

Abstract
Phospholipases A2 (PLA2s) represent one of the largest groups of lipid-modifying enzymes. Over the years, significant advances have been made in understanding their potential physiological and pathological functions. Depending on their calcium requirement for activation, PLA2s are classified into calcium dependent and independent. This paper mainly focuses on brain calcium-independent PLA2 (iPLA2) and on the mechanisms by which they influence neuronal function and regulate synaptic plasticity. Particular attention will be given to the iPLA2 γ isoform and its role in the regulation of synaptic glutamate receptors. In particular, the paper discusses the possibility that brain iPLA2 γ deficiencies could destabilise normal synaptic operation and might contribute to the aetiology of some brain disorders. In this line, the paper presents new data indicating that iPLA2 γ deficiencies accentuate AMPA receptor destabilization and tau phosphorylation, which suggests that this iPLA2 isoform should be considered as a potential target for the treatment of Tau-related disorders.
1. Introduction
The nervous system is formed by integrated neuronal circuits which all require constant adaptation for stabilizing their activities in the face of perturbations that alter, for instance, neuronal excitability. Phenomena that conform to this definition include the activity-dependent regulation of intrinsic neuronal firing properties [1, 2], pre- and postsynaptic forms of excitatory synaptic plasticity, such as synaptic scaling, that adjust all of a neuron's excitatory synapses up or down in the right direction to stabilize firing [3, 4]; the balancing of excitation and inhibition within neuronal networks [5, 6]; compensatory changes in synapse number [7]; apposition of presynaptic and postsynaptic elements [4] and metaplastic mechanisms that adjust long-term changes in synaptic operation [8, 9]. In general, it is believed that the final refinements of neuronal circuits rely on the stabilization of functionally appropriate connections and the elimination of inappropriate ones.
While the molecular mechanisms of synapse formation have been extensively studied, very little is known about the molecular mechanisms that are responsible for stabilization of synaptic connections. Over the recent years, however, it has been proposed that the level of AMPA subtype of glutamate receptors found at neuronal connections might be a crucial component controlling both stabilization of presynaptic inputs and postsynaptic spine morphogenesis (see [10]). In the present paper, we will focus on the possibility that a specific PLA2 isoform can interact with AMPA receptor properties to contribute to synaptic stabilization. We will, in this line, present some new information indicating that iPLA2 γ deficiency might undermine the normal stabilizing mechanisms underlying memory formation in the hippocampus and contribute to Alzheimer's disease pathology.
2. iPLA2 Isoforms, Long-Term Potentiation, and Memory Stabilization
Phospholipases A2 (PLA2s) constitute a large and diverse group of enzymes with broad biological functions, ranging from membrane synthesis and turnover to the generation of signaling molecules. So far, more than 20 isoforms of PLA2 with diverse characteristics, including calcium requirement and subcellular localization, have been identified. Based on nucleotide sequences as well as other properties, PLA2s have been categorized into 15 groups (I–XV) [11, 12]. Several types of released small PLA2s (~14 kDa) require millimolar amounts of calcium for optimal activation. These enzymes have historically been called the secreted forms of PLA2 (or sPLA2). The remaining groups are larger proteins, localized in intracellular compartments, and are either calcium dependent or independent.
The first intracellular PLA2 to be cloned was a protein of 85-kDa, classified as group IV PLA2 [13, 14]. This enzyme, now designated as cytosolic PLA2 α (cPLA2 α), is under the influence of extracellular signals likely to induce calcium mobilization and phosphorylation reactions [13]. Another group of PLA2 (group VI), which does not require calcium variations for activity, has been cloned [15–17]. This PLA2 isoform has been designated as calcium-independent PLA2 (iPLA2) (Table 1) [14, 18], and according to numerous lines of biochemical evidence may account for most PLA2 activity under resting conditions. Whereas cPLA2 and sPLA2 are commonly believed to be preferentially involved in AA release, emerging evidence indicates that iPLA2 activity can contribute to docosahexaenoic acid (DHA) release from brain phospholipids. Pharmacologically, iPLA2 activity is markedly reduced by bromoenol lactone (BEL), a suicide substrate, which is not an effective inhibitor of sPLA2 or cPLA2 at comparable concentrations [15, 19]. Several interesting reviews have considered the physiological and pathological functions of PLA2 enzymes [19–30]. In this paper, we describe new and unique functional roles of iPLA2 in the regulation of brain glutamate receptor functions, neuronal plasticity, and neurodegenerative processes.
Table 1.
Calcium-independent group VI phospholipase A2 (iPLA2).
| Group | Source | Molecular mass (kDa) | Feature | Alternate names |
|---|---|---|---|---|
| VIA-1 | Human/Murine | 84-85 | 8 ankyrin repeats | iPLA2 |
| VIA-2 | Human/Murine | 88–90 | 7 ankyrin repeats | iPLA2 β |
| VIB | Human/Murine | 88–91 | Membrane-bound | iPLA2 γ |
| VIC | Human/Murine | 146 | Integral membrane protein | iPLA2 δ |
| VID | Human | 53 | Acylglycerol transacylase, triglycerol lipase | iPLA2 ε |
| VIE | Human | 57 | Acylglycerol transacylase, triglycerol lipase | iPLA2 ξ |
| VIF | Human | 28 | Acylglycerol transacylase, triglycerol lipase | PLA2 η |
Among PLA2 enzymes, group IV (cPLA2) and group VI (iPLA2) families represent intracellular enzymes with a catalytic serine in their lipase consensus motif. Various studies, including gene targeting, have indicated that group IVA cPLA2 (cPLA2α), which is regulated by calcium-dependent membrane translocation and mitogen-activated protein kinase- (MAPK-) dependent phosphorylation, is essential for stimulus-dependent eicosanoid biosynthesis [31, 32]. On the other hand, group VIA iPLA2 (iPLA2β) and group VIB iPLA2 (iPLA2 γ) isoforms mainly exhibit phospholipase activity, whereas the other iPLA2 isoforms δ, ε, ξ, and η display triglyceride lipase and transacylase activities (see Table 1) [33, 34]. Members of this family share a protein domain discovered initially in patatin, the most abundant protein of the potato tuber. Patatin (also called iPLA2α) is a lipid hydrolase with an unusual folding topology that differs from classical lipases. The catalytic dyad (Ser-Asp) rather than the catalytic triad (Ser-His-Asp) is found in classical lipases and does not contain a lid domain usually required for interfacial activation [35]. The catalytic dyad is located in a conserved catalytic domain that shows 40% homology with patatin/iPLA2α. Adjacent to the catalytic center, there is a conserved nucleotide binding motif. The iPLA-type enzymes (i.e., iPLA2β and iPLA2γ) typically possess a long N-terminal domain, which may be involved in protein-protein interaction, distinct translation, and membrane spanning. Like numerous proteins containing the uptake-targeting Ser-Lys-Leu (SKL) sequence, it has been found that iPLA2g tightly associated with peroxisomal membranes [36]. The lipase-type enzymes (i.e., iPLA2 isoforms δ, ε, ξ and η) lack the N-terminal domain and are thought to act primarily on triglycerides or other neutral lipids in lipid droplets [37]. Very little is known about the developmental brain expression of iPLA2 in the brain. In mouse, expression of iPLA2 enzymes has been reported in sagittal sections at embryonic day 14.5 [38]. At this stage, the strongest expression seen in the brain is in the alar plate of the developing hindbrain with prominent expression also in an analogous region of the midbrain. IPLA2s also appear to be expressed in the developing diencephalon and telencephalon of the forebrain. In situ hybridization studies have revealed that across several stages of human embryonic and early fetal development, iPLA2s show a dynamic expression pattern both in terms of the location of expression and the differentiation state of expressing cells. In brief, iPLA2s are expressed in forebrain and midbrain before it is detectable in hindbrain. Throughout the developing brain, iPLA2s are mainly expressed in proliferative zones, suggesting that these enzymes are important for early neuronal development [38]. The precise pattern of expression of both group VIA iPLA2 (iPLA2β) and group VIB iPLA2 (iPLA2 γ) enzymes still unclear, and one important priority for future studies will be the precise identification of iPLA2 isoforms responsible for brain development and stabilization.
