

Abstract
Synaptic plasticity, the cellular basis of learning and memory, involves the dynamic trafficking of AMPA receptors (AMPARs) into and out of synapses. One of the remaining key unanswered aspects of AMPAR trafficking is the mechanism by which synaptic strength is preserved despite protein turnover. In particular, the identity of AMPAR scaffolding molecule(s) involved in the maintenance of GluA2-containing AMPARs is completely unknown. Here we report that the synaptic scaffolding molecule (S-SCAM; also called membrane-associated guanylate kinase inverted-2 and atrophin interacting protein-1) plays the critical role of maintaining synaptic strength. Increasing S-SCAM levels in rat hippocampal neurons led to specific increases in the surface AMPAR levels, enhanced AMPAR-mediated synaptic transmission, and enlargement of dendritic spines, without significantly effecting GluN levels or NMDA receptor (NMDAR) EPSC. Conversely, decreasing S-SCAM levels by RNA interference-mediated knockdown caused the loss of synaptic AMPARs, which was followed by a severe reduction in the dendritic spine density. Importantly, S-SCAM regulated synaptic AMPAR levels in a manner, dependent on GluA2 not GluA1, sensitive to N-ethylmaleimide-sensitive fusion protein interaction, and independent of activity. Further, S-SCAM increased surface AMPAR levels in the absence of PSD-95, while PSD-95 was dependent on S-SCAM to increase surface AMPAR levels. Finally, S-SCAM overexpression hampered NMDA-induced internalization of AMPARs and prevented the induction of long term-depression, while S-SCAM knockdown did not. Together, these results suggest that S-SCAM is an essential AMPAR scaffolding molecule for the GluA2-containing pool of AMPARs, which are involved in the constitutive pathway of maintaining synaptic strength.
Introduction
Efficient synaptic transmission relies on the precise organization of various proteins including neurotransmitter receptors, ion channels, signaling enzymes, and cytoskeletal elements (Okabe, 2007; Sheng and Hoogenraad, 2007). Furthermore, activity-dependent modification of synaptic strength, or synaptic plasticity, demands a fine-tuned orchestration of recruitment and removal of proteins at synapses. For example, long-term potentiation (LTP) and long-term depression (LTD) are mediated by activity-dependent trafficking of AMPA receptors (AMPARs) to and out of excitatory synapses (Collingridge et al., 2004; Derkach et al., 2007; Shepherd and Huganir, 2007; Kessels and Malinow, 2009). Synaptic scaffolding proteins (S-SCAMs) play important roles in the assembly of the macro-signaling complexes and trafficking of synaptic proteins. The membrane-associated guanylate kinase (MAGUK) family of proteins is one of the most well studied synaptic scaffolds at the postsynaptic density (PSD). Among the PSD-MAGUKs, PSD-95 (SAP-90) represents the prototypical member and is involved in the various aspects of excitatory synaptic transmission, including synapse maturation, AMPAR trafficking, and synaptic plasticity (Elias and Nicoll, 2007; Xu, 2011).
S-SCAM was first identified as a protein interacting with SAPAP (also called GKAP; Kim et al., 1997; Hirao et al., 1998). S-SCAM is also known as membrane-associated guanylate kinase inverted-2 (MAGI-2) (Wu et al., 2000) or atrophin interacting protein-1 (Wood et al., 1998). The molecular organization of S-SCAM is in an inverse configuration to PSD-95, and is composed of six PDZ domains, one guanylate kinase (GK) domain, and two WW domains. Studies have shown that S-SCAM interacts with numerous PSD-95-binding proteins including NMDA receptor (NMDAR) (Hirao et al., 1998), ErbB4 (Buxbaum et al., 2008), Neuroligin (Iida et al., 2004), Kif1B (Mok et al., 2002), and transmembrane AMPAR regulatory proteins (TARPs) (Deng et al., 2006). S-SCAM also binds proteins that have no known interaction with PSD-95, which include Axin (Hirabayashi et al., 2004), β-catenin (Nishimura et al., 2002), β-dystroglycan (Sumita et al., 2007), and neuroligin 2 (Sumita et al., 2007). These overlapping protein–protein interaction profiles suggest that S-SCAM may play both similar and distinct roles, compared with PSD-95, in the molecular organization of PSDs and, in particular, AMPAR regulation. There are three S-SCAM isoforms of S-SCAM-α, -β, and -γ, which are generated by differential translational initiations from multiple sites (Hirao et al., 2000). Mice lacking the longest variant S-SCAM-α died within 24 h after birth (Iida et al., 2007), indicating that S-SCAM is an essential protein. Interestingly, hippocampal culture neurons prepared from these mutant mice showed abnormal elongated dendritic spines (Iida et al., 2007), suggesting that S-SCAM may function in the dendritic spine dynamics.
In addition to the probable role of S-SCAM in the molecular organization of PSDs and synaptic transmission, recent genetic studies uncovered S-SCAM gene mutations in patients with neurological diseases such as schizophrenia (Walsh et al., 2008) and infantile spasms (IS) (Marshall et al., 2008). Despite the potential importance of S-SCAM in synaptic biology and neurological diseases, little is currently known for the function of S-SCAM. Here, we report a novel and essential role of S-SCAM in the organization of PSDs and excitatory synaptic transmission.
Materials and Methods
Expression and RNAi construct.
Myc- or GFP-tagged S-SCAM expression constructs were obtained from Dr. Y. Hata (Tokyo Medical and Dental University, Tokyo, Japan). GluA1 and GluA2 shRNAs were as described previously (Lee et al., 2004). S-SCAM shRNA construct was created by cloning the following hairpin sequence GTACAGAACCTGAGCCATATTCAAGAGATATGGCTCAGGTTCTGTAC into pSUPER or pSUPER.neo+gfp vectors (OligoEngine). S-SCAM rescue construct was generated by introducing silent mutations in S-SCAM RNAi targeting sequence via site-directed mutagenesis and confirmed by nucleotide sequencing. PSD-95 shRNA is as described previously (Nakagawa et al., 2004).
Neuron transfection and immunocytochemistry.
Dissociated hippocampal neuron culture was prepared from E18 embryos of either sex as described previously (Lee et al., 2002) and grown in Neurobasal media supplemented with B27. Neuron transfection was performed at DIV 14 using Lipofectamine 2000 reagent (Invitrogen) as described previously (Lee et al., 2004). Cold methanol (−20°C for 10 min) fixation was used for the staining of PSD-95, Shank, GluN1, GluN2B, and GKAP. For costaining of β-Gal (or GFP) and S-SCAM, GKAP, or PSD-95, neurons were incubated first in 2% formaldehyde/4% sucrose/1 × PBS for 2 min followed by cold methanol for 10 min. Surface AMPAR staining was performed as described previously (Lee et al., 2004). Rabbit polyclonal anti-S-SCAM antibodies were prepared by injecting rabbits with purified recombinant protein covering the WW domains of S-SCAM (amino acids 303–405) (Hirao et al., 1998), affinity purified, and used at 0.01 μg/ml concentration for immunocytochemistry or Western blotting. We also tested a commercial rabbit polyclonal anti-MAGI-2 antibody (1:1000 dilution; Sigma) that is prepared from different antigens (amino acids 554–571), which is specific to S-SCAM/MAGI-2. Both antibodies produced very similar immunostaining patterns. However, to eliminate any potential cross-reactivity problem, immunostaining data obtained from the commercial antibody were shown. Primary antibodies and their dilution used for immunocytochemistry are as follows: mouse anti-β-Gal (1:1000; Promega), rabbit anti-β-Gal (1:5000; Abcam), mouse anti-PSD-95 (1:500; clone K28/43, UC Davis/NIH NeuroMab), rabbit anti- GKAP (1:250; Kim et al., 1997), mouse anti-NR2B (1:100, clone N59/36; UC Davis/NIH NeuroMab), mouse anti-pan-SAPAP (1: 250, clone N127/31; UC Davis/NIH NeuroMab), mouse anti-NMDAR1 (1:100; BD PharMingen), mouse anti-Shank (1:250; Naisbitt et al., 1999), mouse anti-Bassoon (1:200; Stressgen), mouse anti-synaptophysin (SVP-38, 1:1000; Sigma), rabbit anti-HA (1:100; Santa Cruz Biotechnology), mouse anti-HA (1:400, clone 12CA5; Roche), mouse anti-myc (1:100, clone 9E10; Santa Cruz Biotechnology), rabbit anti-myc (1:100; Cell Signaling Technology), rabbit anti-GluA1 (5 μg/ml; Oncogene), and mouse anti-GluA2 (5 μg/ml; Millipore Bioscience Research Reagents). Bound primary antibodies were visualized by Alexa Fluor 488 (1:250; Invitrogen), Cy3-conjugated (1:500; Jackson ImmunoResearch Laboratories), and/or Alexa Fluor 647 (1:200; Invitrogen) secondary antibodies.
Internalization assay.
