

Abstract
CD1d is a MHC class-like molecule that presents glycolipids to natural killer T (NKT) cells, then regulates innate and adaptive immunity. The regulation of CD1d gene expression in solid tumors is still largely unknown. Gene expression can be epigenetically regulated by DNA methylation and histone acetylation. We found that histone deacetylase inhibitors, trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA), induced CD1d gene expression in human (A549 and NCI-H292) and mouse (TC-1 and B16/F0) cancer cells. Simultaneous knockdown of HDAC1 and 2 induced CD1d gene expression. Sp1 inhibitor mitramycin A (MTM) blocked TSA- and SAHA-induced CD1d mRNA expression and Sp1 luciferase activity. Co-transfection of GAL4-Sp1 and Fc-luciferase reporters demonstrated that TSA and SAHA induced Sp1 luciferase reporter activity by enhancing Sp1 transactivation activity. The binding of Sp1 to CD1d promoter and histone H3 acetylation on Sp1 sites were increased by TSA and SAHA. These results indicate that TSA and SAHA could up-regulate CD1d expression in tumor cells through inhibition of HDAC1/2 and activation of Sp1.
Keywords: CD1d, histone deacetylase inhibitor, Sp1
Introduction
CD1d, a major histocompatibility complex (MHC) class I-like molecule, is mainly expressed in hematopoietic cells and antigen-presenting cells (APC) such as macrophages, dendritic cells, splenic B cells and T cells.1 CD1d presents glycolipids to an unique group of T cells, the natural killer T (NKT) cells, that regulates innate and adaptive immune responses, thus playing an important role in mediating anti-bacterial, anti-tumor, and auto-immune responses.2 CD1d is also expressed in certain tumor types, such as hematopoietic malignancies, prostate cancers and some neurological tumors. Although the function of CD1d on tumor cells is not well understood, increasing evidences suggest that it acts as a target for NKT-mediated cell killing.3 However, most human and mouse solid tumors are CD1d-negative.4-8 Therefore, understanding how CD1d expression is regulated in solid tumor cells will facilitate the development of effective cancer therapy.
Human CD1d gene has TATA box-less dual promoters with multiple transcription initiation sites. The dual (proximal and distal) promoters may be responsible for tissue and cell-specific expression of CD1d.9 Transcription factors Sp1 and lymphoid enhancer-binding factor-1 (LEF-1) plays crucial roles in the function of the proximal and distal promoter, respectively.9,10 The promoter region within 200 bp from the translational start site of mouse CD1d gene exhibits cell-type specific promoter activity. Analysis of this region revealed an Ets binding site crucial for CD1d promoter activity.11 Moreover, all-trans-retinoic acid (RA), the active metabolite of vitamin A, increases CD1d mRNA in human and rodent monocytic cells. The RA-responsive element (RARE) within the distal 5′-flanking region is responsive for binding of nuclear retinoid receptor.12
Epigenetic mechanisms, which involve DNA methylation and histone modifications, result in the heritable regulation of gene expression without a change of DNA sequence.13 DNA is methylated by DNA methyltransferase (DNMTs) at C5-cytosine of CpG dinucleotides. Approximately 50% of genes are associated with clusters of CpG dinucleotides (CpG islands) in their promoter regions. Methylation of CpG islands inhibits gene expression.14 DNA is wrapped around histone cores to form nucleosomes. The post-translational modifications of histone tails, such as acetylation by histone acetyltransferases (HATs) and methylation by lysine methyltransferases, influence how tightly or loosely the chromatin is compacted, then regulating gene expression.15 It is widely accepted that histone modification and DNA methylation are intricately interrelated, working together to determine the status of gene expression and to decide cell fate.16
In this study, we provide evidences that HDAC inhibition by TSA and SAHA induced CD1d gene expression in solid tumor cells through inhibition of HDAC1/2. The histone H3 acetylation on Sp1-responsive elements within CD1d promoter was induced, leading to the increase of Sp1 transactivation and transcriptional activities. Our results provide the molecular mechanism for the regulation of CD1d expression in solid tumor cells.
