

Abstract
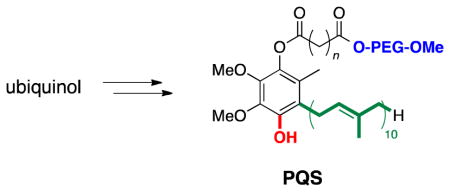
Described herein are newly developed, straightforward entries to “PQS” (Polyethyleneglycol UbiQuinol Succinate, n = 2), a designer surfactant that serves as precursor to micelle-forming, covalently bound catalysts for metal-catalyzed transformations in water with in-flask catalyst recycling.
INTRODUCTION
Catalysis is now considered among the major milestones of last centuries’ developments in organic chemistry. Catalyst recycling, in particular in the case of homogeneous catalysis, despite its many advantages (e.g., reduced waste and/or more efficient energy usage) still remains challenging. This is especially true when costly ligands and/or precious metals are involved. Aqueous biphasic systems have become an important industrial technique and offer one solution: the catalyst can be recovered from the aqueous phase although the overall transformation occurs in organic solvent.1,2 We have previously shown that a variety of transition metal-catalyzed transformations can be carried out within micellar media, generated by simply mixing a selected surfactant in water.3 Organic solvents, therefore, are no longer needed, as the self-assembled micelles provide the lipophilic interior that serves, in essence, as the solvent for catalysis; i.e., as nanoreactors in water. Moreover, since only 1–2 wt % of surfactant in water easily exceeds the required critical micelle concentration (CMC), a catalytic amount of the amphiphile is sufficient. While other surfactants typically contain only lipophilic and hydrophilic subsections, PQS (Polyethyleneglycol Ubiquinol Sebacate) was originally designed to accommodate a third component: an internally connected catalyst (Figure 1).4 Hence, the three major subsections of PQS consist of (1) the readily available hydroquinone, ubiquinol (i.e., the reduced form of CoQ10), that supplies the lipophilicity in its 50-carbon side-chain within which organic substrates are to be dissolved. Importantly, it also bears a second phenolic handle enabling covalent attachment of a catalyst that will reside inside the nanoreactor of its micellar array; (2) polyethyleneglycol (PEG-2000), which ensures water solubility; and (3) a linker unit (either sebacic acid or succinic acid) that connects the lipophilic interior (i.e., ubiquinol) to the hydrophilic exterior (i.e., PEG).
Figure 1.

Designed components of PQS.
The concept of in-flask catalyst recycling was successfully demonstrated on ring-closing- and cross-metathesis reactions in water employing Grubbs-Hoveyda’s first- and second-generation catalysts, which were independently covalently attached to PQS (4: PQS-GH-1 and 5: PQS-GH-2, Figure 2).4 A study of up to 10 recycles without significant loss of catalyst activity highlighted the potential of the PQS platform. Likewise, a related system based on PQS that does not focus on transition metal catalysts, e.g. PQS-proline (7) for use in organocatalysis, has recently appeared.5
Figure 2.

PQS as a platform for covalent attachment of a catalyst.
Although a first generation synthesis (i.e., for 1) gave satisfactory results, limited selectivity in each coupling step using sebacoyl chloride resulted in purification issues.4 Moreover, scale-up experiments suggested that removal of impurities would be impractical through chromatographic means. Therefore, a synthetically more attractive route to the PQS platform was sought. Herein we describe two novel and improved entries to the designer surfactant PQS-1 (1). We also disclose a synthesis of PQS-3 (3), a third generation surfactant, employing a succinic acid linker that enhances significantly the availability of the PQS platform.
RESULTS AND DISCUSSION
Initial attempts towards 1 focused on commercially available sebacic acid (8), which after anhydride (9) formation,6 was treated with M-PEG-2000 to obtain PEGylated sebacic acid 10. Subsequent coupling with ubiquinol, either through its acid chloride or via EDCI/DMAP led to PQS-1 (1) in high isolated yield and of good purity. To obtain 1 of higher quality, a modified approach from sebacoyl dichloride (11) was investigated. Formation of mono-benzylated sebacic acid 12 (57% yield after chromatography) and then coupling and deprotection steps gave mono-PEGylated sebacic acid 10 in high purity following simple precipitation from Et2O. Although the overall yield for PQS-1 (1) was generally lower using route B, the quality of the material was consistently higher than that obtained via route A.7,8
The linker unit between ubiquinol and PEG in PQS-1 (1) is derived from 10-carbon sebacic acid (8), which underwent both mono- and (undesired) diacylation due to the open chain nature of sebacoyl dichloride. Replacement of the C10 unit by a C4 fragment originating from succinic acid anhydride would eliminate opportunities for impurities due to reactions at both termini. Thus, starting with (inexpensive) M-PEG-2000 (14) and succinic anhydride, mono PEGylated succinic acid 15 was formed in excellent yield (Scheme 2). EDCI-mediated coupling of acid 15 and N-hydroxysuccinimide led to activated ester 16. Neither of these two steps required product purification. Lastly, ester 16 was coupled with ubiquinol9 in THF to provide PQS-3 (3) after straightforward purification on silica gel.8 This streamlined, cleaner procedure could be successfully repeated on various scales10 in up to 64% overall yield.