AMPAR internalization assay was performed using GluA2 antibody-recognizing extracellular epitope (MAB397; Millipore), and data analyses were done as described previously (Lee et al., 2004). Briefly, after live labeling surface GluA2 with the antibody (10 μg/ml) in the conditioned medium for 10 min at 37°C, neurons were incubated for 2 min with conditioned medium (control) or conditioned medium plus NMDA (50 μm) or AMPA (100 μm). After a brief wash in Neurobasal medium, neurons were further incubated for 8 min in the conditioned medium before fixation and staining.
Confocal microscopy and image analyses.
Image acquisition was performed using a Nikon C1 plus laser scanning confocal microscope. Acquired z-series stack images were converted to projection images (with maximal projection option) for analysis using MetaMorph software (Molecular Devices) or in-house custom software. Images were analyzed in a double-blind manner. To measure the puncta number per given length of dendrites, per image, five dendritic segments (∼5 to 10 μm in length each) were selected from transfected and neighboring nontransfected neurons, respectively. After applying threshold, only puncta with >4 pixel sizes were counted. All data collected were transferred to Microsoft Excel for computation.
Statistical analysis.
All values represent means ± SEM, unless otherwise indicated. All transfection experiments were repeated at least three times. Statistical significance for pairs was analyzed by the Student's t test (unpaired, two-tailed, assuming unequal variance), unless otherwise indicated in the figure legends. ANOVA with Tukey's post hoc test was used for group comparisons. Cumulative plot data were analyzed by Kolmogrov–Smirnov test (K-S test). p < 0.05 was considered significant.
Electrophysiology.
For miniature EPSC (mEPSC) measurement, we used dissociated culture hippocampal neurons (plated at the density of 75 k cells per coverslip), which were transfected at DIV 14 with GFP alone (pEGFP-C1), GFP + S-SCAM, or pSUPER.neo+gfp plasmid-expressing S-SCAM shRNA. At 3 d post-transfection, transfected pyramidal neurons were identified by GFP fluorescence and morphological inspection. All recordings were performed at 25°C. Whole-cell patch recordings were performed by voltage-clamping neurons at −70 mV in bathing solution containing the following (in mm): 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, 10 HEPES, pH7.4, and ∼300 mOsm, and containing tetrodotoxin (1 μm; Tocris Bioscience) and bicuculline (20 μm; Tocris Bioscience), continuously perfused at the rate of ∼0.5 ml/min. Internal solution was composed of the following (in mm): 140 K·gluconate, 5 KCl, 2 MgCl2, 4 Mg·ATP, 0.3 Na2·GTP, 0.2 EGTA, 10 HEPES, and adjusted to pH7.2 and ∼290 mOsm. Micropipettes with tip resistance of 4–7 MΩ were used. mEPSCs were acquired through a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 kHz, using the “gap-free” protocol. mEPSCs were detected and analyzed with MiniAnalyses software (Synaptosoft) by setting the amplitude threshold to 5 pA (usually root mean square × three values lower than 4), and further filtered by selecting mini events of 10–90% rise time <3 ms. Cumulative probability plots were generated by combining mini events from all recorded neurons and analyzed for statistical significance by the K-S test.
For whole-cell paired recording experiments, we used slices culture prepared from the hippocampi of 7-d-old Sprague Dawley rats of either sex. Slices of 400 μm thickness were prepared using a tissue chopper, transferred onto MilliCell culture plate inserts (Millipore), and cultured for 4–6 d in MEM supplemented with 1 μg/ml insulin, 0.0012% ascorbic acid, 20% horse serum, 1 mm l-glutamine, 1 mm CaCl2, 30 mm HEPES, 13 mm d-glucose, and 5.2 mm NaHCO3 with Pen/Strep. The media was changed every other day. Sindbis virus-expressing GFP-S-SCAM (a gift from Dr. Yutaka Hata) (Nishimura et al., 2002) was injected into the CA1 region of hippocampal slices using a Toohey Spritzer microinjection system. Electrophysiological recordings were performed 15–24 h postinfection. A single slice was removed from the insert and placed in the recording chamber and continuously perfused with artificial CSF containing the following (in mm): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 11 glucose, 26 NaHCO3, 4 MgCl2, 4 CaCl2, and 290 mOsm and 50 μm picrotoxin and 2 μm chloroadenosine, bubbled with a mixture of 5% CO2 and 95% O2. Stimulating electrodes (two conductor platinum/iridium cluster microelectrode, 25 μm diameter; FHC) were placed ∼200 μm on either side of the recording cell. One pathway was used to induce LTD, the other served as a control pathway. The patch pipette was filled with internal solution containing (in mm): 115 CsMeSO3, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 10 Na-phosphocreatine, 0.6 EGTA, 5 QX314, pH 7.2, and 290 mOsm, and had tip resistance of 3–6 MΩ. For rectification experiments, spermine (0.1 mm) was added in the internal solution. AMPA EPSCs were recorded at −60 mV and measured as peak inward current within a 60 ms window after stimulation. NMDA EPSCs were recorded at +40 mV and measured 60–100 ms after the initiation of the EPSC. Rectification index was calculated by dividing peak AMPA amplitudes measured at −60 mV by those at +40 mV. Stimulation pulses were provided at 0.3 Hz. Sixty traces were averaged for AMPA and NMDA EPSCs. For LTD experiments, at least 8 min of stable baseline responses were collected before LTD induction. LTD-inducing stimulus consisted of 200 stimulation pulses at 1 Hz while holding cells at −40 mV. For paired pulse ratio (PPR), two consecutive stimuli were given 25 ms, 50 ms, 100 ms, and 200 ms apart with a 2 s interval between time points. At least 60 traces were averaged.
Results
Increasing S-SCAM levels promotes the enlargement of dendritic spines
To study the function of S-SCAM in excitatory synaptic transmission, we first overexpressed S-SCAM in hippocampal neurons and examined the effect on the dendritic spines. We used the longest variant S-SCAM-α that produced mostly S-SCAM-α (>80%) but also -β, and -γ isoforms when expressed in hippocampal neurons (data not shown). Transfected S-SCAM showed strong enrichment in all dendritic spines identified (Fig. 1A). Quantification of spine density indicated that S-SCAM overexpression reduced spine density to ∼70% of control β-Gal-only transfected neurons (53.1 ± 2.2 vs 37.6 ± 1.6 spines per 100 μm, β-Gal vs S-SCAM; p < 0.001) (Fig. 1B).
Figure 1.
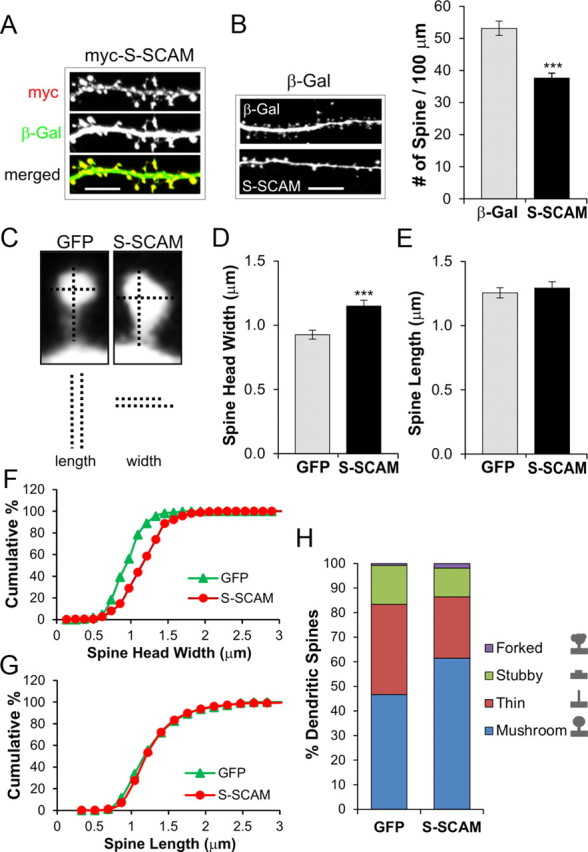
Effect of S-SCAM overexpression on the dendritic spines of hippocampal neurons. Hippocampal neurons were cotransfected with S-SCAM (myc-tagged) and β-gal or GFP, and their dendritic spines were analyzed. A, Representative images of dendrites from hippocampal neurons cotransfected with S-SCAM (red) and β-Gal (green), showing synaptic localization of S-SCAM. Scale bar, 5 μm. B, Effect of S-SCAM overexpression on dendritic spine density. Representative images of dendrites from neurons transfected with β-Gal alone (β-Gal, top) and S-SCAM + β-Gal (S-SCAM, bottom) are shown on the left and quantified spine density data are on the right. n > 20 per condition. ***p < 0.001. C, Representative images of dendritic spines from control (GFP only) or S-SCAM-transfected neurons. Vertical and horizontal dotted lines represent the length and width of dendritic spines, respectively. D, E, Effect of S-SCAM overexpression on the width (D) and length (E) of dendritic spines. F, G, Cumulative plot analyses of dendritic spine width (F) and length (G). p < 0.001 for width and p = 0.35 for length (K-S test). H, Changes in the morphology of dendritic spines after S-SCAM overexpression. Dendritic spines from >10 neurons per group were analyzed.
