

Abstract
Sortases anchor surface proteins to the cell wall of Gram-positive pathogens through recognition of specific motif sequences. Loss of sortase leads to large reductions in virulence, which identifies sortase as a target for the development of antibacterials. By screening 135,625 small molecules for inhibition, we report here that aryl (β-amino)ethyl ketones inhibit sortase enzymes from staphylococci and bacilli. Inhibition of sortases occurs through an irreversible, covalent modification of their active site cysteine. Sortases specifically activate this class of molecules via β-elimination, generating a reactive olefin intermediate that covalently modifies the cysteine thiol. Analysis of the three-dimensional structure of Bacillus anthracis sortase B with and without inhibitor provides insights into the mechanism of inhibition and reveals binding pockets that can be exploited for drug discovery.
The emergence of bacterial strains resistant to antibiotic therapy is a major public health threat (1). Of particular concern is Staphylococcus aureus, because this Gram-positive pathogen is the leading cause of infections in the bloodstream, lower respiratory tract, skin, and soft tissue in the United States (2). S. aureus strains exhibiting resistance against multiple antibiotics, such as methicillin-resistant S. aureus, are isolated in 30–60% of community and >80% of hospital infections with this pathogen (3). Vancomycin or other glycopeptides are considered last-resort therapies for methicillin-resistant S. aureus; however, S. aureus strains with intermediate or full resistance to vancomycin can cause infections for which antimicrobial treatment may no longer be effective (4).
Surface proteins of Gram-positive bacteria play important roles during pathogenesis (5). Sortases anchor these polypeptides to the bacterial cell wall envelope (6). For example, S. aureus sortase A recognizes proteins destined for the cell surface via an LPXTG motif in their C-terminal sorting signal (7). Following cleavage between the threonine and the glycine residues, an acyl-enzyme intermediate captures cleaved substrate at the active site thiol of sortase (8). Nucleophilic attack of the amino group of the pepti-doglycan precursor lipid II (C55-PP-MurNAc-(L-Ala-D-iGln-L-Lys(NH2-Gly5)-D-Ala-D-Ala)-GlcNAc) at the thioester intermediate resolves the acyl enzyme and forms an amide bond between the C-terminal threonine of surface protein and pentaglycine cross-bridges (9). Lipid II-linked polypeptide is subsequently incorporated into the cell wall envelope of staphylococci (10). The final product of this pathway, protein linked to cell wall pentaglycine cross-bridges, is displayed on the bacterial surface and enables interactions between the pathogen and tissues of its host.
Surface protein anchoring to the cell wall envelope is thought to be an essential strategy for bacterial survival during infection, because mutants lacking genes for one or more sortase enzymes are attenuated in virulence (11). Inhibition of sortases by small molecules may therefore function as a therapeutic strategy for bacterial infections. Previous work described several sortase inhibitors, including methane-thiosulfonates (12), peptide substrate-derived affinity labels (13), natural compounds (14–16), vinyl sulfones (17), diarylacrylonitriles (18), bis(indole) alkaloids (19), peptidomimetics (20), isoquinoline alkaloids (16), and threonine analogues (21). However, most of these compounds are either of low activity, lack specificity, or display undesirable structural features that confound therapeutic use. To overcome these obstacles, we have screened a library of small molecules and identified aryl (β-amino)ethyl ketones as mechanism-based inactivators of sortases.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Reagents
S. aureus sortase A (SrtA),2 Bacillus anthracis sortase A, SrtB, and SrtC were purified from Escherichia coli strain BL21(DE3) using Ni2+-nitrilotriacetic acid affinity chromatography (8). Reagents were obtained from Sigma-Aldrich unless otherwise noted. Compounds AAEK1 (5927860) and AAEK2 (5927943) were purchased from Chembridge (San Diego, CA), and their structures were validated by 1H NMR and liquid chromatography-MS.
Identification of Sortase Inhibitors
The NSRB at Harvard Medical School provided compound libraries for sortase inhibitor studies. Purified S. aureus SrtA (2 μM) in black polystyrene 384-well plates was incubated with 1 μl of test compound (10 ng) in Me2SO for 1 h at 25 °C, followed by addition of 2-amino-benzoyl-LPETG-diaminopropionic acid-dinitrophenyl-NH2 (a-LPETG-d) to 2 μM in 50 μl of reaction buffer (5 mM CaCl2, 150 mM NaCl2, 50 mM Tris-HCl, pH 7.5) and further incubated for 24 h at 37 °C. Fluorescence was measured using an Envision plate reader (excitation λ = 320 nm, emission λ = 420 nm, Fig. 1A, inset). This assay yielded a Z′ of 0.94 (22). Sixteen wells of positive control (no compounds added) and sixteen of negative control (no compound or sortase added) wells were performed per plate. Final Me2SO concentrations were ≤2.5% (v/v), a concentration shown to have no effect on control fluorescence levels (data not shown).
FIGURE 1. HTS of sortase and cheminformatics.
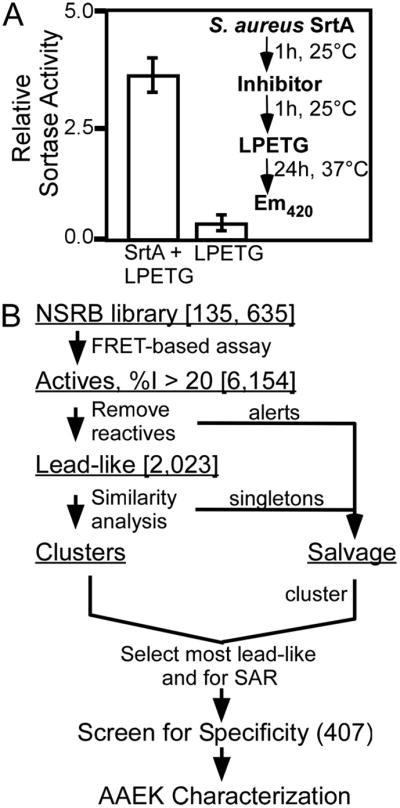
A, a fluorescence resonance energy transfer-based assay using a-LPETG-d as sortase substrate in a 384-well plate format was used in this HTS (Z′-factor of 0.94). The inset demonstrates the assay design used to identify sortase inhibitors. B, compounds displaying inhibition ≥20% of mean positive control (6,154) were computationally filtered to remove reactive inhibitors, promiscuous inhibitors, etc. (see “Experimental Procedures”). Filtered molecules were placed in a Salvage set, and remaining Lead-like actives were clustered and sampled based on lead-likeness and potential for later SAR analysis, with emphasis on the most active compounds. Approximately 210 “clean” compounds were selected from the Lead-like Clusters. To allow for discovery of chemical tools as well (54), these were combined with representatives from the Salvage set (after their re-clustering) to yield a final set of 407 compounds to take into the secondary screen for specificity.
