

The very large group of negative-strand RNA viruses* includes some of the most serious and notorious pathogens of great medical and economic importance (1). These viruses are subdivided into those with segmented, negative-strand genomic RNAs [the Orthomyxoviridae (e.g., influenza viruses), Bunyaviridae (>300 viruses, including Bunyamwera), and Arenaviridae (e.g., Lassa fever)] and those with nonsegmented, negative-strand RNA genomes [Rhabdoviridae (e.g., vesicular stomatitis virus (VSV) and rabies), Paramyxoviridae (e.g., measles and respiratory syncytial virus), and Filoviridae (Marburg and Ebola viruses). The paper by Bridgen and Elliott (2) in this issue of the Proceedings is an important milestone because it reports the first complete recovery of a segmented, negative-strand RNA virus, the Bunyamwera virus, starting from DNA copies of the genome segments. Interestingly, in achieving this recovery, the authors applied a methodology that was developed for the nonsegmented viruses rather than the helper virus methodology developed for the segmented influenza virus. This system will now allow detailed molecular genetic analysis of all aspects of bunyavirus replication and could prove important in vaccine development as well. The rescue of Bunyamwera virus from DNA has its roots in a technical revolution that has occurred in negative-strand RNA virology over the past several years. Understanding of the rescue system requires a brief history of the baroque methods already applied to recovery of other negative-strand RNA viruses from DNA copies.
Analysis of both positive- and negative-strand RNA viruses using directed mutagenesis requires that one be able to recover the virus from a DNA copy of the RNA genome. This breakthrough came early with positive-strand RNA viruses, where the genomic RNA itself acts as mRNA and is infectious. Plasmid DNAs encoding positive-strand RNA genomes of the bacteriophage Qβ or poliovirus yielded RNAs that would regenerate complete infectious viruses (3, 4), and this has now been achieved with many other positive-strand RNA viruses. In contrast, the negative-strand RNA viruses lagged behind and presented a unique problem, because neither their genomic RNAs nor their antigenome complements serve as mRNAs, and neither can be used directly to recover infectious virus. The minimal infectious unit for these viruses is the RNA complexed with nucleocapsid and RNA-dependent RNA polymerase proteins in a ribonucleoprotein (RNP) complex.
The initial breakthrough came with influenza virus, where it was shown that single negative-strand RNA segments could be generated from DNA copies, assembled into RNPs in vitro, and then transfected into cells already infected with influenza virus. These transfected RNPs were replicated, and reassortant viruses containing one of the eight influenza RNA segments derived from the transfected RNP could be obtained if one had a good selection (see refs. 5–7). This methodology has proven extremely useful for the analysis of influenza virus, but was not applicable to the nonsegmented negative-strand viruses. These viruses have genomes that are extremely large (11–18 kb), and it has not been possible to assemble their RNAs into infectious RNPs in vitro.
Before the recovery of complete, infectious negative-strand RNA viruses from DNA, several laboratories had developed systems that allowed replication of small defective negative-strand RNA genomes or RNA minigenomes derived from DNA copies (8–15). These systems used either wild-type viruses to supply the nucleocapsid and polymerase proteins or generated expression of these proteins from plasmids with T7 promoters after infection of cells with vaccinia virus encoding the bacteriophage T7 RNA polymerase (16). Several laboratories had attempted to recover complete, infectious nonsegmented negative-strand viruses using these or similar systems, but none was successful starting with the negative-strand genome RNA.
Schnell et al. (17) then reported what turned out to be key to recovery of infectious rabies virus as well as other nonsegmented negative-strand RNA viruses from plasmid DNA. They found that if they used the vaccinia/T7 system to generate the full-length rabies antigenome RNA (positive strand) in cells along with the mRNAs encoding the nucleocapsid and polymerase proteins, then rabies virus could be recovered, albeit at very low efficiency, with only ≈1 in 107 transfected cells yielding infectious rabies virus. Because of the low efficiency, it was not obvious that this would work with other viruses, but in fact it has now worked with VSV (18, 19), measles (20), Sendai (21), and respiratory syncytial viruses (22), all of which have been recovered starting with the positive-strand antigenome.