Increasing S-SCAM levels also influenced dendritic spine morphology. Following S-SCAM expression, as shown in Figure 1C–E, there was a specific increase in spine head width by 24% (0.93 ± 0.03 vs 1.15 ± 0.05 μm, GFP control vs S-SCAM; p < 0.001) with no significant change in the spine length (1.26 ± 0.04 vs 1.29 ± 0.05 μm for GFP vs S-SCAM; p = 0.57). The cumulative plot of spine head width showed a uniform right shift, indicating that S-SCAM overexpression targeted all spines rather than a specific population (Fig. 1F; p < 0.001). Cumulative plot analysis of dendritic spine length did not show a significant difference between GFP control and S-SCAM overexpressed neurons (Fig. 1G; p = 0.35). The increase of spine head width after S-SCAM overexpression was accompanied by the increased proportion of mushroom-type spines (46% for GFP vs 62% for S-SCAM; p < 0.05) while decreasing the proportion of thin-type spines (37% for GFP vs 24% for S-SCAM; p < 0.05; Fig. 1H). There was no significant difference in stubby-type (16% GFP vs 12% S-SCAM; p = 0.33) and forked-type spines (1% GFP vs 2% S-SCAM; p = 0.17) following S-SCAM transfection. These results collectively indicate that increasing S-SCAM levels in hippocampal neurons promote the maturation of dendritic spines.
S-SCAM overexpression changes the composition of PSD scaffolding proteins
S-SCAM binds to numerous proteins and can potentially influence the composition of synaptic proteins by selectively recruiting and/or stabilizing a set of interacting proteins at synapses. To study the scaffolding function of S-SCAM, we examined the effect of increasing S-SCAM levels on the synaptic accumulation of three key PSD scaffolding proteins. First we examined a direct S-SCAM-binding protein, GKAP. S-SCAM overexpression drastically increased GKAP puncta intensity at dendritic spines by >2.5-fold (Fig. 2A,B), when compared with nontransfected (Non-txf) neighboring neurons (100 ± 3 vs 261 ± 27%, Non-txf vs S-SCAM; p < 0.001). The increase of synaptic GKAP levels after S-SCAM overexpression was accompanied by a small decrease (∼14%) in GKAP puncta density (78 ± 3 vs 67 ± 4 per 100 μm dendrite, Non-txf vs S-SCAM; p < 0.05) (Fig. 2C), which was smaller than the decrease in spine density (∼40% reduction; Fig. 1B). Further analyses indicated that S-SCAM overexpression increased the population of dendritic spines containing multiple GKAP puncta at the expense of single puncta spines (GFP vs S-SCAM, spines with 1 puncta, 84 ± 2 vs 73 ± 2%; p < 0.001; 2 puncta, 15 ± 2 vs 24 ± 2%; p < 0.001; 3 puncta, 2 ± 1 vs 3 ± 1%; p = 0.04) (Fig. 2D,E). In contrast to GKAP, increased S-SCAM levels led to only a modest (∼1.2-fold) increase of PSD-95 intensity at dendritic spines (100 ± 2 vs 122 ± 5%, Non-txf vs S-SCAM; p < 0.001) (Fig. 2A,B). Surprisingly, despite the large increase in GKAP levels at dendritic spines, the levels of Shank, a GKAP-binding scaffolding protein, were reduced by 17% (100 ± 4 vs 83 ± 4%, Non-txf vs S-SCAM; p < 0.01) (Fig. 2A,B). Furthermore, the puncta densities of both PSD-95 and Shank were greatly reduced (by 44% for PSD-95; by 62% for Shank; p < 0.001 for both proteins) (Fig. 2C), to a degree greater than the spine density reduction (by ∼30%; Fig. 1B).Together, these results demonstrate that increasing S-SCAM levels led to drastic changes in the protein composition of PSD scaffolding proteins.
Figure 2.

S-SCAM overexpression affects synaptic levels of PSD proteins. A, Representative images showing the effect of S-SCAM overexpression on GKAP (top), PSD-95 (middle), and Shank (bottom). Dendrites of nontransfected neighboring neurons (Non-txf) are compared with those of S-SCAM-transfected neurons. B, Quantification of intensity of synaptic puncta normalized to Non-txf neurons. C, Quantification of synaptic puncta density of PSD proteins. n > 25 per condition. D, Representative images demonstrating the effect of S-SCAM overexpression, versus GFP control, on the number of GKAP puncta present in a single dendritic spine. E, Quantification of the number of GKAP puncta per spine. ***p < 0.001, **p < 0.01, *p < 0.05 compared with Non-txf. Scale bars: A, 5 μm; D, 0.5 μm.
S-SCAM overexpression specifically increases synaptic AMPAR levels
S-SCAM was reported to bind TARPs (Deng et al., 2006), but its role in the regulation of AMPARs is unknown. We have found that increasing S-SCAM levels in hippocampal neurons led to a drastic increase in the amount of all three major GluA subunits (GluA1, GluA2, and GluA3) at dendritic spines (Fig. 3A,B) (170 ± 9, 210 ± 9, and 140 ± 9% for GluA1, GluA2, and GluA3, respectively, compared with Non-txf; p < 0.001 for all three GluAs). Surface staining of these proteins indicated that S-SCAM increased the surface expression of GluA1 (sGluA1) and GluA2 (sGluA2) at the dendritic spines as well (Fig. 3C,D) 238 ± 14 and 229 ± 9% for sGluA1 and sGluA2, respectively; p < 0.001 for both sGluAs). However, the total expression levels of these proteins, as measured by the staining intensities in the soma and dendrites, did not change significantly (Fig. 3A; quantified in 3B). This suggests that the increase of AMPARs in the dendritic spine came at the expense of AMPARs localized outside dendritic spines. Since S-SCAM overexpression promoted the maturation of dendritic spines, we also examined the number of dendritic spines lacking sGluA2, which measures silent synapses indirectly. Remarkably, as shown in a Figure 3E, S-SCAM overexpression greatly reduced the percentage of the spines lacking sGluA2 (7 ± 2 vs 25 ± 7%, S-SCAM vs control GFP-transfected neurons; p < 0.001), consistent with synapse maturation. Blocking NMDAR activity with APV (100 μm) did not prevent the S-SCAM-induced increase of sGluA2 levels (Fig. 3F,G; p = 0.42), indicating that the increase of AMPAR by S-SCAM overexpression is an activity-independent process. In contrast to AMPAR, GluN1 or GluN2B levels at dendritic spines were not significantly affected by S-SCAM overexpression (Fig. 3H,I; p > 0.05), indicating that S-SCAM overexpression specifically increased AMPAR levels at synapses. Together, these data indicate that S-SCAM overexpression promoted the accumulation and/or stabilization of AMPARs at dendritic spines.
Figure 3.
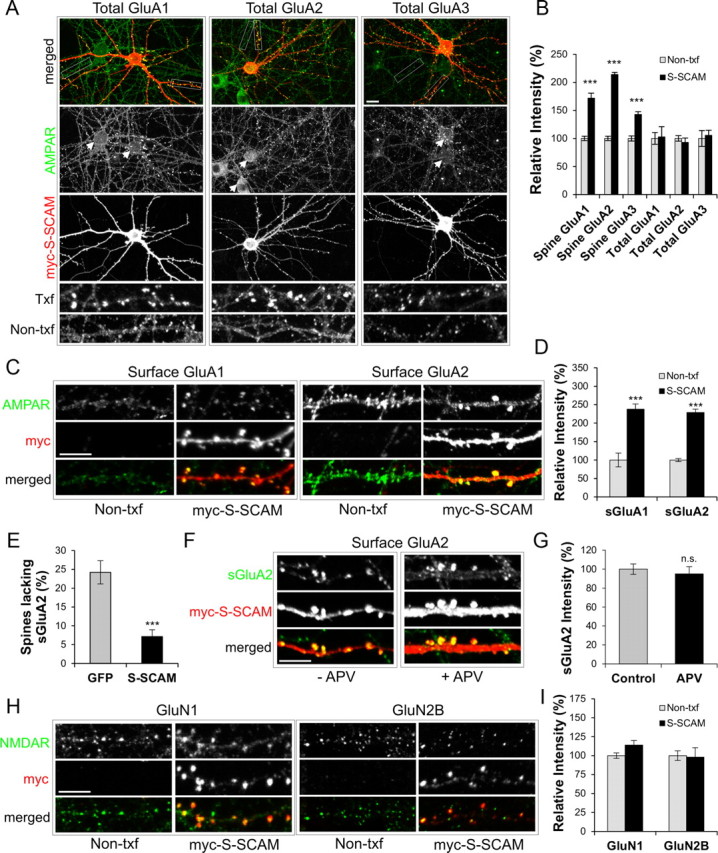
S-SCAM overexpression specifically increases AMPA-type glutamate receptors at synapses. A, Representative images showing the effect of S-SCAM (red) overexpression on total GluA1, GluA2, and GluA3 (green). Boxes indicate selected areas of dendrites used for expanded images of AMPAR staining shown at the bottom two rows. Arrowheads indicate soma of both Non-txf and transfected neurons. B, Quantification of relative intensities of total GluAs in dendritic spines (Spine GluAs) and in soma and dendrites (Total GluAs). C, Representative images showing the effect of S-SCAM (red) overexpression on surface GluA1 (sGluA1) and surface GluA2 (sGluA2). D, Quantification of relative intensities of surface GluRs in dendritic spines. E, Effect of S-SCAM overexpression on the percentage of dendritic spines lacking sGluA2 staining. n > 25 per condition. ***p < 0.001. F, G, Activity-independent increase of sGluA2 by S-SCAM. Representative images (F) showing the effect of S-SCAM overexpression on surface sGluA2 in neurons untreated (−APV) or treated with 100 μm APV (+APV). Quantification of relative intensities of sGluA2 in dendritic spines (G). H, Representative images showing the effect of S-SCAM overexpression on total GluN1 and GluN2B. I, Quantification of relative intensities of GluN1 and GluN2B puncta in Non-txf- and S-SCAM-transfected neurons. n > 25 per condition. ***p < 0.001. Scale bars: (wide view in A) 10 μm; (C,F,H) 5 μm.