Seheme 2a.

a Reagents and conditions: (a) succinic anhydride, NEt3, toluene, 60 °C, 99%. (b) N-hydroxysuccinimide, EDCI, CH2Cl2, rt, 99%. (c) ubiquinol, NaH, THF, 0 °C to rt, 65%.
After lyophilization, PQS-3 (3) appears as a white, fluffy solid, which readily dissolves in water to provide micelles having an average diameter size of 21 nm, as determined by DLS measurements (Figure 3). By contrast, PQS-1 in water consists of particles of ca. 9 nm. The spherical appearance of these self-aggregated nanoparticles, which can appear, in part, to be agglomerated and, therefore, oblong in shape, was confirmed by cryo-TEM measurements (Figure 4).
Figure 3.

Particle size of PQS-3 (3) measured by DLS.
Figure 4.

Cryo-TEM measurements of PQS-3 (3).
To ensure that the minor difference between PQS-1 and PQS-3 (i.e., the 10- vs. 4-carbon linker) does not translate into differences in the observed catalysis, 3 was attached to acid 1711 to yield carbene precursor 18 (Scheme 3). Subsequent insertion of Ru yielded 6 in good overall yield. PQS-3-GH1 (6) appears as a brown solid which is freely soluble in water and forms nano-reactors with an average diameter of 30 nm by DLS (Figure 5).
Seheme 3a.
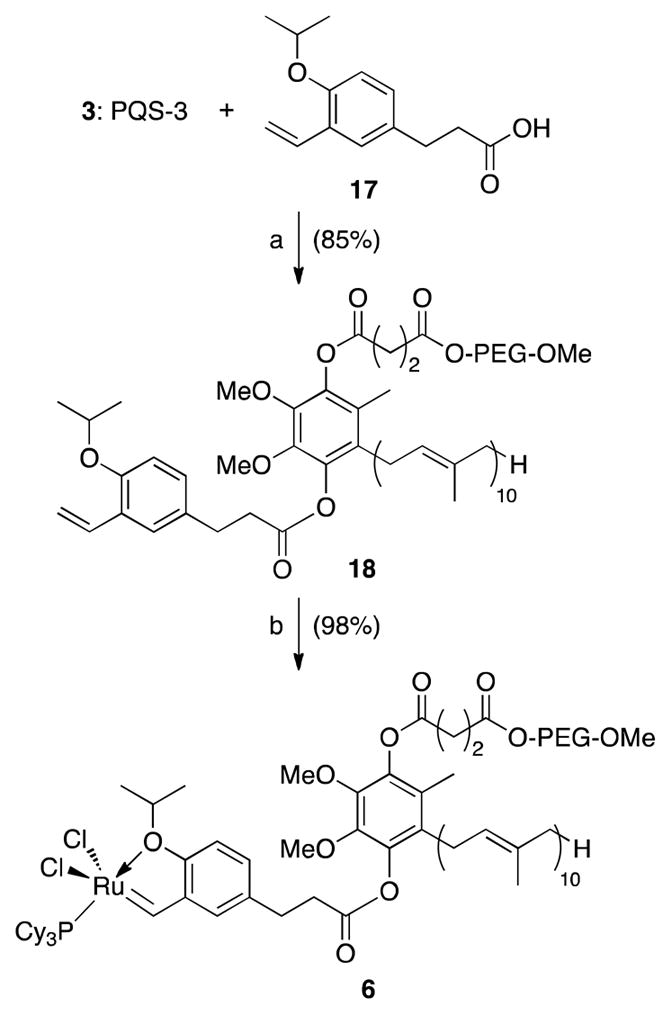
a Reagents and conditions: (a) EDCI, DMAP, NEt3, CH2Cl2, rt, 85%. (b) Grubbs 1st generation, CuCl, CH2Cl2, rt, 98%.
Figure 5.

Particle size of PQS-3-GH1 (6) measured by DLS.