Results
The epigenetic mechanisms controlling the expression of CD1d have not been investigated. To examine whether CD1d expression is epigenetically regulated, human lung cancer A549 cells treated with DNMT inhibitors zebularine (Zeb) and 5-aza-2’-deoxycytidine (5-Aza-CdR) or HDAC inhibitor trichostatin (TSA) were analyzed by RT-PCR. As shown in Figure 1A and 1B, CD1d mRNA expression was significantly induced by Zeb and TSA but only slightly induced by 5-Aza-CdR. Additive effect on CD1d induction was observed in response to combined treatment of Zeb and TSA (Fig. 1B). These results suggested that CD1d expression might be mainly regulated by histone acetylation.

Figure 1. Effects of HDAC and DNMT inhibitors on CD1d mRNA expression. (A) A549 cells were treated the indicated concentration of zebularine (Zeb) or 5-aza-2’-deoxycytidine (5-Aza-CdR) for 48 h. (B) A549 cells were pretreated the indicated concentration of zebularine for 48 h, then treated with TSA (0.5 μM) for 24 h. Total RNA (2 μg) were extracted and RT-PCR was performed. Quantification of each band by densitometry was performed and the CD1d/Actin ratio was indicated. (C) Upper panel: schematic CpG island of CD1d promoter. The region of -270~+620 is the predicted CpG islands. Arrow indicated the translation start site. Lower panel: Bisulfite-treated A549 genomic DNA are amplified by PCR and subcloned into pGEM-T Easy vectors. These DNA clones are directly sequenced.
The above results implied that CD1d might be regulated by DNA methylation. Thus, the methylated pattern of CD1d promoter was further investigated using bisulfite sequencing. Analysis of CD1d promoter and coding regions revealed an 883-bp putative CpG island (-270 to +612) (Fig. 1C, upper panel). After treatment with bisulfite, the cytosine (C) was converted to thymidine (T). However, methylated cytosine (mC) was resistant to bisulfite treatment. CpG islands on CD1d promoter surrounding two Sp1 sites were amplified from A549 genomic DNA. Sequencing analysis of the PCR products indicated that cytosines (blue color) were converted to thymidines (red color) (Fig. 1C), suggesting that predicted CpG islands on CD1d promoter were not methylated.
Since TSA induced CD1d mRNA expression, we further characterized the effect of other HDAC inhibitors on CD1d expression. A549 cells were treated with different HDAC inhibitors including MS-275, sodium butyrate (NaBu), SAHA, oxaflatin (Oxa), TSA and valproic acid (VPA). Although all inhibitors could increase histone H3 acetylation, CD1d mRNA was only induced by TSA and SAHA (Fig. 2A). To further characterize the effects of TSA and SAHA, dose- and time-course experiments were performed. As shown in Figure 2B (upper panel), TSA and SAHA induced CD1d expression in a dose-dependent manner, in which 1 μM TSA or 5 μM SAHA treatment showed the maximum effects. When cells were treated with 1 μM TSA and 5 μM SAHA for the indicated time intervals, the induction of CD1d mRNA was increased and reached saturation at 12 h (Fig. 2B, lower panel). To demonstrate that TSA- and SAHA-induced CD1d expression were universal, human lung cancer cells (NCI), mouse lung cancer (TC-1) and melanoma (B16/F0) cells were also treated with TSA and SAHA. CD1d expression was induced in these tumor cells, similar to the pattern of A549 cells (Fig. 2C). The induction of CD1d mRNA and protein in A549 cells by TSA and SAHA were confirmed by real time PCR (Fig. 2D) and western blot analysis (Fig. 2E).