The knockdown of S-SCAM led to the loss of surface AMPARs and dendritic spines
To examine the role of endogenous S-SCAM, we designed shRNA sequences to specifically knock down S-SCAM levels in hippocampal neurons. When a plasmid expressing short hairpin RNA-targeting S-SCAM (designated S-SCAM RNAi) was cotransfected into COS cells, as shown in Figure 4A, it specifically reduced the protein levels of S-SCAM to <5% of control levels (S-SCAM cotransfected with unrelated Zn-T3 shRNA) (Lee et al., 2004). The S-SCAM RNAi had no significant effect on the level of PSD-95 (Fig. 4A) or GKAP (data not shown), showing specificity of the knockdown. When transfected into hippocampal neurons, S-SCAM RNAi effectively reduced S-SCAM levels to 37% in dendrites (p < 0.001) as determined by immunofluorescent intensity (Fig. 4B,C).
Figure 4.
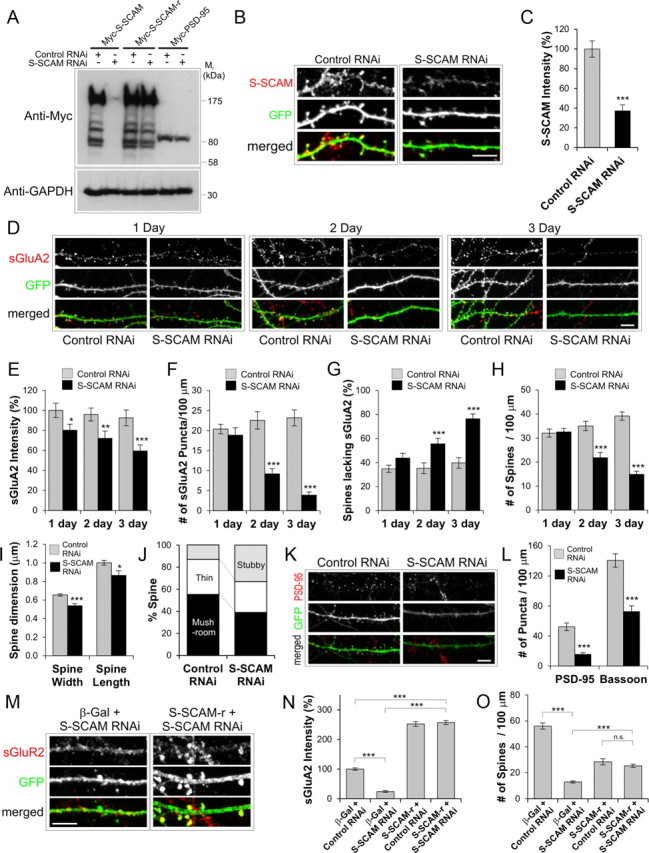
S-SCAM knockdown by RNAi reduces surface AMPARs and led to the loss of dendritic spines. A–C, Specificity and efficacy of S-SCAM RNAi. A, COS cells were cotransfected with myc-S-SCAM, myc-S-CAM-r (RNAi-resistant S-SCAM), or myc-PSD-95 and S-SCAM RNAi or control RNAi (Zn-T3 RNAi). Note that S-SCAM RNAi reduced S-SCAM levels but had no significant effect on the S-SCAM-r and Myc-PSD-95. Anti-GAPDH blot is shown as a loading control. B, C, Efficient knockdown of endogenous S-SCAM by S-SCAM RNAi in hippocampal neurons. GFP (green) was coexpressed from the RNAi expression plasmid and used as transfection marker. Representative images are shown in B, and quantification of data are provided in C. D, Represents images showing the time course of S-SCAM RNAi effect on sGluA2 (red) levels. E–J, Quantification of the effect of S-SCAM RNAi on sGluA2 intensity (E), the density of sGluA2 puncta (F), percentage of dendritic spines lacking sGluA2 staining (G), dendritic spine density (H), dendritic spine dimension (I), and dendritic spine morphology (J). K, L, Effect of S-SCAM RNAi on PSD-95 and Bassoon clusters (red). Representative results are shown in K. Quantification of the S-SCAM RNAi effect on the density of PSD-95 and Bassoon clusters is shown in L. M–O, Rescue experiments with S-SCAM-r. Representative images of neurons cotransfected with S-SCAM RNAi and S-SCAM-r stained for sGluA2 and GFP (M). Quantification of the rescue experiments for sGluA2 intensity (N) and dendritic spine density (O). n > 25 per condition. ***p < 0.001, **p < 0.01, *p < 0.05. Scale bar, 5 μm.
We first examined the effect of S-SCAM RNAi on GluA2, a major subunit of AMPARs in the hippocampal neurons. S-SCAM RNAi reduced both the intensity and number of sGluA2 puncta significantly (Fig. 4D–F). The reduction of sGluA2 intensity started to show as early as 1 d post-transfection of S-SCAM RNAi (100 ± 7 vs 80 ± 6%, control vs S-SCAM RNAi; p < 0.05) and reached 60% of control levels 3 d post-transfection (Fig. 4E). The reduction of sGluA2 puncta density became statistically significant 2 d post-transfection (23 ± 2 vs 9 ± 1 per 100 μm dendrite, control vs S-SCAM RNAi; p < 0.001) (Fig. 4F) and was reduced to 17% of control level after 3 d (23 ± 2 vs 4 ± 1 per 100 μm dendrite, control vs S-SCAM RNAi; p < 0.001). The reduction in sGluA2 was accompanied by a significant increase in the proportion of dendritic spines lacking sGluA2 staining (after 3 d, 40 ± 4 vs 76 ± 4%, control vs S-SCAM RNAi; p < 0.001) (Fig. 4G). These data clearly indicate that S-SCAM RNAi removes sGluA2 from dendritic spines.
S-SCAM RNAi also had a dramatic effect on dendritic spines Fig. 4B,D; GFP channels). Knockdown of S-SCAM reduced the dendritic spine density (at 3 d post-transfection, 39 ± 2 vs 15 ± 1 per 100 μm dendrite, control vs S-SCAM RNAi; p < 0.001) (Fig. 4H) and the size of dendritic spines (p < 0.001 for width and p < 0.05 for length) (Fig. 4I). Furthermore, S-SCAM knockdown increased the proportion of stubby spines (13 vs 33%, control vs S-SCAM RNAi; p < 0.001) at the expense of mushroom-type mature spines (55 vs 39% control vs S-SCAM RNAi; p < 0.05) (Fig. 4J). These data suggest that losing S-SCAM from synapses promotes the collapse of mushroom-type dendritic spines to stubby ones, which are eliminated eventually. Consistent with the reduction in the number of dendritic spines, S-SCAM RNAi greatly reduced the puncta density of PSD-95 (52 ± 5 vs 16 ± 2 per 100 μm dendrite, control vs S-SCAM RNAi; p < 0.001) (Fig. 4K,L) and Bassoon (141 ± 9 vs 73 ± 8 per 100 μm dendrite, control vs S-SCAM RNAi; p < 0.001) (Fig. 4L), indicating the reduction of overall synapse numbers.
To further confirm the specificity of S-SCAM RNAi effect, we performed a “rescue” experiment with S-SCAM RNAi-resistant S-SCAM (Fig. 4A, designated S-SCAM-r). When rescuing S-SCAM-r was coexpressed with S-SCAM RNAi, the amount of sGluR2 was increased to a level similar to S-SCAM overexpression (257 ± 7 vs 252 ± 8%, S-SCAM RNAi + S-SCAM-r vs S-SCAM-r + Control RNAi; p = 0.67) (Fig. 4M,N) and the number of dendritic spines was similarly restored (25 ± 1 vs 28 ± 2 per 100 μm dendrite, S-SCAM RNAi + S-SCAM-r vs S-SCAM-r + Control RNAi; p = 0.26) (Fig. 4O). Thus, the effect of S-SCAM RNAi on AMPARs and dendritic spines was specifically related to the loss of S-SCAM proteins and not caused by nonspecific effect of RNAi. Together, these data point out that S-SCAM is an essential scaffolding molecule for the stabilization/maintenance of AMPARs and dendritic spines.