Group VIA iPLA2 β, the most extensively studied iPLA2 isoform, has been implicated in various cellular events, such as phospholipid remodelling [18, 39], eicosanoid formation [40], cell growth [41, 42], apoptosis [43], and activation of store-operated channels and capacitative calcium influx [44]. Disruption of the iPLA2 β gene causes impaired sperm motility [45], mitigated insulin secretion [46, 47], and neuronal disorders with iron dyshomeostasis [48]. Group VIB iPLA2 γ is a membrane-bound iPLA2 enzyme with unique features, such as the utilization of distinct translation initiation sites producing different sizes of enzymes with distinct subcellular localizations [36, 49–53] and phospholipid selectivity in terms of sn-1/sn-2 positional specificity, which differs among substrates [54]. iPLA2 γ has a mitochondrial localization signal in the N-terminal region and a peroxisomal localization signal near the C-terminus, and the 88-kDa full-length and 63-kDa translation products of iPLA2 γ are preferentially distributed in mitochondria and peroxisomes, respectively [49–51]. In brain, iPLA2 constitutes the predominant phospholipase activity under resting conditions [55, 56]. Reverse transcription-polymerase chain reaction experiments have revealed that rat brain constitutively expresses mRNAs for at least 3 calcium-independent PLA2 isoforms, iPLA2 β, iPLA2 γ and cPLA2 γ [16, 57, 58]. These isoforms are characterized by different sensitivity to PLA2 inhibitors, including different enantiomers of an inhibitor; Jenkins et al. [59] established that the (S)-enantiomer of BEL selectively reduces iPLA2 β activity, while its (R)-enantiomer blocks the iPLA2 γ isoform more efficiently.
Although little is known about iPLA2 functions in neurons, a growing body of evidence suggests their involvement in hippocampal long-term potentiation (LTP) of excitatory synaptic transmission [55, 60]. Hippocampal LTP, first described by Bliss and Lomo in 1973, is commonly regarded as a functional model of synaptic adaptation (i.e., plasticity) that likely participates in certain forms of learning and memory [61–63]. PLA2 activities are increased in membranes prepared from the dentate gyrus after LTP induction in anaesthetized rats [64]; it has been proposed that PLA2 could be involved in hippocampal LTP expression by elevating the production of arachidonic acid (AA) that could retrogradely increase transmitter release at glutamatergic synapses [65, 66]. The hypothesis that facilitation of transmitter release by PLA2s occurs during LTP is reinforced by the fact that iPLA2 activity plays an important role in membrane fusion processes required for exocytosis [21, 67].
The notion that iPLA2 activity may facilitate LTP expression by increasing glutamate release is contradicted, however, by a number of reports demonstrating that synaptic potentiation, at least in area CA1 of hippocampus, is not dependent on changes in transmitter release, but rather is mediated by upregulation of postsynaptic responses mediated by alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors at glutamatergic synapses [68, 69]. Several alterations have been reported at postsynaptic sites during LTP, including faster kinetics of receptor ion channels [70, 71], redistribution of existing receptors within the postsynaptic density [72], and insertion of new receptors at synapses [73, 74]. Consistent with these observations, we recently demonstrated that pretreatment of hippocampal slices with the iPLA2 inhibitor BEL completely abolishes AMPA receptor translocation in synaptic membranes and expression of CA1 hippocampal LTP [75]. Interestingly, both LTP and AMPA receptor translocation display enantio-selective impairment by the iPLA2 γ blocker (R)-BEL but not by the iPLA2 β inhibitor (S)-BEL, suggesting that iPLA2 γ represents the crucial isoform governing hippocampal synaptic stability. iPLA2 γ mRNAs and proteins are enriched with the endoplasmic reticulum (ER)-Golgi apparatus in several cell types [57], where they may be essential for diverse intracellular trafficking pathways, such as retrograde movement from the Golgi complex to the ER, transport of material from the trans-Golgi network to the plasma membrane, or recycling of membranes and receptors through endocytic pathways [21]. In particular, Péchoux et al. [76] reported that iPLA2 inhibition slowed down the transport of caseins from the ER to the Golgi apparatus and from the trans-Golgi network to the plasma membrane, indicating that iPLA2 could participate in membrane trafficking events leading to the secretion of milk proteins. Since AMPA receptors trafficking from the ER-Golgi complex to postsynaptic membranes is critically involved in LTP [77], the iPLA2 γ isoform may be well suited to facilitate AMPA receptor translocation from intracellular pools to synaptic membranes during LTP.
Animal experiments have revealed that PLA2 inhibition resulted in synaptic plasticity impairment and decreased performance in memory tasks. For instance, intracerebral injection of wide-spectrum PLA2 inhibitors into chick intermediate medial hyperstriatum ventrale curbs the learning of a passive avoidance task [78], while intraperitoneal injection in rats impedes spatial learning in the Morris water maze [79]. Likewise, intracerebroventricular or intrahippocampal injection of specific iPLA2 inhibitors impairs spatial working memory in rodents [80]. In addition, acquisition of 1-trial step-down inhibitory avoidance in rats was shown to be correlated with increased iPLA2 activity in hippocampus, while bilateral injection of iPLA2 inhibitors in region CA1 of the dorsal hippocampus prior to training hindered both short-term and long-term memory [81]. In a modified protocol developed to test memory retrieval, the same group recently showed that injection of the dual cPLA2 and iPLA2 inhibitor palmitoyl trifluoromethylketone in region CA1 of the rat dorsal hippocampus before performance testing impaired trained behaviour in the step-down inhibitory avoidance task [82]. Importantly, memory retrieval was re-established after recovery of PLA2 activity, indicating that these PLA2s are indeed necessary for memory stabilization. Hence, intact iPLA2 activity seems to be critical for proper memory acquisition as well as retrieval. However, the identity of iPLA2 isoforms involved in memory acquisition and retrieval remains to be determined.
3. iPLA2 and Neuronal Cell Death Mechanisms
Recently, evidence from studies with nonneuronal cells has suggested that iPLA2 enzymes may have diverse effects on cell death. First, constitutive iPLA2 activity may contribute to cell death since iPLA2 β overexpression amplifies thapsigargin-induced apoptosis in INS-1 insulinoma cells [83] and accelerates U937 cell death after long-term exposure to hydrogen peroxide [84]. iPLA2 has been shown to play a pivotal role in oxidative damage of astrocytes [85], and its blockade by BEL dampens oligomeric amyloid-β- (Aβ1-42-) induced mitochondrial membrane potential loss and reactive oxygen species production in these cells [86]. Moreover, iPLA2 inhibition reduces the size of infarcts produced by global ischemia [87]. On the other hand, iPLA2 activity has also been shown to protect against cell death, as inhibition of iPLA2 accentuates oxidant-induced cell death in renal proximal tubule cells and astrocytes [88, 89]. Likewise, iPLA2 activity may also have deleterious or beneficial effects on neurons. For instance, acute inhibition of iPLA2 activity by racemic BEL has been found to be neuroprotective in organotypic hippocampal slices exposed to oxygen-glucose deprivation [90]. In contrast, immature cultures of primary cortical neurons exposed for several days to BEL showed decreased cellular viability and neuritic growth [91, 92]. Moreover, iPLA2 β knockout mice exhibit abnormal motor behaviors possibly related to the appearance of vacuoles and ubiquitin-positive axonal swelling (spheroids) in many brain regions [93, 94], suggesting that iPLA2 β dysfunction leads to axonal dystrophy.
While the reported impact of iPLA2 on cell viability is mostly attributable to iPLA2 β, involvement of the iPLA2 γ isoform is much less clear. A previous report demonstrated that iPLA2 γ localized in mitochondria catalyzed AA liberation that mediated mitochondrial permeability transition, a key control point for apoptosis [95]. On the other hand, iPLA2 γ expression may exert cytoprotective effects during complement-mediated glomerular epithelial cell injury [96]. In addition, recent findings from our laboratory have revealed that constitutive iPLA2 γ activity might represent an important neuroprotective system capable of limiting brain excitotoxic damage. In particular, we showed that iPLA2 γ inhibition by the enantio-specific inhibitor (R)-BEL makes cultured hippocampal slices more vulnerable to AMPA-mediated excitotoxicity [97]. Overactivation of N-methyl-D-aspartic acid (NMDA) or AMPA receptors results in a massive entry of calcium into cells, leading to the activation of a number of enzymes, including ATPases, lipases, proteases, and endonucleases that, in turn, deplete energy stores or damage cell membranes, cytoarchitecture or nuclear components, respectively. Excitotoxicity has been reported to contribute to a variety of neuropathological disorders, including ischemic stroke, epilepsy, amyotrophic lateral sclerosis, and Alzheimer's disease (AD) [98, 99].