Cryo-TEM measurements of PQS-3-GH1 (6) confirmed the size of these particles and showed their spherical appearance (Figure 6).
Figure 6.

Cryo-TEM measurements of PQS-3-GH1 (6)
Diene 19 was subjected to RCM to yield the cyclized product 20 using 6 as the micelle forming catalyst (Scheme 4). Compared to results obtained previously with 4,4a the newly developed 3rd generation platform (i.e., 6) functions virtually identically.
Seheme 4.

New systems, such as PQS-(R)-BINAP (21) are currently under investigatation for applications to asymmetric Rh- and Pd-catalyzed transformations in water, providing similar options for straightforward catalyst recycling (Figure 7).
Figure 7.

PQS-(R)-BINAP (21) for Rh and/or Pd catalysis.
CONCLUSIONS
Two alternative synthetic entries to PQS-1 (1) are presented. A further improved, third-generation designer surfactant in the PQS series, PQS-3 (3), has been developed, the synthesis of which is straightforward, higher yielding than existing routes, and potentially scalable. Both surfactants function equally well when covalently derivatized with ruthenium catalysts, thereby enabling olefin metathesis reactions to be carried out under homogeneous conditions in water at room temperature with in-flask catalyst recycling.
EXPERIMENTAL SECTION
Materials and Methods
Unless otherwise noted, all reactions were performed in oven-dried glassware under an atmosphere of Argon. Poly(ethylene glycol) methyl ether (13, M-PEG-2000) was obtained from Aldrich (catalog # 202509). 1H and 13C spectra were recorded at 22°C on a 400 MHz or 500 MHz NMR spectrometer. Chemical shifts in 1H NMR spectra are reported in parts per million (ppm) on the δ scale from an internal standard of residual chloroform (7.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in hertz (Hz), and integration. Chemical shifts of 13C NMR spectra are reported in ppm from the central peak of CDCl3 (77.23 ppm) on the δ scale.
Route A:7 Synthesis of mono-PEGylated sebacic acid 10
A 250 mL round bottom flask containing a strong magnetic stir bar was charged with poly(ethylene glycol) methyl ether (14, 10.00 g, 5.00 mmol, typical Mn 2.000), sebacic acid derivative6 (1.38 g, 7.50 mmol), 4-dimethylaminopyridine (90 mg, 0.75 mmol) and pyridine (25 mL). After attaching a reflux condenser, the system was purged with argon and heated to 100°C in an oil bath for 4 h whereas all solids dissolved. After cooling the clear solution to room temperature the reaction mixture was acidified with 1.5 N aq. HCl until pH <7 and extracted with CH2Cl2 (3x). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, concentrated and placed overnight under high vacuum to give 10 as a white waxy solid (10.84 g, 4.96 mmol, 99% isolated yield). Alternatively the product can be purified by the following procedure: after concentration and high vacuum (~1 h), the crude product mixture was dissolved in a minimal amount of CH2Cl2, a strong stir bar was added and under continuous stirring an excess of Et2O (~1 L) was added to precipitate out the product (if necessary cooling to −20 °C using an NaCl/ice bath helps the precipitation process). Stirring was continued for 1 h at −20 °C and the formed white precipitate was filtered to yield 10 as a white crystalline solid. Special care needs to be taken during the filtration step: water has to be excluded and a glass frit should be used. A regular empty glass column (4.5 × 30 cm) with frit was used for this purpose. The cooled suspension was poured into the column and a positive pressure of dried argon was applied immediately. The filtrate was washed twice with cold Et2O and dried under a stream of argon. To finally dry the product, the stopcock of the column was closed and high vacuum was applied. The product 10 was isolated as a white crystalline solid.
Synthesis of PQS-1 (1)
An oven dried 50 mL round bottom flask (A) containing a magnetic stir bar was charged with MeOPEG sebacic acid 10 (3.08 g, 1.41 mmol), a reflux condenser was attached and the system purged with argon. Thionyl chloride (5 mL) was added through the reflux condenser and heated to reflux for 2 while the evolving gas was bubbled through water and aq. NaOH. The reaction mixture was cooled to room temperature under argon and excess thionyl chloride was taken off using an aspirator. Further drying on high vacuum over night gave a pale orange solid that was dissolved in dry THF (10 mL, distilled from Na/benzophenone) and used in the next step without further purification. In a glove box an oven dried 250 mL round bottom flask (B) was loaded with a strong magnetic stir bar, ubiquinol (4.89 g, 5.65 mmol) and sodium hydride (170 mg, 4.24 mmol, 60% w/w in mineral oil). Outside the glove box, under an atmosphere of argon, dry THF (10 mL, distilled from Na/benzophenone) was added and the suspension was stirred at room temperature for 1 h and cooled to 0 °C. The solution of MeOPEG sebacic chloride 10 in THF (flask A) was added via canulla dropwise to flask B at 0 °C. Flask A was rinsed with additional dry THF (5–10 mL) and transferred to flask B. Under continuous stirring, the orange suspension was slowly warmed to room temperature overnight. The reaction was quenched by the dropwise addition of water and subsequent addition of 1N aq. HCl until pH<7. The mixture was extracted using CH2Cl2 (1 × 100 mL, 2 × 50 mL), the combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated to yield a pale orange oil which can be purified by two different procedures.