S-SCAM levels regulate the AMPAR component of excitatory synaptic transmission
To measure directly the effect of changing S-SCAM levels on AMPAR-mediated synaptic transmission, we measured AMPAR mEPSCs after transfecting dissociated hippocampal culture neurons with S-SCAM or S-SCAM RNAi constructs. Increasing S-SCAM levels enhanced AMPA mEPSC amplitudes (17.4 ± 0.9 vs 23.4 ± 1.0 pA, GFP control vs S-SCAM; p < 0.001) (Fig. 5A–C), while S-SCAM RNAi reduces AMPA mEPSC amplitudes (14.5 ± 0.9 pA; p < 0.01 compared with GFP control) (Fig. 5A–C). We did not observe significant changes in the AMPA mEPSC frequency after S-SCAM overexpression (3.9 ± 0.5 vs 4.6 ± 1.0 Hz, GFP control vs S-SCAM; p = 0.35) (Fig. 5D), despite the reduction in dendritic spine density. This is likely due to the “unsilencing” of silent synapses by acquiring AMPAR (Fig. 3E). In contrast, S-SCAM RNAi greatly reduced mEPSC frequency (1.7 ± 0.4 Hz; p < 0.005), consistent with the reduction in synapse numbers after S-SCAM RNAi (Fig. 4H). Therefore, S-SCAM levels directly influence synaptic AMPAR levels.
Figure 5.
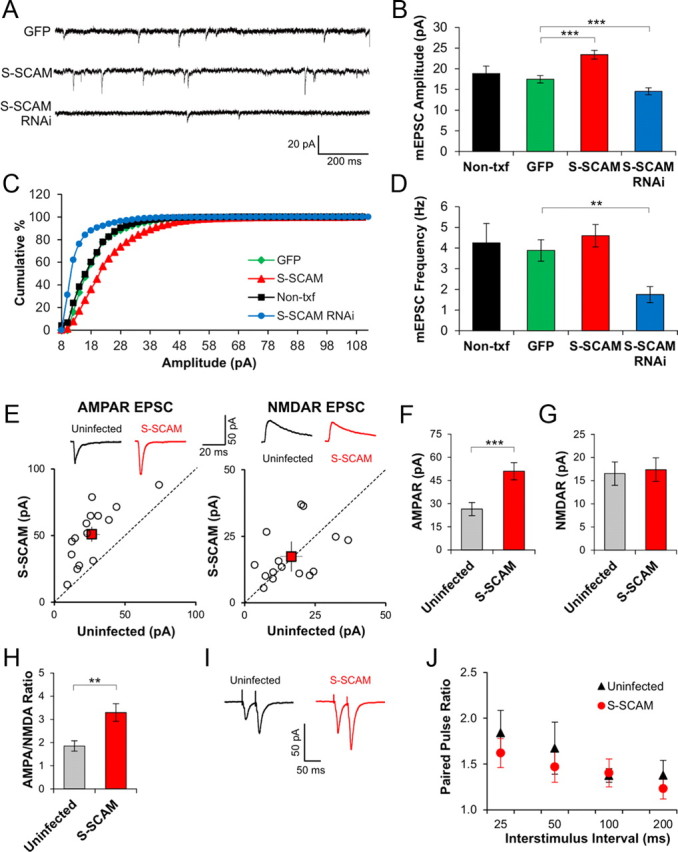
Changing S-SCAM levels modulate AMPAR-mediated synaptic transmission. A, Representative traces of AMPAR mEPSCs measured after transfecting dissociated hippocampal culture neurons with GFP, S-SCAM, or S-SCAM RNAi. B, Bar graph showing average mEPSC amplitudes of each group. ***p < 0.001 and n > 10 per condition. C, Cumulative histograms of mEPSC amplitudes in Non-txf, GFP-, S-SCAM-, or S-SCAM RNAi-transfected neurons. p = 0.46 for Non-txf vs GFP and p < 0.001 for GFP versus S-SCAM and GFP versus S-SCAM RNAi (K-S test). D, Bar chart showing average mEPSC frequency of each group. **p < 0.01, n > 10 per condition. E–H, Enhanced AMPAR-mediated EPSCs after S-SCAM overexpression in pyramidal CA1 neurons of cultured hippocampal slices. Synaptic currents were simultaneously recorded from neurons infected with sindbis virus-expressing GFP-S-SCAM (S-SCAM) and uninfected nearby pyramidal neurons (uninfected) held at −60 or +40 mV. E, Top, Sample recording traces mediated by AMPAR and NMDAR. Quantification of AMPAR EPSC amplitudes and NMDAR EPSC amplitudes are plotted for each pair of S-SCAM-infected and -uninfected nearby cells. Each open circle represents a single pair of recording. Mean ± SEM is shown by closed red squares. Bar graphs for AMPA EPSC (F) and NMDAR EPSC (G). ***p < 0.001 and n = 15 per group. H, Bar graph of AMPA/NMDA ratio showing an increased AMPA/NMDA ratio in S-SCAM-infected neurons.**p < 0.005 (paired t test). I, J, PPR of S-SCAM-infected and -uninfected neurons measured using pairs of stimulation pulses separated by various intervals. Representative traces of PPR at 50 ms interval are shown in I and quantified PPR data are shown in J. n = 10 per group.
S-SCAM overexpression in CA1 pyramidal neurons of hippocampal slices cultured by sindbis virus infection drastically increased AMPAR-mediated synaptic transmission measured at −60 mV (26.5 ± 4.2 vs 51.0 ± 5.5 pA, uninfected neighboring neuron vs S-SCAM-infected; p < 0.001) (Fig. 5E,F). In contrast, we did not detect significant changes in the NMDAR-mediated responses measured at +40 mV (16.5 ± 2.5 vs 17.4 ± 2.6 pA, uninfected vs S-SCAM-infected; p = 0.77) (Fig. 5E,G), indicating that S-SCAM overexpression did not affect NMDAR-mediated synaptic transmission significantly. Importantly, S-SCAM-infected neurons showed a significant increase in the AMPA/NMDA ratio (1.9 ± 0.2 vs 3.3.0 ± 0.4, uninfected vs S-SCAM-infected; p < 0.005) (Fig. 5H), indicating that S-SCAM overexpression specifically increased the AMPA component of excitatory synaptic transmission. We did not find a significant difference in the PPR of S-SCAM-infected CA1 neurons from uninfected neighboring neurons (1.7 ± 0.3 vs 1.5 ± 0.2 at 50 ms interval, 1.4 ± 0.1 vs 1.4 ± 0.2 at100 ms interval, uninfected vs S-SCAM infected; p > 0.4) (Fig. 5I,J), indicating that S-SCAM overexpression did not change presynaptic function significantly.
S-SCAM regulates synaptic AMPAR independently of PSD-95
Our data and literature indicate that both S-SCAM and PSD-95 can regulate synaptic AMPAR levels. Do they play redundant or independent roles? To address this question, we performed “reciprocal rescuing” experiments. When S-SCAM or PSD-95 was overexpressed in hippocampal neurons (with a GFP txf marker), both proteins increased sGluA1 levels comparably (Fig. 6A,B) (100 ± 8 vs 235 ± 18 or 221 ± 43%, non-txf vs S-SCAM or PSD-95; p < 0.001). Surface GluA2 levels were also increased by both S-SCAM and PSD-95 overexpression, although S-SCAM was more effective than PSD-95 (100 ± 15 vs 184 ± 19 or 134 ± 11%, non-txf vs S-SCAM or PSD-95, p < 0.001; S-SCAM vs non-txf, p < 0.05; S-SCAM vs PSD-95). On the other hand, S-SCAM RNAi and PSD-95 RNAi reduced both sGluA levels drastically (for sGluA1, 41 ± 9 vs 57 ± 11%; for sGluA2, 23 ± 10 vs 46 ± 7%, S-SCAM RNAi vs PSD-95 RNAi; p < 0.001 compared with GFP control for both RNAi). Importantly, overexpression of PSD-95 in the presence of S-SCAM RNAi did not increase or restore either sGluA subunit level at synapses (18 ± 5% for GluA1, 6 ± 3% for GluA2). In sharp contrast, overexpression of S-SCAM in the presence of PSD RNAi not only rescued the loss of PSD-95 but also increased both sGluA levels at dendritic spines comparable to S-SCAM overexpression alone (sGluA1, 196 ± 18 vs 235 ± 18%; sGluA2, 184 ± 19 vs 180 ± 22%,;PSD-95 RNAi + S-SCAM vs S-SCAM; for both sGluAs, p < 0.001 and p > 0.1, compared with PSD-95 RNAi and to S-SCAM alone, respectively) (Fig. 6A,B). Quantification of relative intensities of PSD-95 and S-SCAM verified that both protein overexpressions reached >5-fold higher levels than the endogenous protein levels, regardless of the presence of RNAi (Fig. 6C). Therefore, S-SCAM increased surface AMPARs independently of PSD-95, while PSD-95 was dependent on S-SCAM to exert its effect on AMPARs. These data suggest that S-SCAM is an indispensable scaffolding molecule for AMPARs.
Figure 6.