Interestingly, iPLA2 γ inhibition-induced enhancement of AMPA-mediated toxicity is associated with selective phosphorylation and upregulation of the AMPA receptor GluR1 but not GluR2 subunits in synaptic membrane fractions [97, 98, 100]. In hippocampus, AMPA receptors generally form heterodimers containing 2 copies of each of the GluR1 and GluR2 subunits. It is now well-recognized that the presence of GluR2 subunits render AMPA receptors impermeable to calcium. Consequently, its presence or absence plays a critical role in cellular calcium homeostasis and in determining susceptibility to excitotoxicity [101, 102]. Hence, iPLA2 γ inhibition, by promoting surface expression of GluR1 over GluR2 subunits (which is reflected by a rise in the GluR1/GluR2 ratio in the membrane fraction), could exacerbate excitotoxic cell death through the increased formation of GluR2-lacking AMPA receptors that would allow adverse Ca2+ influx upon prolonged AMPA receptor activation. Consistent with this possibility, the greater cell death observed following iPLA2 γ inhibition is prevented by GluR1/3-specific AMPA receptor antagonists [97]. How inhibition of iPLA2 γ influences the expression of AMPA receptor subtypes in synaptic membranes remains an open question. As mentioned earlier, this may be the result of an effect of the lipase on protein transport through intracellular secretory pathways [76]. There are other circumstances in which GluR1 subunits are selectively upregulated in hippocampal neurons, such as after neuronal activity inhibition elicited by prolonged blockade of AMPA receptors [103] or by tumor necrosis factor-alpha receptor activation [104]. In the latter case, it has been proposed that upregulation of GluR1 homomeric receptors could be produced by a reserve pool of non-GluR2-containing AMPA receptors located near the membrane. Independently of the exact mechanism, these observations raise the possibility that constitutive iPLA2 γ activity may be a crucial mechanism to maintain synaptic stability and constitute a molecular device to prevent neuronal dysfunctions.
4. iPLA2 Dysfunction and Neurodegenerative Disorders
As previously described, cPLA2 and sPLA2 are commonly believed to be preferentially involved in AA release; emerging evidence indicates that iPLA2 activity can contribute to docosahexaenoic acid (DHA) release from brain phospholipids [105]. The first suggestion that brain iPLA2 activity may be crucial for DHA release came from a study by Strokin et al. [106] who showed that racemic BEL inhibited DHA release from astrocytes. Later, using siRNA silencing procedures, the same group demonstrated that DHA release from astrocytic phospholipids was mainly dependent on iPLA2 γ activity [107]. DHA is one of the most abundant omega-3 polyunsaturated fatty acids (PUFA) present in phospholipids of mammalian brain [108], where it is recognized to be important for the maintenance of neural membranes and brain function integrity [109]. Deficiency in dietary intake of DHA has been associated with lower performance in learning tasks in rodents [110–112]. On the other hand, DHA dietary supplementation was shown to decrease the risk of developing AD [113–115] and to exert neuroprotective actions in a mouse model presenting numerous aspects of Parkinson's disease [116], while high-fat consumption combined with low omega-3 PUFA intake promoted AD-like neuropathology [117]. Both iPLA2 activity and DHA levels have been reported to be decreased in the plasma of AD patients [118, 119]. Lower iPLA2 activity has also been reported in AD brains [120, 121]. Whether or not decreased iPLA2 γ activity is a factor contributing to AD pathology remains to be established. Numerous neurobiological studies have demonstrated that DHA may be acting in different cellular pathways to counteract several molecular manifestations of AD. There are, for instance, strong indications that DHA release in the brain may diminish oxidative stress [122, 123] and glutamate-induced toxicity [124]. In this line, DHA-induced reduction of excitotoxic damage in hippocampus might be dependent on internalization of AMPA receptors [125]. The potential ability of DHA to reduce caspase activation [114, 115], Aβ peptide accumulation, and Tau hyperphosphorylation [126, 127] also strongly supports the notion that DHA deficiency, as a result of iPLA2 deficiency, could represent a precursor event that could initiate the cellular manifestations of AD pathology.
Normally, Tau predominantly localizes to neuronal axons where it modulates the stability and assembly of microtubules [128, 129]. In so doing, Tau generates a partially stable, but still dynamic, state in microtubules that is important for axonal growth and effective axonal transport [130]. In addition to binding microtubules, some but not all studies provide evidence that Tau can interact, either directly or indirectly, with actin and affect actin polymerization as well as the interaction of actin filaments with microtubules [131, 132]. Furthermore, Tau appears to interact with the plasma membrane and with several proteins involved in signal transduction [133–141]. From a pathological perspective, Tau dysfunction resulting from biochemical defects (i.e., aberrant phosphorylation, truncation, and glycosylation) has been proposed to be an important factor contributing to the initiation and development of several neuropathological conditions such as AD [142–147]. As discussed above, lower iPLA2 activity has been observed in AD brains and considering our hypothesis that iPLA2γ is an important factor controlling AMPA-mediated toxicity in the hippocampus, we anticipated that defect in iPLA2 γ activity can contribute to enhance Tau phosphorylation. Here, we are presenting the first experimental evidence that Tau become hyperphosphorylated after selective inhibition of iPLA2 γ. We first examined Tau phosphorylation levels at Ser199 residue following treatment of hippocampal slices with R-BEL and S-BEL, which preferentially block iPLA2 γ or iPLA2β, respectively (see chemical structures; Figure 1(a)). In initial experiments, we observed that hippocampal tissues were strongly and consistently stained with an antibody recognizing the phosphorylated Ser199 epitope of a Tau isoform of 62 kDa (Figure 1(b), top panels). As shown in Figure 1, staining for this hyperphosphorylated Tau isoform increased following iPLA2 γ inhibition by R-BEL. When the results were normalized with staining levels of Tau-5 (an antibody that recognizes Tau independent phosphorylation), it appears that R-BEL elevated levels of phosphorylated Tau at all concentrations tested, with a maximal increase of 120 ± 10% over control values in slices preincubated for 3 hr. However, the same analysis showed that phosphorylation of Tau at Ser199 was not altered by exposure to the iPLA2 β inhibitor S-BEL. It is noteworthy that levels of Tau-5 immunoreactivity in the hippocampal slices were not significantly changed by treatments with either R- or S-BEL, indicating that iPLA2 γ inhibition-induced increases in Ser199 phosphorylation do not depend on Tau synthesis and/or degradation. De-Paula and collaborators recently showed that injection of the dual cPLA2 and iPLA2 inhibitor methyl arachidonyl fluorophosphonate (MAFP) induced Tau phosphorylation at Ser214 [148]. In contrast to our results, however, they reported that Tau hyperphosphorylation was associated with a reduction in levels of total Tau [149], suggesting that inhibition of both cPLA2 and iPLA2 might influence several biochemical aspects of Tau proteins. Accordingly, recent experimental results have provided evidence that cPLA2 and iPLA2 activities can play divergent roles during spinal cord injuries [150]. We recently tested the effect of R-BEL-mediated iPLA2 γ inhibition on Tau subcellular localisation in CA1 pyramidal cells. Using organotypic hippocampal slice cultured from transgenic mice expressing human Tau, we observed that treatment with the specific iPLA2 γ inhibitor (R)-BEL for up to 12 h resulted in increases in Tau phosphorylation at the Thr231 site. High-resolution imaging showed that hyperphosphorylated Tau was primarily localized in the cell bodies and dendrites of hippocampal pyramidal neurons (see Figure 2).
Figure 1.

