

Abstract
We report the discovery of antibacterial leads, keto- and diketo-acids, targeting two prenyl transferases: undecaprenyl diphosphate synthase (UPPS) and dehydrosqualene synthase (CrtM). The leads were suggested by the observation that keto- and diketo-acids bind to the active site Mg2+/Asp domain in HIV-1 integrase, and similar domains are present in prenyl transferases. We report the X-ray crystallographic structures of one diketo-acid and one keto-acid bound to CrtM, which supports the Mg2+ binding hypothesis, together with the X-ray structure of one diketo-acid bound to UPPS. In all cases, the inhibitors bind to a farnesyl diphosphate substrate-binding site. Compound 45 had cell growth inhibition MIC90 values of ∼250–500 ng/mL against Staphylococcus aureus, 500 ng/mL against Bacillus anthracis, 4 μg/mL against Listeria monocytogenes and Enterococcus faecium, and 1 μg/mL against Streptococcus pyogenes M1 but very little activity against Escherichia coli (DH5α, K12) or human cell lines.
Keywords: antibacterials, isoprenoid biosynthesis, HIV integrase, undecaprenyl diphosphate synthase, dehydrosqualene synthase
There is currently an urgent need for new types of antibacterials exhibiting novel modes of action, due to the rapid rise in drug resistance,1 and isoprenoid biosynthesis2,2b is one attractive target. For example, cell wall biosynthesis can be inhibited by targeting farnesyl diphosphate synthase (FPPS) or undecaprenyl diphosphate synthase (UPPS), involved in lipid I biosynthesis (Figure 1). In addition, in Staphylococcus aureus, the formation of the virulence factor staphyloxanthin3 can be blocked by inhibiting dehydrosqualene synthase (CrtM), resulting in a lowering of the antioxidant shield to host derived reactive oxygen species (ROS)4 (Figure 1). The bisphosphonate class of drugs such as zoledronate (1, Chart 1) are potent, low nanomolar inhibitors of FPPS, but 1 has little antibacterial activity (presumably due to lack of cell penetration), although more lipophilic bisphosphonates such as 2 (BPH-210, Chart 1) have modest activity (IC50 ∼ 30 μM) against Escherichia coli.5 More lipophilic bisphosphonates also potently target UPPS,6 as well as CrtM,4 but again, they have essentially no activity in bacteria. Replacing one phosphonate group by a sulfonate to form a phosphonosulfonate results, however, in potent CrtM inhibitors (e.g., 3, BPH-652, Chart 1, IC50 ∼ 7.9 μM, Ki ∼ 80 nM) that also blocks carotenoid pigment formation in cells (IC50 ∼ 110 nM).7 In addition, there has recently been interest in developing phosphorus-free prenyl transferase inhibitors, which might have even more druglike properties. For example, Jahnke et al. reported a series of FPPS inhibitors, dicarboxylic acids, that bound to a novel, allosteric site.8 In addition, other species such as tetramic acid UPPS inhibitors have been described (e.g., 4, Chart 1),9 but to date, their X-ray structures have not been reported, although an allosteric model has been proposed.10
Figure 1.
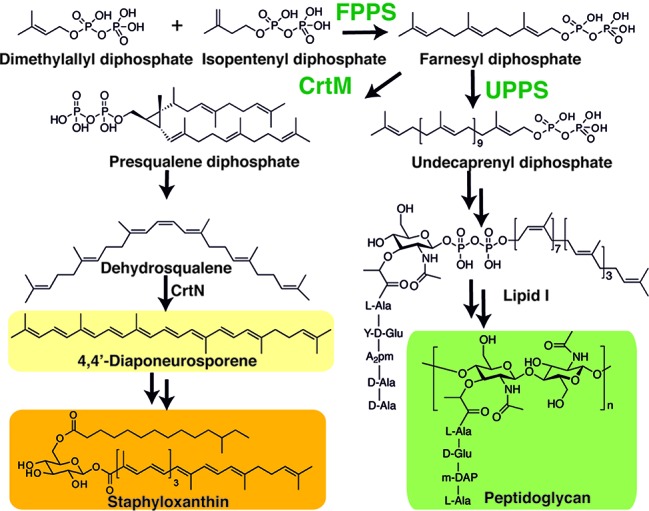
Biosynthetic reactions catalyzed by CrtM and UPPS, with the end products of the pathways shown.