Purification A
Short path column. An empty 240g single step column (Thomson Instrument Company) was slurry packed with ~240g of silica gel in Et2O. The pale orange oil was evenly loaded onto the column using a minimal amount of CH2Cl2 and the column was carefully closed using a head space frit. Using a Biotage SP4 system the column was flashed with Et2O (500 mL, flow rate 45 mL/min), CH2Cl2 (500 mL, flow rate 40 mL/min) and eluted with 10% to 15% MeOH/CH2Cl2 over 2 L (flow rate 30 mL/min). Fractions containing PEG-ylated material were combined, evaporated and dried on high vacuum over night to yield PQS-1 (1) as an amber solid (3.45 g, 1.12 mmol, 86% pure by NMR). Obtained spectral data matched previously reported compound.4a
Purification B
Precipitation in ether. The pale orange oil was dissolved in a minimal amount of CH2Cl2, a strong stir bar was added and under continuous stirring an excess of Et2O (~1 L) was added and if necessary cooled to −20 °C using an NaCl/ice bath. Stirring was continued for 1 h to yield a white suspension of PEG-ylated material in Et2O. Due to the fine particle size all attempts for filtration failed and centrifugation was used instead. Unfortunately the centrifuge used for this purpose could not be cooled during the centrifugation process and led to significant loss of material. Furthermore the centrifuge could only be operated with 6 × 10 mL and was therefore not suited for scale up purposes. An analytical sample (~50 mg) confirmed the purity of PQS.
Route B: Synthesis of mono-benzylated sebacic acid 12
A 100 mL round bottom flask was charged with sebacoyl chloride (10, 8.42 g, 35.2 mmol) and closed with a rubber septum. Et2O (25 mL) was added and the solution cooled to −78 °C. Benzyl alcohol (2.51 g, 23.2 mmol) and NEt3 (4.9 mL, 35.2 mmol) were added slowly, the cooling bath was removed and the reaction warmed to rt. After addition of 1 N HCl, the aqueous phase was extracted with Et2O (3 × 25 mL), the combined organic layers were washed with brine, dried and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel, eluting with hexanes to 1:1 EtOAc/hexanes gradient to afford mono benzylated sebacic acid 12 (3.90 g, 57%) as a white solid; mp 52–53 °C (recrystalized from hexanes); IR (thin film): 2934, 2914, 2849, 1737, 1699, 1463, 1412, 1299, 1225, 1194, 1172, 940, 906, 745 cm−1; 1H NMR (500 MHz, CDCl3): δ7.39-7.31 (m, 5H), 5.12 (s, 2H), 2.36 (t, J = 7.0 Hz, 2H), 2.35 (t, J = 7.0 Hz, 2H), 1.67-1.60 (m, 4H), 1.34-1.27 (m, 8H); 13C NMR (125 MHz, CDCl3): δ 180.0, 173.9, 136.3, 128.7, 128.39, 128.38, 66.3, 34.5, 34.2, 29.22, 29.21, 29.15, 25.1, 24.8; MS (EI) m/z (%): 292 (M, 1), 274 (6), 264 (13), 185 (19), 107 (41), 98 (29), 91 (100); HRMS (EI) calcd for C17H24O4 [M]+ = 292.1675, found 292.1671.