S-SCAM regulates AMPAR levels independently of PSD-95. Hippocampal neurons were transfected with GFP alone, GFP + S-SCAM, GFP + PSD-95, S-SCAM RNAi alone, PSD-95 RNAi alone, S-SCAM RNAi + PSD-95, or PSD-95 RNAi + S-SCAM. Transfected neurons were visualized by GFP fluorescence (RNAi vectors coexpress GFP) and analyzed for sGluA1 or sGluA2 staining. Scale bar, 5 μm. A, Representative images from each group showing sGluA (red) and GFP staining (green). B, Quantification of relative intensities of surface GluAs in dendritic spines. C, Quantification of relative intensities of PSD-95 and S-SCAM in dendritic spines. n > 20 per group. ***p < 0.001, *p < 0.05.
S-SCAM controls the GluA2-containing pool of AMPARs
PSD-95 regulates AMPARs mainly through GluA1 subunits that are inserted into synapses during high activity (Ehrlich and Malinow, 2004; Xu, 2011). Does S-SCAM also exhibit such AMPAR subunit-specific regulation? To examine the question, we used AMPAR subunit-specific RNAi (Lee et al., 2004). When neurons were transfected with GluA1 RNAi, as shown in Figure 7A,B, the level of sGluA1 was greatly reduced regardless of S-SCAM overexpression (<25% of β-Gal + Control RNAi; p < 0.001), showing the effectiveness of the RNAi. GluA2 RNAi exhibited similar effectiveness (<23% of β-Gal + Control RNAi; p < 0.001). In hippocampal neurons cotransfected with GluA1 RNAi and S-SCAM, sGluA2 levels were significantly increased to a level comparable to that of S-SCAM + Control RNAi (226 ± 18 vs 198 ± 18%, S-SCAM + GluA1 RNAi vs S-SCAM + Control RNAi, p = 0.28; p < 0.001 compared with β-Gal + Control RNAi). Therefore, GluA1 was not required for the S-SCAM-induced increase of surface AMPAR levels. On the other hand, surprisingly, S-SCAM failed to increase sGluA1 levels in neurons cotransfected with GluA2 RNAi (106 ± 9 vs 293 ± 14%, S-SCAM + GluA2 RNAi vs S-SCAM + Control RNAi; p < 0.001). These results suggest that S-SCAM increases surface AMPAR levels in a manner dependent on GluA2 subunit.
Figure 7.
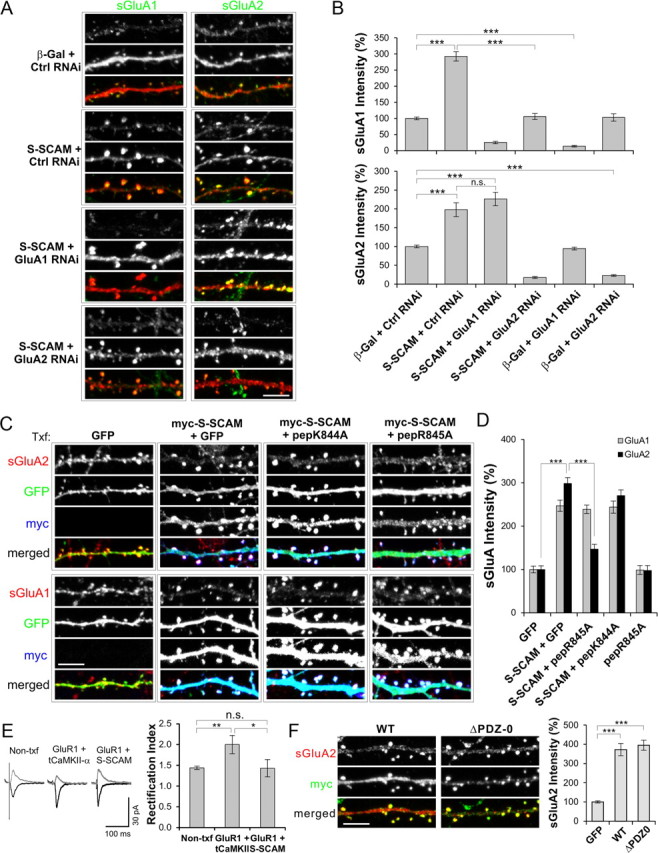
S-SCAM regulates AMPARs through GluA2 subunit. A, B, Effect of GluA1- or GluA2-specific RNAi on S-SCAM overexpression-induced increase of AMPAR. Hippocampal neurons were transfected with β-Gal + Control RNAi, S-SCAM + Control RNAi, S-SCAM + GluA1 RNAi, or S-SCAM + GluA2 RNAi and immunostained for sGluA1 and sGluA2. Representative images are shown in A and quantification is provided as bar graphs in B. Note that S-SCAM failed to increase sGluA1 levels in the presence of GluA2 RNAi, while S-SCAM increased sGluA2 levels in the presence of GluA1RNAi. ***p < 0.001 and n > 20 per condition. C, D, Effect of NSF-interaction blocking peptides on S-SCAM-induced increase of surface AMPAR levels. Hippocampal neurons were transfected with GFP alone, S-SCAM + GFP, S-SCAM + GFP-pepK844A, or S-SCAM + GFP-pepR845A and examined for sGluA1 and sGluA2 levels. Representative images are shown in C and quantified data are shown in D. Scale bar, 5 μm. ***p < 0.001 and n >25 per condition. E, Bar graph showing rectification index of neurons transfected with GluR1 + S-SCAM or GluR1 + tCaMKII-α. Control represents untransfected neighboring neurons. **p < 0.01, *p < 0.05. n >15 per condition. F, Effect of PDZ-0 domain deletion on sGluA2 levels in hippocampal neurons. ***p < 0.001, n = 30 per condition.
To further corroborate that S-SCAM exerts its influence through GluA2, we took advantage of N-ethylmaleimide-sensitive fusion protein (NSF)-interaction blocking peptide that targets GluA2 containing AMPARs and causes the rundown of AMPAR mEPSCs (Song et al., 1998; Lüscher et al., 1999; Lüthi et al., 1999; Lee et al., 2002; Evers et al., 2010). The NSF peptide-sensitive AMPARs are thought to represent the constitutively cycling pool of AMPARs involved in the maintenance of synaptic strength. We used peptides fused to enhanced green fluorescent protein for easier identification of transfection (Lee et al., 2002). Coexpressing pepR845A peptide that specifically blocks NSF interaction with GluA2 greatly attenuated S-SCAM-induced increase of sGluA2 (299 ± 13 vs 147 ± 11%, S-SCAM + GFP vs S-SCAM + pepR845A; p < 0.001) (Fig. 7C,D). In contrast, the negative control peptide pepK844A that does not interfere with NSF or AP2 interaction had no significant effect (299 ± 13 vs 270 ± 13%, S-SCAM + GFP vs S-SCAM + pepK844A; p = 0.14). Consistent with previous results (Lee et al., 2002), the expression of pepR845A alone did not change sGluA2 levels significantly (100 ± 8 vs 98 ± 11%, GFP vs pepR845A; p = 0.87). This is perhaps due to the fact that surface staining of AMPAR, unfortunately, cannot distinguish between synaptic and extrasynaptic AMPAR in the dendritic spines (Lee et al., 2002). Thus, pepR845A specifically targeted the sGluA2 pool related to S-SCAM overexpression. The effect of pepR845 was specific for sGluA2, as it did not show a significant effect on the sGluA1 levels increased by S-SCAM (247 ± 13 vs 239 ± 10%, S-SCAM + GFP vs S-SCAM + pepR845A; p = 0.61).
To further support the GluA2-specific effect of S-SCAM, we performed electrophysiological experiments measuring rectification index (measured by peak AMPA amplitude ratios at −60/+40 mV). If S-SCAM can support the incorporation of AMPAR to synapse in a GluA1-dependent manner, S-SCAM coexpression with GluA1 should increase the rectification index (overexpressed GluA1 forms inward rectifying homomeric receptors) (Shi et al., 2001). As shown in Figure 7E, GluA1 coexpressed with S-SCAM did not change the rectification index of transfected neurons (1.44 ± 0.1 vs 1.43 ± 0.2, Non-txf vs S-SCAM + GluA1, p = 0.95), while GluA1 cotransfected with a truncated α-CaMKII (tCaMKII-α) clearly increased the rectification index as expected (1.48 ± 0.1 vs 2.0 ± 0.2, Non-txf vs GluA1 + tCaMKII-α; p < 0.01). These data further support our notion that S-SCAM is regulating AMPAR through GluA2, not via GluA1.
In Caenorhabditis elegans, MAGI-1L regulates AMPAR through the PDZ-0 domain (Emtage et al., 2009). However, the deletion of PDZ-0 of S-SCAM did not impair the S-SCAM's ability to increase sGluA2 (372 ± 32 vs 395 ± 27%, S-SCAM WT vs ΔPDZ-0; p = 0.61) (Fig. 7F).
