


Abstract
Methyltransferases (MTases) form a major class of tRNA-modifying enzymes needed for the proper functioning of tRNA. Recently, RNA MTases from the TrmN/Trm14 family that are present in Archaea, Bacteria and Eukaryota have been shown to specifically modify tRNAPhe at guanosine 6 in the tRNA acceptor stem. Here, we report the first X-ray crystal structures of the tRNA m2G6 (N2-methylguanosine) MTase TTCTrmN from Thermus thermophilus and its ortholog PfTrm14 from Pyrococcus furiosus. Structures of PfTrm14 were solved in complex with the methyl donor S-adenosyl-l-methionine (SAM or AdoMet), as well as the reaction product S-adenosyl-homocysteine (SAH or AdoHcy) and the inhibitor sinefungin. TTCTrmN and PfTrm14 consist of an N-terminal THUMP domain fused to a catalytic Rossmann-fold MTase (RFM) domain. These results represent the first crystallographic structure analysis of proteins containing both THUMP and RFM domain, and hence provide further insight in the contribution of the THUMP domain in tRNA recognition and catalysis. Electrostatics and conservation calculations suggest a main tRNA binding surface in a groove between the THUMP domain and the MTase domain. This is further supported by a docking model of TrmN in complex with tRNAPhe of T. thermophilus and via site-directed mutagenesis.
INTRODUCTION
RNA molecules are often highly post-transcriptionally modified, with over 100 different chemical modifications known to date. The majority and largest variety of modified nucleotides are found in transfer RNA (tRNA) (1). These modifications play different structural and functional roles and contribute to (i) the proper folding and stability of tRNA, (ii) the correct codon–anticodon recognition at the decoding center of the ribosome and (iii) the recognition of the tRNA by its cognate aminoacyltransferase (2). Methylation constitutes by far the most abundant kind of nucleotide modification, and has been reported on the 2′-O-atom of ribose (3), and at various positions of the nucleotide bases on carbon and nitrogen atoms (4). These base methylations can be part of a biosynthetic pathway, leading to more complex hyper-modifications that are often present in the anticodon stem–loop. Examples of such methyltransferases (MTases) that have attracted a lot of attention recently are the Trm9/Trm112 complex, involved in the biosynthesis of methoxycarbonylmethyl-5-uridine (mcm5U) (5) and TYW5, involved in the synthesis of the hypermodified nucleoside wybutosine (6). On the other hand, the methyl group is often the end product of the base modification, as even simple methylations are known for their ability to stabilize the tertiary structure of tRNA (7,8).
One abundant type of methylation is the m2G modification, which is well characterized at positions G10, G26 and G27 of various tRNAs (9,10). However, m2G modifications are also known to exist at positions 6, 7, 9 and 18 (11). Trm11 was shown in yeast to facilitate the m2G10 modification in complex with the ‘hub’ protein Trm112, whereas in Archaea a single polypeptide TrmG10 is required for the same modification (12,13). Trm1 is involved in the production of 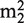 G26 (N2,N2-dimethylguanosine) in most eukaryotic and archaeal tRNAs and in the production of
G26 (N2,N2-dimethylguanosine) in most eukaryotic and archaeal tRNAs and in the production of 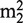 G26 and m2G27 in the bacterium Aquifex aeolicus (9,10,14).
G26 and m2G27 in the bacterium Aquifex aeolicus (9,10,14).
Recently, Trm14 was identified in Archaea as the tRNA MTase that catalyzes the formation of the m2G modification at position 6 in the acceptor stem of tRNA (15). In Methanocaldococcus jannaschii, it modifies tRNACys using S-adenosyl-l-methionine (SAM) as methyl donor. Independently, we identified this enzyme in Bacteria and showed that Thermus thermophilus TrmN (ortholog of the archaeal Trm14; nomenclature according to bacterial tRNA MTases) catalyzes formation of m2G6 in tRNAPhe (16). Orthologs of Trm14 are also found in Eukaryota, but their activity has not been experimentally tested yet.
Known RNA MTases can be classified into four superfamilies, including Rossmann-fold (RFM), SPOUT (SpoU and TrmH), radical-SAM and FAD/NAD(p)-dependent MTases [reviewed in (17)]. RFM enzymes are the largest superfamily of MTases. They share a common structure with a seven-stranded mixed β-sheet that is a variation on the classical (di-)nucleotide binding Rossmann fold (18). Crystal structures of representatives of all four classes have been solved (19,20), while structures in complex with tRNA or a tRNA mimic are available only for representatives of the RFM and SPOUT superfamily (21,22).
While some MTases catalyze the methyl transfer reaction using a catalytic domain alone, others are fused to one of the various RNA binding domains (23). One of these domains is the THUMP domain (named after THioUridine synthase, MTase and Pseudouridine synthase), which was initially proposed to be an ancient RNA binding domain on the basis of bioinformatics analyses (24). It was proposed that the THUMP domain consists of a minimal core, which is often fused to a so-called N-terminal ferredoxin-like domain (NFLD domain) (25). Relatively, few structures of proteins containing a THUMP domain have been solved to date. The crystal structures of ThiI, which is involved in the s4U modification, show the THUMP domain linked to a sulfur transfer catalytic domain (25), while in the cytidine deaminase CDAT8, it is fused to a deaminase domain (26). PUS10, on the other hand, has a core-THUMP domain linked to a Psi synthase domain, involved in the formation of pseudouridine (27). Examples of tRNA-modifying enzymes containing a THUMP domain fused to an RFM domain are described in literature, like Trm11 and PAB1283, involved in the m2G10 and 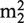 G10 modification (12,13,28), and Trm14, involved in the formation of m2G on position 6 of certain tRNAs (15). Although these proteins are well described, their structures and mode of interactions with the RNA substrates remain unknown. It has been proposed that the THUMP domain is mainly involved in the modification of nucleotides in the core of tRNA (13), but very little is known about its exact roles in (t)RNA binding and in catalysis of the modification reactions.
G10 modification (12,13,28), and Trm14, involved in the formation of m2G on position 6 of certain tRNAs (15). Although these proteins are well described, their structures and mode of interactions with the RNA substrates remain unknown. It has been proposed that the THUMP domain is mainly involved in the modification of nucleotides in the core of tRNA (13), but very little is known about its exact roles in (t)RNA binding and in catalysis of the modification reactions.
Here, we present the X-ray crystal structures of the bacterial tRNA:m2G6 MTase TTCTrmN from Thermus thermophilus (TTC1157) and its archaeal ortholog PfTrm14 from Pyrococcus furiosus (PF1002) in complex with the methyl donor SAM, the product S-adenosyl-homocysteine (SAH) and the inhibitor SFG (sinefungin). This study provides the first detailed crystallographic structure analysis of tRNA-modifying enzymes consisting of an MTase domain fused to a THUMP domain. Analysis of electrostatics combined with conservation of the surface amino acids suggest a binding patch for the substrate tRNA. A docking model of tRNA with TTCTrmN proposes the binding of tRNA in a groove between the two domains. This binding model is supported by site-directed mutagenesis in the proposed binding region.
MATERIALS AND METHODS
Protein expression and purification
PfTrm14 was cloned in a pET30 vector containing a C-terminal histidine tag and TTCTrmN was cloned in a pET28 vector containing an N-terminal histidine tag, as described (29). The proteins were expressed in Escherichia coli Rosetta (DE3) (PfTrm14) or E. coli BL21 (DE3) cells (TTCTrmN). Cells were initially grown at 310 K to an OD600 of 0.5. The strain expressing Se–Met-PfTrm14 was grown in minimal medium supplemented with selenomethionine (30). After induction with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), the cells grew at 288 K and were harvested the next day. Proteins were subsequently purified via nickel affinity chromatography as described earlier (29).