Synthesis of benzyl protected PEG sebacic acid 13
Mono benzylated sebacic acid 12 (2.00 g, 6.85 mmol) was dissolved and refluxed in SOCl2 (4 mL). After 3 h, excess SOCl2 was removed through distillation under reduced pressure. The residue was taken up in dry CH2Cl2 (6 mL). A solution of poly(ethylene glycol) monomethyl ether-2000 (6.17 g, 3.08 mmol) and NEt3 (1.0 mL, 6.85 mmol) in dry CH2Cl2 (6 mL) was added dropwise and stirred over night at rt. After addition of 1N HCl, the aqueous phase was extracted with CH2Cl2 (3 × 20 mL), and the combined organic layers were dried and concentrated in vacuo. The residue was taken up in a minimal amount of CH2Cl2 and an excess of ice cold Et2O was added to form a white precipitate. After further cooling for 1 h, the precipitate was filtered to yield 13 (6.45 g, 92%) as a white solid. IR (neat): 3484, 2885, 2740, 2695, 1734, 1469, 1414, 1360, 1344, 1280, 1235, 1147, 1118, 1062, 947, 844 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.36-7.28 (m, 5H), 5.09 (s, 2H), 4.21-4.19 (m, 2H), 3.77-3.46 (m, PEG), 3.36 (s, 3H), 2.33 (t, J = 7.5 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.64-1.56 (m, 4H), 1.27 (br s, 8H); 13C NMR (125 MHz, CDCl3): δ 173.9, 173.7, 136.3, 128.7, 128.3, 72.1, 70.75, 70.70, 70.65, 69.3, 66.2, 63.5, 59.2, 34.4, 34.3, 29.2, 25.03, 24.97; MS (ESI): m/2z ~ 1167 (M + 2Na)+2.
Synthesis of mono-PEGylated sebacic acid 10
A round bottom flask was charged with 13 (6.41 g, 2.82 mmol) and Pd/C (0.60 g, 0.28 mmol, 5% Pd/C, Fluka) and closed with a rubber septum. MeOH (30 mL) was added and an atmosphere of H2 was provided (balloon). After stirring overnight, the reaction was filtered through Celite, washed with MeOH, and concentrated in vacuo. The residue was taken up in a minimal amount of CH2Cl2 and an excess of ice cold Et2O was added to form a white precipitate. After further cooling for 1 h, the precipitate was filtered to afford 10 (6.05 g, 98%) as a white solid. IR (neat): 3527, 2887, 1735, 1639, 1467, 1346, 1281, 1243, 1148, 948, 843 cm−1; 1H NMR (500 MHz, CDCl3): δ 4.24-4.21 (m, 2H), 3.79-3.48 (m, PEG), 3.38 (s, 3H), 2.33 (t, J = 7.5 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.64-1.58 (m, 4H), 1.31 (br s, 8H); 13C NMR (125 MHz, CDCl3): δ175.8, 173.5, 71.7-69.0 (m, PEG), 63.1, 58.8, 33.9, 33.7, 28.87, 28.84, 28.79, 24.6; MS (ESI): m/2z ~ 1122 (M + 2Na)+2.
Route C: Synthesis of mono-PEGylated succinic acid 15
To a solution of poly(ethylene glycol) monomethyl ether-2000 (14, 15.00 g, 7.50 mmol) and succinic anhydride (1.50 g, 15.00 mmol) in toluene (7.5 mL), Et3N (0.53 mL, 3.75 mmol) was added at rt with stirring, and the stirring was continued at 60 °C for 8 h. Water was added to the reaction mixture and extracted with CH2Cl2. The combined organic layers were washed with 1N HCl (3 × 50 mL), brine (2 × 30 mL), dried over anhydrous Na2SO4 and concentrated in vacuo to afford the poly(ethylene glycol) monomethyl ether-2000 succinate 15 (15.6 g, 99%) as a white solid. IR (thin-film): 3512, 2874, 1734, 1647, 1468, 1349, 1284, 1250, 1109, 949, 844 cm−1; 1H NMR (400 MHz, CDCl3): δ 4.28-4.25 (m, 2H), 3.83-3.46 (m, PEG), 3.38 (s, 3H), 2.69-2.61 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 173.1, 171.7, 71.4-68.5 (m, PEG), 63.2, 58.5, 28.6, 28.2; MS (ESI): m/4z ~ 551 (M + 4Na)+4.
Synthesis of activated PEGylated succinic acid 16
Poly(ethylene glycol) monomethyl ether-2000 succinate 15 (2.10 g, 1.00 mmol) was dissolved in CH2Cl2 (10 mL) and cooled to 0 °C. N-hydroxysuccinimide (0.14 g, 1.20 mmol), and 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide (EDCI, 0.25 g, 1.30 mmol), were then directly added in succession to the mixture as solids. The resulting mixture was stirred at rt for 12 h. Water was added to the reaction mixture and extracted with CH2Cl2. The combined organic layers were washed with water, brine, dried, and concentrated in vacuo to afford 16 (2.17 g, 99%) as a white waxy solid. IR (thin-film): 2883, 1814, 1784, 1739, 1645, 1468, 1360, 1280, 1234, 1205, 1147, 1116, 1062, 947, 843 cm−1; 1H NMR (400 MHz, CDCl3): δ4.29-4.27 (m, 2H), 3.83-3.46 (m, PEG), 3.38 (s, 3H), 2.97 (t, J = 7.2 Hz, 2H), 2.84 (br s, 4H), 2.79 (t, J = 7.2 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 170.9, 168.9, 167.6, 71.8-68.8 (m, PEG), 64.1, 59.0, 28.6, 26.2, 25.5; MS (ESI): m/4z ~ 576 (M + 4Na)+4.