We all know that a positive attitude can often be helpful, but why was the positive strand better than the negative? After all, the viruses have the negative strand as their genome. In retrospect it seems obvious. If a naked, negative-strand RNA genome is produced in the cytoplasm of cells also producing naked complementary mRNAs encoding viral proteins, the two can hybridize and prevent the critical assembly of the genome into the RNP, the template for transcription and replication. The negative-strand viruses in fact always keep their genomes in RNP form, probably in part to avoid this antisense problem. When one starts with the positive-strand antigenome, this RNA can form an RNP without any interference from the mRNAs encoding the nucleocapsid and polymerase proteins. Once in RNP form, the positive strand can then be replicated to form full-length negative-strand RNPs that are wrapped into RNPs as nascent RNA chains.
The magnitude of the antisense problem is illustrated in recent work from a group in Japan. This group reported that they can actually obtain recoveries even starting with the negative-strand RNA genome of Sendai virus (23), but at very low efficiencies. The efficiency with the positive strand was in the range of one recovery per 104–105 transfected cells, the highest efficiency yet reported for any nonsegmented negative-strand virus, but recovery with the negative-strand construct was 100-fold lower. In this system, the negative-strand genome was synthesized much better than the positive strand by T7 RNA polymerase, so the differential is probably even greater than 100-fold.
Members of the Bunyaviridae have three negative-strand genomic RNAs termed L, M, and S. These segments are in RNP form and often appear circular due to base pairing of nucleotides at the 5′ and 3′ ends of the RNPs (24, 25). After viral entry, these RNPs are transcribed in the cytoplasm by the viral polymerase to generate naked subgenomic mRNAs encoding the polymerase (L), membrane glycoproteins (M), and nucleocapsid (S) proteins. They are also replicated into full-length positive-strand RNPs and more full-length negative-strand RNPs. Budding of the virus occurs at intracellular membranes (26) and probably involves a direct interaction between the negative-strand RNPs and cytoplasmic tails of the glycoproteins.
The complicated system used by Bridgen and Elliott (2) to recover Bunyamwera virus from DNA is diagrammed in Fig. 1. Six different plasmids are transfected into cells that have already been infected with vaccinia virus encoding the bacteriophage T7 RNA polymerase. The first three plasmids transfected contain T7 promoters driving expression of mRNAs encoding the Bunyamwera polymerase, membrane glycoproteins, and nucleocapsid proteins, as well as two nonstructural proteins of unknown function. Subsequently, three more plasmids encoding each of the three positive-strand bunyavirus antigenomic RNAs are transfected into the cells. The positive strands were used to avoid the antisense problem. These antigenomic RNAs have terminal sequences that extend beyond those in the mRNAs, and they cannot be translated efficiently. The mRNAs must be therefore be supplied separately. Also the antigenomic RNAs initially transcribed include the hepatitis delta virus antigenomic ribozyme (27), which cleaves itself from the RNA to generate the precise 3′ end of the antigenome. Use of this ribozyme was based on earlier findings with VSV, which indicated that it was critical to generate a precise 3′ end to obtain RNA replication (11). The Bunyamwera antigenome segments are presumably wrapped in nucleocapsid protein to form RNPs and then replicated by the viral polymerase to generate full-length RNPs containing the genome segments. Because the glycoproteins are also made in this system, these RNPs can likely be packaged directly into virus particles by budding into the Golgi apparatus. Subsequent release of infectious particles into the medium completes the rescue.
Figure 1.
