

Abstract
Neuromuscular junction (NMJ) formation requires proper interaction between motoneurons and muscle cells. β-Catenin is required in muscle cells for NMJ formation. To understand underlying mechanisms, we investigated the effect of β-catenin gain of function (GOF) on NMJ development. In HSA-β-catflox(ex3)/+ mice, which express stable β-catenin specifically in muscles, motor nerve terminals became extensively defasciculated and arborized. Ectopic muscles were observed in the diaphragm and were innervated by ectopic phrenic nerve branches. Moreover, extensive outgrowth and branching of spinal axons were evident in the GOF mice. These results indicate that increased β-catenin in muscles alters presynaptic differentiation. Postsynaptically, AChR clusters in HSA-β-catflox(ex3)/+ diaphragms were distributed in a wider region, suggesting that muscle β-catenin GOF disrupted the signal that restricts AChR clustering to the middle region of muscle fibers. Expression of stable β-catenin in motoneurons, however, had no effect on NMJ formation. These observations provide additional genetic evidence that pre- and postsynaptic development of the NMJ requires an intricate balance of β-catenin activity in muscles.
Keywords: Neuromuscular junction, β-Catenin, AChR, Axon outgrowth, Retrograde signals, Mouse
INTRODUCTION
The neuromuscular junction (NMJ), a synapse between motoneurons and muscle fibers, is crucial for control of muscle contraction. This classic chemical synapse has served as an informative model of synapse function and synaptogenesis (Wu et al., 2010). NMJ formation is regulated by interactions between motoneurons and muscles (Sanes and Lichtman, 2001; Wu et al., 2010). Motoneurons release agrin, which binds directly to LRP4 and thus activates MuSK, both of which are required for NMJ formation (McMahan, 1990; DeChiara et al., 1996; Gautam et al., 1996; Glass et al., 1996; Weatherbee et al., 2006; Kim, N. et al., 2008; Zhang et al., 2008; Zong et al., 2012). By contrast, activation of muscles by acetylcholine (ACh) from motoneurons suppresses the expression of acetylcholine receptors (AChRs) and dissembles AChR clusters (Schaeffer et al., 2001; Misgeld et al., 2002; Brandon et al., 2003; Chen et al., 2007).
Less is known about retrograde signals from muscle fibers for presynaptic differentiation. Muscles are known to produce necessary factors for motoneurons (Hamburger, 1934). Several factors have been implicated in motoneuron survival or synapse elimination including glial cell line-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF) (Oppenheim et al., 1995; Nguyen et al., 1998; Keller-Peck et al., 2001; Lu and Je, 2003). Transforming growth factor β (TGFβ), fibroblast growth factor (FGF), laminin and collagen appear to play a role in orchestrating presynaptic development at the NMJ (McCabe et al., 2003; Rawson et al., 2003; Nishimune et al., 2004; Fox et al., 2007; Feng and Ko, 2008; Nishimune et al., 2008). Previously, we demonstrated that phrenic nerve branches are mislocated and synaptic vesicle release is compromised in mice lacking β-catenin in muscles (Li et al., 2008), suggesting a necessary role of muscle β-catenin for presynaptic differentiation or function.
In this study, we investigated the GOF effects of β-catenin on NMJ formation. When a conditional GOF mutation of β-catenin was introduced by HSA Cre in muscle cells, both presynaptic and postsynaptic deficits were observed. Intriguingly, some phenotypes were similar whereas others were converse, compared with those in muscle-specific β-catenin loss-of-function (LOF) mutants. However, when the GOF mutation of β-catenin was introduced by HB9 Cre in motoneurons, it had no apparent effect on NMJ development. We studied the effects of muscle β-catenin by multiple approaches. Results provide additional genetic evidence that an intricate balance of muscle β-catenin activity is crucial for both pre- and postsynaptic differentiation during NMJ development.
MATERIALS AND METHODS
Generation and genotyping of mouse lines
β-Cateninflox(ex3) loxP P85 (PGK reverse) [β-catflox(ex3)] mice harbor two loxP sites flanking exon 3 of the β-catenin gene (Ctnnb1 – Mouse Genome Informatics) (Harada et al., 1999). This exon encodes a domain that contains crucial Ser/Thr residues phosphorylation of which promotes the degradation of β-catenin (Fig. 1A). β-Catflox(ex3) mice were crossed with human skeletal α-actin (HSA)-Cre and HB9-Cre mice, which express Cre specifically in skeletal muscles and motoneurons, respectively (Miniou et al., 1999; Yang et al., 2001; Li et al., 2008), to generate HSA-β-catflox(ex3)/+ and HB9-β-catflox(ex3)/+. The Wnt signaling TOP-EGFP reporter mice were purchased from RIKEN BioResource Center (RBRC02229) (Moriyama et al., 2007) and crossed with β-catflox(ex3) and HSA-Cre mice to produce TOP-EGFP;β-catflox(ex3)/+ and TOP-EGFP;HSA-β-catflox(ex3)/+ mice. Genotyping was performed as described in the legend to supplementary material Fig. S1. Animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Georgia Health Sciences University.
Fig. 1.

Increased stability and activity of β-catenin in skeletal muscles in HSA-β-catflox(ex3)/+ mice. (A) Schematic of the wild-type and targeted β-catenin allele. E, exon; red triangle, loxP sequence; green arrows, primers for genotyping. (B) Specific expression of exon 3-deleted β-catenin (arrow) in skeletal muscles of HSA-β-catflox(ex3)/+ mice, but not in other tissues. (C) Increased stability of exon 3-deleted β-catenin compared with wild type. HEK293 cells were transfected with HA-tagged wild-type (asterisk) and mutant (arrow) β-catenin, and treated with 20 μg/ml CHX for various times. β-Actin was used as a loading control. (D) Quantification of data in C (mean ± SEM, n=3). (E) Schematic of the left hemi-diaphragm. Red dots, AChR clusters; rectangle, area shown in F; D, dorsal; V, ventral; L, lateral; M, medial. (F) Expression of TOP-EGFP in diaphragm muscles by β-catenin GOF mutant. Muscles of control (TOP-EGFP;β-catflox(ex3)/+) and TOP-EGFP;HSA-β-catflox(ex3)/+ mice were stained with Alexa Fluor 594-conjugated α-BTX (red). Image was taken by confocal fluorescence microscope. Area shown is indicated by rectangle in E. (G) Neonatal mice of indicated genotypes.
Reagents, constructs and antibodies
Chemicals were purchased from Sigma-Aldrich unless otherwise indicated. Alexa Fluor 594-conjugated α-bungarotoxin (α-BTX) was purchased from Invitrogen (B-13423; 1:3000 for staining). HA-tagged mouse Ctnnb1 (pKH3-β-catenin) was constructed as described previously (Kim, C. H. et al., 2008). Ctnnb1 exon3 deletion mutant, β-catΔex3, was generated by using the Quick Change Site-Directed Mutagenesis Kit (Stratagene). The authenticity of all constructs was verified by DNA sequencing. Antibodies used were as follows [antigen (company, catalog number; dilution)]: neurofilament (NF) (Millipore, AB1991; 1:1000 for staining); synaptophysin (Dako, A0010; 1:2000 for staining); SV2 (Developmental Studies Hybridoma Bank, SV2; 1:500 for staining); β-catenin (BD Biosciences, 610154; 1:2000 for western); α-Tubulin (Santa Cruz, sc-23948; 1:3000 for western); β-actin (Novus, NB600-501; 1:3000 for western). Rabbit anti-rapsyn (2741) and anti-MuSK antibodies were described previously (1:1000 for western) (Luo et al., 2002). Antibodies raised against AChRα-subunit (mAb35) and β-subunit (mAb124) were gifts from Dr Richard Rotundo (Miller School of Medicine, Miami, FL, USA) and Dr Jon Lindstrom (Perelman School of Medicine, Philadelphia, PA, USA), respectively (1:1000 for western). Rabbit anti-HB9 (C-terminal 307-403) antibody was a gift from Dr Samuel Pfaff (The Salk Institute, La Jolla, CA, USA) (1:4000 for staining) (Thaler et al., 1999). Alexa Fluor 488-conjugated goat anti-rabbit IgG was purchased from Invitrogen (A-11034, 1:1000 for staining); horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (32260) and goat anti-mouse IgG (32230) antibodies were purchased from Pierce (Thermo Scientific) (1:3000 for western).
Western blotting
Western blotting was performed as described previously (Luo et al., 2002; Zhu et al., 2006).
Analysis of NMJ morphology and function
Whole-mount staining of muscles for AChR, nerve terminals and acetylcholinesterase (AChE); muscle section staining; analysis of NMJs in individual muscle fibers; and electrophysiological recordings were performed as described previously (Li et al., 2008).
Analysis of motoneuron survival
To study the effect of muscle β-catenin on motoneuron survival, spinal cords between C3 and C5 were dissected from postnatal day (P)0 mice, fixed in 4% paraformaldehyde in PBS (pH 7.3) overnight and immersed in 0.1 M phosphate buffer (pH 7.3) containing 30% sucrose for 24 hours. Tissues were embedded in OCT compound (TissueTek), and sectioned on a cryostat. Motoneurons were identified by staining spinal cord sections (14 μm) with anti-HB9 antibody as described previously (Arber et al., 1999). For quantification, HB9-positive motoneurons from hemiventral columns in every fourth section were counted by individuals blind to the genotypes.