Synthesis of PQS-3 (3)
NaH (0.026 g, 0.65 mmol, 60% suspension in mineral oil) was added to a stirred solution of ubiquinol (0.52 g, 0.60 mmol) in THF (5.0 mL) at 0 °C. After addition, the reaction mixture was stirred at 22 °C for 1 h. A solution of 16 (1.10 g, 0.50 mmol) in THF (5.0 mL) was added to the mixture at 0 °C, and the stirring was continued for 30 min. The mixture was then stirred for another 8 h at rt. It was then cooled to 0 °C and saturated aqueous NH4Cl was added and then extracted with CH2Cl2. The combined organic layers were washed with water, brine, dried, and concentrated in vacuo affording a yellowish liquid, which was purified by flash column chromatography on silica gel eluting with a CH2Cl2 to 1:19 MeOH/CH2Cl2 gradient to afford 3 (0.95 g, 65%, mixture of two regioisomers) as a white waxy solid. IR (thin-film): 3518, 2885, 2740, 1761, 1738, 1663, 1467, 1360, 1343, 1280, 1242, 1147, 1114, 1062, 964, 843 cm−1; 1H NMR (400 MHz, CDCl3): δ5.78 (s, 0.3H), 5.74 (s, 0.7H), 5.12-5.06 (m, 9H), 4.98-4.93 (m, 1H), 4.27-4.24 (m, 2H), 3.89 (s, 3H), 3.78 (s, 3H), 3.70-3.44 (m, PEG), 3.37 (s, 3H), 3.31 (d, J = 6.4 Hz, 1.4H), 3.16 (d, J = 6.4 Hz, 0.6H), 2.94-2.89 (m, 2H), 2.80-2.75 (m, 2H), 2.11-1.96 (m, 39H), 1.74 (s, 2.1H), 1.72 (s, 0.9H), 1.66 (s, 3H), 1.58-1.56 (m, 27H); 13C NMR (100 MHz, CDCl3): δ 172.04, 171.96, 170.9, 170.7, 145.4, 145.0, 142.1, 141.9, 137.8, 137.6, 135.3, 135.1, 135.0, 134.9, 134.8, 131.1, 128.4, 124.9, 124.4, 124.2, 124.1, 124.0, 121.9, 121.6, 117.9, 71.9-69.0 (m, PEG), 63.9, 60.9, 60.8, 60.6, 60.5, 59.0, 39.7, 29.0, 28.8, 28.7, 26.7, 26.6, 26.0, 25.7, 25.3, 17.7, 16.3, 16.2, 16.0, 12.0, 11.3; MS (ESI): m/4z ~ 763 (M + 4Na)+4.