Chart 1. Chemical Structures of Selected Compounds and the Inhibition of CrtM, E. coli UPPS, and S. aureus UPPS by 7–9, 41, 42, 44, and 45.

A key component of the active site of most prenyl transferases is a Mg2+/Asp motif that interacts with a substrate's diphosphate group. We reasoned that HIV-1 integrase (IN) inhibitors11 might provide clues for new prenyl transferase inhibitors, since IN contains a similar Asp/Mg2+ motif12 and IN inhibitors such as 5 (L-708,906, Chart 1)13 and 6 (elvitegravir, Chart 1),14 diketo-acids and keto-acids, respectively, are thought to bind at or near the Mg2+/Asp motif in the IN active site.15,15b In addition, many other IN inhibitors like raltegravir, dolutegravir, MK2048, etc. (structures not shown) have been found to bind Mg2+.15b−16b
We thus made a small screening library (38 compounds) of IN inhibitor-inspired molecules and their structures, and inhibition of S. aureus CrtM, E. coli UPPS, and S. aureus UPPS are shown in Figure S1 in the Supporting Information. Most compounds were amide-diketo acids (7–40, class I, Figure S1 in the Supporting Information) and were conveniently prepared from the synthon (Z)-2,2-dimethyl-5-carboxymethylene-1,3-dioxolan-4-one17 by amine coupling. Among these compounds, 7 (Chart 1) inhibited CrtM with IC50 ∼ 24 μM, Ki ∼ 250 nM (for comparison, Ki of 3 ∼ 70 nM7) and blocked staphyloxanthin pigment formation (IC50 = 4 μM). Inhibitors of class II were keto-acids, dihydropyridone-3-carboxylates, and were based on 6 (Elvitegravir) and dihydroquinoline-3-carboxylic acid IN inhibitors,18 which again are thought to bind via their carboxyl and carbonyl oxygens to Mg2+/Asp.19 We made two analogues, 41 and 42 (Chart 1), with alkoxy-aryl tails to mimic the substrate farnesyl diphosphate (FPP). The longer chain species 42 had no activity, but the shorter chain species 41 had a CrtM IC50= 45 μM, Ki = 450 nM, and a loss of pigmentation IC50 = 33 μM.
To see how these inhibitors bound to CrtM, we carried out cocrystallization and soaking experiments with 7 (class I) and 41 (class II) and obtained crystals (by soaking) that diffracted to 2.3 and 1.9 Å, respectively. Full X-ray crystallographic data and structure refinement details are given in the Supporting Information, Table S1. Electron density results for 7 are shown in Figure 2a and indicate the presence of 7 in addition to one molecule of farnesyl monophosphate (FMP) that copurified with the protein. The identity of FMP was further confirmed by LC-MS (Supporting Information, Figure S2) and the electron density results (Figure 2a). The diphenyl ether fragment in 7 (cyan) binds into the CrtM S1 site20 and is shown in Figure 2b superimposed on one of the S-thiolo-farnesyl diphosphate (FSPP) inhibitors (in yellow, green) whose structures were reported previously.4 This binding mode is similar to that seen with the phosphonosulfonate 3 (Figure 2c), with the diketo-acid headgroup interacting with two of the three Mg2+ (Mg2+B,C) seen in the CrtM-FSPP structure (Figure 2d). The farnesyl side chain in FMP bound to the S2 site and had a 0.8 Å rmsd from the S2 FSPP reported previously.4 With 41, the ligand electron density is again well-defined (Figure 3a), and the crystallographic results show that the side chain binds in S2, similar to the farnesyl side chain in the FSPP structures (Figure 3b), as well as the phosphonoacetamide analogue of 7 (43, BPH-830,21 Figure 3c). There are three Mg2+ in the X-ray structure. However, these are not the Mg2+ABC seen in most prenyl transferases22 but rather Mg2+BCD. That is, there is a new Mg2+ binding site, Mg2+D. The dihydropyridone side chain interacts with Mg2+CD but, surprisingly, via the two ring oxygens, not the carboxylate (Figure 3d), which interacts with two water molecules (Supporting Information, Figure S3). These inhibition and structural results for 7 and 41 clearly support the Mg2+ binding hypothesis, at least for CrtM.