Hippocampal Tau phosphorylation at Ser199 residue is accentuated by R-BEL. Hippocampal slices (350 μm) were pre-incubated at 32°C for 3 h with DMSO alone (control) or with increasing concentrations of the iPLA2 γ inhibitor R-BEL or the iPLA2 β inhibitor S-BEL (chemical structures of both compounds are presented in (a)). (b) Phosphorylation and Tau protein levels were determined by Western blotting of cell extracts (40 μg of proteins) obtained from acute hippocampal slices. Phosphorylated Tau levels, expressed relative to total Tau (i.e., Tau-5; AbCam, Cambridge, MA, USA. Dilution 1 : 500), were measured using antibodies raised against Tau phosphorylated at Ser199 (AbCam, Cambridge, MA, USA. Dilution 1 : 1,000). The data were expressed as percentage of control values and are means ± SEM of 3 measurements per cell extract obtained from 7 different rats. Statistical analysis was performed by one-way ANOVA followed by Neuman-Keuls' post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, drug-treated versus control.
Figure 2.

Inhibition of iPLA2 γ induces Tau phosphorylation in area CA1 of hippocampus. Cultured hippocampal slices from P301L Tau transgenic mice were treated with the iPLA2 γ inhibitor (R)-BEL. Slices were then processed for confocal immunofluorescence microscopy with an antibody recognizing Tau phosphorylation at Threonine 231 epitopes (AT231, in green) (AbCam, Cambridge, MA, USA. Dilution 1 : 750). When compared to controls (upper panel), immunostaining revealed increased phosphorylation in the CA1 region of cultured hippocampal slices incubated with 3 μM (R)-BEL for a period of 12 h (lower panel). DAPI (in blue) was included in the mounting medium to label nuclei. This observation was qualitatively reproduced in hippocampal slices obtained in 3 different cultures. Scale bar = 25 μm.
One of the central hypotheses for AD pathogenesis is that the production of cytotoxic Aβ peptides impairs neuronal activity and leads to a decline in memory and cognition [151]. Some PLA2 enzymes may exacerbate Aβ cytotoxicity, as Aβ peptides stimulate cPLA2 α activity in neuronal cultures [86] and primary cortical astrocytes [152]; in addition, Aβ-induced learning and memory deficits in a transgenic mouse model of AD are prevented by genetic ablation of cPLA2 α activity in brain [152]. On the other hand, it has been well established that iPLA2 activity is essential for maintaining membrane phospholipid integrity by reducing peroxidative damage, especially that originating in mitochondria. In this regard, iPLA2 expression prevents the loss of mitochondrial membrane potential and attenuates the release of cytochrome c as well as of other apoptotic proteins, and ultimately reduces apoptosis in INS-1 cells exposed to staurosporine [153]. Furthermore, Kinsey et al. [95, 154] reported that a major component of PLA2 activity in mitochondria of rabbit renal proximal tubular cells is provided by iPLA2 γ and is of critical importance for the prevention of basal lipid peroxidation and maintenance of mitochondrial viability. Based on recent studies, it has been proposed that Aβ-induced neurotoxicity might derive from mitochondrial defects. Indeed, in vitro experiments have shown that Aβ peptides can be internalized by cells, imported into mitochondria and ultimately elicit mitochondrial dysfunctions [155]. Given its localization, it is thus tempting to propose that iPLA2 γ might represent an important cellular component that prevents mitochondrial dysfunctions. Experiments are required to determine whether iPLA2 γ overexpression activity might exert protective effects against Aβ peptide-induced mitochondrial dysfunctions.
From a pathological perspective, it has been demonstrated that iPLA2 activity is upregulated in the hippocampus of patients suffering from schizophrenia [156]. The precise implication of this iPLA2 dysfunction in the development of schizophrenia-related symptoms remains unknown. However, the results presented above would predict that upregulation of iPLA2 γ activity could eventually lead to reduction in GluR1-containing receptors. Interestingly, GluR1 downregulation has been reported to evoke striatal hyperdopaminergic activity [157], a well-established biological defect involved in schizophrenia-related symptoms. The potential relationship between iPLA2s and the dopaminergic system is reinforced by the fact that iPLA2 inhibition or knockdown in rat striatum, motor cortex and thalamus results in the apparition of Parkinson-related manifestations [158], which are also known to depend on dopamine dysfunction. Of course, future experiments will be required to establish the potential role of iPLA2 enzymes in stabilizing dopamine-mediated responses.
5. Conclusion
Here, we have summarized growing evidence linking iPLA2 γ activity to the stabilization of synaptic AMPA receptor properties in hippocampal neurons. First, it appears evident that without appropriate levels of iPLA2 γ activity in area CA1 of hippocampal slices synaptic stabilization of AMPA receptors, which is required for the expression of long-term changes in synaptic strength (i.e., LTP), is compromised. As mentioned previously, iPLA2 γ mRNAs and proteins are enriched with the endoplasmic reticulum (ER)-Golgi apparatus in several cell type, where they will be essential for diverse intracellular trafficking pathways, such as retrograde movement from the Golgi complex to the ER, transport of material from the trans-Golgi network to the plasma membrane, or recycling of membranes and receptors through endocytic pathways [21]. Since AMPA receptors trafficking from the ER-Golgi complex to postsynaptic membranes is critically involved in LTP [77], the iPLA2 γ isoform may be well suited to facilitate AMPA receptor translocation from intracellular pools to synaptic membranes during LTP. However, given their biochemical properties and localization, future experiments will be required to determine how the effects of iPLA2γ on LTP might derive from alterations of other cellular processes controlling synaptic stability such as regulation of arachidonic acid release, membrane fusion events, receptor trafficking pathways, and protein kinase activities. Besides, we also documented that iPLA2γ deficiency can destabilize synaptic GluR1 subunits of AMPA receptors in hippocampl membranes and accentuate glutamate-induced toxicity. In this line, iPLA2γ-null mice have been generated [159, 160] and were found to exhibit phenotypic abnormalities that include altered mitochondrial morphology, function, and lipid composition associated with hippocampal degeneration. Interestingly, we provided here preliminary evidence showing that iPLA2 γ activity appears to be important for stabilizing Tau phosphorylation in hippocampal pyramidal neurons, suggesting that downregulation of iPLA2 activity may contribute to the development of tauopathies in AD [161]. A putative biochemical model that could account for the potential influence of iPLA2 γ on Tau pathology is presented in Figure 3. Indeed, considering the growing evidence relating the importance of iPLA2 γ in physiological and pathological conditions, targeting iPLA2 γ activity may represent a potentially new therapeutic strategy to address several neurological disorders characterized with destabilisation of synaptic properties.
Figure 3.
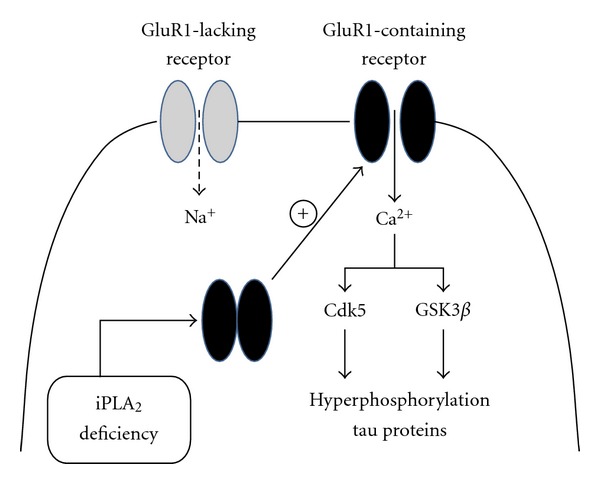
A putative model illustrating the potential implication of iPLA2 γ in Alzheimer's disease. In this simplified model, iPLA2 dysfunction leads to excessive delivery of GluR1-containing receptors to neuronal membranes. These receptors are more likely to be calcium-permeable and therefore to stimulate calcium influx and, eventually, Tau phosphorylation by calcium-dependent protein kinases such as Cdk5 and GSK-3β.