The PfTrm14 loop mutants were constructed by the use of four oligonucleotides (Supplementary Table S1), directing the desired deletion/insertion. Site-directed mutagenesis was performed according to the protocol described in the QuikChange site-directed mutagenesis kit (Stratagene). Incorparation of the mutations and integrity of the rest of the open reading frame was confirmed via sequencing. The used oligonucleotides are outlined in Supplementary Table S1.
Preparation of tRNA and in vitro assays
The in vitro transcript of tRNAPhe of T. thermophilus was generated according to a method described earlier (31). The tRNA MTase assay was based on the procedure described previously (32). The reaction mixture for the tRNA-MTase assay (400 μl) consisted of 50 mM Tris–HCl pH 8, 5 mM MgCl2, 106 cpm of radioactive [α32P] GTP tRNAPhe transcript, 500 μM SAM and variable amounts of purified protein. After 30 min of incubation at 60°C for TTCTrmN or 70°C for PfTrm14, the reaction was stopped by phenol extraction and the tRNA was ethanol precipitated. The recovered radioactive tRNA was then digested completely by nuclease P1 (1 μg), in the presence of 5 μg total yeast tRNA as carrier. Conversion of pG to pm2G was analyzed by 1D-thin layer chromatography (TLC) on cellulose plates (Merck) in solvent A (isobutyric acid/concentrated NH4OH/water; 66/1/33; v/v/v). The migration pattern was visualized by autoradiography.
For band shift assays, 104 cpm of radioactive [α32P] GTP tRNAPhe transcript from T. thermophilus was incubated in the presence of increasing amounts of enzyme in buffer B (20% glycerol, 50 mM Tris pH 8) at 60°C for TTCTrmN or 70°C for PfTrm14 for 30 min. The binding reaction (total volume of 20 μl) was stopped by the addition of 2 μl of stop solution (0.05% bromophenol blue in 30% glycerol) and the mixture was separated by 6% PAGE (190 mm × 160 mm × 1.5 mm) in TB buffer at room temperature. The PAGE was subjected to a voltage of 180 V until the samples entered the gel and further at 150 V until the end of the run (∼2 h).
Crystallization, data collection and structure determination
Wild-type unlabeled PfTrm14 was crystallized in a buffer consisting of 50 mM Tris–HCl pH 8, 10 mM MgCl2, 500 mM NaCl, 280 mM imidazole, 1 mM DTT by 1 : 1 mixing with crystallization solution (100 mM Tris–acetate pH 8, 32% polyethylene glycol (PEG) 4000, 15% glycerol) in a hanging drop vapor diffusion setup. Wild-type unlabeled TTCTrmN was crystallized in a buffer consisting of 50 mM Tris–HCl pH 8, 250 mM NaCl and 350 mM imidazole by 1 : 1 mixing with crystallization solution (100 mM citrate/phosphate pH 3.5, 15% PEG 6000, 200 mM NaCl, 100 mM sodium citrate) in a hanging drop vapor diffusion setup (29). For the phasing, selenomethionine (Se–Met)-derivatized PfTrm14 was crystallized in a crystallization solution containing 100 mM Tris–acetate pH 8, 32% PEG 4000, 15% glycerol, after streak seeding from a native PfTrm14 crystal. All crystals were flash frozen in liquid nitrogen using either the crystallization buffer (PfTrm14) or using crystallization buffer containing 20% glycerol as cryoprotectant (TTCTrmN). Crystals of protein–ligand complexes of PfTrm14 were obtained by overnight soaking, using mother liquor containing 1 mM SFG or 1 mM SAM. After 24-h incubation crystals were flash frozen in liquid nitrogen using the mother liquor containing 20% glycerol as cryoprotectant.
The diffraction data of Se–Met-derivatized PfTrm14 were collected at 100 K at the ID14-4 beamline (ESRF, Grenoble) using a single wavelength of 0.97936 Å, corresponding to the selenium absorption peak. The statistics of the data collection and processing are summarized in Table 1. The raw data were processed and scaled using the XDS suite (33). The structure was solved using the SAS protocol of Auto-Rickshaw: the EMBL-Hamburg automated crystal structure determination platform (34). The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs of the CCP4 suite (35). Heavy atom structure factor (FA) values were calculated using the program SHELXC (36). Twelve heavy atoms were found using the program SHELXD (37). The correct hand for the substructure was determined using the programs ABS (38) and SHELXE (39). Initial phases were then calculated after density modification using the program SHELXE (39). The initial phases were improved using density modification and phase extension using the program DM (40). The model was partially built using the program ARP/wARP (41,42). Further model building was done manually using Coot (43) and the structure refinement was carried out using refmac5 (44).
Table 1.
Data-collection, refinement and validation statistics of the structures of PfTrm14 and TTCTrmN
| Data set |
PfTrm14 |
TTCTrmN Apo form | |||
|---|---|---|---|---|---|
| Se-SAD peak | SAH bound | SAM bound | SFG bound | ||
| Data-collection | |||||
| X-ray source | ESRF ID 14-4 | ESRF ID23-2 | SLS PX-III | SLS PX-III | ESRF ID23-2 |
| X-ray wavelength (Å) | 0.97936 | 0.8726 | 1.00150 | 0.98000 | 0.8726 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Space group | P21 | P21 | P21 | P21 | P3221 |
| Unit-cell parameter (Å, °) | a = 82.2, b = 45.0, c = 120.7, β = 91.3 | a = 85.5, b = 45.3, c = 122.2, β = 91.2 | a = 82.1, b = 45.2, c = 121.4, β = 91.8 | a = 82.3, b = 45.1, c = 121.1, β = 91.8 | a = b = 65.9, c = 144.6 |
| Resolution range (Å) | 50–2.3 (2.4–2.3) | 50–2.2 (2.3–2.2) | 50–1.95 (2.0–1.95) | 50–2.27 (2.4–2.27) | 50–2.05 (2.16–2.05) |
| Total/unique reflections | 581 156/77 726 | 162 685/46 452 | 462 285/65 283 | 306 409/79 652 | 502 157/23 534 |
| Rmerge (%)a | 11.4 (61.2) | 10.8 (57.7) | 7.3 (54.4) | 14.7 (64.3) | 10.1 (52.5) |
| Rmeas (%)b | 12.3 (65.7) | 12.7 (68.4) | 7.8 (59.1) | 17.1 (74.8) | 10.4 (53.7) |
| Data completeness (%) | 99.7 (99.3) | 99.7 (99.7) | 99.5 (98.9) | 98.7 (92.3) | 100 (100) |
| Average I/σ | 14.0 (3.8) | 9.3 (2.7) | 18.3 (3.6) | 8.6 (2.1) | 22.4 (8.1) |
| Redundancy | 7.4 (7.6) | 3.5 (3.5) | 7.1 (6.6) | 3.8 (3.7) | 21.3 (22.1) |
| Refinement | |||||
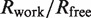 c c
|
18.6/22.0 | 16.5/19.7 | 20.5/21.9 | 15.7/21.2 | |
| Total number | |||||
| Amino acid residues | 728 | 728 | 735 | 332 | |
| Water molecules | 373 | 383 | 253 | 196 | |
| Ligand atoms | 56 (2 × SAH, 1 × ACT) | 54 (2 × SAM) | 54 (2 × SFG) | 5 (PO4) | |
| rmsd | |||||
| Bond length (Å) | 0.015 | 0.015 | 0.015 | 0.016 | |
| Bond angles (°) | 1.6 | 1.4 | 1.6 | 1.5 | |
| Ramachandran Plot (%) | |||||
| Favored regions | 98.1 | 98.3 | 98.1 | 97.9 | |
| Allowed regions | 1.9 | 1.7 | 1.9 | 1.8 | |
| Disallowed regions | 0 | 0 | 0 | 0.3 (Glu88A) | |
| PDB code | 3TLJ | 3TM4 | 3TM5 | 3TMA | |
Number in parentheses are statistics in the highest resolution shell.
aRmerge = 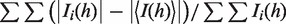 .