Role of S-SCAM in NMDAR-dependent hippocampal LTD formation
Both S-SCAM overexpression and RNAi-mediated knockdown data suggest that S-SCAM maintains AMPARs at synapses. Does S-SCAM support activity-dependent changes in the AMPA levels at synapses? To examine the role of S-SCAM in synaptic plasticity, we first examined the effect of S-SCAM on AMPAR internalization, which is a molecular mechanism underlying hippocampal LTD (Lee et al., 2002). Both Non-txf and GFP-transfected hippocampal neurons briefly treated with NMDA (2 min, 50 μm NMDA) showed a great increase in the internalized AMPAR levels as visualized by anti-GluA2 antibody (>3- to 5-fold increase compared with untreated control condition; p < 0.001) (Fig. 8A,B). In contrast, S-SCAM overexpressed neurons did not show such NMDA-induced internalization at all. Further, the basal level of AMPAR internalization (untreated) in S-SCAM overexpressed neurons was decreased (1.0 ± 0.09 vs 0.5 ± 0.05, Non-txf vs S-SCAM; p < 0.01). However, S-SCAM-transfected neurons showed normal AMPA-induced internalization (>6.5-fold; p < 0.001), which is comparable to Non-txf and GFP-transfected neurons (>4-fold and >7-fold, respectively). Therefore, S-SCAM overexpression resulted in the failure of AMPAR internalization specifically related to NMDA receptor activation. These results suggested that S-SCAM overexpression impairs the formation of hippocampal LTD. Consistent with this idea, hippocampal CA1 neurons infected with S-SCAM-virus indeed showed no induction of LTD (Fig. 8C,D) (80 ± 7 vs 91 ± 14% of baseline after 25–30 min, LTD vs unpaired pathway; n = 10; p = 0.42). In contrast, neighboring uninfected CA1 neurons exhibited a pathway-specific robust and stable LTD (49 ± 7 vs 90 ± 6% of baseline after 25–30 min, LTD vs unpaired pathway; n = 10; p < 0.001). Inclusion of APV in the bathing medium completely abolished the LTD formation (95 ± 7 vs 89 ± 10% of baseline after 25–30 min, LTD vs unpaired pathway; n = 6; p = 0.6) (Fig. 8E), indicating that the LTD is NMDAR dependent. CA1 neurons transfected with S-SCAM RNAi showed normal LTD indistinguishable from Non-txf neighboring neurons (57 ± 5 vs 53 ± 6% of baseline after 25–30 min, S-SCAM RNAi vs Non-txf; n = 10, p = 0.61) (Fig. 8E,F). Together, these results suggest that S-SCAM is not involved in the regulation of LTD-forming AMPAR pool.
Figure 8.

S-SCAM overexpression blocks NMDA-induced AMPAR internalization and induction of hippocampal LTD. A, B, Effect of S-SCAM on AMPAR internalization. GFP or S-SCAM was transfected into hippocampal neurons and their effect on GluA2 internalization was measured by a fluorescence-based antibody-feeding assay. Neurons were stimulated for 2 min with conditioned media (Control), 50 μm NMDA, or 100 μm AMPA. Representative images are shown in A and quantified data are presented in B. Scale bar, 10 μm. n > 25 per condition. ***p < 0.001, **p < 0.01. Scale bar represents 10 μm. C, D, Effect of S-SCAM overexpression on hippocampal LTD. CA1 neurons were infected with sindbis virus-expressing GFP or GFP-S-SCAM. Paired recordings were performed in infected and uninfected nearby neurons. LTD was induced by pairing 200 pulses of 1 Hz stimulation and postsynaptic depolarization at −40 mV. Normalized EPSC traces are shown in C and quantified in D. While uninfected control neurons show normal pathway-specific LTD (LTD vs unpaired pathway, ***p < 0.001, n = 10), LTD induction was blocked in neurons infected with SCAM (p = 0.42, n = 10). E, F, Effect of APV and S-SCAM knockdown on the hippocampal LTD formation. Normalized EPSC traces are shown in E and bar graphs are in F (n = 6 for APV and n = 10 for other conditions).
Discussion
In this study, we elucidate a novel and essential function of S-SCAM as a synaptic AMPAR scaffold. From overexpression experiments, we found that increasing S-SCAM levels promotes enlargement of dendritic spines, changes the molecular composition of PSD proteins, enhances AMPAR-mediated synaptic transmission, and blocks the induction of hippocampal LTD. Conversely, knockdown studies demonstrated that the loss of S-SCAM decreases synaptic AMPAR levels, reduces the size of dendritic spines, and severely reduces excitatory synapse numbers. Therefore, S-SCAM is an important scaffolding molecule involved in the synaptic protein organization and the control of synaptic AMPAR.
The most significant finding here is the novel and essential role of S-SCAM in the maintenance of AMPAR at synapses. One of the prevailing hypotheses in the AMPAR trafficking is that two separate pools of AMPARs contribute to differential aspects in AMPAR trafficking: constitutive and regulated pathways (Shi et al., 2001; Derkach et al., 2007; Shepherd and Huganir, 2007; Kessels and Malinow, 2009). The former is important for the maintenance of synaptic strength during protein turnover, while the latter is involved in changing synaptic AMPAR levels during synaptic plasticity. In this model, different AMPAR subunits play leading roles in the differential phases of AMPAR trafficking: GluA1 plays a dominant role in the activity-dependent insertion of AMPAR during LTP, while GluA2 is involved in the maintenance of synaptic AMPAR levels and activity-dependent removal of AMPAR from synapses during LTD. Accumulating evidence supports that PSD-95 is important for the regulated pathways. For example, PSD-95 overexpression strongly enhanced AMPA receptor-mediated synaptic transmission by molecular mechanisms similar to LTP, in that it drives GluA1-containing receptors to synapses, converts silent synapses to functional synapses, occludes LTP, and enhances LTD (El-Husseini et al., 2000; Schnell et al., 2002; Béïque and Andrade, 2003; Stein et al., 2003; Ehrlich and Malinow, 2004; Nakagawa et al., 2004; Elias et al., 2006). On the other hand, knockdown of PSD-95 reduced synaptic AMPAR levels (Nakagawa et al., 2004; Prange et al., 2004), and impaired LTD (Ehrlich et al., 2007; Xu et al., 2008) without affecting synapse numbers significantly (Béïque and Andrade, 2003; Elias et al., 2006).
In contrast to the regulated pathway, the identity of scaffolding protein(s) involved in the constitutive pathway has remained unclear. Our results suggest that S-SCAM is the scaffolding molecule important for the constitutive pathway. Multiple lines of evidence support this notion: First, S-SCAM increases synaptic AMPAR levels through a GluA2-dependent mechanism, as shown by GluA subunit-specific knockdown and NSF-peptide experiments (Fig. 5). Second, the increase of AMPAR by S-SCAM is an activity-independent process (Fig. 3E,F), consistent with the delivery mechanism of GluA2 receptors replacing GluA1 (Shi et al., 2001). Third, S-SCAM is required for maintaining AMPAR levels even in the presence of PSD-95 overexpression, as indicated by knockdown experiments (Figs. 4, 6). Fourth, the knockdown of S-SCAM led to the loss of excitatory synapses (Fig. 4H), while S-SCAM overexpression did not increase the number of excitatory synapses (PSD-95 overexpression increases it). Fifth, S-SCAM does not support activity-dependent changes of synaptic AMPA receptor levels, as demonstrated by the blockade of LTD after overexpression and by normal LTD after knockdown (Fig. 8). Thus, a pool of synaptic AMPARs anchored by S-SCAM is expected to play a “house-keeping” role during synaptic plasticity. Together, we propose a model in which S-SCAM stabilizes a maintenance pool of AMPARs at synapses and PSD-95 (together with other PSD-MAGUKs) regulates a plasticity-related pool of AMPAR.
How does S-SCAM serve a differential function from PSD-95 in the regulation of AMPA receptors and dendritic spines? We speculate that it arises from the overlapping but differential protein–protein interaction profile and/or post-translational modifications of S-SCAM protein. For example, S-SCAM does not have a palmitoylation site and lacks AKAP interaction, both of which were shown to be important for activity-dependent AMPAR internalization and LTD (El-Husseini et al., 2002; Bhattacharyya et al., 2009). In addition, S-SCAM interacts with N-cadherins through β-catenin (Nishimura et al., 2002; Okabe et al., 2003), which may provide a mechanism for its stabilization at synapses during plastic changes. It is intriguing that S-SCAM promoted dendritic spine enlargements in the absence of increased synaptic levels of Shank (Fig. 2C) and SPAR (data not shown), critical molecules involved in dendritic spine enlargement/maturation (Pak et al., 2001; Sala et al., 2001). S-SCAM may increase dendritic spines through GluA2 subunits, whose extracellular interaction with N-cadherin mediates dendritic spine enlargement (Passafaro et al., 2003; Saglietti et al., 2007). Further studies are necessary to examine these possibilities.
Our finding that S-SCAM plays an important role in the regulation AMPAR is not without precedents. In C. elegans, the interaction of MAGI-1L (long form of MAGI-1) with the AMPA-like subunit GLR-2 has shown to be important for the synaptic localization of GLR-1/2 (Emtage et al., 2009). However, the molecular details of the action are different from the vertebrate system. While GLR-1/2 directly binds to MAGI-1L through PDZ interaction, vertebrate GluAs interact with S-SCAM indirectly via TARP (Deng et al., 2006), like PSD-95. Furthermore, the first PDZ domain (PDZ-0) is important for the synaptic GLR regulation by MAGI-1L in C. elegans, but PDZ-0 is not important for AMPAR regulation (Fig. 7F). Further detailed studies on the S-SCAM–TARP interaction are necessary to establish molecular mechanisms underlying S-SCAM-mediated regulation of AMPARs.