References
- 1.Marder E, Prinz AA. Current compensation in neuronal homeostasis. Neuron. 2003;37(1):2–4. doi: 10.1016/s0896-6273(02)01173-x. [DOI] [PubMed] [Google Scholar]
- 2.Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nature Reviews Neuroscience. 2003;4(11):885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- 3.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 4.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nature Reviews Neuroscience. 2004;5(2):97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron. 2006;49(4):563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 6.Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nature Neuroscience. 2004;7(12):1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- 7.Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. Journal of Neurophysiology. 2006;96(4):2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- 8.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends in Neurosciences. 1996;19(4):126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 9.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. Journal of Neuroscience. 1982;2(1):32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ripley B, Otto S, Tiglio K, Williams ME, Ghosh A. Regulation of synaptic stability by AMPA receptor reverse signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):367–372. doi: 10.1073/pnas.1015163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovascular Drugs and Therapy. 2009;23(1):49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. Journal of Lipid Research. 2009;50:S237–242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leslie CC. Properties and regulation of cytosolic phospholipase A2 . Journal of Biological Chemistry. 1997;272(27):16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 14.Dennis EA. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends in Biochemical Sciences. 1997;22(1):1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- 15.Balboa MA, Balsinde J, Jones SS, Dennis EA. Identity between the CA2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. Journal of Biological Chemistry. 1997;272(13):8576–8580. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 16.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. Journal of Biological Chemistry. 1997;272(13):8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 17.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. Pancreatic islets express a CA2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. Journal of Biological Chemistry. 1997;272(17):11118–11127. [PubMed] [Google Scholar]
- 18.Balsinde J, Dennis EA. Function and inhibition of intracellular calcium-independent phospholipase A2 . Journal of Biological Chemistry. 1997;272(26):16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 19.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins and Other Lipid Mediators. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 20.Bazan NG, Allan G, Rodriguez De Turco EB. Role of phospholipase A2 and membrane-derived lipid second messengers in membrane function and transcriptional activation of genes: implications in cerebral ischemia and neuronal excitability. Progress in Brain Research. 1993;96:247–257. doi: 10.1016/s0079-6123(08)63271-9. [DOI] [PubMed] [Google Scholar]
- 21.Brown WJ, Chambers K, Doody A. Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic. 2003;4(4):214–221. doi: 10.1034/j.1600-0854.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 22.Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukotrienes and Essential Fatty Acids. 2004;71(3):161–169. doi: 10.1016/j.plefa.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Farooqui AA, Ong WY, Horrocks LA. Biochemical aspects of neurodegeneration in human brain: involvement of neural membrane phospholipids and phospholipases A2 . Neurochemical Research. 2004;29(11):1961–1977. doi: 10.1007/s11064-004-6871-3. [DOI] [PubMed] [Google Scholar]
- 24.Kolko M, Prause JU, Bazan NG, Heegaard S. Human secretory phospholipase A2, group IB in normal eyes and in eye diseases. Acta Ophthalmologica Scandinavica. 2007;85(3):317–323. doi: 10.1111/j.1600-0420.2006.00809.x. [DOI] [PubMed] [Google Scholar]
- 25.Leslie CC. Regulation of arachidonic acid availability for eicosanoid production. Biochemistry and Cell Biology. 2004;82(1):1–17. doi: 10.1139/o03-080. [DOI] [PubMed] [Google Scholar]
- 26.Phillis JW, O’Regan MH. A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Research Reviews. 2004;44(1):13–47. doi: 10.1016/j.brainresrev.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cellular Signalling. 2005;17(9):1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Hooks SB, Cummings BS. Role of CA2+-independent phospholipase A2 in cell growth and signaling. Biochemical Pharmacology. 2008;76(9):1059–1067. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun GY, Xu J, Jensen MD, et al. Phospholipase A2 in astrocytes: responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Molecular Neurobiology. 2005;31(1-3):27–41. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]
- 30.Sun GY, Xu J, Jensen MD, Simonyi A. Phospholipase A2 in the central nervous system: implications for neurodegenerative diseases. Journal of Lipid Research. 2004;45(2):205–213. doi: 10.1194/jlr.R300016-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2 . Nature. 1997;390(6660):622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 32.Uozumi H, Kume K, Nagase T, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390(6660):618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 33.Quistad GB, Barlow C, Winrow CJ, Sparks SE, Casida JE. Evidence that mouse brain neuropathy target esterase is a lysophospholipase. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(13):7983–7987. doi: 10.1073/pnas.1232473100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. Journal of Biological Chemistry. 2004;279(47):48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 35.Rydel TJ, Williams JM, Krieger E, et al. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry. 2003;42(22):6696–6708. doi: 10.1021/bi027156r. [DOI] [PubMed] [Google Scholar]
- 36.Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium- independent phospholipase A2 . Journal of Biological Chemistry. 2000;275(14):9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 37.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Progress in Lipid Research. 2011;50(2):152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Polster B, Crosier M, Lindsay S, Hayflick S. Expression of PLA2G6 in human fetal development: implications for infantile neuroaxonal dystrophy. Brain Research Bulletin. 2010;83(6):374–379. doi: 10.1016/j.brainresbull.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI CA2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. Journal of Biological Chemistry. 1997;272(46):29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 40.Tay HK, Melendez AJ. FcγRI-triggered generation of arachidonic acid and eicosanoids requires iPLA2 but not cPLA2 in human monocytic cells. Journal of Biological Chemistry. 2004;279(21):22505–22513. doi: 10.1074/jbc.M308788200. [DOI] [PubMed] [Google Scholar]
- 41.Sun B, Zhang X, Yonz C, Cummings BS. Inhibition of calcium-independent phospholipase A2 activates p38 MAPK signaling pathways during cytostasis in prostate cancer cells. Biochemical Pharmacology. 2010;79(12):1727–1735. doi: 10.1016/j.bcp.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Herbert SP, Walker JH. Group VIA calcium-independent phospholipase A2 mediates endothelial cell S phase progression. Journal of Biological Chemistry. 2006;281(47):35709–35716. doi: 10.1074/jbc.M600699200. [DOI] [PubMed] [Google Scholar]
- 43.Atsumi GI, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudot I. Fas-induced arachidonic acid release is mediated by CA2+- independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. Journal of Biological Chemistry. 1998;273(22):13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 44.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nature Cell Biology. 2004;6(2):113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 45.Bao S, Miller DJ, Ma Z, et al. Male mice that do not express Group VIA Phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. Journal of Biological Chemistry. 2004;279(37):38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao S, Song H, Wohltmann M, et al. Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. Journal of Biological Chemistry. 2006;281(30):20958–20973. doi: 10.1074/jbc.M600075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. Effects of stable suppression of group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. Journal of Biological Chemistry. 2006;281(1):187–198. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan NV, Westaway SK, Morton JEV, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nature Genetics. 2006;38(7):752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinsey GR, McHowat J, Beckett CS, Schnellmann RG. Identification of calcium-independent phospholipase A2 γ in mitochondria and its role in mitochondrial oxidative stress. American Journal of Physiology. 2007;292(2):F853–F860. doi: 10.1152/ajprenal.00318.2006. [DOI] [PubMed] [Google Scholar]