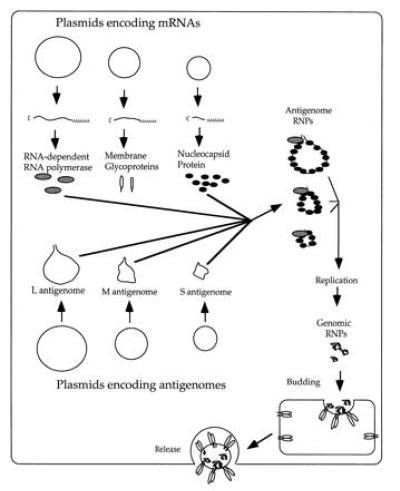
Recovery of infectious Bunyamwera virus from plasmid DNAs. The diagram represents the cytoplasm of a cell that has been transfected with six different plasmids (circles), three encoding the viral proteins and three encoding the antigenome segments of the virus. All of the plasmids contain promoters for the bacteriophage T7 RNA polymerase, which is supplied before infection of the cells with a vaccinia virus/T7 recombinant. The antigenomes are incorporated into RNPs, replicated to form genomic RNPs, and then bud into the Golgi apparatus before release from the cell.
Students or postdoctoral associates daring to venture into negative-strand virus rescue should be well aware of the large efforts and years of work that were required to set up these systems before recoveries were obtained. The systems can now be made to work but often just barely at first; this is molecular virology on the edge. Bridgen and Elliott (2) point out that their success (and that of other laboratories) was critically dependent on developing a good minigenome reporter system (chloramphenicol acetyltransferase or green fluorescent protein in their case), so they could have some idea of feasibility and scale required for the final push back to the virus. It is likely that this general approach will also work for the Arenaviridae with two genome segments and perhaps even for the Orthomyxoviridae with their six to nine genome segments.
We are just beginning to see the applications of recombinant negative-strand viruses, and surely the best is yet to come. For example, in the influenza system, numerous mutations have been introduced into the genome to study protein structure and function, foreign epitopes have been introduced into influenza glycoproteins, and attenuating mutations have been developed (see ref. 7 for review). In the nonsegmented viruses, results published from our own and two other groups (28–30) have demonstrated the ease of insertion and expression of foreign genes due to the modular nature of the genome. Much to everyone’s surprise, these extra genes are stably maintained in the recombinants. This contrasts with positive-strand RNA viruses, where extra genes are typically discarded rapidly, probably because of strict packaging limits within capsids of fixed size. The essentially linear nucleocapsids of negative-strand viruses like VSV can simply grow longer to accomodate extra genes and have not yet revealed any packaging limits (31).
It is possible to exchange the VSV glycoprotein gene for that of another serotype (18), and Bridgen and Elliott (2) accomplish the same feat for Bunyamwera virus by substituting one plasmid to generate a recombinant with the M segment from another bunyavirus. Recently, our group has reported that foreign cellular or viral membrane glycoproteins expressed from extra genes in VSV recombinants are often incorporated at extremely high levels into the virus particle (31). Because VSV can be grown to extremely high titers and in very large amounts, such recombinants could be very useful in development of killed or live vaccines or in specific targeting of viruses to cells. Now that we have direct access to these negative-strand genomes, the applications seem positively limitless.
Footnotes
Negative-strand RNA viruses have single-stranded genomic RNAs that are complementary to the viral mRNAs.