.
bRmeas = 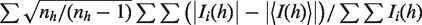 .
.
cRwork = 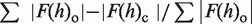 , F(h)o and F(h)c are observed and calculated structure factor amplitudes, respectively. A random subset of data (5%) was used for the Rfree calculation
, F(h)o and F(h)c are observed and calculated structure factor amplitudes, respectively. A random subset of data (5%) was used for the Rfree calculation
Diffraction data of native PfTrm14 either non-soaked (containing endogenous SAH) or soaked with SFG and SAM were collected at 100 K at the ID23-2 (ESRF, Grenoble) or PX-III (SLS, Villingen) beamlines, respectively. The diffraction data of the native TTCTrmN were collected at the ID23-2 beamline (ESRF, Grenoble). The statistics of the data collection and processing are summarized in Table 1. The PfTrm14-SAH, -SFG, -SAM and TTCTrmN structures were solved by molecular replacement using PhaserMR (45). In the case of the TTCTrmN structure, the individual THUMP (without the residues 47–63), and RFM domains of the Se–Met PfTrm14 structure were used as search models in molecular replacement. In the case of the soaked PfTrm14 structures, the full-length Se–Met PfTrm14 structure was used as a search model. Model building and refinement were performed as for the Se–Met PfTrm14 structure. TLS refinement was implemented in the refinement protocol for TTCTrmN, using four individual TLS groups determined by TLSMD (46,47). Stereochemical validation of all models was done using the Molprobity server (48). The figures were prepared using Pymol (http://www.pymol.org). The statistics of structure refinement and final structure models are summarized in Table 1.
Computational analysis, modeling of nucleotide binding and tRNA docking
Electrostatic surface calculations were prepared with PDB2PQR (49) using the PARSE force field and APBS (50). Amino acid conservation was calculated through the ConSurf server (51). Omit maps were generated using omit (52). Modeling of SAM into the active site pocket of TTCTrmN and of guanosine into the PfTrm14-SAM and TTCTrmN structures were done using the program Epitope Match (53). For the modeling of SAM, PfTrm14 was used as epitope model. For the modeling of guanosine into the PfTrm14-SAM structure and TTCTrmN structure, the crystal structure of RsmC (PDB code 3DMH) was used as epitope model (54).
In order to predict the structure of the TTCTrmN-tRNAPhe complex, we used the computational docking method. Since an unbound structure of T. thermophilus tRNAPhe is not available, we have built its homology model with ModeRNA (55) using a structure of E. coli tRNAPhe as a template (PDB code 3L0U). In a separate docking experiment, we have also used the coordinates of T. thermophilus tRNAPhe taken from the cocrystal structure with the cognate phenylalanyl-tRNA synthetase (PDB code 2IY5). For prediction of the complex structure, we used a combination of procedures described in the references (28,56). Briefly, a low-resolution method GRAMM (57) was used to generate 30 000 alternative models (decoys) with physically reasonable geometric compatibility between protein and RNA structures. We have filtered these decoys using FILTREST3D (58) to retain structures with a distance between the target residue G6 in tRNA and the methyl group of SAM in the protein <20 Å. These selected decoys were scored with the Decoys As the Reference State (DARS-RNP) potential (56) and clustered according to their mutual similarity, to retain groups of very similar decoys. The overall best-scored complex, as well as a representative of the largest cluster of well-scored decoys, was selected for further consideration.
RESULTS AND DISCUSSION
Overall structure of P. furiosus Trm14 and T. thermophilus TrmN
The structure of PfTrm14 was solved by single wavelength anomalous dispersion (SAD) using selenomethionine-labeled protein and was refined to 2.3 Å resolution. With this initial structure, the structures of PfTrm14 in complex with SAH, SFG and SAM were solved by molecular replacement to a resolution of 2.2 Å, 1.95 Å and 2.27 Å, respectively. While the substrate SAM and the inhibitor SFG were soaked into the crystals, the product SAH co-purified with PfTrm14. PfTrm14 was crystallized in the space group P21 with two protein molecules per asymmetric unit related by a 2-fold non-crystallographic symmetry axis. The contact interface of the two molecules within the asymmetric unit only buries a solvent accessible surface area of 432 Å2, indicating that these molecules do not correspond to a biological dimer (59). Additionally, the generation of all possible symmetry-related molecules in the crystal did not result in a biologically significant interface (biggest interface area: 515 Å2). This matches with the results of a size exclusion chromatography experiment, which clearly indicates that the protein behaves as a monomer in solution (Supplementary Figure S1). Overall, the three PfTrm14 structures are highly similar with an rmsd of 0.4 Å (calculated over 730 backbone atoms) between the SAM- and SAH-bound structures and an rmsd of 1.2 Å or 1.3 Å (over 727 backbone atoms) between the SFG bound and SAH- or SAM-bound structures, respectively. Clear electron density is visible for most of the 367 amino acids, and some amino acids from the C-terminal his-tag are also visible. One loop spanning the residues 298–303 is highly flexible in the PfTrm14-SAH and PfTrm14-SAM structures and could not be modeled. These residues are located in the RFM domain of PfTrm14, lining the SAM-binding pocket. Interestingly, this region became rigid when SFG was bound in the active site (see further).
The structure of TTCTrmN was solved in the apo state by molecular replacement to a resolution of 2.05 Å. TTCTrmN was crystallized in the space group P3221 with one monomer per asymmetric unit. Application of crystal symmetry operators yielded a symmetrical dimeric arrangement, with an accessible surface area buried in the interface of 799 Å2 (about 5.1% of the total accessible surface area). This value is just below the threshold area used for biological relevant interfaces (59). Moreover, this interface is highly hydrated and made up of completely non-conserved residues, indicating that this interface is formed by crystal packing. Size exclusion experiments with the wt TTCTrmN are not conclusive, since this protein interacts with the column matrix and hence elutes at a volume corresponding to a smaller molecular weight (Supplementary Figure S1). This can be partially circumvented by decreasing the high positive charge density of the protein and the K270E/R300E variant of TTCTrmN (see further) elutes at a volume corresponding to a monomer. Together this suggests that TTCTrmN, like PfTrm14, is a monomer in solution. In the TTCTrmN structure, the same loop as in the PfTrm14 structure (amino acids 266–269) is disordered and did not show any electron density. Details of the data collection and refinement statistics are shown in Table 1.