- 50.Mancuso DJ, Jenkins CM, Sims HF, Cohen JM, Yang J, Gross RW. Complex transcriptional and translational regulation of iPLA2 γ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. European Journal of Biochemistry. 2004;271(23-24):4709–4724. doi: 10.1111/j.1432-1033.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- 51.Murakami M, Masuda S, Ueda-Semmyo K, et al. Group VIB CA2+-independent phospholipase A2 γ promotes cellular membrane hydrolysis and prostaglandin production in a manner distinct from other intracellular phospholipases A2 . Journal of Biological Chemistry. 2005;280(14):14028–14041. doi: 10.1074/jbc.M413766200. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka H, Takeya R, Sumimoto H. A novel intracellular membrane-bound calcium-independent phospholipase A2 . Biochemical and Biophysical Research Communications. 2000;272(2):320–326. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Han X, Gross RW. Identification of hepatic peroxisomal phospholipase A2 and characterization of arachidonic acid-containing choline glycerophospholipids in hepatic peroxisomes. FEBS Letters. 2003;546(2-3):247–250. doi: 10.1016/s0014-5793(03)00581-7. [DOI] [PubMed] [Google Scholar]
- 54.Yan W, Jenkins CM, Han X, et al. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2 γ: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. Journal of Biological Chemistry. 2005;280(29):26669–26679. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 55.Wolf MJ, Izumi Y, Zorumski CF, Gross RW. Long-term potentiation requires activation of calcium-independent phopholipase A2 . FEBS Letters. 1995;377(3):358–362. doi: 10.1016/0014-5793(95)01371-7. [DOI] [PubMed] [Google Scholar]
- 56.Yang HC, Mosior M, Ni B, Dennis EA. Regional distribution, ontogeny, purification, and characterization of the CA2+-independent phospholipase A2 from rat brain. Journal of Neurochemistry. 1999;73(3):1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- 57.Kinsey GR, Cummings BS, Beckett CS, et al. Identification and distribution of endoplasmic reticulum iPLA2 . Biochemical and Biophysical Research Communications. 2005;327(1):287–293. doi: 10.1016/j.bbrc.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 58.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. A novel calcium-independent phospholipase A2, cPLA2-γ, that is prenylated and contains homology to cPLA2 . Journal of Biological Chemistry. 1998;273(34):21926–21932. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 59.Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) β, and not iPLA2 γ, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. Journal of Biological Chemistry. 2002;277(36):32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 60.Fujita S, Ikegaya Y, Nishikawa M, Nishiyama N, Matsuki N. Docosahexaenoic acid improves long-term potentiation attenuated by phospholipase A2 inhibitor in rat hippocampal slices. British Journal of Pharmacology. 2001;132(7):1417–1422. doi: 10.1038/sj.bjp.0703970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang XH, Poo MM. Progress in neural plasticity. Science China Life Sciences. 2010;53(3):322–329. doi: 10.1007/s11427-010-0062-z. [DOI] [PubMed] [Google Scholar]
- 62.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 63.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 64.Clements MP, Bliss TVP, Lynch MA. Increase in arachidonic acid concentration in a postsynaptic membrane fraction following the induction of long-term potentiation in the dentate gyrus. Neuroscience. 1991;45(2):379–389. doi: 10.1016/0306-4522(91)90235-g. [DOI] [PubMed] [Google Scholar]
- 65.Drapeau C, Pellerin L, Wolfe LS, Avoli M. Long-term changes of synaptic transmission induced by arachidonic acid in the CA1 subfield of the rat hippocampus. Neuroscience Letters. 1990;115(2-3):286–292. doi: 10.1016/0304-3940(90)90470-t. [DOI] [PubMed] [Google Scholar]
- 66.Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341(6244):739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- 67.Takuma T, Ichida T. Role of CA2+-independent phospholipase A2 in exocytosis of amylase from parotid acinar cells. Journal of Biochemistry. 1997;121(6):1018–1024. doi: 10.1093/oxfordjournals.jbchem.a021688. [DOI] [PubMed] [Google Scholar]
- 68.Massicotte G. Modification of glutamate receptors by phospholipase A2: its role in adaptive neural plasticity. Cellular and Molecular Life Sciences. 2000;57(11):1542–1550. doi: 10.1007/PL00000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 70.Ambros-Ingerson J, Lynch G. Channel gating kinetics and synaptic efficacy: a hypothesis for expression of long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(16):7903–7907. doi: 10.1073/pnas.90.16.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ambros-Ingerson J, Xiao P, Larson J, Lynch G. Waveform analysis suggests that LTP alters the kinetics of synaptic receptor channels. Brain Research. 1993;620(2):237–244. doi: 10.1016/0006-8993(93)90161-f. [DOI] [PubMed] [Google Scholar]
- 72.Xie X, Liaw JS, Baudry M, Berger TW. Novel expression mechanism for synaptic potentiation: alignment of presynaptic release site and postsynaptic receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(13):6983–6988. doi: 10.1073/pnas.94.13.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu WY, Man HY, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29(1):243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- 74.Pickard L, Noël J, Duckworth JK, et al. Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology. 2001;41(6):700–713. doi: 10.1016/s0028-3908(01)00127-7. [DOI] [PubMed] [Google Scholar]
- 75.Martel MA, Patenaude C, Ménard C, Alaux S, Cummings BS, Massicotte G. A novel role for calcium-independent phospholipase A2 in α-amino-3-hydroxy-5-methylisoxazole-propionate receptor regulation during long-term potentiation. European Journal of Neuroscience. 2006;23(2):505–513. doi: 10.1111/j.1460-9568.2005.04565.x. [DOI] [PubMed] [Google Scholar]
- 76.Péchoux C, Boisgard R, Chanat E, Lavialle F. Ca2+-independent phospholipase A2 participates in the vesicular transport of milk proteins. Biochimica et Biophysica Acta. 2005;1743(3):317–329. doi: 10.1016/j.bbamcr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 77.Broutman G, Baudry M. Involvement of the secretory pathway for AMPA receptors in NMDA-induced potentiation in hippocampus. Journal of Neuroscience. 2001;21(1):27–34. doi: 10.1523/JNEUROSCI.21-01-00027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holscher C, Rose SPR. Inhibitors of phospholipase A2 produce amnesia for a passive avoidance task in the chick. Behavioral and Neural Biology. 1994;61(3):225–232. doi: 10.1016/s0163-1047(05)80005-6. [DOI] [PubMed] [Google Scholar]
- 79.Holscher C, Canevari L, Richter-Levin G. Inhibitors of PLA2 and NO synthase cooperate in producing amnesia of a spatial task. NeuroReport. 1995;6(5):730–732. doi: 10.1097/00001756-199503270-00006. [DOI] [PubMed] [Google Scholar]
- 80.Fujita S, Ikegaya Y, Nishiyama N, Matsuki N. CA2+-Independent phospholipase A2 inhibitor impairs spatial memory of mice. Japanese Journal of Pharmacology. 2000;83(3):277–278. doi: 10.1254/jjp.83.277. [DOI] [PubMed] [Google Scholar]
- 81.Schaeffer EL, Gattaz WF. Inhibition of calcium-independent phospholipase A2 activity in rat hippocampus impairs acquisition of short- and long-term memory. Psychopharmacology. 2005;181(2):392–400. doi: 10.1007/s00213-005-2256-9. [DOI] [PubMed] [Google Scholar]
- 82.Schaeffer EL, Gattaz WF. Requirement of hippocampal phospholipase A2 activity for long-term memory retrieval in rats. Journal of Neural Transmission. 2007;114(3):379–385. doi: 10.1007/s00702-006-0585-4. [DOI] [PubMed] [Google Scholar]
- 83.Ramanadham S, Hsu FF, Zhang S, et al. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2 β) and suppressed by inhibition of iPLA2 β . Biochemistry. 2004;43(4):918–930. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pérez R, Melero R, Balboa MA, Balsinde J. Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. Journal of Biological Chemistry. 2004;279(39):40385–40391. doi: 10.1074/jbc.M402562200. [DOI] [PubMed] [Google Scholar]