Synthesis of carbene precursor 18
3 (0.50 g, 0.17 mmol) was dissolved in CH2Cl2 (2.2 mL) and cooled to 0 °C. 1-(p-Isopropoxy-m-vinylphenyl) propionic acid (17)11 (0.05 g, 0.22 mmol), 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide (EDCI) (0.04 g, 0.26 mmol), and DMAP (0.008 g, 0.07 mmol) were then directly added in succession to the mixture as solids. Et3N (0.04 mL, 0.30 mmol) was added through a syringe. The resulting mixture was stirred at 22 °C for 20 h. Water was added to the reaction mixture and extracted with CH2Cl2. The combined organic layers were washed with saturated NaHCO3, water, brine, dried and concentrated in vacuo affording a colorless liquid, which was purified by flash column chromatography on silica gel, eluting with Et2O, followed by CH2Cl2 to 1:12 MeOH/CH2Cl2 gradient afforded the compound 18 (0.45 g, 85%, mixture of two regioisomers) as a white foam. IR (thin-film): 2885, 1765, 1737, 1467, 1360, 1344, 1280, 1242, 1147, 1113, 1061, 963, 843 cm−1; 1H NMR (400 MHz, CDCl3): δ7.39-7.37 (m, 1H), 7.12-7.08 (m, 1H), 7.03 (dd, J = 18.0, 11.2 Hz, 1H), 6.84-6.81 (m, 1H), 5.74 (dt, J = 18.0, 1.6 Hz, 1H), 5.23 (d, J = 11.2 Hz, 1H), 5.13-5.06 (m, 9H), 4.97-4.94 (m, 1H), 4.50 (sep, J = 6.4 Hz, 1H), 4.28-4.25 (m, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.72-3.45 (m, PEG), 3.38 (s, 3H), 3.19-3.14 (m, 2H), 3.06-3.00 (m, 2H), 2.97-2.87 (m, 4H), 2.82-2.76 (m, 2H), 2.10-1.94 (m, 39H), 1.72-1.58 (m, 33H), 1.34 (d, J = 6.4 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 171.0, 170.9, 169.9, 169.7, 169.4, 169.3, 152.9, 142.7, 142.5, 139.9, 139.8, 139.6, 139.5, 134.6, 134.0, 133.8, 131.4, 131.1, 130.1, 127.8, 127.4, 127.0, 125.6, 125.57, 124.0, 123.9, 123.6, 123.5, 123.2, 120.7, 113.8, 113.7, 113.1, 71.2, 70.9-69.3 (m, PEG), 68.2, 63.1, 59.8, 59.6, 58.1, 39.0, 38.9, 34.9, 29.4, 28.2, 28.1, 26.1, 25.9, 25.5, 25.0, 21.5, 17.0, 15.6, 15.4, 11.4, 11.3; MS (ESI): m/4z ~ 839 (M + 4Na)+4.
Synthesis of PQS-3-GH1 Catalyst (6)
18 (0.44 g, 0.137 mmol) was weighed into a 25 mL round-bottom flask and dissolved in 6.5 mL of CH2Cl2. (PCy3)2Cl2Ru=CHPh (0.14 g, 0.17 mmol) and CuCl (0.018 g, 0.18 mmol) were added directly to this solution as solids. The mixture was stirred for a period of 4 h at 22 °C, during which time the original purple solution turned dark brown. The following workup procedures were conducted in air with reagent grade solvents. The mixture was concentrated at reduced pressure and passed through a short column of silica gel eluting with CH2Cl2 followed by Et2O. Finally, the column was flushed with 8% MeOH/CH2Cl2, at which point the product elutes (brown band). Solvent removal afforded the catalyst 6 (0.49 g, 98%, mixture of two regioisomers) as dark brown foam. IR (thin-film): 2883, 1764, 1737, 1644, 1468, 1449, 1359, 1344, 1280, 1234, 1146, 1113, 1061, 946, 843 cm−1; 1H NMR (400 MHz, CDCl3): δ17.40 (d, JPH = 4.4 Hz, 1H), 7.60-7.58 (m, 1H), 7.56-7.52 (m, 1H), 7.02-7.00 (m, 1H), 5.25 (sep, J = 6.4 Hz, 1H), 5.13-5.10 (m, 9H), 5.00-4.95 (m, 1H), 4.29-4.25 (m, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 3.70-3.47 (m, PEG), 3.38 (s, 3H), 3.22-3.16 (m, 2H), 2.97-2.87 (m, 4H), 2.82-2.76 (m, 2H), 2.36-2.28 (m, 2H), 2.10-1.98 (m, 39H), 1.81-1.58 (m, 66H), 1.42-1.24 (m, 6H); 13C NMR (200 MHz, CDCl3): δ 279.1, 171.95, 171.86, 170.8, 170.7, 170.3, 170.2, 151.5, 144.0, 143.4, 143.3, 143.2, 140.6, 140.4, 140.3, 135.8, 135.1, 134.8, 134.43, 134.39, 131.1, 129.45, 129.37, 128.4, 128.3, 125.0, 124.9, 124.4, 124.2, 124.0, 123.9, 122.5, 121.1, 116.0, 113.3, 75.5, 71.9, 70.5-69.0 (m, PEG), 63.90, 63.87, 60.6, 59.0, 39.7, 39.6, 35.9, 35.8, 35.7, 35.6, 31.7, 30.1, 29.7, 29.0, 28.8, 28.7, 27.74, 27.7, 26.9, 26.7, 26.67, 26.64, 26.3, 26.24, 26.16, 26.1, 25.7, 22.1, 22.0, 17.7, 16.34, 16.3, 16.0, 12.1; MS (ESI): m/4z ~ 927 (M + 4Na)+4.