Overall, the structures of PfTrm14 and TTCTrmN are very similar (Figures 1 and 2) with an rmsd of 2.0 Å, calculated over all main chain atoms (286 residues). Globally, PfTrm14 and TTCTrmN adopt a cylindrical shape with an approximate size of 66.0 × 35 × 35 Å3 (Figure 2). The structures consist of two globular domains. An N-terminal THUMP domain spanning approximately the residues 1–183 in PfTrm14 and 1–151 in TTCTrmN, and a C-terminal RFM domain spanning approximately the residues 193–367 in PfTrm14 and the residues 160–335 in TTCTrmN. Both domains are connected via a long linker containing several positively charged amino acids. The orientation of the THUMP and RFM domains toward each other is similar in PfTrm14 and TTCTrmN.
Figure 1.

Sequence alignment of PfTrm14 and TTCTrmN with the secondary structures, as deduced from the crystal structures, indicated below the sequence. Since the sequence identity of the two THUMP domains is rather low (22%), the sequence alignment was entirely based on the superposition of the two structures using CCP4 superpose. β-strands are indicated by lighter colored arrows, α-helices by darker colored bars. Secondary structures of the RFM domain are shown in blue. Secondary structures of the THUMP domain are shown in red for the core-THUMP subdomain and in yellow for the NFLD subdomain. The β-strand shared by these two subdomains is shown in orange. Squares depict residues in the active site of the methyltransferase domain interacting with the ligand. Residues that were mutated in this study are indicated by stars.
Figure 2.

Crystal structures of PfTrm14 and TTCTrmN. (A and C) show the structure of PfTrm14 in cartoon representation (A) and as a topology diagram (C); (B and D) show the structure of TTCTrmN in cartoon representation (B) and as a topology diagram (D). β-strands are shown in red, yellow and blue for the core-THUMP, NFLD and RFM (sub)domains, respectively. α-Helices are shown in dark red, yellow and blue. The β-strand shared by the core-THUMP and NFLD subdomains is shown in orange. The two additional β-strands in the PfTrm14 structure are named βi and βii. The structure of PfTrm14 contains sinefungin bound in the active site, represented in ball and stick.
Structural similarity searches with PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/) (60) revealed three protein structures that align over nearly the total length with the TTCTrmN protein (Supplementary Table S2): a possible methylase from C. difficile (PDB code 3LDU, Tan,K., Wu,R., Buck,K. and Joachimiak, A., unpublished data), a predicted N6 adenine DNA methylase from L. monocytogenes (PDB code 3K0B, Nocek,B., Xu,X., Cui,H., Ng,J., Savchenko,A., Edwards,A. and Joachimiak,A., unpublished data) and the putative MTase SMU.422 from Streptococcus mutans (PDB code 3LDG, Wang,K.-T. and Su,X.-D., unpublished data). These three proteins belong to the ‘putative RNA methylase family UPF0020’ (PFAM nomenclature) and exhibit the same domain architecture and orientation as TrmN/Trm14. No publications describing these structures or their function was available at the time of the writing of this article. Moreover, the surface-exposed residues in the THUMP domain exhibit relatively low similarity to TTCTrmN, suggesting that substrate specificity is not necessarily conserved. For these three structures, it should be noted that they contain an insertion of two α-helices in their MTase domain compared to the MTase domain of TTCTrmN and PfTrm14, between α-helix 7 and β-strand 9 (corresponding to TrmN/Trm14 numbering).
The PDBeFold server was also used to search for structural similarities of the isolated THUMP and RFM domains of PfTrm14 and TTCTrmN. The RFM domain of TTCTrmN is not particularly similar to any experimentally characterized enzyme other than the three structures mentioned above. Instead, it exhibits highest similarity to various RFM domains of putative MTases with experimentally uncharacterized functions and unknown substrate specificities (Supplementary Table S2). These RFM domains are often fused to other domains, unrelated to THUMP. The THUMP domain is generally less conserved than the RFM domain, which is reflected in the relatively lower Z-scores of the top matches (Supplementary Table S2). The closest homologs of the THUMP domain of PfTrm14 and TTCTrmN (with or without NFLD subdomain, see further) are proteins involved in different reactions and fused to other catalytic domains, like the ThiI enzyme involved in the formation of the s4U modification in tRNA (25). Two RNA modification enzymes with known structures and possessing a THUMP domain, human pseudouridine synthase Pus10 (PDB code 2V9K) (27) and archaeal cytidine deaminase CDAT8 (PDB code 3G8Q) (26) were not reported in PDBeFold searches. CDAT8 possesses both the NFLD and core-THUMP subdomains of the THUMP domain (see further), while Pus10 lacks the NFLD subdomain. ThiI, Pus10 and CDAT8 possess all unrelated catalytic domains fused to the THUMP domain. The superposition of these structures onto TTCTrmN using the core-THUMP subdomains reveals that the catalytic domains assume different orientations (Supplementary Figure S2). Only the unrelated catalytic domains of TTCTrmN and ThiI assume relatively similar orientations with respect to the THUMP domain. At first sight, this might be related to the fact that TTCTrmN and ThiI modify bases at similar positions in the tRNA (position 8 in ThiI versus position 6 in TTCTrmN). However, also CDAT8 targets cytosine at position 8 but does not show the same domain orientation.
The THUMP domains of PfTrm14 and TTCTrmN differ by the insertion of an anti-parallel β-sheet
The N-terminal THUMP domain of TTCTrmN adopts a globular α/β structure with a total of seven β-strands and four α-helices (Figure 2B and D) of which helix 3 is kinked. The seven β-strands of this domain are arranged in two β-sheets that are connected by one long kinked β-strand that continues from one β-sheet to the other. The mixed β-sheet, consisting of the four C-terminal β-strands together with the two α-helices that are packed to one side of this β-sheet, corresponds to the core-THUMP domain as initially predicted by Aravind and Koonin (24). This core-THUMP domain is intimately associated with the N-terminal four-stranded anti-parallel β-sheet. This latter β-sheet, together with two α-helices, has previously been referred to as the NFLD domain (based on a topology similar to the ferredoxin-like fold) (25,26). However, from this and previous structures of the THUMP domain, it appears that both regions are subdomains of one globular domain fold and therefore both domains could also be regarded as one autonomously foldable THUMP domain (25,26,28).
The THUMP domains of PfTrm14 and TTCTrmN are structurally very similar (backbone rmsd of 2.3 Å) and they are also very similar to the THUMP domains of ThiI (rmsd of 2.7 Å and 3.2 Å for superposition on TTCTrmN and PfTrm14, respectively) and the cytidine deaminase CDAT8 (rmsd of 2.8 Å and 2.9 Å for superposition on TTCTrmN and PfTrm14, respectively) (25,26).
One minor difference between the THUMP domains from Trm14 and TrmN, and also with the THUMP domains of ThiI and CDAT8, concerns the angle relating the β-sheets of the core-THUMP subdomain and the NFLD subdomain. These two β-sheets are somewhat more perpendicular to each other in PfTrm14 compared to TTCTrmN and the THUMP domains of ThiI and CDAT8. Whether or not this change is relevant to the fine-tuning of the interaction with the tRNA substrate remains to be investigated.