Data Analysis and Hit Selection
Data were qualified for analysis by evaluation of within- and across-plate biases and other systematic errors using Spotfire Decision® (SpotFire U.S., Somerville, MA). Percent activities (%A, the ratio of fluorescence from a test compound well to the plate positive control mean, multiplied by 100) and percent inhibition (%I = 100 − %A) were computed. Compounds were rank ordered according to %I. 6,154 compounds (4.5% of initial libraries) were active and displayed %I ≥ 20 (Fig. 1B). Structures of hit compounds were examined using custom sub-structure search routines in SARNavigator® 1.2 (Tripos, St. Louis, MO) to “filter” reactive and promiscuous inhibitors (23–26), known mutagens and genotoxics (27, 28), and molecules lacking good physicochemical hit/lead characteristics (23, 26, 29, 30). The alert molecules were separated into a salvage set, generating a set of 2,023 compounds (1.5% of initial) that were assigned lead-like activities. Analysis of these compounds for structural similarity based on computed Tanimoto distances (SARNavigator or Accord® 6.1, Accelrys, San Diego, CA (31)) was followed by visual inspection to identify “clean” lead-like clusters and singletons (a molecule/chemotype without a second example) (26, 31). These clusters were sampled with emphasis on compounds with %I ≥ 50 and potential for providing SAR in subsequent studies. Analysis of these for structural similarity identified >80 clusters with >1150 molecules (median size, 3; mean, 14; and range, 2–63) and >850 putative singletons. Approximately 210 clean compounds were selected for secondary assays. The alerts salvage set was combined with the singletons and other weakly active clean molecules, and this combined set was clustered and examined visually. Clusters were again sampled on the basis of activity, properties, and potential to provide SAR, and the best representatives and the most active and lead-like singletons were then added to the 210 to yield a final set of 407 compounds with potential both as research tools and as therapeutic candidates. These 407 compounds were subjected to a secondary screen using B. anthracis SrtA and papain and data analyzed as described for HTS of S. aureus SrtA.
IC50 Determination
S. aureus and B. anthracis sortases (8 μM) were incubated with increasing concentrations of either AAEK1 or AAEK2 (0.01–3200 μM) for 1 h at 37 °C, followed by addition of a-LPETG-d (SrtA), a-KTDNPKTGDEA-d (SrtB), or a-GEKLPNTASNN-d (SrtC) in 300 μl of reaction buffer for 15 min at 37 °C, and fluorescence was quantified as described above. IC50 values were determined by fitting data to a default four parameter variable slope sigmoidal function in GraphPad Prism® 4.0c using a nonlinear least squares algorithm.
Michaelis-Menten Kinetics
S. aureus SrtA (4 μM) was incubated with or without AAEK1 (50 μM) or AAEK2 (200 μM) in the presence of various concentrations of substrate (a-LPETG-d, 0–68 μM) for 15 min at 37 °C in reaction buffer and 300-μl volume, followed by fluorescence measurements. Kinetic constants were determined by Lineweaver-Burke analysis. The amount of cleaved substrate was <5% for all velocity determinations. For dialysis experiments, S. aureus SrtA (4 μM) was incubated with or without AAEK1 or AAEK2 (0.8 mM) for 2 h at 25 °C, followed by dialysis against 1 liter of reaction buffer for 24 h at 4 °C. Substrate (8 μM a-LPETG-d) was added, and reactions were incubated at 37 °C for 12 h, followed by fluorescence measurements.
MS of Sortase-Inhibitor Complexes
Wild-type or C184A S. aureus SrtA (10 μM) was incubated with or without AAEK1 (800 μM) at 25 °C for 1 h in reaction buffer. Trypsin (5 μg) was added, and sortase inhibitor complexes were digested at 37 °C for 12 h. Reaction aliquots (500 μl) were subjected to reversed-phase high-performance liquid chromatography on C18 column (2 × 250 mm, C18). Cleaved peptides were eluted with a linear gradient of acetonitrile in 0.1% trifluoroacetic acid (100 min, 0.5 ml min−1 fractions collected). Peak fractions (Abs.215) were dried, dissolved in 15 μl of 30% acetonitrile, 0.1% trifluoroacetic acid mixed with saturated α-cyano-4-hydroxycinnamic acid suspensions, spotted onto sample plates, and air-dried. For MALDI-TOF experiments, samples were ionized with an N2 UV laser in a Reflectron time-of-flight spectrometer (Applied Biosystems) in reflectron mode. Two hundred laser shots were conducted at an accelerating voltage of 25,000 V and laser intensity of 2,075 (repetition rate, 3 Hz). The instrument was calibrated using bovine serum albumin as an internal standard control, and scans were processed using Biosystems Voyager 6004 software. Peptides with m/z 1555.74 and m/z 1693.78 were subjected to tandem mass spectrometry.
Synthesis of Olefin Intermediates
Inhibition intermediate 12, an α, β-unsaturated olefin, was prepared in two steps from commercial thiophene-2-carbaldehyde by procedures summarized here. The comparable 1-(thiophen-3-yl) products were prepared from their corresponding aryl aldehydes. Chromatographically pure products were isolated by Still-type isocratic 60-Å SiO2 adsorption chromatography at medium pressure (0.7 MPa, Biotage SP1™ system, KP-SIL™, 40–63 μM), using 3–15% linear gradients of EtOAc in hexanes with 2- to 4-column volume initial and final isocratic steps. Subsequent spectroscopic data, pulsed Fourier transform 1H and 13C NMR, electrospray ionization-MS, and Fourier transform IR, were consistent with the structures reported and literature data available. Purified products were stored under argon at −20 °C in the dark until use. rac-1-(Thiophen-2-yl)prop-2-en-1-ol (11) (32) was prepared from vacuum-distilled thiophene-2-carbal-dehyde by treatment with 1.10–1.20 equivalents of vinylmagnesium chloride in anhydrous Fluka tetrahydrofuran at 15–20 °C for 3–4 h (reaction 0.7 M in aldehyde, scale 3.4–15 mmol) (33). After weakly acidic aqueous workup, tert-butyl-methyl ether extraction, and standard drying, chromatographic isolation gave the indicated pure product (yields, 74–92%). 1-(Thiophen-2-yl)prop-2-en-1-one (12) (34) was prepared from purified 11 or directly from the crude propenol extract of 11 after drying. The extract was treated with 1.50 equivalent of the oxidant N-methylmorpholine N-oxide and 0.15 equivalent of the Fluka catalyst tetra-N-propylammonium perruthenate in anhydrous Fluka dichloromethane at 0–5 °C for 15–17 h (reaction 0.10 M in alcohol, scale 0.6–1.8 mmol) (35). After catalyst adsorption/filtration (ICN GmbH SiliTech SiO2, 32–63 μM/Celite 545®), filtrate evaporation, and extraction of the residue with hexanes, chromatographic isolation gave the indicated pure product (yields: 59% on 63% conversion from pure 11, 24% direct from extract). Compounds were diluted to 0.57–0.58 M in 50% aqueous Me2SO containing sortase reaction buffer (see above). Compounds were further diluted to assay concentrations of 5 or 50 μM, and assays were performed as described for the DTT experiments.
Effect of DTT on AAEK1- and AAEK2-mediated Inhibition of Sortase
Reactions included 4 μM S. aureus SrtA, 8 μM a-LPETG-d, inhibitor (50 μM AAEK1 or 200 μM AAEK2), and increasing concentrations of DTT (2–500 μM) and were incubated at 37 °C for 1 h, followed by fluorescence measurements.