Analysis of axons in brachial plexus
To examine axon projection from spinal cords, whole-mount staining was performed on embryos as previously described (Qiu et al., 1997). Briefly, embryos were fixed in methanol:DMSO (4:1) overnight at 4°C, bleached in methanol:DMSO:30% H2O2 (4:1:1) for 4-5 hours at 25°C, and rehydrated by incubation in 50% methanol, 15% methanol and PBS (30 minutes each). After being incubated in PBSMT (2% instant skim milk powder, 0.1% Triton X-100 in PBS) for 2 hours at 25°C, they were incubated with anti-NF antibody (1:1000) in PBSMT at 4°C overnight. Embryos were washed twice (1 hour each) in PBSMT at 4°C and three times at 25°C then incubated at 4°C with HRP-conjugated goat anti-mouse IgG (Thermo Fisher, 32230; 1:1000) in PBSMT. After washing, embryos were incubated at 25°C in PBT (0.2% BSA, 0.1% Triton X-100 in PBS) for 20 minutes and in PBT containing 0.3 mg/ml diaminobenzidine tetrahydrochloride (DAB) for 30-60 minutes. Nerve axons were visualized by addition of 0.0003% H2O2. Embryos were then rinsed in PBT to stop the reaction, dehydrated through a methanol series (30%, 50%, 80% and 100% for 30-60 minutes each) and cleared in benzyl alcohol:benzyl benzoate (1:2). They were photographed using a Zeiss stemi 200 dissecting microscope (Carl Zeiss).
Electron microscopy
Muscles were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer for 1 hour at 25°C and further fixed in sodium cacodylate-buffered (pH 7.3) 1% osmium tetroxide for 1 hour at 25°C. After washing three times with phosphate buffer (10 minutes each) tissues were dehydrated through a series of ethanol: 30%, 50%, 70%, 80%, 90%, 100%; the 100% ethanol step was followed with three changes of 100% propylene oxide and embedded in plastic resin (EM-bed 812, EM Sciences). Serial thick sections (1-2 μm) of tissue blocks were stained with 1% Toluidine Blue for light microscopy, from which sections with phrenic nerves were identified and cut into ultra-thin sections. They were mounted on 200-mesh unsupported copper grids and stained with uranyl acetate (3% in 50% methanol) and lead citrate (2.6% lead nitrate and 3.5% sodium citrate, pH 12.0). Electron micrographs were taken using a JEOL 100CXII operated at 80 KeV.
Electrophysiology
Neonatal mice diaphragm with ribs and intact phrenic nerves were dissected in oxygenated (95% O2/5% CO2) Ringer’s solution (136.8 mM NaCl, 5 mM KCl, 12 mM NaHCO3, 1 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2 and 11 mM d-glucose, pH 7.3), and pinned on Sylgard gel in a dish perfused with oxygenated Ringer’s solution (Li et al., 2008). Microelectrodes, 20-50 MΩ when filled with 3 M KCl, were pierced into the center of muscle fibers. To elicit endplate potential (EPP), phrenic nerves were sucked into a suction electrode and stimulated (0.1 msecond, 2-5 V) with the resting potential between –45 and –55 mV (Liu et al., 2008). Data were collected with Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), digitized with Digidata 1322A (Molecular Devices) and analyzed using pClamp 9.2 (Molecular Devices).
Quantitative RT-PCR analysis
Quantitative RT-PCR analysis was performed as described previously (Liu et al., 2011). Primers used for analysis are listed in supplementary material Table S1.
Statistical analysis
Data are presented as mean ± s.e.m. and analyzed using Student’s t-test or one-way ANOVA analysis, as appropriate. In the hypertonic sucrose-evoked mEPP experiment, the variability of frequency was analyzed using repeated measures ANOVA analysis. Graphs were generated by GraphPad Prism 5.0 software. P<0.05 was considered to be significant.
RESULTS
Expression of stable β-catenin in skeletal muscles
To study the effect of β-catenin GOF in skeletal muscles on NMJ formation, we introduced a conditional mutation of stable β-catenin in muscle cells by crossing HSA-Cre mice with β-catflox(ex3) mice, in which exon 3 was floxed (Harada et al., 1999) (Fig. 1A). Resulting HSA-β-catflox(ex3)/+ mice express a β-catenin mutant that lacks the critical phosphorylation residues encoded by exon 3 in skeletal muscles, but not in other tissues, including spinal cord (Fig. 1B). The mutant β-catenin is believed to be resistant to degradation mediated by GSK3β-dependent phosphorylation and is able to activate constitutively Wnt canonical signaling (Rubinfeld et al., 1997; Iwao et al., 1998). In agreement, we found that exon3-deleted β-catenin was more stable compared with wild-type β-catenin in cells treated with cycloheximide (CHX) (Fig. 1C,D). To demonstrate that β-catenin-dependent transcription activity was increased in HSA-β-catflox(ex3)/+ muscles, we crossed them with TOP-EGFP mice, a reporter line of Wnt canonical signaling (Moriyama et al., 2007). As shown in Fig. 1F, EGFP was increased in muscles of TOP-EGFP;HSA-β-catflox(ex3)/+ mice, compared with control mice, indicating the GOF effect of β-catenin in HSA-β-catflox(ex3)/+ mice. Unless otherwise indicated, β-catflox(ex3)/+ littermates were used as control in the present study. They were viable and fertile and showed no difference in life span, general behavior, or NMJ morphology and function, compared with wild-type controls (data not shown). HSA-β-catflox(ex3)/+ mice were also viable at birth, but they were smaller in body size compared with control littermates (Fig. 1G), and appeared to have subcutaneous hemorrhage at the posterior fontanel site of the skull (Fig. 1G).
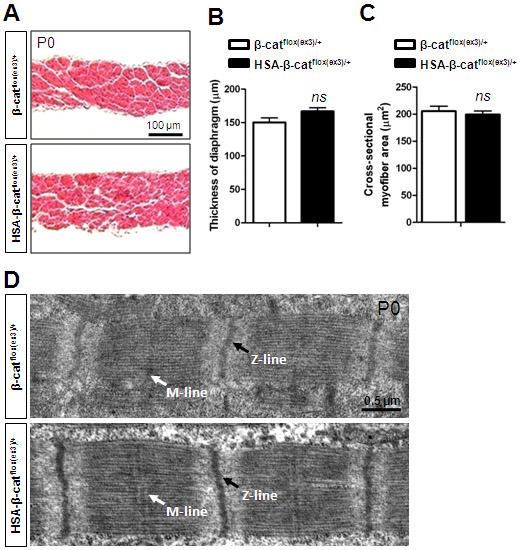
β-Catenin is localized on the muscle membrane without specific enrichment at the NMJ in both control and HSA-β-catflox(ex3)/+ mice, except at higher levels in the latter (supplementary material Fig. S1). β-Catenin GOF seemed to have no effect on diaphragm thickness and the cross-sectional area of muscle fibers (supplementary material Fig. S2A-C). Moreover, the nuclei of muscle fibers in HSA-β-catflox(ex3)/+ mice were located beneath the membrane, not in the central region, suggesting that muscle fibers were not regenerated (supplementary material Fig. S2A). Finally, electron microscopic studies indicated that muscle contractile units in HSA-β-catflox(ex3)/+ mice were similar, compared with controls (supplementary material Fig. S2D). These results suggest that muscle β-catenin GOF did not change the overall structure of muscle fibers. Nevertheless, to avoid potential long-term effect on muscle health, we focused on muscles of P0 neonatal mice.
Defasciculation and arborization of axons in HSA-β-catflox(ex3)/+ diaphragms
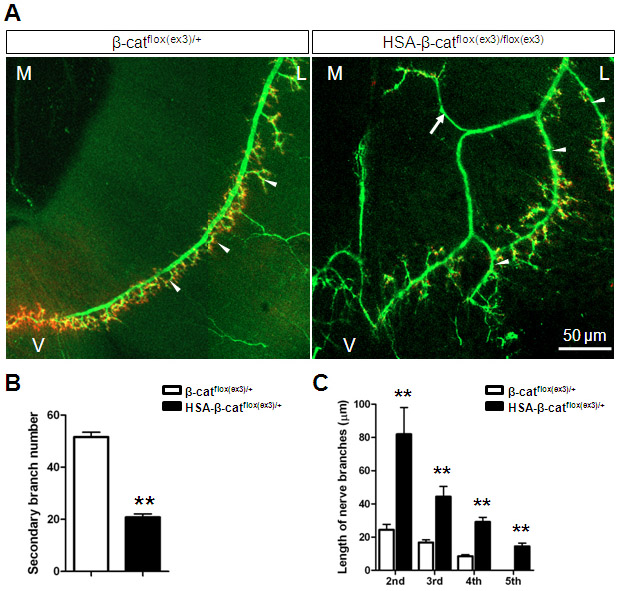
Whole-mount diaphragms were stained with a mixture of antibodies against neurofilament (NF) and synaptophysin to label both nerve branches and terminals (Li et al., 2008). In control diaphragms, primary phrenic branches traveled in the middle region of muscle fibers and sent out numerous, but short, secondary or intramuscular branches that form NMJs close the primary branches (Fig. 2A). In HSA-β-catflox(ex3)/+ diaphragms, however, the nerve branches were extensively defasciculated, which generated a net of axons covering a broader central region (Fig. 2A). The length of secondary branches was increased but their number was reduced in HSA-β-catflox(ex3)/+ mice (Fig. 2B,C). By contrast, tertiary and quaternary branches, which were few and short in control diaphragms, were apparent and long in HSA-β-catflox(ex3)/+ mice. The 5th branches, which did not exist in control diaphragms, were observable in HSA-β-catflox(ex3)/+ diaphragms (Fig. 2C). Similar deficits were observed in diaphragms of HSA-β-catflox(ex3)/flox(ex3) homozygous mice (supplementary material Fig. S3).