Another obvious difference of the THUMP domain of PfTrm14 in comparison to TTCTrmN and other known THUMP domains concerns an insertion of two anti-parallel β-strands between β-strand 3 and helix 2 of the NFLD subdomain of the THUMP domain of PfTrm14 (Figure 2A and C). This two-stranded anti-parallel β-sheet is extending away from the THUMP domain on the opposite side of the presumed tRNA binding surface and the SAM binding pocket of the RFM domain (see further). Sequence analysis using a BLAST search (61) showed that this insertion is conserved only in Archaea, with over 50 matches found. No comparable insertions were found in THUMP domains of Bacteria and Eukaryota. Interestingly, these two β-strands are made up mainly of conserved positively charged (five lysines or arginines) and aromatic residues (Y51, Y52), resulting in one side of the β-sheet having a positively charged surface (Figure 4A). To investigate a possible role of these two β-strands in tRNA binding and catalysis of the m2G6 modification, we made two different deletion variants of PfTrm14. In a first variant, the peptide region spanning the inserted β-sheet (residues Y51 to E61) was replaced by two glycines that should be sufficient to span the distance between β-strand 3 and helix 2 of the THUMP domain (PfTrm14_2G variant). In a second variant, this region was replaced by the corresponding loop region of TTCTrmN (R41 to G45) (PfTrm14_TTC variant). The G6 MTase activity and tRNA binding affinity of both variants were compared with the wild-type PfTrm14 (Supplementary Figure S3A and B). Interestingly, only small changes in affinity towards tRNA and in the catalytic rate were observed in vitro upon deletion. This might suggest that the β-strands are a conserved ornament of the basic NFLD fold or a relic of a previous activity. Alternatively, this element could be involved in another, yet undiscovered, function of archaeal Trm14.
Figure 4.

(A and B) Electrostatic surface potential mapped on the solvent accessible surface of PfTrm14 (A) and TTCTrmN (B). kb = Boltzmann’s constant, T = temperature, ec = charge of an electron. (C and D) Mapping of conservation scores of amino acids on the surface of PfTrm14 (C) and TTCTrmN (D). The color code of the conservation score is indicated in the legend. The bound SFG (PfTrm14) and modeled SAM (TTCTrmN) in the active site of the RFM domain are shown in ball and stick representation.
Substrate binding and ligand-induced conformational changes in the RFM domain
The C-terminal MTase domains of PfTrm14 and TTCTrmN both adopt a topology that is typical for the RFM superfamily of MTases (Figure 2C and D). Here, the C-terminal (7th) β-strand of a central seven-stranded β-sheets is inserted in an anti-parallel way between strands 5 and 6 of the sheet, and the β-sheet is packed on both sides by a total of six α-helices. The RFM domains of TTCTrmN and PfTrm14 are very similar and superpose with an rmsd of 1.3 Å (using the main chain atoms of 155 residues).
While the TTCTrmN structure was solved in the apo form, the PfTrm14 was solved with the RFM domain either bound to SAH (PfTrm14-SAH), SAM (PfTrm14-SAM) or SFG (PfTrm14-SFG). One loop region, connecting β-strand 4 and helix 5, is highly flexible and could not be observed in the electron density of the crystal structures of TTCTrmN and PfTrm14-SAH and PfTrm14-SAM. Only when SFG was bound to PfTrm14 the loop was structured and became visible in the density map. Since both SAM and SFG were soaked into pre-existing crystals of PfTrm14, this loop movement is probably not caused by crystal packing interactions, but concerns a ligand-induced conformational change. Although SAM and SFG have the same net charge (Supplementary Figure S4), only SFG seems to evoke this conformational change. This might be due to the position of the positive charge, which is located on the Sγ in SAM and on the Nε in SFG (62). Assuming a dissociative SN2 reaction mechanism, one could speculate that the positive charge on the ε position would mimic an intermediate along the methyl transfer reaction pathway where the Sγ-Cε bond in SAM is partially broken. Also the large loop region, connecting the THUMP domain to the first α-helix of the RFM domain, interacts with the methionine moiety of the bound co-factor and this loop undergoes some structural rearrangements in the SFG-bound form, moving the backbone 1.5 Å away from the ligand.
The adenosine moieties of the ligands SAH, SAM and SFG are bound to PfTrm14 in a pocket located at the first β–α–β–α–β motif of the Rossmann-fold and the cross-over towards the second β–α–β motif, as is often seen in other nucleotide binding proteins (63). The pocket for the methionine moiety is formed by the loop region connecting the THUMP domain to the RFM domain together with the first two α-helices of the RFM domain. Omit maps of the binding pocket of PfTrm14 (Figure 3) show that the ligand SAM is bound in an extended conformation (dihedral C4′, C5′, S, Cγ = −70.7°) which is common for SAM-dependent MTases (4,62). The reaction product SAH is also bound in an extended conformation but with a dihedral angle of −34.6°. The dihedral angle of the bound inhibitor SFG is −101.1°.
Figure 3.

Active site of PfTrm14 bound to (A) SAH, (B) SAM and (C) SFG. The 2Fo − Fc omit density map is shown for the ligands. The map is contoured at 2 σ within 1.6 Å of the ligand. The ligands are shown with their C atoms colored green, the amino acid residues are shown with their C atoms colored pink (interacting with adenine), orange (interacting with ribose) or cyan (interacting with the homocysteine part). Hydrogen bonds are indicated by dashed lines.
The SAM-binding cleft is mainly lined by residues belonging to the conserved motifs I, II, III, IV, V and X that exist in related N-MTases (9,62,64). The three ligands SAH, SAM and SFG are engaged in a similar set of hydrogen-bonding and van der Waals interactions with PfTrm14. Since TTCTrmN was solved in the apo state only, modeling of SAM into the TTCTrmN structure was performed using Epitope Match (rmsd 1.7 Å, 26 residues in the epitope) (Supplementary Figure S5). The model shows that the binding pocket is conserved in TTCTrmN and chemically related residues are involved in the binding of SAM.
SAH is bound to the RFM domain by a total of 10 hydrogen bonds and several van der Waals interactions (Figure 3A). The adenine moiety of the co-factor is stacked between the side chains of Met225 and Lys249 of PfTrm14 using van der Waals interactions. The N6 exocyclic amine of the adenine base forms a hydrogen bond to Asp276 (Asp243 in TTCTrmN), while its N1 is within hydrogen bonding distance to the main chain amine of Ala277. Asp276 is highly conserved and is part of the MTase motif III (P/G,QTXD276AXXC/C/L), while Met225 is part of motif I (M225XGXG).
The ribose moiety of the co-factor is hydrogen bonded with both its 2′- and 3′ hydroxyls to the side chain of the highly conserved Glu248 of motif II (Asp216 in TTCTrmN). The 3′-OH is also involved in a hydrogen bond with His198.
The homocysteine/methionine moieties of SAH and SAM interact via their α- with Asn293 (part of motif IV) and via their α-COO− with Ser228 (side and main chain) and Thr230 which are located in or next to motif I, and with the main chain of Leu202, located in the proximity of motif X.
with Asn293 (part of motif IV) and via their α-COO− with Ser228 (side and main chain) and Thr230 which are located in or next to motif I, and with the main chain of Leu202, located in the proximity of motif X.
Motif IV is part of a possible guanosine binding pocket
The typical motif IV of RFM enzymes acting on exocyclic amine groups consists of the consensus sequence D/N-P/L-P-Y/F/H (65). In PfTrm14 and TTCTrmN, this corresponds to N293LPY and N260PPH, respectively. These residues are located right next to the flexible loop that is missing in our structures and line a pocket that leads towards the εCH3 of SAM. Asn293 is located in close proximity to the methyl group of SAM (3.9 Å). This pocket will likely accommodate the G6 of the tRNA that is modified by PfTrm14 and TTCTrmN and provide the catalytic residues for the methyl transfer reaction. A crystal structure of the 16 S rRNA MTase RsmC in complex with SAM and guanine indeed shows that the residues of motif IV interact with the guanine substrate (54). In the latter structure, Phe308 (of the N305PPF motif) interacts via base stacking with guanine, while Asn305 forms a hydrogen bond with the exocyclic amine group on position 2.