Figure 2. Effects of HDAC inhibitors on CD1d mRNA expression in various tumor cells. (A) A549 cells were treated with TSA (1 μM), SAHA (5 μM), MS-275 (1 μM), NaBu (1 mM), oxaflatin (Oxa, 1 μM), or VPA (1 mM) overnight. Total RNA (2 μg) was used for RT-PCR (upper panel) and total cell lysates were subjected to western blot analysis using antibody specific for acetylated-histone H3 (lower panel). The blot was representative for at least three independent experiments. (B) A549 cells were treated with the indicated dose of TSA or SAHA overnight, or treated with TSA (1 μM) or SAHA (5 μM) for the indicated time. Total RNA (2 μg) was used for RT-PCR. (C) A549, NCI-H292, TC-1 and B16/F0 cells were treated with TSA (1 μM) or SAHA (5 μM) for 16 h, then total RNA (2 μg) were used for RT-PCR. (D) A549 cells were treated with SAHA (3 or 5 μM) or TSA (0.5 or 1 μM) for 16 h. Total RNA were used for real time PCR. Relative CD1d mRNA expression was presented as the mean ± S.D., *:p < 0.05, as compared with basal. Three independent experiments were performed. (E) A549 cells were treated with TSA (1 μM) or SAHA (5 μM) for 24 h, then total cell lysates were subjected to western blot analysis using antibody specific for CD1d and Actin. The blot was representative for at least three independent experiments.
To elucidate which HDAC isoform was responsible for the induction of CD1d gene expression, A549 cells were treated with inhibitors specific for HDAC6 (tubacin) and HDAC8 (PCI-34051), or transfected with siRNAs for HDAC1, 2 or 3. Only knockdown of HDAC1 or 2 alone slightly induced CD1d mRNA expression (Fig. 3A and 3B). Simultaneous inhibition of HDAC1 and 2 increased the levels of CD1d mRNA in A549 and TC-1 cells (Fig. 3C). MS-275 is reported to be a HDAC1-selective inhibitor but it can also inhibit HDAC2 activity with mild efficacy.17 Indeed, higher doses (5 and 10 μM) of MS-275 could induce CD1d mRNA expression (Fig. 3D). These results suggested that CD1d gene expression was regulated by both HDAC1 and 2.

Figure 3. Effects of HDAC1/2 inhibition on CD1d mRNA expression. (A) A549 cells were treated with TSA (1 μM), SAHA (5 μM), tubacin (20 μM), or PCI-34051 (5 μM) for 16 h, then total RNA (2 μg) were used for RT-PCR. (B) A549 cells were transfected with siRNA specific for HDAC1, 2 or 3 for 72 h. Total cell lysate were subjected to western blot analysis using antibody specific for HDAC1, 2 or 3 (upper panel). Total RNA (2 μg) was used for RT-PCR (lower panel). Each blot was representative for at least three independent experiments. (C) A549 and TC-1 cells were transfected with HDAC1 and/or HDAC2 siRNA for 72 h. Total cell lysates were subjected to western blot analysis using antibody specific for HDAC1 or HDAC2 (upper panel). Total RNA (2 μg) was used for RT-PCR (lower panel). (D) A549 cells were treated with the indicated concentration of MS-275 and TSA for 24 h, then total RNA (2 μg) were used for RT-PCR. Each blot was representative for at least three independent experiments.
To dissect the molecular mechanism(s) of CD1d expression by HDAC inhibition, A549 and TC-1 cells were pre-treated with different kinds of signaling inhibitors including PKC inhibitor (Ro318220), ROCK inhibitor (Y27632), CaM Kinase II inhibitor (KN-62), p38 inhibitor (SB203580), MEK inhibitor (PD98059), JNK inhibitor (SP600125), PI3K inhibitors (LY294002 and wortmannin), mTOR inhibitor (rapamycin) and Akt inhibitor (SH-5) for 1 h and then exposed to 1 μM TSA for 16 h. The TSA-induced CD1d expression was not altered by these inhibitors (Fig. S1). We next analyzed the cis-elements within the CD1d promoter region. RARE- and Sp1-luciferase reporters were used and SAHA dramatically increased RARE luciferase activity to 160-fold (Fig. 4B, upper panel). However, two retinoic acids, ATRA and 9cRA, only induced 6- and 5-fold reporter activities, and CD1d mRNA levels were not induced in response to these two drugs (Fig. 4B). Since TK-promoter of 3xRARE reporter construct contains two Sp1 binding sites (Fig. 4A), the Sp1 sites might be important for TSA- and SAHA-induced CD1d expression. Indeed, Sp1 inhibitor, mithramycin (MTM) blocked TSA- and SAHA-induced RARE luciferase activities (Fig. 4C, upper panel). Consistently, MTM dose-dependently reduced SAHA-induced CD1d mRNA expression (Fig. 4C, lower panel).