Fig. 2.

Aberrant innervation of motor axons in HSA-β-catflox(ex3)/+ mice. (A) P0 left hemi-diaphragms of indicated genotypes. Phrenic nerves and terminals were stained with anti-NF/synaptophysin antibodies, which were visualized with Alexa Fluor 488-conjugated goat anti-rabbit antibodies. (B) Decreased number of secondary/intramuscular nerve branches in HSA-β-catflox(ex3)/+ muscles (**P<0.01, n=7, t-test). (C) Increased length of secondary, tertiary, quaternary and 5th branches in HSA-β-catflox(ex3)/+ muscles (**P<0.01, n=6, one-way ANOVA). (D) E13 left hemi-diaphragms of indicated genotypes. (E) Increased secondary branch length in developing HSA-β-catflox(ex3)/+ embryos (*P<0.05, **P<0.01, n=5, one-way ANOVA). (F) Developmental changes of secondary branches in HSA-β-catflox(ex3)/+ embryos (**P<0.01, n=5, one-way ANOVA). (G) Increased endplate band width in E16.5 and E18.5 HSA-β-catflox(ex3)/+ embryos (**P<0.01, n=10, one-way ANOVA). In A and D, arrowheads indicate secondary nerve branches; arrow indicates ectopic axon. D, dorsal; V, ventral; L, lateral; M, medial. Error bars indicate s.e.m.
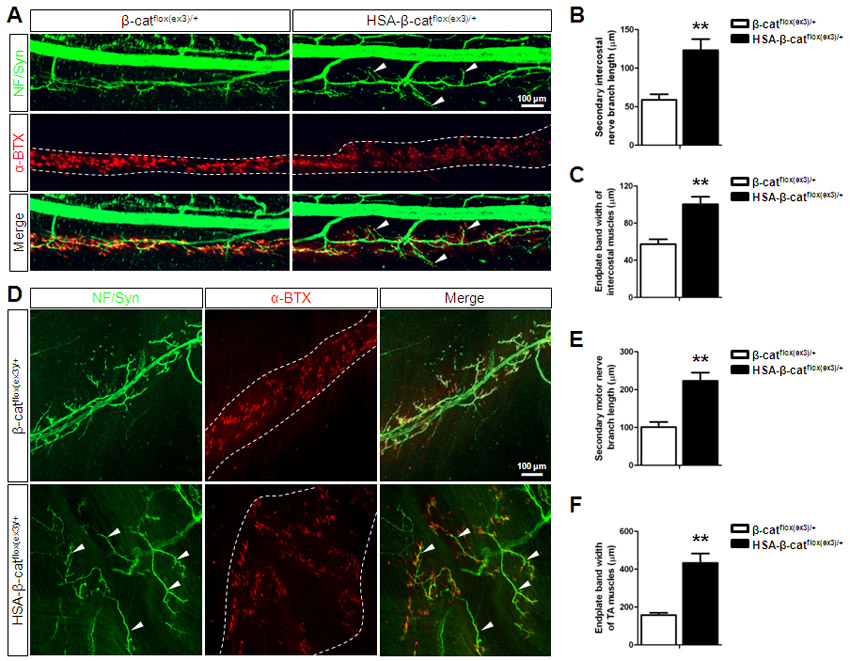
To determine when the arborization of phrenic nerve terminals occurred, we examined diaphragms at different embryonic ages. At embryonic day (E)13, the primary branches were located in the middle region of muscle fibers in HSA-β-catflox(ex3)/+ embryos, suggesting no problem in axon projection. Compared with controls, in which secondary branches were hardly detectable, branches were readily visible in HSA-β-catflox(ex3)/+ diaphragms (Fig. 2D, arrowheads). The length of secondary branches and the endplate band width continued to increase in both control and HSA-β-catflox(ex3)/+ mice during development, but the rate was significantly faster for the latter (Fig. 2E,G). The number of secondary branches increased more rapidly in control than in HSA-β-catflox(ex3)/+ embryos (Fig. 2F). These results demonstrate that increased β-catenin activity in muscle fibers leads to arborization or defasciculation of phrenic nerve terminals. Similar phenotypes, including wider central endplate band and axon arborization, were observed in intercostal muscles and tibialis anterior muscles in HSA-β-catflox(ex3)/+ mice (supplementary material Fig. S4).
Formation and innervation of ectopic muscles in HSA-β-catflox(ex3)/+ mice
Although the Wnt/β-catenin-dependent pathway has been implicated in muscle development (Borello et al., 1999; Parker et al., 2003; Perez-Ruiz et al., 2008; Gros et al., 2009), the in vivo effect of muscle β-catenin GOF remained unclear. Intriguingly, new muscles were found in the central tendoneous region of HSA-β-catflox(ex3)/+ diaphragms, a region that usually is clean and transparent in wild-type and control mice. Quantitatively, HSA-β-catflox(ex3)/+ mice formed at least one, and as many as three to four, ectopic muscles (Fig. 3D). These results indicate that β-catenin expression under the control of the HSA promoter promotes the formation of ectopic muscles in tendoneous regions.
Fig. 3.

Formation and innervation of ectopic muscles in HSA-β-catflox(ex3)/+ mice. (A) Schematic of left hemi-diaphragm with ectopic muscles. Square frame indicates parts of diaphragms analyzed in B-D. D, dorsal; V, ventral; M, medial; L, left. (B-D) Staining of diaphragms of different genotypes at indicated ages (NF/synaptophysin, green; AChR, red). White dashed lines indicate the edge of diaphragms. Red dashed lines encircle ectopic muscles. Arrowheads indicate extensive and longer secondary branches. Arrows indicate secondary nerve branches. (E) Enlarged image of an ectopic muscle. Arrow, ectopic axons; arrowheads, AChR clusters.
The ectopic muscles were innervated by ectopic branches of phrenic nerves (Fig. 2A, Fig. 3A). In developing diaphragms, phrenic nerves send branches towards the ribs and rarely (if any) towards the central cavity. However, axon branches were evident towards the central cavity in HSA-β-catflox(ex3)/+ diaphragms as early as E14.5, when ectopic muscle mass was barely visible in the central tendoneous region (Fig. 3B). By E16.5, the overshooting phrenic axons arrived at ectopic muscles in the central tendoneous region (Fig. 3C). At birth, the terminals of ectopic branches were fully arborized and formed NMJs (AChR clusters) on ectopic muscle fibers (Fig. 3D,E). These results demonstrate that ectopic muscles in the central tendoneous region were able to attract phrenic nerves to form ectopic branches for innervation, suggesting the existence of an axon attraction signal by muscle β-catenin.
Axon attraction activity from muscles expressing stable β-catenin
To determine whether muscle β-catenin alters axon extension, we examined the primary ventral phrenic branch of developing left hemi-diaphragms, a branch with better spatial resolution. As shown in Fig. 2D and Fig. 3B, it extended more towards the ventral region in E13 and E14.5 HSA-β-catflox(ex3)/+ mice compared with controls (Fig. 2D, Fig. 3B), suggesting that increased β-catenin activity in muscles produces a signal that promotes motor axon extension or outgrowth. To examine this hypothesis further, we looked at nerve projections from early embryonic spinal cords. In brachial plexus of control embryos with 45 somites, radial and axillary nerves (RN and AN, respectively), but not ulnar nerves (UN), were visible (Qiu et al., 1997; Arber et al., 1999; Vickerman et al., 2011) (Fig. 4A,B). By comparison, RN and AN were more prominent and UN were visible in HSA-β-catflox(ex3)/+ embryos. RN terminals appeared to defasciculate prematurely and extend further in the mutant (Fig. 4B). Immature branching and extension of RN axons were more obvious in embryos with 54 somites (Fig. 4C). Quantitatively, the number of tertiary branch points and lengths of RN branches were increased in embryos with 54 somites (Fig. 4D-F). These results indicate that β-catenin GOF in muscle fibers promotes the outgrowth of spinal nerve axons in HSA-β-catflox(ex3)/+ mice.
Fig. 4.