In order to confirm that essentially the same binding pocket for the G6 nucleoside exists in PfTrm14 and TTCTrmN, an epitope match was performed using the program Epitope Match (53) and using the guanosine binding epitope of the RsmC-guanosine crystal structure (54). For PfTrm14 as well as TTCTrmN, a possible binding pocket was found that could accommodate a guanosine (corresponding to G6) with only minor structural rearrangements (Supplementary Figure S6). In the case of TTCTrmN, the epitope contained 13 residues with an rmsd of 2.7 Å. As expected, His263 of motif IV is in our model stacked with the guanosine base, although a small movement of this residue upon tRNA binding is necessary for perfect accommodation of guanosine. Such movement would be possible, since His263 is located next to the highly flexible loop of PfTrm14. The catalytic Asn263 residue (of the same motif IV) is in our model located in between the N2 atom of guanosine and the methyl group of SAM, bridging a distance of 4 Å between methyl donor and acceptor. In PfTrm14, the epitope contained 13 residues with an rmsd of 2.8 Å. In this case, Tyr296 of motif IV was involved in base stacking to the ribose, as proposed for the corresponding His of TTCTrmN. The catalytic Asn293 is again located between the methyl donor (SAM) and the methyl acceptor (N2 of G6), bridging a distance of 4.9 Å between the donor and the acceptor.
To confirm the catalytic role of this motif in Trm14/TrmN, His263 of TTCTrmN was substituted by an alanine. In agreement with the proposed importance of the His263 residue, the tRNA MTase activity of the TTCTrmN(H263A) variant was decreased to 25% of the wild-type activity. The decrease in activity must be related to the accommodation of G6 in the catalytic pocket, since the affinity towards tRNA was unchanged upon introduction of the point substitution (Supplementary Figure S3C and D).
The tRNA substrate binds on the interface of the RFM and THUMP domains
The surface electrostatics of TTCTrmN and PfTrm14 show a very large patch of positively charged residues leading into the SAM binding pocket of the RFM domain. The location of this positively charged surface overlaps in both proteins, although it is more pronounced in TTCTrmN (Figure 4A and B). Such a positively charged surface matches with the tRNA binding and modification role of both proteins. Moreover, the positively charged residues overlap with the evolutionarily conserved region, suggesting a functional role in substrate tRNA binding (Figure 4C and D). The positively charged residues that form the proposed tRNA binding site are located on both the THUMP and RFM domains. A region of the solvent accessible surface with particularly strong positive electrostatics, due to the presence of several positively charged residues, is located around the interface of the THUMP domain and the RFM domain, building a groove suitable for tRNA accommodation. A significant contribution to the positive charge in this region is provided by the long peptide linker connecting the two domains, suggesting also a functional role for this region. A second region with positive electrostatics is centered around the SAM binding pocket, where the acceptor stem of the tRNA containing the target nucleotide G6 might bind. In PfTrm14 an additional patch of conserved, positively charged residues is present on the 2-stranded β-sheet extension of the THUMP domain, but a functional role in the m2G6 MTase activity in vitro was ruled out (see before).
The THUMP domain has previously been suggested to be an RNA binding domain implicated in the modification of bases in the core of the tRNA molecules (13). However, previous studies with the isolated THUMP domain of the m2G10 MTase PAB1283 have already shown that the isolated THUMP domain is not able to bind the tRNA substrate (28). This is in agreement with our suggestion that the tRNA binding surface of Trm14/TrmN is formed by both the RFM and THUMP domain, and that also the linker region contributes to a large extent. Consequently, it was found that in the case of PfTrm14 neither the isolated RFM nor the THUMP domain is able to bind tRNA or catalyze the methyltransfer reaction. Also an attempt to reconstitute the activity by mixing the individual domains was not successful, suggesting that both domains indeed synergistically collaborate, together with the linker, in the binding of the substrate (16).
To obtain a more detailed image of the binding mode of tRNA to Trm14/TrmN, a docking model was generated for the binding of tRNAPhe of T. thermophilus onto TTCTrmN. An unbound structure of T. thermophilus tRNAPhe is not available. Therefore, we used two variants of T. thermophilus tRNAPhe: a homology model of the unbound conformation based on the coordinates of a homolog from E. coli crystallized in the apo form (PDB code 3L0U) and an experimental model in the bound conformation taken from the cocrystal structure with phenylalanyl-tRNA synthetase (PDB code 2IY5). The unbound conformation corresponds to the unmodified substrate RNA as would be encountered by TTCTrmN in solution. The bound tRNA conformation is altered, in particular in the anticodon loop, by interactions with the tRNA synthetase. However, in both cases docking yields the same two preferred orientations of the tRNAPhe towards TTCTrmN. The best-scored docking model for the unbound tRNA conformation (which is similar to one of the top-scoring and largest clusters obtained for the bound conformation) is shown in Figure 5A and Supplementary Figure S7A. This model seems mechanistically most relevant, as the target nucleoside residue is positioned in such a way that it can be easily rotated out of the tRNA acceptor stem into the catalytic site, like observed in numerous other MTases acting on nucleic acids (66). In an alternative model, that represents the largest cluster for the unbound conformation, the tRNA is rotated by ∼90°(shown in Supplementary Figure S7B). Here, the target nucleoside is positioned unfavorably to enter the active site, suggesting that this orientation of tRNA may not be compatible with the catalysis of the methyl transfer.
Figure 5.

(A) Docking model of tRNAPhe of T. thermophilus onto TTCTrmN. TTCTrmN is shown with the electrostatic potential mapped on its solvent accessibility surface, and the tRNA (a homology model based on the orthologous tRNAPhe from E. coli) is shown in cartoon style. The guanosine on position 6 of the tRNA molecule and SAM, bound to the active site of the RFM domain, are shown in ball-and-stick and stick representation, respectively. Black circles indicate the position of residues that were mutated in this study. (B) Band shift assay showing binding of [α-32P] GTP-labeled T. thermophilus tRNAPhe to increasing amounts of wt TTCTrmN and the K270E/R300E and K129E/R130E/R157E variants. Band shift assays for other TTCTrmN variants are shown in Supplementary Figure S7C. The docking model is available for download from: ftp://ftp.genesilico.pl/iamb/models/RNA.MTases/TrmN/.
In the preferred model (Figure 5A), tRNAPhe binds with its core, formed by the D and T stem and loops, in the groove formed between the RFM and THUMP domain, while a part of the anticodon stem interacts with the THUMP domain. Consistent with a role of Trm14/TrmN in G6 methylation, the tRNA is binding with its acceptor stem directed towards the RFM domain. In this model, the distance between the methyl group of SAM and the N2 exocyclic amine group of G6 is 19.3 Å. The position of the G6 suggests that a flip-out mechanism could bring the guanine base in closer contact to the methyl donor for the reaction to occur. In this model, the tRNA is also binding in the vicinity of the flexible loop, adjacent to motif IV, that harbors the positively charged residues Arg266, Arg271 and Lys272, suggesting some induced fit upon tRNA binding.