Figure 2.
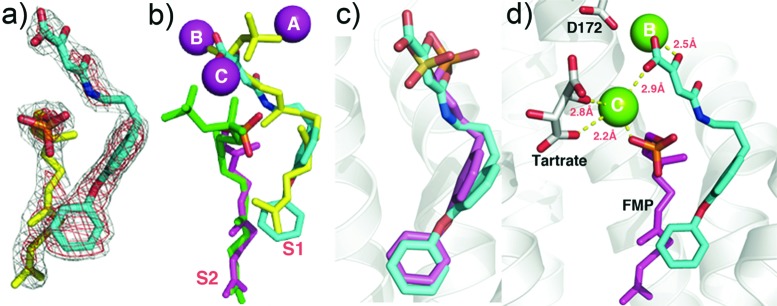
CrtM crystallographic structures. (a) Compound 7 (cyan) plus FMP (yellow) electron density, bound to CrtM. (b) Compound 7 (cyan) bound to CrtM, superimposed on two FSPP molecules (yellow, green; PDB ID code 2ZCP). Also shown is the FMP (magenta) that cocrystallized. The Mg2+ are from the FSPP structure. (c) Comparison between 7 (cyan) and 3 (magenta, PDB ID code 2ZCQ) bound to CrtM. Both diphenyl ether side chains bind in S1. (d) Interactions between 7 (cyan), FMP (magenta), and Mg2+ in CrtM.
Figure 3.
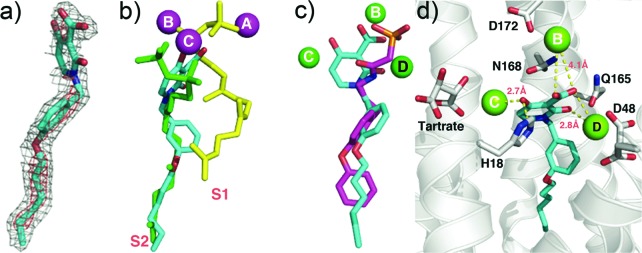
CrtM crystallographic structures. (a) Electron density of 41 bound to CrtM. (b) Structure of 41 (cyan) bound to CrtM shown superimposed on two FSPP molecules (yellow, green). (c) Comparison between 41 (cyan) and the phosphonoacetamide analog of 7 (compound 43, BPH-830)21 (magenta) bound to CrtM (PDB ID code 2ZY1). (d) Interactions between 41 (cyan) and Mg2+ in CrtM.
CrtM is a so-called head-to-head prenyl transferase so we next sought to see if any of the molecules synthesized might also inhibit the head-to-tail prenyl transferase FPPS or the cis-prenyl transferase, UPPS. There was no activity against FPPS (probably due to the lack of a positively charged feature that mimics the carbocation involved in FPP biosynthesis), but most of the amide-diketo acids (class I) were potent UPPS inhibitors with the most active one (8) having an IC50 ∼ 240 nM and Ki ∼ 120 nM, comparable to the most active bisphosphonate UPPS inhibitor BPH-629 (IC50 ∼ 300 nM for E. coli UPPS).6 There are four different ligand-binding sites in UPPS (designated 1–4 in ref (6)) found with bisphosphonate inhibitors. This is not unexpected since the UPPS product, undecaprenyl diphosphate (UPP), contains 55-carbon atoms and is thus much larger than the (C15) FPP substrate. In principle, then, novel inhibitors might occupy multiple binding sites.