Augmented extension and branching of brachial plexuses in HSA-β-catflox(ex3)/+ embryos. (A) Schematic lateral view of mouse embryo. Roman numerals indicate cranial nerves. C, cervical; T, thoracic. (B) Embryos at 45-somite stage stained whole-mount with anti-NF antibody, which was visualized with diaminobenzidine (DAB). Arrows indicate brachial plexus. Areas in rectangles in top panels are enlarged in middle panels. Lower panels show camera lucida drawing of axons. an, axillary nerve; rn, radial nerve; un, ulnar nerve. (C) Embryos at 54-somite stage stained as described in B. Areas in rectangles in top panels are enlarged in middle panels. Lower panels show camera lucida drawing of axons. (D) Schematic of axon branches. Color matches data shown in E. (E) Increased numbers of rn tertiary branch points in HSA-β-catflox(ex3)/+ embryos shown in C. **P<0.01, n=3, one-way ANOVA. (F) Increased length of rn branches in HSA-β-catflox(ex3)/+ embryos shown in C. Shown are ratios of rn branch length over limb bud length. **P<0.01, n=3, one-way ANOVA. Error bars indicate s.e.m.
Motoneurons are known to be regulated by signals from muscle cells (Oppenheim, 1991). To determine whether muscle β-catenin GOF alters motoneurons, we looked at spinal cords at cervical levels (C3-C5), where phrenic nerves arise from. Spinal cord sections were stained for HB9, a motoneuron-specific homeodomain transcription factor (Saha et al., 1997; Arber et al., 1999; Thaler et al., 1999). As shown in supplementary material Fig. S5A, HB9-positive motoneurons were located in the hemi-ventral segment of the spinal cord (Arber et al., 1999) (supplementary material Fig. S5A). The numbers of motoneurons in this segment were similar between control and HSA-β-catflox(ex3)/+ mice (supplementary material Fig. S5B). HB9-positive motoneurons of phrenic nerves were primarily located in the lateral and medial portions of the anterior horn [termed the lateral motor column (LMC) and medial motor column (MMC), respectively] (supplementary material Fig. S5A) (Polleux et al., 2007). Muscle β-catenin GOF did not change the numbers of motoneurons in these two subpopulations (supplementary material Fig. S5C,D). These observations suggest that enhanced β-catenin activity in muscles alters presynaptic differentiation without apparent effect on motoneuron number and distribution.
Postjunctional deficits in HSA-β-catflox(ex3)/+ NMJs
To determine whether aberrant expression of stable β-catenin alters the postsynaptic differentiation, diaphragms were whole-mount stained with α-BTX. In advance of innervation, muscle fibers are ‘pre-patterned’ with aneural AChR clusters that are distributed in the central region and form a band perpendicular to muscle fibers of diaphragms (Lin et al., 2001; Yang et al., 2001). Muscle fiber pre-patterning in E13.5 HSA-β-catflox(ex3)/+ diaphragms was similar to that of littermate controls, suggesting that expression of stable β-catenin does not alter the formation and localization of aneural AChR clusters (data not shown). However, the endplate band width in HSA-β-catflox(ex3)/+ embryos became significantly larger than that in controls as early as E16.5 (Fig. 2G, Fig. 3C). In P0 control diaphragms, almost all AChR clusters were restricted to the central region, forming a narrow endplate band width. However, in HSA-β-catflox(ex3)/+ diaphragms, the band width is 2.6-fold larger (Fig. 5A,B). No change was observed in the areas or length of individual clusters (Fig. 5C,D). Moreover, the total number of AChR clusters was similar between HSA-β-catflox(ex3)/+ and control diaphragms (Fig. 5E), indicating expression of stable β-catenin had no effect on the number of NMJs, but altered their location. In addition, we also determined the number of AChR clusters per muscle fiber and found no difference between the two genotypes (Fig. 5F,G). AChE is accumulated in the synaptic cleft via a distinct mechanism (Arikawa-Hirasawa et al., 2002). Its distribution, assayed by in situ enzymatic activity, was also in a broader central region in HSA-β-catflox(ex3)/+ mice than in control littermates (Fig. 5H). Together, these observations indicate that expression of stable β-catenin in developing muscles alters the location where NMJs form, but does not change the size or the number of AChR clusters per muscle fiber.
Fig. 5.

Postsynaptic deficits of HSA-β-catflox(ex3)/+ NMJs. (A) P0, left hemi-diaphragms were stained for AChR. White dashed lines were drawn to include most AChR clusters. (B) Increased endplate band width in HSA-β-catflox(ex3)/+ diaphragms (**P<0.01, n=6, t-test). (C-F) No change in AChR cluster size (C), length (D) or density (E) in HSA-β-catflox(ex3)/+ diaphragms (P>0.05, n≥5, t-test). (F) Individual muscle fibers with AChR clusters (red). Nuclei were stained with DAPI (blue). (G) Quantitative analysis of data shown in F (P>0.05, n=22, t-test). (H) Scattered AChE clusters in HSA-β-catflox(ex3)/+ diaphragms (P0). V, ventral; L, lateral; M, medial. Error bars indicate s.e.m.
Impairment of synaptic transmission at HSA-β-catflox(ex3)/+ NMJs
To determine whether neurotransmission was altered at HSA-β-catflox(ex3)/+ NMJs, we measured miniature endplate potentials (mEPPs), events generated by spontaneous vesicle release. The amplitudes of mEPPs were similar in control and HSA-β-catflox(ex3)/+ mice (2.74±0.42 mV and 2.50±0.13 mV, respectively; P>0.05, n=6 mice) (Fig. 6A,C), suggesting that AChR density was not altered. This observation is in agreement with results of light microscopic characterization that AChR clusters were apparently normal (Fig. 5C-G) and biochemical studies that expression of postsynaptic proteins was normal in HSA-β-catflox(ex3)/+ muscles, including AChRα- and β-subunits, MuSK and rapsyn (Fig. 6F-J). By contrast, mEPP frequencies were markedly reduced in HSA-β-catflox(ex3)/+ NMJs (Fig. 6B), suggesting potential defects in spontaneous ACh release from presynaptic terminals. Presynaptic vesicle release deficiency might result from a decrease in ready release pool (RRP) or a change in vesicle fusion probability. To address these questions, we determined whether RRP was altered by recording mEPPs in response to hypertonic stimulation with 0.5 M sucrose, which causes rapid release of vesicles in the RRP (Stevens and Tsujimoto, 1995). As shown in Fig. 6D,E, there was no difference in the frequencies of sucrose-evoked mEPPs between the two genotypes (Fig. 6D,E), indicating that the size of RRP was not altered in HSA-β-catflox(ex3)/+ mice. Endplate potentials (EPPs) were similar between the genotypes (data not shown), suggesting normal vesicle release in response to action potentials.
Fig. 6.

Impaired synaptic transmission in HSA-β-catflox(ex3)/+ NMJs. (A) Representative superimposed mEPP sample traces. Traces were recorded from neonatal mice at 2 mM Ca2+ (1-second × 600 traces). (B) Reduced mEPP frequency in HSA-β-catflox(ex3)/+ NMJs (**P<0.01, n=6, t-test). (C) mEPP amplitude was not changed at mutant NMJs (P>0.05, n=6, t-test). (D) Representative mEPP traces in response to 0.5 M sucrose. Shown are traces 7 minutes after the addition of sucrose. Time scale: 1 second upper panel; 1 minute lower panel. (E) Quantitative analysis of hypertonic evoked mEPP frequency shown in D (P>0.05, n=5, repeat measure). (F-J) No change in synaptic proteins in HSA-β-catflox(ex3)/+ muscle lysates. Data are mean ± s.e.m. (P>0.05, n=3 each group, t-test).
Altered NMJ structures in muscle β-catenin LOF, but not GOF, mice
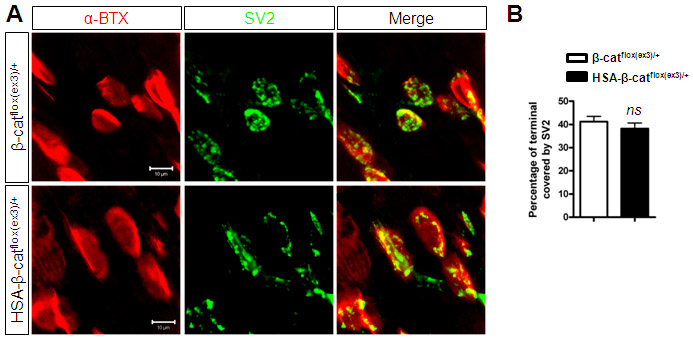
The reduction in mEPP frequency and apparently normal RRP suggest that the reduced ACh release might be due to functional, but not structural, deficits. To test this hypothesis, we determined expression of SV2, a presynaptic vesicle protein, in HSA-β-catflox(ex3)/+ NMJs. SV2 staining was in good registration with AChR clusters (supplementary material Fig. S6A). The areas of AChR clusters covered by SV2 were similar in control and HSA-β-catflox(ex3)/+ NMJs (supplementary material Fig. S6B). These results suggest that presynaptic specialization is normal in HSA-β-catflox(ex3)/+ NMJs. Next, we examined NMJ structures by electron microscopic analysis. As for NMJs in control littermates, axon terminals in HSA-β-catflox(ex3)/+ skeletal muscles were capped by processes of perisynaptic Schwann cells; they were filled with abundant synaptic vesicles (Fig. 7A). Quantitative studies revealed no difference between control and HSA-β-catflox(ex3)/+ NMJs in numbers of nerve terminals, numbers of active zones, vesicle densities, and synaptic vesicle diameters (Fig. 7B-E). These results demonstrate that the presynaptic structures of HSA-β-catflox(ex3)/+ NMJs were normal. Moreover, synaptic clefts had similar width between the two genotypes (Fig. 7F) and were both filled with synaptic basal lamina (Fig. 7A). On the postsynaptic side, muscle membrane folds were observable, and there was no difference in the average length of junctional folds between control and HSA-β-catflox(ex3)/+ (data not shown). Together, these results indicate that NMJ structures were largely normal in HSA-β-catflox(ex3)/+ mice, in support of the functional deficit hypothesis.