Crystallography
Protein expression and purification was carried out as described (36). The construct did not include 36 N-terminal amino acids (signal peptide) and encoded a 242-amino acid SrtB polypeptide with a 24-residue His tag at its N terminus. A 2 mM protein stock solution in 10 mM Tris-HCl, pH 7.4, 20 mM NaCl, and 1 mM DTT was used for crystallization. Selenomethionine-labeled SrtB was prepared using the methionine biosynthesis inhibition method (37). Inhibitors were diluted to 10 and 50 mM in crystallization buffer for AAEK1 and AAEK2, respectively. The SrtB/AAEK adduct structures were obtained using vapor diffusion at 25 °C and crystallized under different conditions as compared with apo-protein. The SrtB/AAEK1 adduct crystallizes in a different space group (see Table 3). The structures of SrtB/AAEK1 and SrtB/AAEK2 adducts were determined by single-wavelength anomalous dispersion phasing using HKL3000 (38) and selenomethionine-labeled enzyme. The structures were auto traced using ARP/wARP and refined with REFMAC against the averaged peak data (39). The initial models of SrtB/AAEK1 and SrtB/AAEK2 were adjusted manually using COOT and refined to the final crystallographic R of 18.7% and Rfree of 25.9% for SrtB/AAEK1, and to the final crystallographic R of 18.6% and Rfree of 22.6% for SrtB/AAEK2, both with zero σ cutoff (see Table 3). The stereochemistry of the structures was examined with PROCHECK and the Ramachandran plot. The final model of SrtB/AAEK1 does not have sufficient density for nine N-terminal residues (64–65 and 85–191) and four internal residues (238–241). The final model of SrtB/AAEK2 does not have sufficient density for four internal residues (236–240). Atomic coordinates have been deposited in the Protein Data Bank.
TABLE 3.
SrtB-AAEK adducts
| Summary of crystal | ||
|---|---|---|
| AAEK1 | AAEK2 | |
| Unit cell | A=34.33 Å, b=73.54 Å, c=88.61 Å, α=90°, β=90°, γ=90° | a=40.49 Å, b=64.96 Å, c=43.65 Å, α=90°, β=105.06°, γ=90° |
| Space group | P212121 | P21 |
| MW Da (residues) | 29,210(254) | 29,210(254) |
| Mol (AU) | 1 | 1 |
| SeMet (AU) | 11 | 11 |
| SAD data collection (Peak) | ||
| AAEK1 | AAEK2 | |
| Wavelength (Å) | 0.9793 | 0.9793 |
| Resolution range (Å) | 2.1 | 1.6 |
| No. of unique reflections | 14492 | 27707 |
| Completeness (%) | 97.13 (80.4) | 99.58 (97.59) |
| R merge (%) | 9.4 (4.5) | 9.4 (4.0) |
| Phasing | ||
| AAEK1 (2OQW) | AAEK2 (2OQZ) | |
| Resolution (Å) | 2.1 | 1.6 |
| No. of inflections* | 9379 | 28794 |
| FOM | 0.22 | 0.25 |
| Density Modification | 0.80 | 0.84 |
| Refinement | ||
| Resolution range (Å) | 56.8–2.1 (2.1–2.15)** | 42.14–1.6 (1.6–1.64)** |
| No. of reflections | 12870 (773)** | 27372 (1947)** |
| R-value (%) | 18.7 (18.9)** | 18.6 (27.6)** |
| Free R-value (%) | 25.9 (29.4)** | 22.6 (29.8)** |
| Rms deviations from ideal geometry | ||
| bond length (1–2) (Å) | 0.014 | 0.013 |
| angle (°) | 1.5 | 1.4 |
| No. of atoms | ||
| Protein | 1782 | 1818 |
| Water | 124 | 230 |
| Mean B-factor (Å2) | ||
| all atoms | 34.17 | 23.04 |
| Ramachandran plot statistics (%) | ||
| Residues in most favored regions | 92.3 | 92.3 |
| Residues in additional allowed regions | 7.7 | 7.7 |
| Residues in disallowed region | 0 | 0 |
Summary of crystal, data collection, and crystallographic statistics for complexes with AAEK1 and AAEK2.
RESULTS
Screening for Sortase Inhibitors
A biochemical assay that monitors S. aureus sortase A cleavage of the fluorescence resonance energy transfer substrate (a-LPETG-d) between the threonine and glycine residues was optimized for HTS (8). Translation of this assay into a 384-well format yielded a Z′ of 0.94, which is well above the value suggested for successful HTS assays (Fig. 1A) (22). Using the NSRB library, 135,625 small molecules were screened in duplicate for inhibition of SrtA (Fig. 1A, inset). Of the compounds assayed, 6,154 displayed percent inhibitions (%I) >20% (Fig. 1B). Of these, ~two-thirds were placed in a salvage set for being reactive, genotoxic, promiscuous (frequent hits in unrelated assays), or lacking in drug-like properties (see “Experimental Procedures”) (23–30). The remaining lead-like activities were clustered on the basis of structure to identify common structural cores (chemotypes) and selected based on SARs. After combining these with the most active compounds from the salvage set, a total of 407 compounds was attained (Fig. 1B).
To examine specificity of inhibition, the 407 compounds were screened for inhibition of B. anthracis SrtA and papain, a eukaryotic protease with an active site thiol (40). Many of the most potent and specific sortase inhibitors belonged to five chemotypes (Table 1): Class I, aryl (β-amino)ethyl ketones; Class II, N-aryl maleamides and aryl fumaramides, and related compounds; Class III, N,4-diaryl-2-aminothiazoles; Class IV, 3-heteroatom-substituted (N-alkyl/aryl)pyrrolidine-2,5-diones; and Class V, variously substituted maleimide-furan Diels-Alder products. Of these, the aryl (β-amino)ethyl ketones (AAEK) were the most active inhibitors of sortases from staphylococci and bacilli (Table 1). These compounds are Mannich bases (41), with a propiophenone (or related heteroaromatic) core, bearing a β-arylamino or β-dialkylamino substituent. Two compounds, AAEK1 and AAEK2, were selected for further characterization because of their substantial inhibition of sortase, limited inhibition of papain, and presentation of heavy atoms to assist in eventual x-ray structure determination (Table 2).
TABLE 1.