Cocrystallization of E. coli UPPS with 9 (IC50 = 560 nM) produced well-formed crystals with E. coli UPPS, and the electron density was well resolved (Figure 4a). As can be seen in Figure 4b, 9 binds to site 1,6 the FPP binding site, and as can be seen in Figure 4c, 9 (in cyan) closely maps the FPP backbone structure (in yellow) with the diketo-acid fragment being located close to two of the three most essential residues in UPPS, D26 and N28 (Figure 4d). We found no evidence for the presence of Mg2+, but this observation is not entirely unexpected since even with the five E. coli UPPS X-ray structures with strong Mg2+ chelators, bisphosphonates (PDB ID codes 2E98, 2E99, 2E9A, 2E9C, and 2E9D),6 Mg2+ was not observed.
Figure 4.
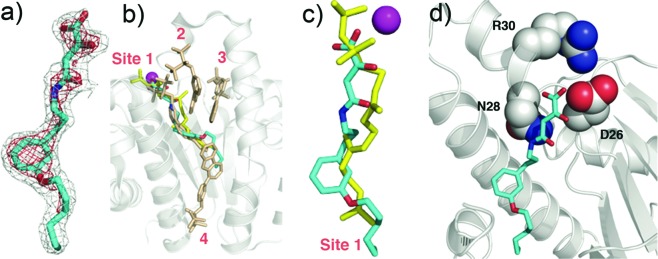
UPPS crystallographic structures. (a) Electron density of 9 bound to UPPS. (b) Structure of 9 (cyan) bound to UPPS, superimposed on FSPP/Mg2+ (from PDB ID code 1X06) and four bisphosphonate inhibitors (PDB ID code 2E98). (c) Superposition of 9 (cyan) on FSPP (yellow) in site 1 in UPPS. The Mg2+ is from the FSPP structure. (d) The diketo-acid headgroup of 9 binds into the active site of UPPS and interacts with D26 and N28.
The amide-diketo acids were not growth suppressive toward S. aureus or E. coli, perhaps due to the instability of the amide bond inside the cells or a lack of cell permeability. However, 44 and 45 (aryldiketo acids, class III) had good activity against S. aureus UPPS (44, IC50 = 0.73 μM, Ki = 230 nM; 45, IC50 = 2.0 μM, Ki = 670 nM), and both were active against the USA300 (MRSA) strain of S. aureus with MIC90 values of 500 (44) and 250–500 ng/mL (45). There was no appreciable activity against the Gram-negative E. coli; however, there was promising activity against other Gram-positives: ∼ 500 ng/mL against Bacillus anthracis str. Sterne, ∼ 4 μg/mL against Listeria monocytogenes and Enterococcus faecium U503, and ∼1 μg/mL for Streptococcus pyogenes M1. While the precise mechanism of action of these compounds in each cell remains to be determined, UPPS inhibition is a likely candidate. In addition, we found low toxicity against a human cell line (MCF-7; IC50 ∼ 30 μM), consistent with poor FPPS inhibition.
These results are important for several reasons. First, we tested the hypothesis that keto- and diketo-acids might inhibit prenyl transferase enzymes, based on the presence of Mg2+/Asp motifs in their active sites—an “integrase inhibitor-inspired” approach. The best CrtM inhibitors had Ki ∼ 250 nM and were active in blocking staphyloxanthin biosynthesis in S. aureus, and we solved two structures of lead compounds bound to CrtM. In both, the inhibitor head groups bound to Mg2+, while the side chains bound to one or the other of the two FPP side chain binding sites. Second, we tested this small library for FPPS and UPPS inhibition. There was no FPPS inhibition, but the most potent UPPS inhibitor had an IC50 = 240 nM, and we determined the structure of one such lead bound to E. coli UPPS—the first UPPS X-ray structure reported for a nonbisphosphonate inhibitor. We also found low toxicity and promising activity against a subset of Gram-positive bacteria with MIC90 values as low as 250–500 ng/mL against USA300 S. aureus and 500 ng/mL against Bacillus anthracis str. Sterne and low activity against E. coli and a human cell line. Overall, these results indicate that integrase-inspired inhibitors may be engineered into drug leads that target isoprenoid biosynthesis.