- 85.Xu J, Yu S, Sun AY, Sun GY. Oxidant-mediated AA release from astrocytes involves cPLA2 and iPLA2 . Free Radical Biology and Medicine. 2003;34(12):1531–1543. doi: 10.1016/s0891-5849(03)00152-7. [DOI] [PubMed] [Google Scholar]
- 86.Zhu D, Lai Y, Shelat PB, Hu C, Sun GY, Lee JCM. Phospholipases A2 mediate amyloid-β peptide-induced mitochondrial dysfunction. Journal of Neuroscience. 2006;26(43):11111–11119. doi: 10.1523/JNEUROSCI.3505-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Williams SD, Gottlieb RA. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochemical Journal. 2002;362(1):23–32. doi: 10.1042/0264-6021:3620023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum CA2+-independent phospholipase A2 in oxidant-induced renal cell death. American Journal of Physiology. 2002;283(3):F492–F498. doi: 10.1152/ajprenal.00022.2002. [DOI] [PubMed] [Google Scholar]
- 89.Peterson B, Knotts T, Cummings BS. Involvement of CA2+-independent phospholipase A2 isoforms in oxidant-induced neural cell death. NeuroToxicology. 2007;28(1):150–160. doi: 10.1016/j.neuro.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 90.Strokin M, Chechneva O, Reymann KG, Reiser G. Neuroprotection of rat hippocampal slices exposed to oxygen-glucose deprivation by enrichment with docosahexaenoic acid and by inhibition of hydrolysis of docosahexaenoic acid-containing phospholipids by calcium independent phospholipase A2 . Neuroscience. 2006;140(2):547–553. doi: 10.1016/j.neuroscience.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 91.Forlenza OV, Mendes CT, Marie SKN, Gattaz WF. Inhibition of phospholipase A2 reduces neurite outgrowth and neuronal viability. Prostaglandins Leukotrienes and Essential Fatty Acids. 2007;76(1):47–55. doi: 10.1016/j.plefa.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 92.Mendes CT, Gattaz WF, Schaeffer EL, Forlenza OV. Modulation of phospholipase A2 activity in primary cultures of rat cortical neurons. Journal of Neural Transmission. 2005;112(10):1297–1308. doi: 10.1007/s00702-004-0271-3. [DOI] [PubMed] [Google Scholar]
- 93.Malik I, Turk J, Mancuso DJ, et al. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. American Journal of Pathology. 2008;172(2):406–416. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shinzawa K, Sumi H, Ikawa M, et al. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. Journal of Neuroscience. 2008;28(9):2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kinsey GR, McHowat J, Patrick KS, Schnellmann RG. Role of CA2+-independent phospholipase A2 γ in CA2+-induced mitochondrial permeability transition. Journal of Pharmacology and Experimental Therapeutics. 2007;321(2):707–715. doi: 10.1124/jpet.107.119545. [DOI] [PubMed] [Google Scholar]
- 96.Cohen D, Papillon J, Aoudjit L, Li H, Cybulsky AV, Takano T. Role of calcium-independent phospholipase A2 in complement-mediated glomerular epithelial cell injury. American Journal of Physiology. 2008;294(3):F469–F479. doi: 10.1152/ajprenal.00372.2007. [DOI] [PubMed] [Google Scholar]
- 97.Ménard C, Chartier C, Patenaude C, et al. Calcium-independent phospholipase A2 influences AMPA-mediated toxicity of hippocampal slices by regulating the GluR1 subunit in synaptic membranes. Hippocampus. 2007;17(11):1109–1120. doi: 10.1002/hipo.20343. [DOI] [PubMed] [Google Scholar]
- 98.Villmann C, Becker CM. Reviews: on the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist. 2007;13(6):594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- 99.Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Current Opinion in Neurobiology. 2006;16(3):281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 100.Ménard C, Patenaude C, Massicotte G. Phosphorylation of AMPA receptor subunits is differentially regulated by phospholipase A2 inhibitors. Neuroscience Letters. 2005;389(1):51–56. doi: 10.1016/j.neulet.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 101.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67(1):11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 102.Geiger JRP, Melcher T, Koh DS, et al. Relative abundance of subunit mRNAs determines gating and CA2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15(1):193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 103.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47(5):725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 104.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α . Journal of Neuroscience. 2005;25(12):3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Green JT, Orr SK, Bazinet RP. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. Journal of Lipid Research. 2008;49(5):939–944. doi: 10.1194/jlr.R700017-JLR200. [DOI] [PubMed] [Google Scholar]
- 106.Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and CA2+ . British Journal of Pharmacology. 2003;139(5):1014–1022. doi: 10.1038/sj.bjp.0705326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Strokin M, Sergeeva M, Reiser G. Prostaglandin synthesis in rat brain astrocytes is under the control of the n-3 docosahexaenoic acid, released by group VIB calcium-independent phospholipase A2 . Journal of Neurochemistry. 2007;102(6):1771–1782. doi: 10.1111/j.1471-4159.2007.04663.x. [DOI] [PubMed] [Google Scholar]
- 108.Glomset JA. Role of docosahexaenoic acid in neuronal plasma membranes. Science’s STKE. 2006;2006(321):p. pe6. doi: 10.1126/stke.3212006pe6. [DOI] [PubMed] [Google Scholar]
- 109.Youdim KA, Martin A, Joseph JA. Essential fatty acids and the brain: possible health implications. International Journal of Developmental Neuroscience. 2000;18(4-5):383–399. doi: 10.1016/s0736-5748(00)00013-7. [DOI] [PubMed] [Google Scholar]
- 110.Catalan J, Moriguchi T, Slotnick B, Murthy M, Greiner RS, Salem N. Cognitive deficits in docosahexaenoic acid-deficient rats. Behavioral Neuroscience. 2002;116(6):1022–1031. doi: 10.1037//0735-7044.116.6.1022. [DOI] [PubMed] [Google Scholar]
- 111.Fedorova I, Salem Jr. N. Omega-3 fatty acids and rodent behavior. Prostaglandins Leukotrienes and Essential Fatty Acids. 2006;75(4-5):271–289. doi: 10.1016/j.plefa.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 112.Takeuchi T, Fukumoto Y, Harada E. Influence of a dietary n-3 fatty acid deficiency on the cerebral catecholamine contents, EEG and learning ability in rat. Behavioural Brain Research. 2002;131(1-2):193–203. doi: 10.1016/s0166-4328(01)00392-8. [DOI] [PubMed] [Google Scholar]
- 113.Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukotrienes and Essential Fatty Acids. 2007;77(5-6):287–293. doi: 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 114.Calon F, Lim GP, Morihara T, et al. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s disease. European Journal of Neuroscience. 2005;22(3):617–626. doi: 10.1111/j.1460-9568.2005.04253.x. [DOI] [PubMed] [Google Scholar]
- 115.Calon F, Lim GP, Yang F, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43(5):633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bousquet M, Saint-Pierre M, Julien C, Salem N, Cicchetti F, Calon F. Beneficial effects of dietary omega-3 polyunsaturated fatty acid on toxin-induced neuronal degeneration in an animal model of Parkinson’s disease. FASEB Journal. 2008;22(4):1213–1225. doi: 10.1096/fj.07-9677com. [DOI] [PubMed] [Google Scholar]
- 117.Julien C, Tremblay C, Phivilay A, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiology of Aging. 2010;31(9):1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 118.Conquer JA, Tierney MC, Zecevic J, Bettger WJ, Fisher RH. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids. 2000;35(12):1305–1312. doi: 10.1007/s11745-000-0646-3. [DOI] [PubMed] [Google Scholar]
- 119.Gattaz WF, Forlenza OV, Talib LL, Barbosa NR, Bottino CMC. Platelet phospholipase A2 activity in Alzheimer’s disease and mild cognitive impairment. Journal of Neural Transmission. 2004;111(5):591–601. doi: 10.1007/s00702-004-0142-y. [DOI] [PubMed] [Google Scholar]
- 120.Ross BM, Turenne S, Moszczynska A, Warsh JJ, Kish SJ. Differential alteration of phospholipase A2 activities in brain of patients with schizophrenia. Brain Research. 1999;821(2):407–413. doi: 10.1016/s0006-8993(99)01123-3. [DOI] [PubMed] [Google Scholar]
- 121.Talbot K, Young RA, Jolly-Tornetta C, Lee VMY, Trojanowski JQ, Wolf BA. A frontal variant of Alzheimer’s disease exhibits decreased calcium-independent phospholipase A2 activity in the prefrontal cortex. Neurochemistry International. 2000;37(1):17–31. doi: 10.1016/s0197-0186(00)00006-1. [DOI] [PubMed] [Google Scholar]
- 122.Yavin E, Brand A, Green P. Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutritional Neuroscience. 2002;5(3):149–157. doi: 10.1080/10284150290003159. [DOI] [PubMed] [Google Scholar]