Fig. 7.

Reduced number of synaptic vesicles and active zones in muscle β-catenin LOF, but not GOF, NMJs. (A) Representative electron micrographic images of NMJs in β-catflox(ex3)/+ control (upper panel) and HSA-β-catflox(ex3)/+ (lower panel) mice. N, nerve terminal; M, muscle fiber; SC, Schwann cell; SVs, synaptic vesicles; SBL, synaptic basal lamina; JFs, junctional folds; Asterisks mark active zones. (B-F) Quantitative analysis showed no difference in nerve terminal numbers (B), active zone numbers per nerve terminal (C), synaptic vesicle density (D), synaptic vesicle diameter (E) or synaptic cleft width (F) (P>0.05, n=10, t-test). (G) Representative electron micrographic images of NMJs in β-catloxP/loxP control (upper panel) and HSA-β-cat–/– (lower panel) mice. (H-L) Quantitative data are shown for nerve terminal numbers (H), synaptic vesicle diameter (K), synaptic cleft width (L), active zone numbers per nerve terminal (I) and synaptic vesicle density (J) (**P<0.01, n=10, t-test). Error bars indicate s.e.m.
Interestingly, mEPP frequency was reduced in muscle-specific β-catenin mutant (HSA-β-cat–/–) mice (Li et al., 2008). Does β-catenin LOF regulate neurotransmission by a similar mechanism as GOF? Unlike control (β-catflox/flox) terminals, which were filled with abundant SVs, HSA-β-cat–/– terminals contained sparsely populated SVs (Fig. 7G). Quantitatively, SV densities and active zones were decreased in HSA-β-cat–/– mutant compared with controls, although the numbers of nerve terminals were similar between the two genotypes (Fig. 7H-J). However, no difference was observed in SV diameters, synaptic cleft width (Fig. 7K,L) or junctional folds (data not shown). These results indicate that β-catenin LOF impairs NMJ development, in particular presynaptic differentiation, and provides a mechanism of compromised neurotransmission. Together with results from studies of GOF NMJs, they indicate distinct mechanisms by which β-catenin LOF and GOF regulate NMJ development.
Changes in gene transcription in HSA-β-catflox(ex3)/+ muscles
Mechanisms by which muscle β-catenin regulates NMJ development could be complex (see Discussion for details). It is possible that some presynaptic deficits occur because of overexpression of a factor(s) by increased β-catenin activity in muscles (Fig. 1F). In an initial step towards its identification, quantitative RT-PCR was performed. Fig. 8 shows increased expression of genes that are known to be β-catenin targets in GOF muscles, including Axin2, cyclin D1 (Ccnd1 – Mouse Genome Informatics), Myc, Lef1 and Dkk1 (He et al., 1998; Tetsu and McCormick, 1999; Hovanes et al., 2001; Yan et al., 2001). This result confirmed the increased β-catenin activity in muscles, as indicated by TOP-EGFP (Fig. 1F). Next, we compared the expression of morphogens, neurotrophic factors and axon guidance molecules between β-catenin GOF muscles and controls. Interestingly, several were found to be increased, including sonic hedgehog (Shh), Wnt7a, Fgf20, Gdnf, Hgf and reelin, whereas others were reduced, including Bmp9 (Gdf2 – Mouse Genome Informatics), Wnt1, Wnt7b and Nt3 (Ntf3 – Mouse Genome Informatics) (Fig. 8). These results are in agreement with the hypothesis that β-catenin GOF alters expression of various secreted factors to regulate motoneuron differentiation.
Fig. 8.

Different gene expression in HSA-β-catflox(ex3)/+ muscles. Total RNA was isolated and subjected to quantitative real-time PCR for indicated genes. mRNA levels were calibrated to Gapdh mRNA levels and normalized to mRNAs from control mice (**P<0.01, *P<0.05, n=3, t-test). Known Wnt/β-catenin target genes are shown in red; morphogens in green; neurotrophic factors in black; and axon guidance molecules in blue. Error bars indicate s.e.m.
Normal NMJ development in motoneuron β-catenin GOF mice
To investigate whether enhanced levels of stable β-catenin in motoneurons alter NMJ development, HB9-β-catflox(ex3)/+ mice were generated by crossing β-catflox(ex3)/+ mice with HB9-Cre mice that specifically express Cre in developing spinal cords from E9.5 (Arber et al., 1999; Li et al., 2008). HB9-β-catflox(ex3)/+ mice expressed the truncated β-catenin specifically in the spinal cord, but not in other tissues, including muscles and the cerebral cortex (supplementary material Fig. S7A). However, HB9-β-catflox(ex3)/+ mice were viable at birth and normal in body size during development to adult. NMJs in HB9-β-catflox(ex3)/+ mice did not show any observable abnormality in phrenic nerves, including primary branch location or secondary branch number and length (supplementary material Fig. S7B-E). No difference was observed in the number or size of AChR clusters, or endplate band width between control and HB9-β-catflox(ex3)/+ mice (supplementary material Fig. S7B,F-H). Finally, mEPP amplitudes and frequencies at HB9-β-catflox(ex3)/+ NMJs were similar to those at control, suggesting normal neurotransmission (data not shown). These results indicate that motoneuron β-catenin GOF had no effect on NMJ formation or function.
DISCUSSION
This study provides evidence that NMJs do not form properly when muscle β-catenin activity is increased. Intriguingly, β-catenin LOF and GOF in muscles generate converse as well as similar NMJ deficits (Table 1). These observations suggest that NMJ formation requires a critical level of β-catenin activity in the muscle.
Table 1.
Comparison of NMJ phenotypes in muscle β-catenin LOF and GOF mice

AChR cluster localization
A similar phenotype between muscle β-catenin LOF and GOF is the increased central area in which AChR clusters are distributed (Fig. 5A,B) (Table 1). How would both muscle β-catenin ablation and expression cause mislocation of AChR clusters? In the simplest scenario, β-catenin regulates cluster localization and this function can be disrupted by either elevated or decreased levels. β-Catenin interacts with cadherins to regulate cytoskeleton (Kemler, 1993; Huber et al., 1996; Orsulic et al., 1999; Nelson and Nusse, 2004), a mechanism that has been implicated in synaptogenesis in the CNS. For example, the N-cadherin-β-catenin complex accumulates at synapses onto dendritic spines and regulates dendritic spine morphogenesis, synaptic vesicle recycling, and recruitment of vesicles to active zones (Murase et al., 2002; Togashi et al., 2002; Bamji et al., 2003; Vitureira et al., 2012). Two major types of cadherins, N- and M-type, are expressed in muscles (Moore and Walsh, 1993; Cifuentes-Diaz et al., 1994; Kaufmann et al., 1999); however, their function in NMJ formation remains unclear.
β-Catenin could interact with rapsyn, which is necessary for agrin-induced AChR clustering (Zhang et al., 2007). Rapsyn is an adapter protein that interacts directly with AChR and is required for AChR clusters (Gautam et al., 1995). The fact that clusters form in both LOF and GOF mice suggests a modulatory role. Moreover, mEPP amplitudes of these mice were similar (Fig. 6C) (Table 1), suggesting that β-catenin might not regulate AChR density in the clusters. Finally, β-catenin-dependent transcription is elevated in entire muscle fibers, as indicated by TOP-EGFP (Fig. 1F); however, most AChR clusters remained in the central, albeit widened, region. These results suggest a scaffold function of β-catenin in controlling where AChR clusters form.
Prior to innervation, muscle fibers are pre-patterned with aneural AChR (Lin et al., 2001; Yang et al., 2001). However, the GOF seemed to have no effect on the pre-patterning (data not shown). Innervation, probably via ACh, disperses AChR clusters in non-synaptic areas (Misgeld et al., 2002; Brandon et al., 2003). This function might be impaired in the LOF and GOF mice because of reduced mEPP frequency. However, the number of AChR clusters per muscle fiber was not altered by the GOF (Fig. 5F,G). Therefore, the phenotype of scattered clusters might not be due to a problem with cluster dispersal. Recently, dihydropyridine receptor (an L-type calcium channel) was shown to control AChR cluster location (Chen et al., 2011). However, this mechanism might not be involved because dihydropyridine receptor mutation impaired mobility, in contrast to β-catenin GOF.