Acknowledgments
We thank Andrew H.-J. Wang of the Institute of Biological Chemistry, Academia Sinica (Taipei, Taiwan), for providing E. coli UPPS plasmids and S. aureus CrtM plasmids.
Glossary
Abbreviations
- CrtM
dehydrosqualene synthase
- UPPS
undecaprenyl diphosphate synthase
- FPPS
farnesyl diphosphate synthase
- FPP
farnesyl diphosphate
- FMP
farnesyl monophosphate
- FSPP
S-thiolo-farnesyl diphosphate
- IN
HIV-1 integrase
Supporting Information Available
X-ray study, synthesis, and characterization of the screening library compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the U.S. Public Health Service (NIH Grant 5R01AI074233-16 to E.O.) and the NIH Director's New Innovator Award Program (DP2 OD008463 to D.A.M.). K.J.M. was supported in part by a NIH Cellular and Molecular Biology Training Grant (T32 GM007283). The Advanced Photon Source was supported by Department of Energy Contract DE-AC02-06CH11357. The Life Science Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor (Grant 085P000817).
The authors declare no competing financial interest.
Author Contributions
○ These authors contributed equally.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Walsh C. T.; Fischbach M. A. Repurposing libraries of eukaryotic protein kinase inhibitors for antibiotic discovery. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 1689–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E. Targeting isoprenoid biosynthesis for drug discovery: Bench to bedside. Acc. Chem. Res. 2010, 43, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield E.; Lin F. Y. Terpene biosynthesis: Modularity rules. Angew. Chem., Int. Ed. Engl. 2012, 51, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. Y.; Essex A.; Buchanan J. T.; Datta V.; Hoffman H. M.; Bastian J. F.; Fierer J.; Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. I.; Liu G. Y.; Song Y.; Yin F.; Hensler M. E.; Jeng W. Y.; Nizet V.; Wang A. H.; Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon A.; Liu L.; Yang Y.; Hudock M. P.; Hall P.; Yin F.; Studer D.; Puan K. J.; Morita C. T.; Oldfield E. Isoprenoid biosynthesis as a drug target: Bisphosphonate inhibition of Escherichia coli K12 growth and synergistic effects of fosmidomycin. J. Med. Chem. 2006, 49, 7331–7341. [DOI] [PubMed] [Google Scholar]
- Guo R. T.; Cao R.; Liang P. H.; Ko T. P.; Chang T. H.; Hudock M. P.; Jeng W. Y.; Chen C. K.; Zhang Y.; Song Y.; Kuo C. J.; Yin F.; Oldfield E.; Wang A. H. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Lin F. Y.; Yin F.; Hensler M.; Rodrigues Poveda C. A.; Mukkamala D.; Cao R.; Wang H.; Morita C. T.; Gonzalez Pacanowska D.; Nizet V.; Oldfield E. Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. J. Med. Chem. 2009, 52, 976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke W.; Rondeau J. M.; Cotesta S.; Marzinzik A.; Pelle X.; Geiser M.; Strauss A.; Gotte M.; Bitsch F.; Hemmig R.; Henry C.; Lehmann S.; Glickman J. F.; Roddy T. P.; Stout S. J.; Green J. R. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat. Chem. Biol. 2010, 6, 660–666. [DOI] [PubMed] [Google Scholar]
- Peukert S.; Sun Y.; Zhang R.; Hurley B.; Sabio M.; Shen X.; Gray C.; Dzink-Fox J.; Tao J.; Cebula R.; Wattanasin S. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): Tetramic, tetronic acids and dihydropyridin-2-ones. Bioorg. Med. Chem. Lett. 2008, 18, 1840–1884. [DOI] [PubMed] [Google Scholar]