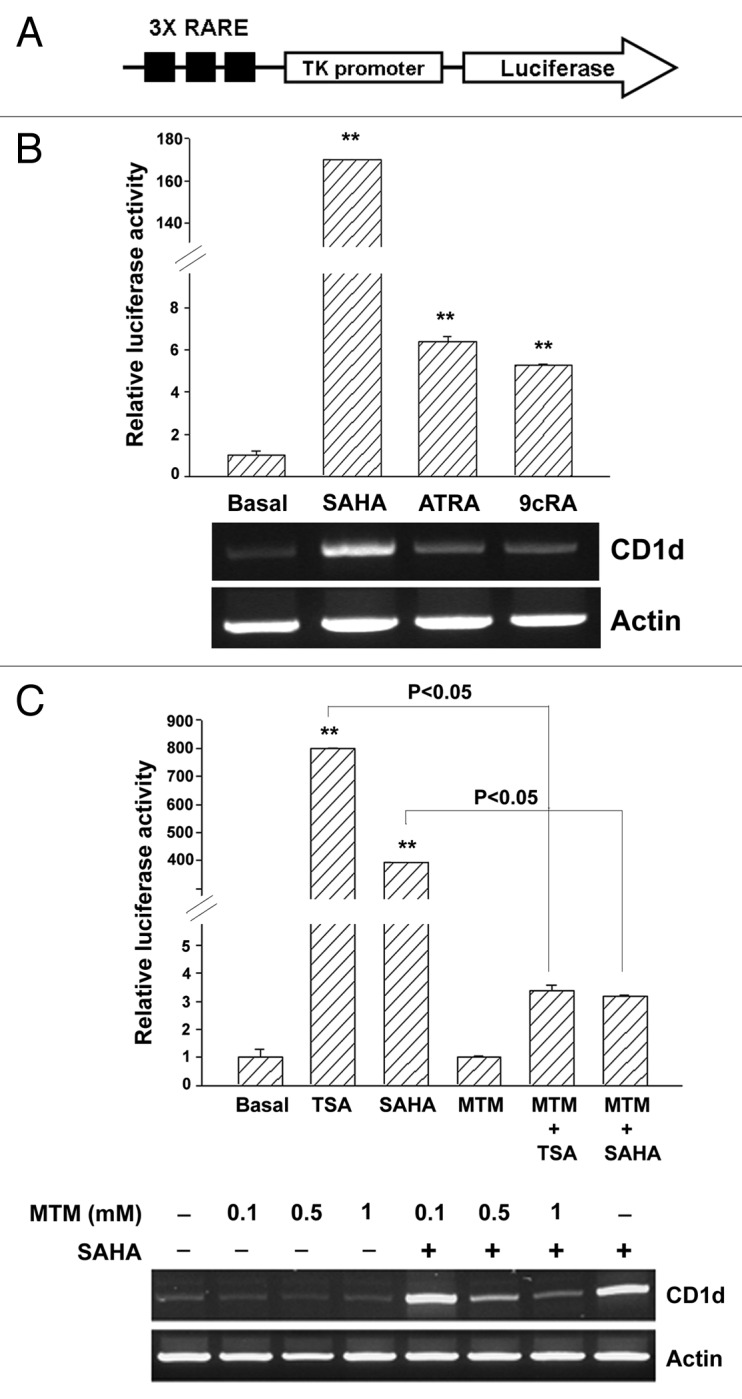
Figure 4. Effects of SAHA, ATRA and 9cRA on RARE3-tk-luc reporter activity in A549 cells. In (A), CD1d promoter contains two Sp1 sites in the proximal region and one RARE site in the distal region. 3xRARE-tk luciferase reporter was used for the promoter activity assay. In (B) and (C), A549 cells were transfected with RARE luciferase reporter and β-galactosidase plasmids. After 24 h transfection, cells were treated with SAHA (5 μM), all-trans RA (30 μM), 9-cis RA (50 μM) for 16 h (B), or pretreated MTM (1 μM) for 1 h, then treated with SAHA (5 μM) for 16 h (C). Luciferase activity was measured (upper panel) and total RNA (2 μg) were used for RT-PCR (lower panel). Relative luciferase activity was presented as the mean ± S.D., **p < 0.01, as compared with basal. At least three independent experiments were performed.