It is remarkable that S-SCAM did not increase sGluA1 levels in neurons with GluA2 knockdown, suggesting a GluA2-dependent mechanism. Since GluA2 plays an important role in the assembly and synaptic incorporation of heteromeric AMPAR (Wenthold et al., 1996; Lu et al., 2009), GluA2 knockdown may have caused a poor assembly and/or trafficking of GluA1-containing receptors to neuronal surface. However, this is unlikely the case in our condition, since GluA2 knockdown showed normal sGluA1 levels (Fig. 7B). Consistent with this, homomeric GluA1 receptors were detected at synapses shortly after GluA2 deletion (Lu et al., 2009). Furthermore, S-SCAM failed to accommodate GluA1 homomeric receptors at the synapses (Fig. 7E). Therefore, these data indicate that S-SCAM regulates AMPAR in a GluA2-dependent manner. At present, the molecular bases of GluA2 dependence of the S-SCAM action are unclear, as TARP binds all GluA subunits. However, one could imagine that GluA2-specific binding proteins such as GRIP, NSF, PICK1, and/or yet unidentified protein(s) may work together with S-SCAM to provide a GluA2-dependent mechanism at synapses. Another intriguing question is what role S-SCAM plays in the control of inhibitory synaptic transmission, since S-SCAM is also present in inhibitory synapses (Sumita et al., 2007).
Previously, mutant mice lacking the expression of S-SCAM-α did not show drastic abnormalities in dendritic spine density, albeit they showed elongated spine length (Iida et al., 2007). However, the S-SCAM-α knock-out mice still express other S-SCAM isoforms of β and γ. S-SCAM-β is the most abundant form expressed in the forebrain (Hirao et al., 2000; Deng et al., 2006). The main difference between the α and β isoform is the presence of PDZ-0 domain. As ΔPDZ-0 mutant increased sGluR2 levels as effectively as WT (Fig. 7F), PDZ-0 domain is not required for the S-SCAM-mediated regulation of AMPAR. Thus, it is highly likely that the remaining S-SCAM-β (and possibly γ) in the S-SCAM-α knock-out mice is sufficient to control AMPAR and dendritic spine.
In addition to uncovering the role of S-SCAM in excitatory synaptic transmission, our study also contributes to the understanding of the pathobiology of neurological diseases, since S-SCAM gene duplication and deletion were found in individuals with schizophrenia and IS, respectively (Marshall et al., 2008; Walsh et al., 2008). Although these discoveries do not establish a causal relationship between S-SCAM and schizophrenia and/or IS per se, our results provide valuable insight into the pathological bases of these diseases. Here we have demonstrated that changing levels of S-SCAM profoundly affects synaptic transmission and synaptic plasticity. For example, increasing levels of S-SCAM enhances AMPAR-mediated synaptic transmission, but, at the same time, renders neurons less adaptable for at least one type of synaptic plasticity, LTD. Conversely, decreased S-SCAM levels profoundly reduce the number of excitatory synapses and weaken synaptic transmission. Incidentally, elevated levels of S-SCAM, a highly likely situation under S-SCAM gene duplication, caused the reduction in the number of dendritic spines, which is consistent with postmortem studies on the brains of patients with schizophrenia (Garey et al., 1998; Glantz and Lewis, 2000; Sweet et al., 2009; Penzes et al., 2011). Furthermore, aberrant Neuregulin-1 (NRG-1)/ErbB4 receptor signaling is one of the prevailing hypotheses in schizophrenia research (Mei and Xiong, 2008). Since S-SCAM also binds to ErbB4 receptors through PDZ interaction (Buxbaum et al., 2008), further studies on the role of S-SCAM in the NRG-1/ErbB4 signaling will provide important clues on a better understanding pathological bases of the disorder.
Footnotes
This work was supported by the Whitehall foundation (S.H.L.) and U.S. National Institute of Health Grants MH078135 (S.H.L.) and AG032320 (N.G.). We thank Dr. Yutaka Hata (Tokyo Medical and Dental University) for providing S-SCAM expression plasmids and Dr. Qing-song Liu for advice on electrophysiology and data analyses.
References
- Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Georgieva L, Young JJ, Plescia C, Kajiwara Y, Jiang Y, Moskvina V, Norton N, Peirce T, Williams H, Craddock NJ, Carroll L, Corfas G, Davis KL, Owen MJ, Harroch S, Sakurai T, O'Donovan MC. Molecular dissection of NRG1-ERBB4 signaling implicates PTPRZ1 as a potential schizophrenia susceptibility gene. Mol Psychiatry. 2008;13:162–172. doi: 10.1038/sj.mp.4001991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Deng F, Price MG, Davis CF, Mori M, Burgess DL. Stargazin and other transmembrane AMPA receptor regulating proteins interact with synaptic scaffolding protein MAGI-2 in brain. J Neurosci. 2006;26:7875–7884. doi: 10.1523/JNEUROSCI.1851-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Emtage L, Chang H, Tiver R, Rongo C. MAGI-1 modulates AMPA receptor synaptic localization and behavioral plasticity in response to prior experience. PLoS One. 2009;4:e4613. doi: 10.1371/journal.pone.0004613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers DM, Matta JA, Hoe HS, Zarkowsky D, Lee SH, Isaac JT, Pak DT. Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nat Neurosci. 2010;13:1199–1207. doi: 10.1038/nn.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Hirabayashi S, Nishimura W, Iida J, Kansaku A, Kishida S, Kikuchi A, Tanaka N, Hata Y. Synaptic scaffolding molecule interacts with axin. J Neurochem. 2004;90:332–339. doi: 10.1111/j.1471-4159.2004.02497.x. [DOI] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A novel multiple PDZ domain-containing molecule interacting with N-methyl-D-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Yao I, Deguchi M, Kawabe H, Mizoguchi A, Takai Y. Three isoforms of synaptic scaffolding molecule and their characterization. Multimerization between the isoforms and their interaction with N-methyl-D-aspartate receptors and SAP90/PSD-95-associated protein. J Biol Chem. 2000;275:2966–2972. doi: 10.1074/jbc.275.4.2966. [DOI] [PubMed] [Google Scholar]
- Iida J, Hirabayashi S, Sato Y, Hata Y. Synaptic scaffolding molecule is involved in the synaptic clustering of neuroligin. Mol Cell Neurosci. 2004;27:497–508. doi: 10.1016/j.mcn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Iida J, Ishizaki H, Okamoto-Tanaka M, Kawata A, Sumita K, Ohgake S, Sato Y, Yorifuji H, Nukina N, Ohashi K, Mizuno K, Tsutsumi T, Mizoguchi A, Miyoshi J, Takai Y, Hata Y. Synaptic scaffolding molecule alpha is a scaffold to mediate N-methyl-D-aspartate receptor-dependent RhoA activation in dendrites. Mol Cell Biol. 2007;27:4388–4405. doi: 10.1128/MCB.01901-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Lüthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron. 1999;24:389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Marshall CR, et al. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet. 2008;83:106–111. doi: 10.1016/j.ajhg.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok H, Shin H, Kim S, Lee JR, Yoon J, Kim E. Association of the kinesin superfamily motor protein KIF1Balpha with postsynaptic density-95 (PSD-95), synapse-associated protein-97, and synaptic scaffolding molecule PSD-95/discs large/zona occludens-1 proteins. J Neurosci. 2002;22:5253–5258. doi: 10.1523/JNEUROSCI.22-13-05253.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44:453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Nishimura W, Yao I, Iida J, Tanaka N, Hata Y. Interaction of synaptic scaffolding molecule and β-catenin. J Neurosci. 2002;22:757–765. doi: 10.1523/JNEUROSCI.22-03-00757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S. Molecular anatomy of the postsynaptic density. Mol Cell Neurosci. 2007;34:503–518. doi: 10.1016/j.mcn.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Okabe T, Nakamura T, Nishimura YN, Kohu K, Ohwada S, Morishita Y, Akiyama T. RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the beta-catenin-N-cadherin and N-methyl-D-aspartate receptor signaling. J Biol Chem. 2003;278:9920–9927. doi: 10.1074/jbc.M208872200. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M. Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature. 2003;424:677–681. doi: 10.1038/nature01781. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci U S A. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumita K, Sato Y, Iida J, Kawata A, Hamano M, Hirabayashi S, Ohno K, Peles E, Hata Y. Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J Neurochem. 2007;100:154–166. doi: 10.1111/j.1471-4159.2006.04170.x. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JD, Yuan J, Margolis RL, Colomer V, Duan K, Kushi J, Kaminsky Z, Kleiderlein JJ, Sharp AH, Ross CA. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci. 1998;11:149–160. doi: 10.1006/mcne.1998.0677. [DOI] [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci U S A. 2000;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr Opin Neurobiol. 2011;21:306–312. doi: 10.1016/j.conb.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]