Representative classes of sortase inhibitors revealed by HTS
| Percent Inhibition, Range [Median] | ||||
|---|---|---|---|---|
| Class | Chemotype [Number, of 407] | S. aureus SrtA | B anthracis SrtA | C. papaya papain |
| I |
[16]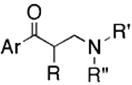
|
44–92 [68] | 71–92 [90] | 0–77 [53] |
| II |
[8]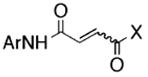
|
22–92 [70] | 63–90 [87] | 0–51 [10] |
| III |
[6]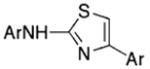
|
34–92 [71] | 27–85 [38] | 2–53 [21] |
| IV |
[18]
|
13–92 [66] | 1–91 [78] | 0–40 [14] |
| V |
[9]
|
27–93 [65] | 19–92 [73] | 0–22 [7] |
Shown are the core structures of five chemotypes of sortase inhibitors with a cluster size ≥6 that demonstrate limited inhibition of papain. The range of percent inhibitions observed for the class and the median (brackets) are shown for sortase A (S. aureus and B. anthracis) and papain (C. papaya). R, R′, and R″ indicate a hydrogen atom or an alkyl-type substituent (alkyl, methylene, or methine); Ar indicates a carbocyclic aromatic or heteroaromatic, including complex arrays; X indicates a heteroatom functional group such as OR, -NHR, etc.; and a dashed line indicates the presence/absence of a ring unsaturation.
TABLE 2.
The structures of ten representatives from the AAEK chemotype class
| Inhibition, Percent of Control | ||||
|---|---|---|---|---|
| No. | AAEK Inhibitor Structure | S. aureus SrtA | B. anthracis SrtA | C. papaya papain |
| 1 |  |
54. 8 | 90.7 | 19.1 |
| 2 |  |
89.6 | 90.4 | 49.3 |
| 3 | 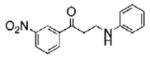 |
66.0 | 87.3 | 34.0 |
| 4 | 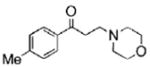 |
56.9 | 88.9 | 31.2 |
| 5 | 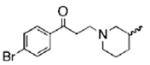 |
56.9 | 83.7 | 33.8 |
| 6 | 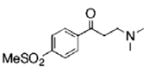 |
69.8 | 91.3 | 37.1 |
| 7 |  |
54.3 | 90.2 | 47.2 |
| 8 |  |
47.2 | 89.1 | 46.6 |
| 9 |  |
84.4 | 90.6 | 50.4 |
| 10 |  |
80.2 | 90.7 | 48.5 |
The mean percent inhibitions of sortase A (S. aureus and B. anthracis) and papain (C. papaya) are presented.
Inhibition of Sortases by AAEKs
Inhibitory concentration (IC50) values of AAEK1 and AAEK2 for sortases from B. anthracis and S. aureus were determined. AAEK1 and AAEK2 were ~10- and 3-fold more potent inhibitors of B. anthracis SrtA than of S. aureus SrtA (Fig. 2, A and B). Inhibition of B. anthracis SrtA was severalfold greater than inhibition of SrtB or SrtC (Fig. 2, B–D). As different sortase enzymes recognize unique substrates, it seems plausible that observed differences in IC50 values may be due to differences in active site configuration for members of this enzyme family. Nevertheless, the data suggest that AAEK1 and AAEK2 function as inhibitors of all four sortases tested.
FIGURE 2. AAEK1 and −2 inhibit different sortases from bacilli and staphylococci.

S. aureus SrtA (A) and B. anthracis SrtA (B), SrtB (C), or SrtC (D) were incubated with increasing concentrations of AAEK1 (●) or AAEK2 (○) followed by the addition of substrate (a-LPETG-d for SrtA, a-KTDNPKTGDEA-d for SrtB, or a-GEKLPNTASNN-d for SrtC) and fluorescence measurements. IC50 is the concentration of compound that inhibits 50% of sortase activity and is displayed in micromolar in the upper right corner. Data were fit using an nonlinear least squares method in GraphPad Prism®, and represent the mean of three independent determinations.
Incubation of S. aureus SrtA in the presence of inhibitor (AAEK1 or AAEK2) with increasing concentrations of peptide substrate caused no alterations in Km but decreased Vmax with an apparent non-competitive profile (supplemental Fig. S1A). Lineweaver-Burke transformation of the data corroborated this notion, revealing Vmax values of 88.8 ± 7.8 (AAEK1) and 58.0 ± 3.6 (AAEK2) compared with 139.0 ± 5.4 observed in the absence of inhibitor (supplemental Table S1). Following a 2-h incubation of SrtA with AAEK1 or AAEK2, reactions were subjected to dialysis and then assayed for activity. Dialysis failed to restore activity, consistent with the notion that these compounds cause irreversible inhibition of sortases even in the absence of substrate (supplemental Fig. S1B). If so, AAEK-mediated inhibition likely precedes enzyme nucleophilic attack at the scissile peptide bond.
Inhibition by AAEKs Occurs at the Active Site Thiol of Sortase
S. aureus sortase A was incubated with or without AAEK1. Enzyme preparations were subjected to tryptic digestion, peptides separated by reversed-phase high-performance liquid chromatography, and fractions yielding absorbance at 215 nm were analyzed by mass spectrometry. A compound with m/z 1555.74 eluted in fraction 50, and its mass measurement was in agreement with the predicted m/z 1555.73 of the tryptic peptide QLTLITCDDYNEK, encompassing the active site residue Cys-184 (Fig. 3A). MS/MS of m/z 1555.74 confirmed the peptide sequence QLTLITCDDYNEK (supplemental Table S2). MS failed to identify a compound with m/z 1555.74 in sortase preparations that had been incubated with AAEK1 (Fig. 3B). In contrast, m/z of 1693.78 was present in sortase preparations (fraction 54) that had been treated with AAEK1 but not in sortase preparations without inhibitor (Fig. 3, C and D). MS/MS of m/z 1693.78 confirmed the peptide sequence QLTLITXD-DYNEK (supplemental Table S2), where X represents Cys-184, which has been chemically modified by the inhibitor and is in agreement with the addition of a thienylpropanone moiety (Fig. 3C, inset, supplemental Table S3). If so, the amine moiety of AAEK1 must first be eliminated in order for the compound to react with thiol in the active site of sortase. The modification did not occur when AAEK1 was incubated with a mutant enzyme carrying an alanine substitution at the active site residue (C184A). Thus, modification of sortase with AAEK1 requires an active site Cys-184 (Fig. 3E).
FIGURE 3. AAEK1generates a covalent adduct with Cys-184 of S. aureus SrtA.
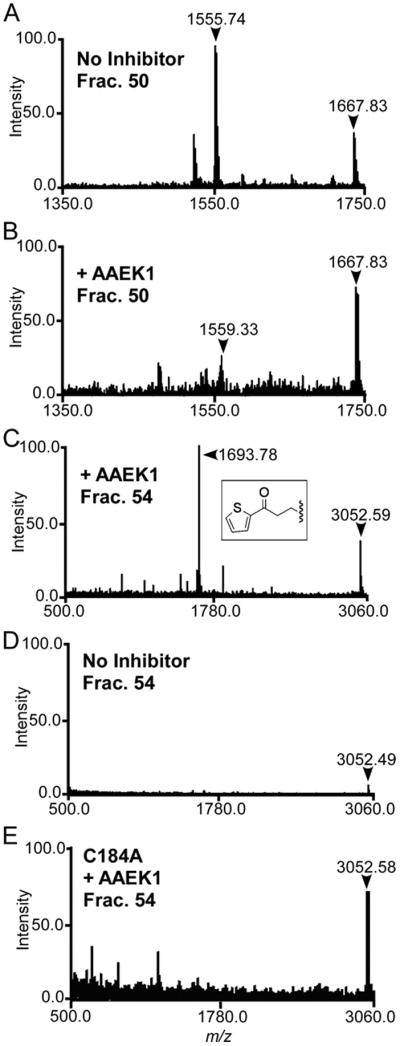
S. aureus wild-type or mutant SrtA (C184A) were incubated with (B, C, and E) or without (A and D) AAEK1, followed by digestion with trypsin. Reaction products were separated by reversed-phase high-performance liquid chromatography and collected fractions subjected to MALDI-TOF MS. Compounds with m/z 1693.78 (fraction 54) and m/z 1555.74 (fraction 50) correspond to tryptic peptides encompassing the modified and unmodified active site Cys-184 of sortase, respectively.