- Lee L. V.; Granda B.; Dean K.; Tao J.; Liu E.; Zhang R.; Peukert S.; Wattanasin S.; Xie X.; Ryder N. S.; Tommasi R.; Deng G. Biophysical investigation of the mode of inhibition of tetramic acids, the allosteric inhibitors of undecaprenyl pyrophosphate synthase. Biochemistry 2010, 49, 5366–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, seeNeamati N.HIV-1 Integrase: Mechanism and Inhibitor Design; John Willey & Sons Inc.: Hoboken, NJ, 2011. [Google Scholar]
- Goldgur Y.; Dyda F.; Hickman A. B.; Jenkins T. M.; Craigie R.; Davies D. R. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda D. J.; Felock P.; Witmer M.; Wolfe A.; Stillmock K.; Grobler J. A.; Espeseth A.; Gabryelski L.; Schleif W.; Blau C.; Miller M. D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [DOI] [PubMed] [Google Scholar]
- Sato M.; Kawakami H.; Motomura T.; Aramaki H.; Matsuda T.; Yamashita M.; Ito Y.; Matsuzaki Y.; Yamataka K.; Ikeda S.; Shinkai H. Quinolone carboxylic acids as a novel monoketo acid class of human immunodeficiency virus type 1 integrase inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [DOI] [PubMed] [Google Scholar]
- Grobler J. A.; Stillmock K.; Hu B.; Witmer M.; Felock P.; Espeseth A. S.; Wolfe A.; Egbertson M.; Bourgeois M.; Melamed J.; Wai J. S.; Young S.; Vacca J.; Hazuda D. J. Diketo acid inhibitor mechanism and HIV-1 integrase: Implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6661–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Smith S. J.; Métifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S.; Cherepanov P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK 1349572). Mol. Pharmacol. 2011, 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Vos A. M.; Clayton R. F.; Thuring J. W.; Cummings M. D.; Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K.; Simpson J. H.; Delaney E. J.; Nugent W. A. Synthesis of Z-5-carboxymethylene-1,3-dioxolan-4-ones: A better way. J. Org. Chem. 2007, 72, 3949–3951. [DOI] [PubMed] [Google Scholar]
- Sechi M.; Rizzi G.; Bacchi A.; Carcelli M.; Rogolino D.; Pala N.; Sanchez T. W.; Taheri L.; Dayam R.; Neamati N. Design and synthesis of novel dihydroquinoline-3-carboxylic acids as HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2009, 17, 2925–2935. [DOI] [PubMed] [Google Scholar]
- Vandurm P.; Cauvin C.; Guiguen A.; Georges B.; Le Van K.; Martinelli V.; Cardona C.; Mbemba G.; Mouscadet J. F.; Hevesi L.; Van Lint C.; Wouters J. Structural and theoretical studies of [6-bromo-1-(4-fluorophenylmethyl)-4(1H)-quinolinon-3-yl)]-4-hydroxy-2-oxo- 3-butenoic acid as HIV-1 integrase inhibitor. Bioorg. Med. Chem. Lett. 2009, 19, 4806–4809. [DOI] [PubMed] [Google Scholar]
- Lin F. Y.; Liu C. I.; Liu Y. L.; Zhang Y.; Wang K.; Jeng W. Y.; Ko T. P.; Cao R.; Wang A. H.; Oldfield E. Mechanism of action and inhibition of dehydrosqualene synthase. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21337–21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Liu C. I.; Lin F. Y.; No J. H.; Hensler M.; Liu Y. L.; Jeng W. Y.; Low J.; Liu G. Y.; Nizet V.; Wang A. H.; Oldfield E. Inhibition of staphyloxanthin virulence factor biosynthesis in Staphylococcus aureus: In vitro, in vivo, and crystallographic results. J. Med. Chem. 2009, 52, 3869–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaron J. A.; Christianson D. W. Trinuclear Metal Clusters in Catalysis by Terpenoid Synthases. Pure Appl. Chem. 2010, 82, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.