References
- 1.Fields B N, Knipe D M, Howley P M. Fields Virology. New York: Lippincott–Raven; 1996. [Google Scholar]
- 2.Bridgen A, Elliott R M. Proc Natl Acad Sci USA. 1996;93:15400–15404. doi: 10.1073/pnas.93.26.15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taniguchi T, Palmieri M, Weissmann C. Ann Microbiol (Milan) 1978;129:535–536. [PubMed] [Google Scholar]
- 4.Racaniello V R, Baltimore D. Science. 1981;214:916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- 5.Luytjes W, Krystal M, Enami M, Pavin J D, Palese P. Cell. 1989;59:1107–1113. doi: 10.1016/0092-8674(89)90766-6. [DOI] [PubMed] [Google Scholar]
- 6.Enami M, Luytjes W, Krystal M, Palese P. Proc Natl Acad Sci USA. 1990;87:3802–3805. doi: 10.1073/pnas.87.10.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Sastre A, Palese P. Annu Rev Microbiol. 1993;47:765–790. doi: 10.1146/annurev.mi.47.100193.004001. [DOI] [PubMed] [Google Scholar]
- 8.Park K H, Huang T, Correia F F, Krystal M. Proc Natl Acad Sci USA. 1991;88:5537–5541. doi: 10.1073/pnas.88.13.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins P L, Mink M A, Stec D S. Proc Natl Acad Sci USA. 1991;88:9663–9667. doi: 10.1073/pnas.88.21.9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calain P, Curran J, Kolakofsky D, Roux L. Virology. 1992;191:62–71. doi: 10.1016/0042-6822(92)90166-m. [DOI] [PubMed] [Google Scholar]
- 11.Pattnaik A K, Ball L A, LeGrone A W, Wertz G W. Cell. 1992;69:1011–1020. doi: 10.1016/0092-8674(92)90619-n. [DOI] [PubMed] [Google Scholar]
- 12.De B P, Banerjee A K. Virology. 1993;196:344–348. doi: 10.1006/viro.1993.1486. [DOI] [PubMed] [Google Scholar]
- 13.Dimock K, Collins P L. J Virol. 1993;67:2772–2778. doi: 10.1128/jvi.67.5.2772-2778.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conzelmann K K, Schnell M. J Virol. 1994;68:713–719. doi: 10.1128/jvi.68.2.713-719.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stillman E A, Rose J K, Whitt M A. J Virol. 1995;69:2946–2953. doi: 10.1128/jvi.69.5.2946-2953.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuerst T R, Niles E G, Studier F W, Moss B. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnell M J, Mebatsion T, Conzelmann K K. EMBO J. 1994;13:4195–4203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawson N D, Stillman E A, Whitt M A, Rose J K. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whelan S P J, Ball L A, Barr J N, Wertz G T W. Proc Natl Acad Sci USA. 1995;92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Billeter M A. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcin D, Pelet T, Calain P, Roux L, Curran T, Kolakofsky D. EMBO J. 1995;14:6087–6094. doi: 10.1002/j.1460-2075.1995.tb00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins P L, Hill M G, Camargo E, Grosfeld H, Chanock R M, Murphy B R. Proc Natl Acad Sci USA. 1995;92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato A, Sakai Y, Shioda T, Kondo T, Nakanishi M, Nagai Y. Genes Cells. 1996;1:569–579. doi: 10.1046/j.1365-2443.1996.d01-261.x. [DOI] [PubMed] [Google Scholar]
- 24.Pettersson R F, vonBonsdorf C H. J Virol. 1975;15:386–392. doi: 10.1128/jvi.15.2.386-392.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hewlett M J, Pettersson R F, Baltimore D. J Virol. 1977;21:1085–1093. doi: 10.1128/jvi.21.3.1085-1093.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Bonsdorff C-H, Saikku P, Oker-Blom N. Acta Virol (Engl Ed) 1970;14:109–114. [PubMed] [Google Scholar]
- 27.Perrotta A T, Been M D. Nature (London) 1991;350:434–436. doi: 10.1038/350434a0. [DOI] [PubMed] [Google Scholar]
- 28.Schnell M J, Buonocore L, Whitt M A, Rose J K. J Virol. 1996;70:2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mebatsion T, Schnell M J, Cox J H, Finke S, Conzelmann K K. Proc Natl Acad Sci USA. 1996;93:7310–7314. doi: 10.1073/pnas.93.14.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bukreyev A, Camargo E, Collins P L. J Virol. 1996;70:6634–6641. doi: 10.1128/jvi.70.10.6634-6641.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnell M J, Buonocore L, Kretzschmar E, Johnson E, Rose J K. Proc Natl Acad Sci USA. 1996;93:11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]