To further validate the docking model, a number of positively charged residues in the proposed interaction surface were substituted by alanine and glutamate, and the ability of the resulting protein variants to bind the tRNA substrate was determined by band-shift assays (Supplementary Figures S5 and S7). The corresponding alanine and glutamate variants show a same tendency of reduced affinity towards tRNAPhe. Yet, the effect of the glutamate substitutions is stronger due to the inversion of charge from positive (Arg or Lys) to negative (Supplementary Figure S7). Apart from direct interactions such as hydrogen bonds and salt bridges, also long-range electrostatics could affect the binding mode and affinity of the tRNA substrate. In the case of a highly positively charged molecule, such as TTCTrmN, the effect of single alanine substitution on these long range interactions could be easily shielded by the surrounding charges.
Arg33 and Arg98 are located on the THUMP domain (respectively on the NFLD and core-THUMP subdomains) and line the highly positively charged groove between the THUMP and RFM domains (Supplementary Figure 5A). Substituting either of these residues by glutamate reduces the affinity for tRNA by at least a factor of 2 (Supplementary Figure S7C). The charged and conserved residues Arg129 and Arg130 are located in the THUMP domain of TTCTrmN and are in the docking model in close proximity to the region between the D-arm and the anticodon arm of the tRNA substrate. R157 is part of the linker region between the THUMP and RFM domain, containing highly conserved positively charged residues, and is involved in the formation of the binding groove. In agreement with the model, a variant with a double substitution K129E/R130E exhibits a reduced affinity towards tRNAPhe by more than 2-fold (Supplementary Figure S7C). Interestingly, the triple substitution K129E/R130E/R157E invokes an even larger effect on affinity and nearly no binding is observed at the highest protein concentration used (∼10-fold effect on KD). These point variants clearly support the docking model and underscore the role of the groove between the THUMP and RFM domains in tRNA binding.
Lys270 and Arg300 are located on the RFM domain close to the SAM binding pocket (Supplementary Figure S5A). The K270E and R300E variants show a clear (∼10-fold) decrease in their affinity to tRNAPhe. In the K270E/R300E variant, no binding could be observed at the highest protein concentration used. These two residues are only in proximity with the tRNA molecule in the preferred docking model and not in the alternative docking model (compare Supplementary Figure S7A with S7B). Consequently, these results further support the docking model in Figure 5A as the biological relevant interaction mode between TTCTrmN and tRNAPhe.
AVAILABILITY
The docking models of TTCTrmN and tRNAPhe discussed in this article are available for download from ftp://ftp.genesilico.pl/iamb/models/RNA.MTases/TrmN/
ACCESSION NUMBERS
Coordinates have been submitted to the RCSB Protein Data Bank with accession codes: 3TLJ for PfTrm14-SAH, 3TM4 for PfTrm14-SAM, 3TM5 for PfTrm14-SFG and 3TMA for TTCTrmN.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–7; Supplementary Tables 1–2.
FUNDING
Fonds Wetenschappelijk Onderzoek (G025909N to W.V.); FWO post-doctoral grant to W.V.; IWT pre-doctoral grant to M.F.; European Social Fund PhD fellowship to I.T.; Polish Ministry of Science and Higher Education (188/N-DFG/2008/0 to J.M.B.); European Research Council (StG grant RNA + P = 123D to J.M.B.); “Ideas for Poland” fellowship from the Foundation for Polish science to J.M.B.; Fonds pour le Recherche Fondamentale Collective (2.4.520.05F to L.D.); Fonds J. Brachet and Fonds D. et A. Van Buuren to L.D. Funding for open access charge: Fonds Wetenschappelijk Onderzoek (G025909N).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the staff at the beamline ID23-2 and ID14-4 of the ESRF in France and the staff of the beamline PX-III of the SLS in Switzerland for assistance during data collection. Authors would also like to thank Stefan Münnich for help during data processing.
REFERENCES
- 1.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopper AK, Phizicky EM. tRNA transfers to the limelight. Genes Dev. 2003;17:162–180. doi: 10.1101/gad.1049103. [DOI] [PubMed] [Google Scholar]
- 3.Gray MW. Analysis of O2′-methylnucleoside 5′-phosphates in snake venom hydrolysates of RNA: identification of O2′-methyl-5-carboxymethyluridine as a constituent of yeast transfer RNA. Can. J. Biochem. 1975;53:735–746. doi: 10.1139/o75-101. [DOI] [PubMed] [Google Scholar]
- 4.Hou YM, Perona JJ. Stereochemical mechanisms of tRNA methyltransferases. FEBS Lett. 2010;584:278–286. doi: 10.1016/j.febslet.2009.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalhor HR, Clarke S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol. Cell Biol. 2003;23:9283–9292. doi: 10.1128/MCB.23.24.9283-9292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato M, Araiso Y, Noma A, Nagao A, Suzuki T, Ishitani R, Nureki O. Crystal structure of a novel JmjC-domain-containing protein, TYW5, involved in tRNA modification. Nucleic Acids Res. 2011;39:1576–1585. doi: 10.1093/nar/gkq919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helm M, Giegé R, Florentz C. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry. 1999;38:13338–13346. doi: 10.1021/bi991061g. [DOI] [PubMed] [Google Scholar]
- 8.Sakurai M, Ohtsuki T, Watanabe K. Modification at position 9 with 1-methyladenosine is crucial for structure and function of nematode mitochondrial tRNAs lacking the entire T-arm. Nucleic Acids Res. 2005;33:1653–1661. doi: 10.1093/nar/gki309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimoto M, Higashijima K, Shirouzu M, Grosjean H, Bessho Y, Yokoyama S. Crystal structure of tRNA N2,N2-guanosine dimethyltransferase Trm1 from Pyrococcus horikoshii. J. Mol. Biol. 2008;383:871–884. doi: 10.1016/j.jmb.2008.08.068. [DOI] [PubMed] [Google Scholar]
- 10.Awai T, Kimura S, Tomikawa C, Ochi A, Bessho Y, Yokoyama S, Ohno S, Nishikawa K, Yokogawa T, et al. Aquifex aeolicus tRNA (N2,N2-guanine)-dimethyltransferase (Trm1) catalyzes transfer of methyl groups not only to guanine 26 but also to guanine 27 in tRNA. J. Biol. Chem. 2009;284:20467–20478. doi: 10.1074/jbc.M109.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purushothaman SK, Bujnicki JM, Grosjean H, Lapeyre B. Trm11p and Trm112p are both required for the formation of 2-methylguanosine at position 10 in yeast tRNA. Mol. Cell Biol. 2005;25:4359–4370. doi: 10.1128/MCB.25.11.4359-4370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armengaud J, Urbonavicius J, Fernandez B, Chaussinand G, Bujnicki JM, Grosjean H. N2-methylation of guanosine at position 10 in tRNA is catalyzed by a THUMP domain-containing, S-adenosylmethionine-dependent methyltransferase, conserved in Archaea and Eukaryota. J. Biol. Chem. 2004;279:37142–37152. doi: 10.1074/jbc.M403845200. [DOI] [PubMed] [Google Scholar]
- 14.Awai T, Ochi A, Sengoku T, Hirata A, Bessho Y, Yokoyama S, Hori H. Substrate tRNA recognition mechanism of a multisite-specific tRNA methyltransferase, Aquifex aeolicus Trm1, based on the X-ray crystal structure. J. Biol. Chem. 2011;286:35236–46. doi: 10.1074/jbc.M111.253641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menezes S, Gaston KW, Krivos KL, Apolinario EE, Reich NO, Sowers KR, Limbach PA, Perona JJ. Formation of m2G6 in Methanocaldococcus jannaschii tRNA catalyzed by the novel methyltransferase Trm14. Nucleic Acids Res. 2011;39:7641–7655. doi: 10.1093/nar/gkr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roovers M, Oudjama Y, Fislage M, Bujnicki JM, Versées W, Droogmans L. The open reading frame TTC1157 of Thermus thermophilus HB27 encodes the methyltransferase forming N2-methylguanosine at position 6 in tRNA. RNA. 2012;18:815–824. doi: 10.1261/rna.030411.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Czerwoniec A, Kasprzak JM, Kaminska KH, Rother K, Purta E, Bujnicki JM. Folds and functions of domains in RNA modification enzymes. In: Grosjean H, editor. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Landes Bioscience, Austin, Texas, USA; 2009. pp. 289–302. [Google Scholar]