To further confirm that TSA and SAHA induced Sp1 transcriptional activity, the 3xSp1 luciferase reporter was used (Fig. 5A). As shown in Figure 5B, TSA and SAHA increased Sp1 luciferase activities (Fig. 5B). Their effects were completely blocked by MTM (Fig. 5C). To examine whether TSA and SAHA induced Sp1 transcriptional activity through an increase of its transactivation ability, GAL4-Sp1 and Fc-luciferase reporter plasmids were transfected into A549 cells, and TSA and SAHA increased the transactivation activities of Sp1 (Fig. 5D and 5E). Therefore, our results indicated that TSA and SAHA induced CD1d expression through Sp1-dependent pathway.

Figure 5. Effects of HDAC inhibitors on Sp1 transcription and transactivation activity in A549 cells. In (A), Sp1 luciferase reporter construct was depicted. In (B) and (C), A549 cells were transfected with the luciferase reporter and β-galactosidase plasmids. After 24 h, cells were exposed with TSA (1 μM), SAHA (5 μM), MS275 (1 μM), NaBu (1 mM), oxaflatin (1 μM) or VPA (1 mM) for 16 h (B), or pretreated with MTM (1 μM) for 1 h, then treated with TSA (1 μM) or SAHA (5 μM) for 16 h (C). In (D), GAL4-Sp1 and Fc-luciferase reporter constructs were depicted. In (E), A549 cells were co-transfected with Gal4-Sp1, Fc-luciferase reporter andβ-galactosidase plasmids. After 24 h, cells were exposed to TSA (1 μM) or SAHA (5 μM) for 16 h. Luciferase activities were measured. Relative luciferase activity was presented as the mean ± S.D., **p < 0.01, as compared with basal. At least three independent experiments were performed.
The Sp1 transactivation is correlated with its DNA binding activity; therefore, the effect of SAHA and TSA on the recruitment of Sp1 to CD1d promoter was examined by chromatin immunoprecipitation (ChIP) assay (Fig. 6A). SAHA and TSA significantly induced Sp1 binding and the histone H3 acetylation (Fig. 6B), indicating that they induced CD1d expression through the induction of histone H3 acetylation on Sp1 response elements and recruitment of Sp1 to these sites. To investigate the importance of each Sp1 site, oligonucleotides harboring mutations of Sp1 sites were used as probes (Fig. 6A) and DNA affinity precipitation assay (DAPA) was performed. As shown in Figure 6C, SAHA increased the affinity of Sp1 protein to wild-type Sp1 sites, which was prevented when either Sp1 site was mutated, suggesting that both Sp1 sites were essential for CD1d induction.