N-Benzoyl-3-pyrroline (20)
Diene 19 (20 mg, 0.10 mmol) and catalyst 6 (7.5 mg, 0.002 mmol) were both added into a Teflon-coated-stir-bar-containing Biotage 2–5 mL microwave reactor vial at room temperature, and sealed with a septum. H2O (1.0 mL) was added via syringe, and the resulting solution was allowed to stir at room temperature for 4 h. The homogeneous reaction mixture was then diluted with EtOAc (2 mL), filtered through a bed of silica gel layered over Celite, and the bed further washed (2 × 4 mL) with EtOAc to collect all of the cyclized material. The volatiles were removed in vacuo to afford the crude product, which was subsequently purified by flash chromatography using silica gel (40% EtOAc/hexanes) to afford the title compound 20 as a colorless liquid (17 mg, 96%). The 1H NMR spectral data obtained was in accord with data previously reported for this compound.4a
Supplementary Material
Scheme 1a.

a Reagents and conditions: (a) Ac2 O, reflux, 76%. (b) HO-PEG-Me, DMAP, pyridine, 100 °C, 99%. (c) SOCl2, reflux, then re-dissolved in THF, ubiquinol, NaH, THF, rt, 80%. (d) EDCI, DMAP, ubiquinol, NEt3, CH2Cl2, rt, 31%. (e) BnOH, NEt3, Et2O, −78 °C to rt, then 1 N HCl, 57%. (f) SOCl2, reflux, then HO-PEG-Me, NEt3, CH2Cl2, rt, 92%. (g) Pd/C, H2, MeOH, rt, 98%.
Acknowledgments
Financial support provided by the NIH (GM 86485) is warmly acknowledged.
Footnotes
Supporting Information. A detailed comparison of Route A vs. B, and spectral data, including copies of 1H and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For biphasic catalysis, see: Cornils B, Herrmann WA. Aqueous Phase Organometallic Catalysis - Concepts and Applications. Wiley-VCH; Weinheim, Germany: 1998.
- 2.For industrial importance of biphasic catalysis, see: Cornils B, Herrmann WA, Eckl RW. Industrial Aspects of Aqueous Catalysis. J Mol Catal A. 1997;116:27–33.Cornils B. Bulk and Fine Chemicals via Aqueous Biphasic Catalysis. J Mol Catal A. 1999;143:1–10.Horváth IT, Joo F, editors. Aqueous Organometallic Chemistry and Catalysis. Kluwer; Dordrecht: 1995. Cornils B. Industrial Aqueous Biphasic Catalysis: Status and Directions. Org Proc Res Dev. 1998;2:121–127.
- 3.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2008;41:59–72.Lipshutz BH, Abela AR, Boskovic ZV, Nishikata T, Duplais C, Krasovskiy A. Top Catal. 2010;53:985–990.Lipshutz BH, Ghorai S, Abela AR, Moser R, Nishikata T, Duplais C, Krasovskiy A, Gaston RD, Gadwood R. J Org Chem. 2011;76:4379–4391. doi: 10.1021/jo101974u.Dwars T, Paetzold E, Oehme G. Angew Chem, Int Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. and references therein.
- 4.(a) Lipshutz BH, Ghorai S. Org Lett. 2009;11:705–708. doi: 10.1021/ol8027829. [DOI] [PubMed] [Google Scholar]; (b) Lipshutz BH, Ghorai S. Tetrahedron. 2010;66:1057–1063. [Google Scholar]
- 5.Lipshutz BH, Ghorai S. Org Lett. 2012;14:422. doi: 10.1021/ol203242r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For a similar procedure, see: Rath P, Gomez-Orellana MI, Vuocolo EA. Aryl Ketone Compounds and Compositions for Delivering Active Agents. U.S. Patent WO/2005/117854. 2005 Dec 15;
- 7.For a detailed comparison of the quality of PQS-1 obtained through route A or B, see SI.
- 8.PQS is usually obtained as a mixture of regioisomers; PQS-1 (1 - Route A) 2:1; PQS-1 (1 - Route B) 2:1; PQS-3 (3 - Route C) 3:1.
- 9.Obtained through Zn/HOAc reduction of coenzyme Q10; for procedure, see: Morgan AC, Graves SS, Woodhouse CS, Sikorska M, Walker R, Wilbur DS, Borowy-Borowski H. Water Soluble Ubiquinone Compositions, Prodrugs, and Methods Relating Thereto. CAN 125:123707 PCT. 1996
- 10.Reactions were performed between 0.25 mmol and 2.2 mmol scales, with consistently good overall yields above 60% in all cases.
- 11.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.