- 123.Wu A, Ying Z, Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. Journal of Neurotrauma. 2004;21(10):1457–1467. doi: 10.1089/neu.2004.21.1457. [DOI] [PubMed] [Google Scholar]
- 124.Wang X, Zhao X, Mao ZY, Wang XM, Liu ZL. Neuroprotective effect of docosahexaenoic acid on glutamate-induced cytotoxicity in rat hippocampal cultures. Neuroreport. 2003;14(18):2457–2461. doi: 10.1097/00001756-200312190-00033. [DOI] [PubMed] [Google Scholar]
- 125.Ménard C, Patenaude C, Gagné A-M, Massicotte G. AMPA receptor-mediated cell death is reduced by docosahexaenoic acid but not by eicosapentaenoic acid in area CA1 of hippocampal slice cultures. Journal of Neuroscience Research. 2009;87(4):876–886. doi: 10.1002/jnr.21916. [DOI] [PubMed] [Google Scholar]
- 126.Green KN, Martinez-Coria H, Khashwji H, et al. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. Journal of Neuroscience. 2007;27(16):4385–4395. doi: 10.1523/JNEUROSCI.0055-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oksman M, Iivonen H, Hogyes E, et al. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiology of Disease. 2006;23(3):563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 128.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(5):1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. Journal of Molecular Biology. 1977;116(2):227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 130.Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cellular and Molecular Life Sciences. 2002;59(10):1668–1680. doi: 10.1007/PL00012495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamauchi PS, Purich DL. Microtubule-associated protein interactions with actin filaments: evidence for differential behavior of neuronal MAP-2 and tau in the presence of phosphatidyl-inositol. Biochemical and Biophysical Research Communications. 1993;190(3):710–715. doi: 10.1006/bbrc.1993.1107. [DOI] [PubMed] [Google Scholar]
- 132.Selden SC, Pollard TD. Phosphorylation of microtubule-associated proteins regulates their interaction with actin filaments. Journal of Biological Chemistry. 1983;258(11):7064–7071. [PubMed] [Google Scholar]
- 133.Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. Journal of Cell Biology. 1995;131(5):1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maas T, Eidenmüller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. Journal of Biological Chemistry. 2000;275(21):15733–15740. doi: 10.1074/jbc.M000389200. [DOI] [PubMed] [Google Scholar]
- 135.Jenkins SM, Johnson GVW. Tau complexes with phospholipase C-γ in situ. NeuroReport. 1998;9(1):67–71. doi: 10.1097/00001756-199801050-00014. [DOI] [PubMed] [Google Scholar]
- 136.Lee G, Todd Newman S, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. Journal of Cell Science. 1998;111(21):3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- 137.Liao H, Li Y, Brautigan DL, Gundersen GG. Protein phosphatase 1 is targeted to microtubules by the microtubule- associated protein tau. Journal of Biological Chemistry. 1998;273(34):21901–21908. doi: 10.1074/jbc.273.34.21901. [DOI] [PubMed] [Google Scholar]
- 138.Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of tau by protein phosphatase 2A. Neuron. 1996;17(6):1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- 139.Agarwal-Mawal A, Qureshi HY, Cafferty PW, et al. 14-3-3 Connects glycogen synthase kinase-3β to tau within a brain microtubule-associated tau phosphorylation complex. Journal of Biological Chemistry. 2003;278(15):12722–12728. doi: 10.1074/jbc.M211491200. [DOI] [PubMed] [Google Scholar]
- 140.Reynolds CH, Garwood CJ, Wray S, et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cγ1, Grb2, and Src family kinases. Journal of Biological Chemistry. 2008;283(26):18177–18186. doi: 10.1074/jbc.M709715200. [DOI] [PubMed] [Google Scholar]
- 141.Vega IE, Traverso EE, Ferrer-Acosta Y, et al. A novel calcium-binding protein is associated with tau proteins in tauopathy. Journal of Neurochemistry. 2008;106(1):96–106. doi: 10.1111/j.1471-4159.2008.05339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang Y, Tian Q, Zhang Q, Zhou X, Liu S, Wang JZ. Hyperphosphorylation of microtubule-associated tau protein plays dual role in neurodegeneration and neuroprotection. Pathophysiology. 2009;16(4):311–316. doi: 10.1016/j.pathophys.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 143.Burack MA, Halpain S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience. 1996;72(1):167–184. doi: 10.1016/0306-4522(95)00546-3. [DOI] [PubMed] [Google Scholar]
- 144.Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Molecular Neurodegeneration. 2009;4:p. 13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maurage CA, Sergeant N, Ruchoux MM, Hauw JJ, Delacourte A. Phosphorylated serine 199 of microtubule-associated protein tau is a neuronal epitope abundantly expressed in youth and an early marker of tau pathology. Acta Neuropathologica. 2003;105(2):89–97. doi: 10.1007/s00401-002-0608-7. [DOI] [PubMed] [Google Scholar]
- 146.Bi X, Haque TS, Zhou J, et al. Novel cathepsin D inhibitors block the formation of hyperphosphorylated tau fragments in hippocampus. Journal of Neurochemistry. 2000;74(4):1469–1477. doi: 10.1046/j.1471-4159.2000.0741469.x. [DOI] [PubMed] [Google Scholar]
- 147.Bi X, Yong AP, Zhou J, Ribak CE, Lynch G. Rapid induction of intraneuronal neurofibrillary tangles in apolipoprotein E-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8832–8837. doi: 10.1073/pnas.151253098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.De-Paula VJ, Schaeffer EL, Talib LL, Gattaz WF, Forlenza OV. Inhibition of phospholipase A2 increases Tau phosphorylation at Ser214 in embryonic rat hippocampal neurons. Prostaglandins Leukotrienes and Essential Fatty Acids. 2010;82(1):57–60. doi: 10.1016/j.plefa.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 149.Schaeffer EL, De-Paula VJ, Da Silva ER, et al. Inhibition of phospholipase A2 in rat brain decreases the levels of total Tau protein. Journal of Neural Transmission. 2011;118(9):1273–1279. doi: 10.1007/s00702-011-0619-4. [DOI] [PubMed] [Google Scholar]
- 150.López-Vales R, Ghasemlou N, Redensek A, et al. Phospholipase A2 superfamily members play divergent roles after spinal cord injury. FASEB Journal. 2011;25(12):4240–4252. doi: 10.1096/fj.11-183186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443(7113):768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 152.Sanchez-Mejia RO, Newman JW, Toh S, et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nature Neuroscience. 2008;11(11):1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. Journal of Biological Chemistry. 2006;281(31):22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kinsey GR, Blum JL, Covington MD, Cummings BS, McHowat J, Schnellmann RG. Decreased iPLA2 γ expression induces lipid peroxidation and cell death and sensitizes cells to oxidant-induced apoptosis. Journal of Lipid Research. 2008;49(7):1477–1487. doi: 10.1194/jlr.M800030-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hansson Petersen CA, Alikhani N, Behbahani H, et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Smesny S, Kinder D, Willhardt I, et al. Increased calcium-independent phospholipase A2 activity in first but not in multiepisode chronic schizophrenia. Biological Psychiatry. 2005;57(4):399–405. doi: 10.1016/j.biopsych.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 157.Wiedholz LM, Owens WA, Horton RE, et al. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ’schizophrenia-related’ behaviors. Molecular Psychiatry. 2008;13(6):631–640. doi: 10.1038/sj.mp.4002056. [DOI] [PubMed] [Google Scholar]
- 158.Lee LY, Ong WY, Farooqui AA, Burgunder JM. Role of calcium-independent phospholipase A2 in cortex striatum thalamus cortex circuitry-enzyme inhibition causes vacuous chewing movements in rats. Psychopharmacology. 2007;195(3):387–395. doi: 10.1007/s00213-007-0912-y. [DOI] [PubMed] [Google Scholar]
- 159.Mancuso DJ, Kotzbauer P, Wozniak DF, et al. Genetic ablation of calcium-independent phospholipase A2 γ leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. Journal of Biological Chemistry. 2009;284(51):35632–35644. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mancuso DJ, Sims HF, Han X, et al. Genetic ablation of calcium-independent phospholipase A2 γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. Journal of Biological Chemistry. 2007;282(48):34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schaeffer EL, Forlenza OV, Gattaz WF. Phospholipase A2 activation as a therapeutic approach for cognitive enhancement in early-stage Alzheimer disease. Psychopharmacology. 2009;202(1–3):37–51. doi: 10.1007/s00213-008-1351-0. [DOI] [PubMed] [Google Scholar]