Motor axon outgrowth, navigation and arborization
In the LOF mice, phrenic nerve bundles are smaller with reduced axon size, but not number (Li et al., 2008) (Table 1). This phenotype suggests a trophic function of muscle β-catenin for motor axons. This notion is supported by increased extension and outgrowth of phrenic nerves (Fig. 2A,D, Fig. 3B-D) and spinal axons (Fig. 4C) in GOF mice. Intriguingly, aberrant activation of β-catenin stimulates the formation of ectopic muscles (Fig. 3), which were apparently able to induce and attract ectopic branches of phrenic nerves (Fig. 2A, Fig. 3C-E). This phenotype suggests that muscle β-catenin GOF might direct a guidance activity for innervation. However, in muscle β-catenin LOF mice, phrenic axons are able to navigate to the middle region, suggesting that attractive signals from muscles are not altered. However, the mislocation of primary branches is more severe in LOF mice compared with that in GOF mice (Fig. 2A, Fig. 3D) (Table 1) (Li et al., 2008).
Converse phenotypes of phrenic axon defasciculation and arborization were observed in LOF and GOF muscles. In the LOF mice, secondary branches are scarce, but elongated (Li et al., 2008), whereas in GOF diaphragms, short secondary branches were abundant in addition to long ones (Fig. 2A, Fig. 3D). GOF also increased the length and/or number of tertiary and quaternary branches (Fig. 2). Arborization was also observed in axons of brachial plexus (Fig. 4). Nevertheless, phrenic terminals were confined within the area where AChR clusters are located (Fig. 3), suggesting that the axon stop signal from muscle fibers is locally expressed or delivered although β-catenin activity was increased in entire muscle fibers (Fig. 1F). Several factors have been implicated in promoting axon branching of various neurons including BDNF, CNTF, netrin 1, Wnt3a and Sema3A (Gurney et al., 1992; Cohen-Cory and Fraser, 1995; Krylova et al., 2002; Bagri et al., 2003; Dent et al., 2004; Tang and Kalil, 2005; Singh et al., 2008). However, their expression was not altered by muscle β-catenin GOF (Fig. 8).
Terminal differentiation and function
The frequency of mEPPs is reduced in both GOF and LOF mice. This superficially similar phenotype is apparently caused by distinct mechanisms. Electron microscopic analysis demonstrates that the numbers of synaptic vesicles and active zones were significantly decreased in β-catenin LOF NMJs (Fig. 7G-J), suggesting that the production and docking of synaptic vesicles were compromised in the absence of muscle β-catenin. By contrast, the ultrastructure of NMJs in GOF mice appears normal (Fig. 7A-F). These observations suggest that neurotransmission deficits in β-catenin LOF NMJs were due to impaired development, whereas those in GOF NMJs were not structural, but functional. In support of this notion, action potentials are able to elicit normal EPPs in GOF, but not LOF NMJs (Table 1) (Li et al., 2008).
Wnt signaling or cadherin signaling in NMJ formation
Recent studies suggest a role of Wnt signaling in synapse formation. In C. elegans, Wnt signaling determines the position of NMJs by inhibiting synaptogenesis (Klassen and Shen, 2007), whereas in Drosophila, Wnt promotes NMJ formation (Packard et al., 2002; Wu et al., 2010; Budnik and Salinas, 2011). The extracellular domain of MuSK contains a cysteine-rich domain (CRD) that is homologous to that in Fz (Valenzuela et al., 1995; Glass et al., 1996). MuSK also interacts with Dvl, which regulates agrin-induced AChR clustering (Luo et al., 2002). APC, an adaptor downstream of Dvl in the canonical pathway, was shown to regulate AChR clustering (Wang et al., 2003). In zebrafish, Wnt11r binds to unplugged, the zebrafish MuSK homolog, to guide motor axons (Jing et al., 2009). In mammalian muscle cells, five Wnts (Wnt9a, Wnt9b, Wnt10b, Wnt11 and Wnt16), presumably by direct binding to MuSK, are able to stimulate AChR clustering, in a manner dependent on LRP4 (Zhang et al., 2012). Recently, WNT4 has been implicated in muscle pre-patterning and to stimulate AChR clustering via activation of MuSK (Strochlic et al., 2012). In addition, agrin-induced AChR clustering was enhanced by WNT3, but reduced by WNT3a (Henriquez et al., 2008; Wang et al., 2008).
β-Catenin-dependent transcription seemed not required for agrin- or Wnt-induced AChR clustering (Zhang et al., 2007) (B. Zhang and L.M., unpublished observations). How muscle β-catenin regulates presynaptic differentiation and/or function remains unclear. It is possible that muscle β-catenin acts by regulating the transcription of genes encoding retrograde factors involved in presynaptic differentiation. In favor of this hypothesis is the observation that various neural morphogens, neurotrophic factors and axon guidance cues are increased in expression (Fig. 8). Overexpression of GDNF has been shown to promote axon growth and delay synapse elimination (Nguyen et al., 1998). However, the number of NMJs per muscle fibers was apparently normal in β-catenin GOF mice (Fig. 5F,G). Sema3A has been implicated in the controlled growth of motor axons into the developing limb territory (Huber et al., 2005); however, its expression was not altered by β-catenin GOF and no apparent deficits in motor axon navigation were observed in β-catenin GOF mice. HGF was shown to promote neurite outgrowth of motoneurons (Ebens et al., 1996; Yamamoto et al., 1997) and, intriguingly, its level was elevated in β-catenin GOF muscles. Further study is needed to determine if HGF is crucial for NMJ formation in vivo.
Whether muscle β-catenin regulates presynaptic differentiation via cadherin signaling remains an outstanding question. N-cadherin could stimulate neurite outgrowth (Matsunaga et al., 1988; Bixby and Zhang, 1990; Payne et al., 1992). The N-cadherin-β-catenin complex was shown to promote dendritic arborization in hippocampal neurons, and the effect does not require β-catenin-dependent transcription (Yu and Malenka, 2003). A recent study reports a trans-synaptic activity of N-cadherin/β-catenin signaling in regulating synapse function in hippocampal neurons (Vitureira et al., 2012). Intriguingly, only disruption of postsynaptic, but not presynaptic, N-cadherin activity modulates vesicle release probability (Vitureira et al., 2012), a phenomenon similar to our findings that only muscle, not motoneuron, β-catenin may be crucial. Notice that exon 3 of the β-catenin gene does not encode the motif critical for interaction with α-catenin. Therefore, the β-catenin GOF mutant might function in a dominant-negative manner to disrupt interaction with α-catenin. However, such a cadherin-dependent mechanism is believed to be infeasible at mature NMJs because of large synaptic clefts (Wu et al., 2010). It is possible that this mechanism is involved in early development of the NMJ. In fact, N-cadherin was recently shown to regulate primary motor axon growth and branching during zebrafish embryonic development (Bruses, 2011). It is worth pointing out that NMJ formation or function is apparently not altered by both β-catenin LOF and GOF in motoneurons (Li et al., 2008) (supplementary material Fig. S7), which suggests that β-catenin in motoneurons is dispensable. Recent studies indicate a redundant function of γ-catenin in motoneurons. Positioning or migration of motoneurons in the spinal cord does not require Wnt canonical signaling, but N-cadherin signaling that can be mediated by either β-catenin or γ-catenin (Demireva et al., 2011; Bello et al., 2012). Phenotypes were observed only when both β-catenin and γ-catenin were lost. However, the loss seemed to have no effect on the ability of axons to select appropriate dorsoventral trajectories upon entering the limb.
In summary, this study provides further genetic evidence that muscle β-catenin is crucial for NMJ development and function. Considering multiple functions of β-catenin, it is likely that different NMJ deficits in LOF as well as GOF mice are caused by complex cell autonomous and non-autonomous mechanisms. Muscle lacking β-catenin and expressing stable β-catenin could provide a niche to identify muscle-derived factors by gene array analysis. However, extensive work (including in vivo rescue) is needed for validation and functional characterization of the candidates.
Supplementary Material
Acknowledgments
We would like to thank Drs Jon Lindstrom, Samuel Pfaff and Richard Rotundo for invaluable antibodies.