X-ray Structure Determination of the Sortase-AAEK Adducts
To gain insights into the mechanism of inhibition by the AAEK class, we solved the three-dimensional structure of B. anthracis sortase B with AAEK1 and AAEK2 by x-ray crystallography and single-wavelength anomalous dispersion. Electron densities were refined to 2.1 Å (AAEK1, R = 18.7%, Rfree = 25.9%) and 1.6 Å (AAEK2, R = 18.6%, Rfree = 22.6%) resolution, respectively (Table 3). The use of experimental phases was important to correctly trace structural changes in the active site. For AAEK1, clear electron density corresponding to the thienylpropanone adduct was observed near Cys-233 (the functional equivalent of Cys-184 in S. aureus SrtA), consistent with covalent modification determined by MS (Fig. 4C). For the SrtB-AAEK2 adduct, ~50% of Cys-233 was modified, most likely due to lower solubility of the compound under crystallization conditions. This fortuitous result allowed us to directly compare bound and unbound structures to determine what changes occur upon inactivation by the AAEK class. Both AAEK1 and AAEK2 modify sortase in a similar fashion, i.e. the β-carbon of the inhibitor is covalently linked to Cys-233 with the aryl group interacting with a critical tyrosine (Tyr-138) (Fig. 4A). Cys-233, Asp-234, and His-140, which make up the catalytic triad, undergo substantial rearrangements upon reaction with the AAEKs. The most important change is Cys-233, which undergoes a rotation of ~180 degrees to accommodate the ligand. In addition, Arg-243 swings away from the active site and is in excellent position in the ligand-free form to stabilize an oxyanion intermediate of the inhibitor, a step that would be required prior to elimination of amine from the AAEK (Fig. 4B). Further, the SrtB-AAEK adduct revealed two binding pockets, one cationic (above) and one anionic (below) the AAEK aryl group, which may be exploited for the engineering of more specific and potent inhibitors (Fig. 5, A and B).
FIGURE 4. Three-dimensional structure of the active site of sortase B modified by AAEK1.
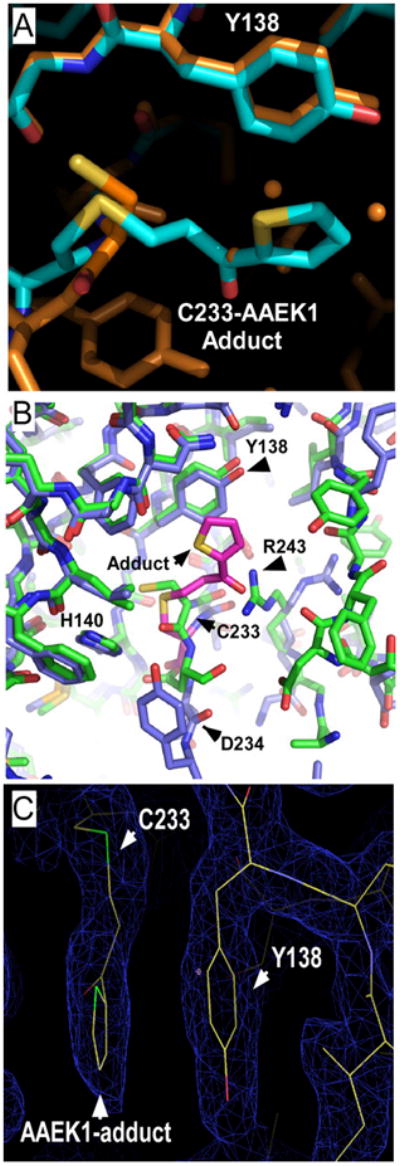
A, thienylpropanone adduct to sortase Cys-233 (blue), the product of the reaction of sortase B with AAEK1, is compared with the sortase B structure at 1.6 Å (orange). The adduct occupies the position of three water molecules (orange spheres) and is stacking against Tyr-138. B, the catalytic triad of sortase B (His140, Asp-234, and Cys-233) and Arg-243 are in close proximity to the inhibitor adduct and undergo substantial structural shifts. The SrtB (green) and SrtB-AAEK adduct (blue) are superimposed. The figure was generated using PyMOL™. C, thienylpropanone modification of the active site of sortase. Electron density map of the B. anthracis sortase B-AAEK1 adduct demonstrating Cys-233, Tyr-138, and the thienylpropanone. Green, sulfur; red, oxygen. The figure was generated using COOT.
FIGURE 5. Surface charge rendering of the sortase-AAEK adduct structures.

AAEK1-SrtB (A) and AAEK2-SrtB (B) adducts revealed modification of the active site thiol of sortase. Electrostatic potential (red for negative and blue for positive) of the active site was generated using GRASP. An ionic pocket is located above and below the aryl group of each adduct. Ligands atoms are color-coded as follows: yellow, sulfur; red, oxygen; green, chloride; and light blue, carbon.
Sortase Converts AAEKs to Reactive Olefin Intermediates
We hypothesized that the β-carbon of AAEK1 or AAEK2 may be linked via Michael-type addition by the sortase thiol to an olefin intermediate generated by amine elimination (42, 43). If so, addition of exogenous thiol would be expected to “capture” the olefin intermediate, thereby preventing the modification of sortase (Fig. 6A). To test this hypothesis, AAEK-mediated inhibition of sortase was examined in the presence of DTT. Enzyme kinetic analysis and mass spectrometry revealed that DTT does not react with AAEK1 or AAEK2 in the absence of sortase (data not shown). However, in the presence of sortase, increasing amounts of DTT prevented inactivation by AAEK1 or AAEK2 and, at 250 μM DTT, AAEK2-mediated inactivation was completely abolished (Fig. 6B). Addition of DTT alone (without inhibitor) did not stimulate sortase activity and could not restore the activity of enzymes whose active site was already modified (Fig. 6B). Taken together, these observations suggest that DTT interferes with modification of the active site by the AAEK inhibitors.
FIGURE 6. An olefin intermediate modifies the active site thiol following sortase activation of AAEK1.

A, proposed structure of the olefin intermediate and its reaction with thiol. B, reactions were performed in the presence or absence of DTT and the degree of peptide cleavage assayed 1 h later, as described for supplemental Fig. S1. The symbols (+ and −) represent the presence or absence of the indicated reagents, whereas the numbers refer to the concentration of DTT. The far right reaction “1hr” refers to the addition of DTT to the sample 1 h after the reaction with inhibitor was started. C, reactions were performed as in B, except that the concentration of AAEK1 and the putative elimination intermediates 11 and 12 were assayed at 5 and 50 μM. The mean ±S.D. of three independent experiments are presented in B and C. Ar, thiophen-2-yl.
