herron C E, Malenka R C. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 36.Mulkey R M, Endo S, Shenolikar S, Malenka R C. Nature (London) 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 37.Bear M, Abraham W. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 38.Errington M L, Bliss T V P, Richter-Levin K, Yenk K, Doyère V, Laroche S. J Neurophysiol. 1995;74:1793–1799. doi: 10.1152/jn.1995.74.4.1793. [DOI] [PubMed] [Google Scholar]
- 39.Heynen A J, Abraham W C, Bear M F. Nature (London) 1996;381:163–166. doi: 10.1038/381163a0. [DOI] [PubMed] [Google Scholar]
- 40.Thiels E, Barrionuevo G, Berger T W. J Neurophysiol. 1994;71:3009–3016. doi: 10.1152/jn.1994.72.6.3009. [DOI] [PubMed] [Google Scholar]
- 41.Mayford M, Wang J, Kandel E, O’Dell T. Cell. 1995;81:1–20. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 42.Mulkey R M, Malenka R C. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 43.Dudek S M, Bear M F. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsumoto T. Prog Neurobiol. 1992;39:209–228. doi: 10.1016/0301-0082(92)90011-3. [DOI] [PubMed] [Google Scholar]
- 45.Bear M F, Kirkwood A. Curr Opin Neurobiol. 1993;3:197–202. doi: 10.1016/0959-4388(93)90210-p. [DOI] [PubMed] [Google Scholar]
- 46.Artola A, Brocher S, Singer W. Nature (London) 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- 47.Kirkwood A, Dudek S M, Gold J T, Aizenman C D, Bear M F. Science. 1993;260:1518–1521. doi: 10.1126/science.8502997. [DOI] [PubMed] [Google Scholar]
- 48.Kirkwood A, Bear M F. J Neurosci. 1994;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castro-Alamancos M A, Donoghue J P, Connors B W. J Neurosci. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frégnac Y, Burke J, Smith D, Friedlander M. J Neurophysiol. 1994;71:1403–1421. doi: 10.1152/jn.1994.71.4.1403. [DOI] [PubMed] [Google Scholar]
- 51.Markram H, Sakmann B. Soc Neurosci Abstr. 1995;21:2007. [Google Scholar]
- 52.Chen W R, Lee S, Kato K, Spencer D D, Sheperd G M, Williamson A. Proc Natl Acad Sci USA. 1996;93:8011–8015. doi: 10.1073/pnas.93.15.8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frégnac Y, Shulz D, Thorpe S, Bienenstock E. Nature (London) 1988;333:367–370. doi: 10.1038/333367a0. [DOI] [PubMed] [Google Scholar]
- 54.Frégnac Y, Shulz D, Thorpe S, Bienenstock E. J Neurosci. 1992;12:1280–1300. doi: 10.1523/JNEUROSCI.12-04-01280.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abraham W, Bear M. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 56.Kirkwood A, Rioult M, Bear M. Nature (London) 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- 57.Clothiaux E E, Bear M F, Cooper L N. J Neurophysiol. 1991;66:1785–1804. doi: 10.1152/jn.1991.66.5.1785. [DOI] [PubMed] [Google Scholar]
- 58.Moser E, Moser M-B, Andersen P. Learning and Memory. 1994;1:53–73. [PubMed] [Google Scholar]
- 59.Bienenstock E L, Cooper L N, Munro P W. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]