Footnotes
Funding
This work was supported in part by grants from the National Institutes of Health [NS056415 to L.M.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.080705/-/DC1
References
- Arber S., Han B., Mendelsohn M., Smith M., Jessell T. M., Sockanathan S. (1999). Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23, 659–674 [DOI] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E., Rossi S. G., Rotundo R. L., Yamada Y. (2002). Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat. Neurosci. 5, 119–123 [DOI] [PubMed] [Google Scholar]
- Bagri A., Cheng H. J., Yaron A., Pleasure S. J., Tessier-Lavigne M. (2003). Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113, 285–299 [DOI] [PubMed] [Google Scholar]
- Bamji S. X., Shimazu K., Kimes N., Huelsken J., Birchmeier W., Lu B., Reichardt L. F. (2003). Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40, 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello S. M., Millo H., Rajebhosale M., Price S. R. (2012). Catenin-dependent cadherin function drives divisional segregation of spinal motor neurons. J. Neurosci. 32, 490–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixby J. L., Zhang R. (1990). Purified N-cadherin is a potent substrate for the rapid induction of neurite outgrowth. J. Cell Biol. 110, 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borello U., Coletta M., Tajbakhsh S., Leyns L., De Robertis E. M., Buckingham M., Cossu G. (1999). Transplacental delivery of the Wnt antagonist Frzb1 inhibits development of caudal paraxial mesoderm and skeletal myogenesis in mouse embryos. Development 126, 4247–4255 [DOI] [PubMed] [Google Scholar]
- Brandon E. P., Lin W., D’Amour K. A., Pizzo D. P., Dominguez B., Sugiura Y., Thode S., Ko C. P., Thal L. J., Gage F. H., et al. (2003). Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J. Neurosci. 23, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruses J. L. (2011). N-cadherin regulates primary motor axon growth and branching during zebrafish embryonic development. J. Comp. Neurol. 519, 1797–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V., Salinas P. C. (2011). Wnt signaling during synaptic development and plasticity. Curr. Opin. Neurobiol. 21, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Qian L., Yang Z. H., Huang Y., Ngo S. T., Ruan N. J., Wang J., Schneider C., Noakes P. G., Ding Y. Q., et al. (2007). Rapsyn interaction with calpain stabilizes AChR clusters at the neuromuscular junction. Neuron 55, 247–260 [DOI] [PubMed] [Google Scholar]
- Chen F., Liu Y., Sugiura Y., Allen P. D., Gregg R. G., Lin W. (2011). Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat. Neurosci. 14, 570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C., Nicolet M., Goudou D., Rieger F., Mege R. M. (1994). N-cadherin expression in developing, adult and denervated chicken neuromuscular system: accumulations at both the neuromuscular junction and the node of Ranvier. Development 120, 1–11 [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S., Fraser S. E. (1995). Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature 378, 192–196 [DOI] [PubMed] [Google Scholar]
- DeChiara T. M., Bowen D. C., Valenzuela D. M., Simmons M. V., Poueymirou W. T., Thomas S., Kinetz E., Compton D. L., Rojas E., Park J. S., et al. (1996). The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85, 501–512 [DOI] [PubMed] [Google Scholar]
- Demireva E. Y., Shapiro L. S., Jessell T. M., Zampieri N. (2011). Motor neuron position and topographic order imposed by beta- and gamma-catenin activities. Cell 147, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent E. W., Barnes A. M., Tang F., Kalil K. (2004). Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J. Neurosci. 24, 3002–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebens A., Brose K., Leonardo E. D., Hanson M. G., Jr, Bladt F., Birchmeier C., Barres B. A., Tessier-Lavigne M. (1996). Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 17, 1157–1172 [DOI] [PubMed] [Google Scholar]
- Feng Z., Ko C. P. (2008). Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J. Neurosci. 28, 9599–9609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M. A., Sanes J. R., Borza D. B., Eswarakumar V. P., Fassler R., Hudson B. G., John S. W., Ninomiya Y., Pedchenko V., Pfaff S. L., et al. (2007). Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129, 179–193 [DOI] [PubMed] [Google Scholar]
- Gautam M., Noakes P. G., Mudd J., Nichol M., Chu G. C., Sanes J. R., Merlie J. P. (1995). Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 377, 232–236 [DOI] [PubMed] [Google Scholar]
- Gautam M., Noakes P. G., Moscoso L., Rupp F., Scheller R. H., Merlie J. P., Sanes J. R. (1996). Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85, 525–535 [DOI] [PubMed] [Google Scholar]
- Glass D. J., Bowen D. C., Stitt T. N., Radziejewski C., Bruno J., Ryan T. E., Gies D. R., Shah S., Mattsson K., Burden S. J., et al. (1996). Agrin acts via a MuSK receptor complex. Cell 85, 513–523 [DOI] [PubMed] [Google Scholar]
- Gros J., Serralbo O., Marcelle C. (2009). WNT11 acts as a directional cue to organize the elongation of early muscle fibres. Nature 457, 589–593 [DOI] [PubMed] [Google Scholar]
- Gurney M. E., Yamamoto H., Kwon Y. (1992). Induction of motor neuron sprouting in vivo by ciliary neurotrophic factor and basic fibroblast growth factor. J. Neurosci. 12, 3241–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V. (1934). The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J. Exp. Zool. 68, 449–494 [Google Scholar]
- Harada N., Tamai Y., Ishikawa T., Sauer B., Takaku K., Oshima M., Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da, Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- Henriquez J. P., Webb A., Bence M., Bildsoe H., Sahores M., Hughes S. M., Salinas P. C. (2008). Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. USA 105, 18812–18817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanes K., Li T. W., Munguia J. E., Truong T., Milovanovic T., Lawrence Marsh J., Holcombe R. F., Waterman M. L. (2001). Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28, 53–57 [DOI] [PubMed] [Google Scholar]
- Huber A. B., Kania A., Tran T. S., Gu C., De Marco Garcia N., Lieberam I., Johnson D., Jessell T. M., Ginty D. D., Kolodkin A. L. (2005). Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48, 949–964 [DOI] [PubMed] [Google Scholar]
- Huber O., Bierkamp C., Kemler R. (1996). Cadherins and catenins in development. Curr. Opin. Cell Biol. 8, 685–691 [DOI] [PubMed] [Google Scholar]
- Iwao K., Nakamori S., Kameyama M., Imaoka S., Kinoshita M., Fukui T., Ishiguro S., Nakamura Y., Miyoshi Y. (1998). Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res. 58, 1021–1026 [PubMed] [Google Scholar]
- Jing L., Lefebvre J. L., Gordon L. R., Granato M. (2009). Wnt signals organize synaptic prepattern and axon guidance through the zebrafish unplugged/MuSK receptor. Neuron 61, 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann U., Martin B., Link D., Witt K., Zeitler R., Reinhard S., Starzinski-Powitz A. (1999). M-cadherin and its sisters in development of striated muscle. Cell Tissue Res. 296, 191–198 [DOI] [PubMed] [Google Scholar]
- Keller-Peck C. R., Feng G., Sanes J. R., Yan Q., Lichtman J. W., Snider W. D. (2001). Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J. Neurosci. 21, 6136–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemler R. (1993). From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 9, 317–321 [DOI] [PubMed] [Google Scholar]
- Kim C. H., Neiswender H., Baik E. J., Xiong W. C., Mei L. (2008). Beta-catenin interacts with MyoD and regulates its transcription activity. Mol. Cell. Biol. 28, 2941–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N., Stiegler A. L., Cameron T. O., Hallock P. T., Gomez A. M., Huang J. H., Hubbard S. R., Dustin M. L., Burden S. J. (2008). Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135, 334–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen M. P., Shen K. (2007). Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell 130, 704–716 [DOI] [PubMed] [Google Scholar]
- Krylova O., Herreros J., Cleverley K. E., Ehler E., Henriquez J. P., Hughes S. M., Salinas P. C. (2002). WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron 35, 1043–1056 [DOI] [PubMed] [Google Scholar]
- Li X. M., Dong X. P., Luo S. W., Zhang B., Lee D. H., Ting A. K., Neiswender H., Kim C. H., Carpenter-Hyland E., Gao T. M., et al. (2008). Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat. Neurosci. 11, 262–268 [DOI] [PubMed] [Google Scholar]
- Lin W., Burgess R. W., Dominguez B., Pfaff S. L., Sanes J. R., Lee K. F. (2001). Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410, 1057–1064 [DOI] [PubMed] [Google Scholar]
- Liu X., Bates R., Yin D. M., Shen C., Wang F., Su N., Kirov S. A., Luo Y., Wang J. Z., Xiong W. C., et al. (2011). Specific regulation of NRG1 isoform expression by neuronal activity. J. Neurosci. 31, 8491–8501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Padgett D., Takahashi M., Li H., Sayeed A., Teichert R. W., Olivera B. M., McArdle J. J., Green W. N., Lin W. (2008). Essential roles of the acetylcholine receptor gamma-subunit in neuromuscular synaptic patterning. Development 135, 1957–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B., Je H. S. (2003). Neurotrophic regulation of the development and function of the neuromuscular synapses. J. Neurocytol. 32, 931–941 [DOI] [PubMed] [Google Scholar]
- Luo Z., Wang Q., Zhou J., Wang J., Liu M., He X., Wynshaw-Boris A., Xiong W., Lu B., Mei L. (2002). Regulation of AChR Clustering by Dishevelled Interacting with MuSK and PAK1. Neuron 35, 489–505 [DOI] [PubMed] [Google Scholar]