We sought to test whether the proposed olefin of AAEK1 can indeed modify sortase and synthesized the predicted intermediate (Fig. 6C, 12). Incubation of sortase with 12 caused concentration-dependent and irreversible enzyme inhibition with a Ki of 69 μM (±21 μM), similar to Ki values observed for its parent AAEK1 compound. No sortase inhibition was observed with a control compound 11, the corresponding alcohol derivative of the olefin intermediate (Fig. 6C). Together, these data corroborate the hypothesis that sortase inhibition by AAEKs proceeds via a reactive olefin intermediate that forms a covalent adduct at the enzyme active site thiol.
DISCUSSION
The emergence of S. aureus strains with broad antibiotic resistance represents a rapidly growing challenge for both hospital- and community-acquired infections. Antibiotic resistance has also been observed in other Gram-positive bacteria, including an agent of bioterrorism, B. anthracis (44). Widespread use of antibiotics for human therapeutic or food industrial purposes is thought to be the primary selection mechanism for the emergence of drug resistance strains (45). Staphylococcal resistance mechanisms have been reported for all known antibiotics, which necessitates identification of new therapeutic targets and development of new drugs that can be used for the treatment of bacterial infections (46). Sortases, a family of transpeptidases that immobilizes polypeptides in the envelope of Gram-positive bacteria, recognize surface protein substrates and attach their C-terminal end to an amino group acceptor of lipid II, which is subsequently incorporated into the cell wall (47). Sortases are required for the pathogenesis of many different bacterial infections and their selective inhibition may be of therapeutic value.
Several recent studies sought to identify inhibitors of sortases. Oh and colleagues (14) evaluated medicinal plant extracts for sortase inhibition; the best results were obtained with Cocculus trilobus extracts. A lipid glucose conjugate from Fritillaria verticillata as well as isoquinoline alkaloids from Coptis chinensis are other inhibitor candidates (15). Isosteres of the scissile bond, i.e. threonine-glycine for sortase A, achieved inhibition when the LPXT peptides were decorated with either diazoketone or chloromethylketone (13). These peptide-derived inhibitors display favorable Ki values, however their rates of inactivation are slow. Vinyl sulfones also react with thiols, but the corresponding peptide vinyl sulfones present even slower rates of sortase inactivation than diazoketone or chloromethyl-ketone derivatives (17). Finally, phosphorous isosteres of peptides are effective transition state analogs and inhibitors of zinc proteases. Peptide mimics carrying replacement of the scissile peptide bond indeed inhibited sortase (20). As with all other peptide-derived compounds, the further development of these types of inhibitors toward a therapeutic use is obstructed by their chemical features, including high molecular weight and undesirable pharmacological properties. Finally, random irreversible inhibition of thiol groups and of sortases can be achieved with small molecules such as methane-thiosulfonates (12, 17). Because such compounds lack specificity, their chemical properties preclude drug development.
Previous studies on small molecule inhibitors of sortases have been limited to the screening of 1000 compounds, which identified diarylacrylonitriles as potential inhibitors (18). In this study, compounds with IC50 values between 10 and 1000 μM were reported, and one compound functioned as competitive inhibitor with very favorable Ki value (18). Diarylacrylonitriles have been proposed to bind to the active site of sortase, although this hypothesis has not yet been supported by experimental evidence (18).
Here we screened a library of 135,625 small molecules for inhibition of S. aureus SrtA with a fluorescence resonance energy transfer assay that measures enzymatic cleavage of the fluorogenic substrate a-LPETG-d between its threonine and glycine residues. By combining a cheminformatic approach with secondary specificity assays, we identified several new classes of sortase inhibitors, each one a distinct series with a common structural core. Class I, AAEKs, display drug-like properties, high relative levels of inhibition of sortase, as well as good specificity. To investigate the mechanism of inhibition by this class, we focused on compounds AAEK1 and AAEK2 due to their high selectivity for inhibition of sortase and presence of heavy atoms for x-ray structural studies. Kinetic studies suggested non-competitive inhibition with substrate and dialysis/re-assay hinted at an irreversible modification. MS confirmed that the active site thiol of sortase was covalently modified with a derivative of AAEK1, implying the parent compound had undergone a change during reaction with sortase.
Comparison of the crystal structures of B. anthracis sortase B-AAEK1 and sortase B-AAEK 2 adducts with the 1.6-Å ligand-free enzyme revealed some remarkable structural changes. Although a majority of the enzyme structure is virtually identical, there are some conformational changes that may be attributed to adduct formation. Remarkably, side chains of all residues in or near the active site that are part of the sortase barrel (loop between β2 and 3, strands β4 and β7) show virtually identical conformation in ligand-free enzyme and in the complex with inhibitors (Asn-102, Leu-106, Phe-121, Asp-123, Arg-125, Tyr-138, and His-140 of the catalytic triad). In contrast, the region of the active site contributed by residues on the loop between α5 and β6 (Phe-189, Tyr-191, Tyr-235, and Arg-243) show much higher mobility and quite different conformation in ligand-free and ligand-bound structures.
In the ligand-free sortase, the region following the Asp-234 is disordered. However, upon AAEK1 adduct formation this region becomes ordered and can be traced up to Leu-237 and for AAEK2 up to Tyr-235. Interestingly, the region near the active site between Thr-186 and Tyr-191 becomes more disordered upon adduct formation. The electron density for AAEK1 is very well defined, and electron density for AAEK2 is good with the aromatic ring and the chlorine atoms well defined (data not shown), although it shows lower occupancy. Both inhibitors react in a very similar manner with sortase B. In both cases the β-carbon is covalently linked to Cys-233 (in agreement with MS data from sortase A) and the aromatic moiety interacts with Tyr-138 and Asn-102. Both adducts are fully accessible to the solvent. As mentioned above, although the majority of protein amino acid side chains are in very similar conformations in both structures, there are some remarkable differences in the residues that make up the sortase catalytic triad. The most important is a change in the conformation of Cys-233, which, to accommodate the inhibitor, must rotate ~180°. Adduct formation swings Arg-243 away from the active site. However, in ligand-free sortase, its guanidinium group is in excellent position to stabilize an oxyanion intermediate of the inhibitor and thus may contribute to AAEK activation.