- 18.Bujnicki JM. Comparison of protein structures reveals monophyletic origin of the AdoMet-dependent methyltransferase family and mechanistic convergence rather than recent differentiation of N4-cytosine and N6-adenine DNA methylation. In Silico Biol. 1999;1:175–182. [PubMed] [Google Scholar]
- 19.Nishimasu H, Ishitani R, Yamashita K, Iwashita C, Hirata A, Hori H, Nureki O. Atomic structure of a folate/FAD-dependent tRNA T54 methyltransferase. Proc. Natl Acad. Sci. USA. 2009;106:8180–8185. doi: 10.1073/pnas.0901330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arragain S, Garcia-Serres R, Blondin G, Douki T, Clemancey M, Latour JM, Forouhar F, Neely H, Montelione GT, Hunt JF, et al. Post-translational modification of ribosomal proteins: structural and functional characterization of RimO from Thermotoga maritima, a radical S-adenosylmethionine methylthiotransferase. J. Biol. Chem. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 2003;22:2593–2603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S. Crystal structure of archaeal tRNA(m(1)G37)methyltransferase aTrm5. Proteins. 2008;72:1274–1289. doi: 10.1002/prot.22019. [DOI] [PubMed] [Google Scholar]
- 23.Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aravind L, Koonin EV. THUMP—a predicted RNA-binding domain shared by 4-thiouridine, pseudouridine synthases and RNA methylases. Trends Biochem. Sci. 2001;26:215–217. doi: 10.1016/s0968-0004(01)01826-6. [DOI] [PubMed] [Google Scholar]
- 25.Waterman DG, Ortiz-Lombardía M, Fogg MJ, Koonin EV, Antson AA. Crystal structure of Bacillus anthracis ThiI, a tRNA-modifying enzyme containing the predicted RNA-binding THUMP domain. J. Mol. Biol. 2006;356:97–110. doi: 10.1016/j.jmb.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Randau L, Stanley BJ, Kohlway A, Mechta S, Xiong Y, Söll D. A cytidine deaminase edits C to U in transfer RNAs in Archaea. Science. 2009;324:657–659. doi: 10.1126/science.1170123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCleverty CJ, Hornsby M, Spraggon G, Kreusch A. Crystal structure of human Pus10, a novel pseudouridine synthase. J. Mol. Biol. 2007;373:1243–1254. doi: 10.1016/j.jmb.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 28.Gabant G, Auxilien S, Tuszynska I, Locard M, Gajda MJ, Chaussinand G, Fernandez B, Dedieu A, Grosjean H, Golinelli-Pimpaneau B, et al. THUMP from archaeal tRNA:m22G10 methyltransferase, a genuine autonomously folding domain. Nucleic Acids Res. 2006;34:2483–2494. doi: 10.1093/nar/gkl145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fislage M, Roovers M, Münnich S, Droogmans L, Versées W. Crystallization and preliminary X-ray crystallographic analysis of putative tRNA-modification enzymes from Pyrococcus furiosus and Thermus thermophilus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011;67:1432–1435. doi: 10.1107/S1744309111036347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doublié S. Preparation of selenomethionyl proteins for phase determination. Meth. Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 31.Reyes VM, Abelson J. A synthetic substrate for tRNA splicing. Anal. Biochem. 1987;166:90–106. doi: 10.1016/0003-2697(87)90551-3. [DOI] [PubMed] [Google Scholar]
- 32.Droogmans L, Grosjean H. 2′-O-methylation and inosine formation in the wobble position of anticodon-substituted tRNA-Phe in a homologous yeast in vitro system. Biochimie. 1991;73:1021–1025. doi: 10.1016/0300-9084(91)90143-o. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D Biol. Crystallogr. 2005;61:449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 35.The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Sheldrick GM, Hauptman HA, Weeks CM, Miller R, Uson I. International tables for crystallography: Crystallography of biological macromolecules. Vol. F. Dordrecht: Kluwer Academic Publishers; pp. 333–345. [Google Scholar]
- 37.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 38.Hao Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Crystallogr. 2004;37:498–499. [Google Scholar]
- 39.Sheldrick GM. Macromolecular phasing with SHELXE. Zeitschrift für Kristallographie. 2002;217:644–650. [Google Scholar]
- 40.Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on protein Crystallography. 31, 34–38. [Google Scholar]
- 41.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 42.Morris RJ, Zwart PH, Cohen S, Fernandez FJ, Kakaris M, Kirillova O, Vonrhein C, Perrakis A, Lamzin VS. Breaking good resolutions with ARP/wARP. J. Synchrotron Radiat. 2004;11:56–59. doi: 10.1107/s090904950302394x. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 47.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 48.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vellieux FMD, Dijkstra BW. Computation of Bhat’s OMIT maps with different coefficients. J. Appl. Crystallogr. 1997;30:396–399. [Google Scholar]
- 53.Jakuschev S, Hoffmann D. A novel algorithm for macromolecular epitope matching. Algorithms. 2009;2:498–517. [Google Scholar]
- 54.Demirci H, Gregory ST, Dahlberg AE, Jogl G. Crystal structure of the Thermus thermophilus 16 S rRNA methyltransferase RsmC in complex with cofactor and substrate guanosine. J. Biol. Chem. 2008;283:26548–26556. doi: 10.1074/jbc.M804005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rother M, Rother K, Puton T, Bujnicki JM. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. 2011;39:4007–4022. doi: 10.1093/nar/gkq1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuszynska I, Bujnicki JM. DARS-RNP and QUASI-RNP: new statistical potentials for protein-RNA docking. BMC Bioinformatics. 2011;12:348. doi: 10.1186/1471-2105-12-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vakser IA, Aflalo C. Hydrophobic docking: a proposed enhancement to molecular recognition techniques. Proteins. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- 58.Gajda MJ, Tuszynska I, Kaczor M, Bakulina AY, Bujnicki JM. FILTREST3D: discrimination of structural models using restraints from experimental data. Bioinformatics. 2010;26:2986–2987. doi: 10.1093/bioinformatics/btq582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ponstingl H, Henrick K, Thornton JM. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 60.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 61.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 63.Versées W, Decanniere K, Pellé R, Depoorter J, Brosens E, Parkin DW, Steyaert J. Structure and function of a novel purine specific nucleoside hydrolase from Trypanosoma vivax. J. Mol. Biol. 2001;307:1363–1379. doi: 10.1006/jmbi.2001.4548. [DOI] [PubMed] [Google Scholar]
- 64.Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995;253:618–632. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- 65.Bujnicki JM. Phylogenomic analysis of 16S rRNA:(guanine-N2) methyltransferases suggests new family members and reveals highly conserved motifs and a domain structure similar to other nucleic acid amino-methyltransferases. FASEB J. 2000;14:2365–2368. doi: 10.1096/fj.00-0076com. [DOI] [PubMed] [Google Scholar]
- 66.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.