- Matsunaga M., Hatta K., Nagafuchi A., Takeichi M. (1988). Guidance of optic nerve fibres by N-cadherin adhesion molecules. Nature 334, 62–64 [DOI] [PubMed] [Google Scholar]
- McCabe B. D., Marques G., Haghighi A. P., Fetter R. D., Crotty M. L., Haerry T. E., Goodman C. S., O’Connor M. B. (2003). The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39, 241–254 [DOI] [PubMed] [Google Scholar]
- McMahan U. J. (1990). The agrin hypothesis. Cold Spring Harb. Symp. Quant. Biol. 55, 407–418 [DOI] [PubMed] [Google Scholar]
- Miniou P., Tiziano D., Frugier T., Roblot N., Le Meur M., Melki J. (1999). Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 27, e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T., Burgess R. W., Lewis R. M., Cunningham J. M., Lichtman J. W., Sanes J. R. (2002). Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36, 635–648 [DOI] [PubMed] [Google Scholar]
- Moore R., Walsh F. S. (1993). The cell adhesion molecule M-cadherin is specifically expressed in developing and regenerating, but not denervated skeletal muscle. Development 117, 1409–1420 [DOI] [PubMed] [Google Scholar]
- Moriyama A., Kii I., Sunabori T., Kurihara S., Takayama I., Shimazaki M., Tanabe H., Oginuma M., Fukayama M., Matsuzaki Y., et al. (2007). GFP transgenic mice reveal active canonical Wnt signal in neonatal brain and in adult liver and spleen. Genesis 45, 90–100 [DOI] [PubMed] [Google Scholar]
- Murase S., Mosser E., Schuman E. M. (2002). Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35, 91–105 [DOI] [PubMed] [Google Scholar]
- Nelson W. J., Nusse R. (2004). Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q. T., Parsadanian A. S., Snider W. D., Lichtman J. W. (1998). Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science 279, 1725–1729 [DOI] [PubMed] [Google Scholar]
- Nishimune H., Sanes J. R., Carlson S. S. (2004). A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432, 580–587 [DOI] [PubMed] [Google Scholar]
- Nishimune H., Valdez G., Jarad G., Moulson C. L., Muller U., Miner J. H., Sanes J. R. (2008). Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 182, 1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim R. W. (1991). Cell death during development of the nervous system. Annu. Rev. Neurosci. 14, 453–501 [DOI] [PubMed] [Google Scholar]
- Oppenheim R. W., Houenou L. J., Johnson J. E., Lin L. F., Li L., Lo A. C., Newsome A. L., Prevette D. M., Wang S. (1995). Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature 373, 344–346 [DOI] [PubMed] [Google Scholar]
- Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. (1999). E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell. Sci. 112, 1237–1245 [DOI] [PubMed] [Google Scholar]
- Packard M., Koo E. S., Gorczyca M., Sharpe J., Cumberledge S., Budnik V. (2002). The Drosophila Wnt, wingless, provides an essential signal for pre- and postsynaptic differentiation. Cell 111, 319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M. H., Seale P., Rudnicki M. A. (2003). Looking back to the embryo: defining transcriptional networks in adult myogenesis. Nat. Rev. Genet. 4, 497–507 [DOI] [PubMed] [Google Scholar]
- Payne H. R., Burden S. M., Lemmon V. (1992). Modulation of growth cone morphology by substrate-bound adhesion molecules. Cell Motil. Cytoskeleton 21, 65–73 [DOI] [PubMed] [Google Scholar]
- Perez-Ruiz A., Ono Y., Gnocchi V. F., Zammit P. S. (2008). beta-Catenin promotes self-renewal of skeletal-muscle satellite cells. J. Cell Sci. 121, 1373–1382 [DOI] [PubMed] [Google Scholar]
- Polleux F., Ince-Dunn G., Ghosh A. (2007). Transcriptional regulation of vertebrate axon guidance and synapse formation. Nat. Rev. Neurosci. 8, 331–340 [DOI] [PubMed] [Google Scholar]
- Qiu Y., Pereira F. A., DeMayo F. J., Lydon J. P., Tsai S. Y., Tsai M. J. (1997). Null mutation of mCOUP-TFI results in defects in morphogenesis of the glossopharyngeal ganglion, axonal projection, and arborization. Genes Dev. 11, 1925–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson J. M., Lee M., Kennedy E. L., Selleck S. B. (2003). Drosophila neuromuscular synapse assembly and function require the TGF-beta type I receptor saxophone and the transcription factor Mad. J. Neurobiol. 55, 134–150 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B., Robbins P., El-Gamil M., Albert I., Porfiri E., Polakis P. (1997). Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 275, 1790–1792 [DOI] [PubMed] [Google Scholar]
- Saha M. S., Miles R. R., Grainger R. M. (1997). Dorsal-ventral patterning during neural induction in Xenopus: assessment of spinal cord regionalization with xHB9, a marker for the motor neuron region. Dev. Biol. 187, 209–223 [DOI] [PubMed] [Google Scholar]
- Sanes J. R., Lichtman J. W. (2001). Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2, 791–805 [DOI] [PubMed] [Google Scholar]
- Schaeffer L., de Kerchove d’Exaerde A., Changeux J. P. (2001). Targeting transcription to the neuromuscular synapse. Neuron 31, 15–22 [DOI] [PubMed] [Google Scholar]
- Singh K. K., Park K. J., Hong E. J., Kramer B. M., Greenberg M. E., Kaplan D. R., Miller F. D. (2008). Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat. Neurosci. 11, 649–658 [DOI] [PubMed] [Google Scholar]
- Stevens C. F., Tsujimoto T. (1995). Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc. Natl. Acad. Sci. USA 92, 846–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strochlic L., Falk J., Goillot E., Sigoillot S., Bourgeois F., Delers P., Rouviere J., Swain A., Castellani V., Schaeffer L., et al. (2012). Wnt4 participates in the formation of vertebrate neuromuscular junction. PLoS ONE 7, e29976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Kalil K. (2005). Netrin-1 induces axon branching in developing cortical neurons by frequency-dependent calcium signaling pathways. J. Neurosci. 25, 6702–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O., McCormick F. (1999). Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- Thaler J., Harrison K., Sharma K., Lettieri K., Kehrl J., Pfaff S. L. (1999). Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687 [DOI] [PubMed] [Google Scholar]
- Togashi H., Abe K., Mizoguchi A., Takaoka K., Chisaka O., Takeichi M. (2002). Cadherin regulates dendritic spine morphogenesis. Neuron 35, 77–89 [DOI] [PubMed] [Google Scholar]
- Valenzuela D. M., Stitt T. N., DiStefano P. S., Rojas E., Mattsson K., Compton D. L., Nunez L., Park J. S., Stark J. L., Gies D. R., et al. (1995). Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 15, 573–584 [DOI] [PubMed] [Google Scholar]
- Vickerman L., Neufeld S., Cobb J. (2011). Shox2 function couples neural, muscular and skeletal development in the proximal forelimb. Dev. Biol. 350, 323–336 [DOI] [PubMed] [Google Scholar]
- Vitureira N., Letellier M., White I. J., Goda Y. (2012). Differential control of presynaptic efficacy by postsynaptic N-cadherin and beta-catenin. Nat. Neurosci. 15, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Jing Z., Zhang L., Zhou G., Braun J., Yao Y., Wang Z. Z. (2003). Regulation of acetylcholine receptor clustering by the tumor suppressor APC. Nat. Neurosci. 6, 1017–1018 [DOI] [PubMed] [Google Scholar]
- Wang J., Ruan N. J., Qian L., Lei W. L., Chen F., Luo Z. G. (2008). Wnt/beta-catenin signaling suppresses Rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J. Biol. Chem. 283, 21668–21675 [DOI] [PubMed] [Google Scholar]
- Weatherbee S. D., Anderson K. V., Niswander L. A. (2006). LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 133, 4993–5000 [DOI] [PubMed] [Google Scholar]
- Wu H., Xiong W. C., Mei L. (2010). To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137, 1017–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Livet J., Pollock R. A., Garces A., Arce V., deLapeyriere O., Henderson C. E. (1997). Hepatocyte growth factor (HGF/SF) is a muscle-derived survival factor for a subpopulation of embryonic motoneurons. Development 124, 2903–2913 [DOI] [PubMed] [Google Scholar]
- Yan D., Wiesmann M., Rohan M., Chan V., Jefferson A. B., Guo L., Sakamoto D., Caothien R. H., Fuller J. H., Reinhard C., et al. (2001). Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta-catenin signaling is activated in human colon tumors. Proc. Natl. Acad. Sci. USA 98, 14973–14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Arber S., William C., Li L., Tanabe Y., Jessell T. M., Birchmeier C., Burden S. J. (2001). Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30, 399–410 [DOI] [PubMed] [Google Scholar]
- Yu X., Malenka R. C. (2003). Beta-catenin is critical for dendritic morphogenesis. Nat. Neurosci. 6, 1169–1177 [DOI] [PubMed] [Google Scholar]
- Zhang B., Luo S., Dong X. P., Zhang X., Liu C., Luo Z., Xiong W. C., Mei L. (2007). Beta-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J. Neurosci. 27, 3968–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Luo S., Wang Q., Suzuki T., Xiong W. C., Mei L. (2008). LRP4 serves as a coreceptor of agrin. Neuron 60, 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Liang C., Bates R., Yin Y., Xiong W. C., Mei L. (2012). Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol. Brain 5, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D., Xiong W. C., Mei L. (2006). Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J. Neurosci. 26, 4841–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y., Zhang B., Gu S., Lee K., Zhou J., Yao G., Figueiredo D., Perry K., Mei L., Jin R. (2012). Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 26, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}