The most significant structural finding is that the AAEK adducts are situated in a crevice between two pockets, each opposite in charge character. The anionic pocket proximal to the carbonyl oxygens may be what draws these compounds into the active site through interaction with the electropositive ammonium moiety. Further, the aryl rings are adequately positioned such that substituent changes could present the appropriate opposite charges to foster interactions that would add potency and selectivity. This principal is partially demonstrated by the sortase-AAEK2 adduct, where the presence of two chlorine atoms shifts the aryl group closer to the cationic pocket. A preliminary investigation using para-substituted AAEKs suggests there is a correlation between inhibition of sortase and increasing anionic character of the substituent, perhaps reflecting interactions with the cationic pocket.3
MS and x-ray crystallographic studies of sortase-AAEK adducts revealed a thienylpropanone covalently tethered via its β-position to the active site cysteine (Fig. 4). The adduct differs from the parent compound by the absence of a di-methylamine moiety. Elimination of this group from an AAEK would generate an aryl vinyl ketone, an electrophilic olefin that would be expected to react with an available thiol (41, 48). Indeed, excess thiol DTT prevented inactivation of sortase by the AAEKs, and the putative olefin elimination product, i.e. intermediate 12, was also a covalent inactivator, thereby corroborating the hypothesis. Thus, a general model for sortase inhibition by the AAEK class can be proposed (Fig. 7). The aqueous milieu, the surface accessibility of the active site, and the orientation of the adducts in it suggest an initial interaction between the ammonium form of the AAEKs and regions of negative electrostatic potential on the sortases (Fig. 5, pocket 2). This attraction of the AAEKs to the sortase active site could be followed by further stabilizing interactions with the active site tyrosine and other residues. The path to alkylation of the cysteine by the AAEKs most likely then proceeds via an elimination-addition mechanism (41, 48–50).
FIGURE 7. Model for the mechanism of inhibition of sortase by AAEKs.

Deprotonation of the α carbon is conjectured to occur via a base in the active site of sortase. This generates enolate 13, which may be stabilized in a manner similar to the oxyanion intermediate of sortase during catalysis (e.g. by the guanidinium of a conserved active site arginine). This intermediate eliminates an amine, here dimethylamine, from the β-position to generate 14. The elec-trophilic nature of 14 allows it to serve as an acceptor in a Michael-type conjugate addition by the thiol of the active site cysteine. This resulting enolate might also be stabilized by the guanidinium moiety; subsequent protonation by enzyme or medium would then generate the stable AAEK thioether adduct observed.
Following deprotonation of AAEKs by an active site base, enolate 13 is formed. This enolate could be stabilized by the same active site features (the guanidinium of the conserved arginine, shown here with 13) that stabilize the oxyanion that forms during sortase cleavage of substrate (Fig. 7) (45). Following β-elimination (13 → 14), the vinyl group of the aryl vinyl ketone is a suitable electrophile for the sulfur nucleophile of the active site thiol (here presented as the thiolate anion) (41, 48). The Michael-type conjugate addition of the thiol to the vinyl group then renders sortase inactive.
The model presented herein suggests the AAEK compounds are mechanism-based inhibitors, which are a class of inactivators with examples among commercial drugs, and so have clear therapeutic potential (51). Although there are no AAEKs among these, previous work revealed that AAEKs display diverse in vivo properties, including anti-inflammation and glutathione reduction in mammalian organisms (52). Of note, AAEK2 has been demonstrated to possess antimicrobial activity against Gram-positive but not against Gram-negative bacteria (53). We have observed similar effects for AAEK1 and −2 against both S. aureus and B. anthracis.3 Whether this antimicrobial property of AAEKs relates specifically to the inhibition of sortases has not yet been determined, although this hypothesis is being explored. Evaluation of the in vivo inhibition of sortases in S. aureus and B. anthracis requires development of assays with improved sensitivity and specificity over those that are currently available. Preliminary results on the toxicity of AAEKs for mammalian cells suggest that AAEK1 and AAEK2 display 70- and 40-fold higher growth inhibitory activity toward microbial cells.3 Collectively, these results suggest that AAEK compounds are suitable for further development. We seek to eventually interrogate such lead compounds for their therapeutic potential as sortase inhibitors in animal models of anthrax or staphylococcal disease.
Supplementary Material
Acknowledgments
We acknowledge Sue Chiang and other members of the NSRB for technical assistance during HTS. We thank D. J. Burns and Y. C. Martin (Abbott GPD) for cheminformatic advice and John Wilmes (Reed College) for programming support. M.-E. D. wishes to thank V. H. Rawal (The University of Chicago (UC)) for sponsorship as a UC Visiting Fellow.
Footnotes
This work was supported by National Institutes of Health Grants AI38897 and AI057153 (to O. S.), GM074942 and GM62414 (to A. J.), and GM08043 and National Science Foundation Grant 9351490 (to M.-E. D. and Chicago State University) and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357 (to A. J.). The authors acknowledge membership within and support from the Region V “Great Lakes” Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (GLRCE, NIAID-NIH Award 1-U54-AI-057153).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and Tables S1–S3.
The atomic coordinates and structure factors (code 2OQW and 2OQZ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: SrtA, sortase A; SrtB, sortase B; SrtC, sortase C; AAEK, aryl (β-amino)ethyl ketone; DTT, dithiothreitol; SAR, structure-activity relationship; HTS, high-throughput screen; NSRB, National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease; MS, mass spectrometry; MS/MS, tandem mass spectrometry; a-LPETG-d, 2-aminobenzoyl-LPETG-diaminopropionic acid-dinitrophenyl-NH2; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight.
A. W. Maresso and O. Schneewind, unpublished observation.
References
- 1.Lowy FD. J Clin Invest. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M. Clin Infect Dis. 2001;32:S114–S132. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan SL, Hulten KG, Gonzalez BE, Hammerman WA, Lamberth L, Versalovic J, Mason EOJ. Clin Infect Dis. 2005;40:1785–1791. doi: 10.1086/430312. [DOI] [PubMed] [Google Scholar]
- 4.Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ, Cardo D, Fridkin SK. N Engl J Med. 2003;348:1342–1347. doi: 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- 5.Navarre WW, Schneewind O. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 7.Schneewind O, Mihaylova-Petkov D, Model P. EMBO J. 1993;12:4803–4811. doi: 10.1002/j.1460-2075.1993.tb06169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O. Proc Natl Acad Sci U S A. 1999;96:12424–12429. doi: 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneewind O, Fowler A, Faull KF. Science. 1995;268:103–106. doi: 10.1126/science.7701329. [DOI] [PubMed] [Google Scholar]
- 10.Ton-That H, Labischinski H, Berger-Bachi B, Schneewind O. J Biol Chem. 1998;273:29143–29149. doi: 10.1074/jbc.273.44.29143. [DOI] [PubMed] [Google Scholar]
- 11.Mazmanian SK, Liu G, Jensen ER, Lenoy E, Schneewind O. Proc Natl Acad Sci U S A. 2000;97:5510–5515. doi: 10.1073/pnas.080520697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ton-That H, Schneewind O. J Biol Chem. 1999;274:24316–24320. doi: 10.1074/jbc.274.34.24316. [DOI] [PubMed] [Google Scholar]
- 13.Scott CJ, McDowell A, Martin SL, Lynas JF, Vandenbroek K, Walker B. Biochem J. 2002;366:953–958. doi: 10.1042/BJ20020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim SW, Chang IM, Oh KB. Biosci Biotechnol Biochem. 2002;66:2751–2754. doi: 10.1271/bbb.66.2751. [DOI] [PubMed] [Google Scholar]
- 15.Kim SH, Shin DS, Oh MN, Chung SC, Lee JS, Chang IM, Oh KB. Biosci Biotechnol Biochem. 2003;67:2477–2479. doi: 10.1271/bbb.67.2477. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Shin DS, Oh MN, Chung SC, Lee JS, Oh KB. Biosci Biotechnol Biochem. 2004;68:421–424. doi: 10.1271/bbb.68.421. [DOI] [PubMed] [Google Scholar]
- 17.Frankel BA, Bentley M, Kruger RG, McCafferty DG. J Am Chem Soc. 2004;126:3404–3405. doi: 10.1021/ja0390294. [DOI] [PubMed] [Google Scholar]
- 18.Oh KB, Kim SH, Lee J, Cho WJ, Lee T, Kim S. J Med Chem. 2004;47:2418–2421. doi: 10.1021/jm0498708. [DOI] [PubMed] [Google Scholar]
- 19.Oh KB, Mar W, Kim S, Kim JY, Oh MN, Kim JG, Shin D, Sim CJ, Shin J. Bioorg Med Chem Lett. 2005;15:4927–4931. doi: 10.1016/j.bmcl.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 20.Kruger RG, Barkallah S, Frankel BA, McCafferty DG. Bioorg Med Chem. 2004;12:3723–3729. doi: 10.1016/j.bmc.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 21.Jung ME, Clemens JJ, Suree N, Liew CK, Pilpa R, Campbell DO, Clubb RT. Bioorg Med Chem Lett. 2005;15:5076–5079. doi: 10.1016/j.bmcl.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 22.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 23.Rishton GM. Drug Discov Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 24.Seidler J, McGovern SL, Doman TN, Shoichet BK. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 25.Davis AM, Keeling DJ, Steele J, Tomkinson NP, Tinker AC. Curr Top Med Chem. 2005;5:421–439. doi: 10.2174/1568026053828411. [DOI] [PubMed] [Google Scholar]
- 26.Martin YC. 232nd National Meeting of the American Chemical Society; San Francisco, CA: American Chemical Society; 2006. p. Abstract MEDI-313. [Google Scholar]
- 27.Sun H. J Chem Inf Comput Sci. 2004;44:1506–1514. doi: 10.1021/ci049917y. [DOI] [PubMed] [Google Scholar]
- 28.Wunberg T, Hendrix M, Hillisch A, Lobell M, Meier H, Schmeck C, Wild H, Hinzen B. Drug Discov Today. 2006;11:175–180. doi: 10.1016/S1359-6446(05)03700-1. [DOI] [PubMed] [Google Scholar]
- 29.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 30.Muegge I. Med Res Rev. 2003;23:302–321. doi: 10.1002/med.10041. [DOI] [PubMed] [Google Scholar]
- 31.Perez JJ. Chem Soc Rev. 2005;34:143–152. doi: 10.1039/b209064n. [DOI] [PubMed] [Google Scholar]
- 32.Morrill C, Beutner GL, Grubbs RH. J Org Chem. 2006;71:7813–7825. doi: 10.1021/jo061436l. and Suppl. Inf. I, p. S10. [DOI] [PubMed] [Google Scholar]
- 33.Trost BM, Fraisse PL, Ball ZT. Angew Chem Int Ed Engl. 2002;114:1101–1103. doi: 10.1002/1521-3773(20020315)41:6<1059::aid-anie1059>3.0.co;2-5. and Suppl. Inf. I, p. 34. [DOI] [PubMed] [Google Scholar]
- 34.Kang SK, Ho PS, Yoon SK, Lee JC, Lee KJ. Synthesis. 1998;1998:823–825. [Google Scholar]
- 35.Ali A, Thompson CF, Balkovec JM, Graham DW, Hammond ML, Quraishi N, Tata JR, Einstein M, Ge L, Harris G, Kelly TM, Mazur P, Pandit S, Santoro J, Sitlani A, Wang C, Williamson J, Miller DK, Thompson CM, Zaller DM, Forrest MJ, Carballo-Jane E, Luell S. J Med Chem. 2004;47:2441–2452. doi: 10.1021/jm030585i. [DOI] [PubMed] [Google Scholar]
- 36.Zhang R, Wu R, Joachimiak G, Mazmanian SK, Missiakas DM, Gornicki P, Schneewind O, Joachimiak A. Structure (Camb) 2004;12:1147–1156. doi: 10.1016/j.str.2004.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Acta Crystallogr Sect D Biol Crystallogr. 1999;55:1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 38.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 39.Morris RJ, Perrakis A, Lamzin VS. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 40.Zong Y, Bice TW, Ton-That H, Schneewind O, Narayana SV. J Biol Chem. 2004;279:31383–31389. doi: 10.1074/jbc.M401374200. [DOI] [PubMed] [Google Scholar]
- 41.Tramontini M, Angiolini L. Tetrahedron. 1990;46:1791–1837. [Google Scholar]
- 42.Naidu BN, Sorenson ME, Connolly TP, Ueda Y. J Org Chem. 2003;68:10098–10102. doi: 10.1021/jo034762z. [DOI] [PubMed] [Google Scholar]
- 43.Davioud-Charvet E, McLeish MJ, Veine DM, Giegel D, Arscott LD, Andricopulo AD, Becker K, Muller S, Schirmer RH, Williams CH, Jr, Kenyon GL. Biochemistry. 2003;42:13319–13330. doi: 10.1021/bi0353629. [DOI] [PubMed] [Google Scholar]
- 44.Bryskier A. Clin Microbiol Infect. 2002;8:467–478. doi: 10.1046/j.1469-0691.2002.00527.x. [DOI] [PubMed] [Google Scholar]
- 45.Levy SB, FitzGerald GB, Macone AB. Nature. 1976;260:40 – 42. doi: 10.1038/260040a0. [DOI] [PubMed] [Google Scholar]
- 46.Tenover FC, McDougal LK, Goering RV, Killgore G, Projan SJ, Patel JB, Dunman PM. J Clin Microbiol. 2006;44:108–118. doi: 10.1128/JCM.44.1.108-118.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marraffini LA, DeDent AC, Schneewind O. Microbiol Mol Biol Rev. 2006;70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith AB, March J. March’s Advanced Organic Chemistry. Wiley Interscience; New York: 2007. [Google Scholar]
- 49.Mollica JA, Smith JB, Nunes IM, Govan HK. J Pharm Sci. 1970;59:1770–1774. doi: 10.1002/jps.2600591213. [DOI] [PubMed] [Google Scholar]
- 50.Andrisano R, Angeloni AS, De Maria P, Tramontini M. J Chem Soc C. 1967;43:2307–2311. [Google Scholar]
- 51.Copeland R. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. Wiley Interscience; New York: 2005. [PubMed] [Google Scholar]
- 52.Gul M, Gul HI, Das U, Hanninen O. Arzneimittel-Forschung. 2005;55:332–337. doi: 10.1055/s-0031-1296868. [DOI] [PubMed] [Google Scholar]
- 53.Gul HI, Denizci AA, Erciyas E. Arzneimittel-Forschung. 2002;52:773–777. doi: 10.1055/s-0031-1299965. [DOI] [PubMed] [Google Scholar]
- 54.Lipinski C, Hopkins A. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.