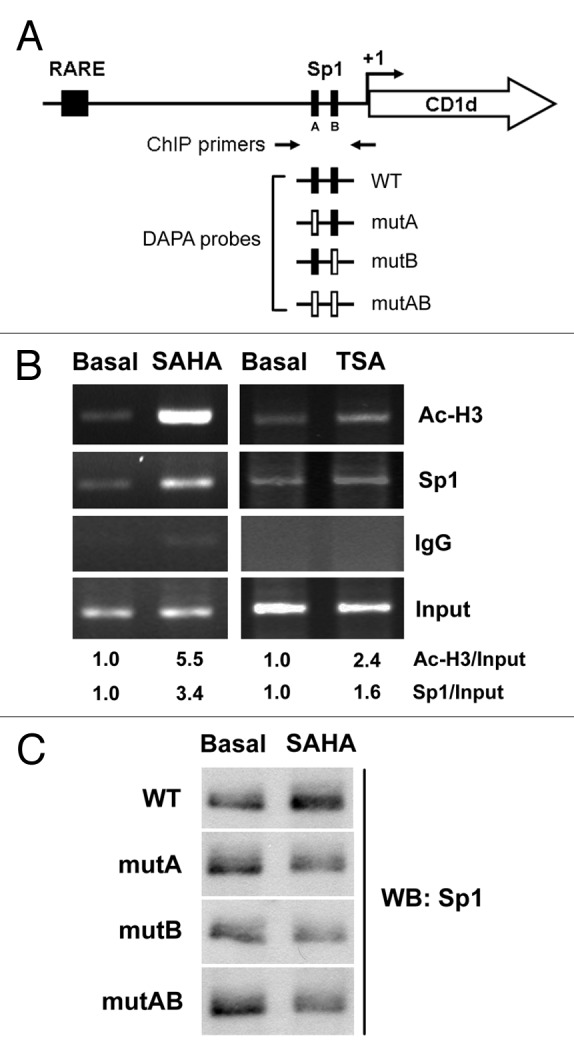
Figure 6. Binding of acetylated histone-H3 and Sp1 to the CD1d promoter. In (A), schematic illustration of RARE and two Sp1 sites on the CD1d promoter was depicted. The position for Sp1 sites was marked as A for GGCGGG (-73 to -68) and B for GGGCGG (-40 to -45). Arrows indicate the primers used for the amplification of the region -364/+21 bp covered the Sp1 sites. The probes for DAPA were listed. WT Sp1 In (B), A549 cells were treated with SAHA (5 μM) and TSA (1 μM) for 16 h, and ChIP assays were performed using anti-acetylated histone H3, anti-Sp1 antibody, or control IgG. Quantification of each band by densitometry was performed and the Ac-H3/Actin and Sp1/Actin ratio were indicated. In (C), A549 cells were treated with SAHA (5 μM) for 16 h, and nuclear extracts were used for DAPA analysis.
Discussion
Overexpression of HDACs induced epigenetic silence of tumor suppressor genes, and HDAC inhibitors are now regarded as a new category of anti-tumor agents.18 HDAC inhibitors can regulate certain gene expression by increasing the level of histone acetylation, and then selectively kill cancer cells by inducing apoptosis, cell cycle arrest, and differentiation.19 Moreover, HDAC inhibitors can inhibit angiogenesis and enhance the sensitivity of chemotherapy for cancers.20,21 However, the effect of HDAC inhibitors on immune responses is rarely investigated. It has been shown that HDAC inhibitors can upregulate the expression of MHC class I/II and co-stimulatory/adhesion molecules such as CD40, CD80, CD86, and intercellular adhesion molecule 1 (ICAM-1) in tumor cells, then, in turn, increasing immunogenicity.22,23 They also increase the expression of MHC class I related molecules MICA and MICB in tumor cells, and killing them by NK cells-mediated mechanisms.24,25 In the present study, we found that HDAC inhibitors induced CD1d expression in human and mouse solid tumor cells. Therefore, regulation of genes with immune functions by HDAC inhibitors may facilitate their antitumor effects.
The transcriptional and transactivation activities of Sp1 were increased by TSA and SAHA. Acetylated histone H3 correlated with the increased binding of Sp1 around the Sp1-responsive elements on CD1d promoter was found. The human CD1d gene contains two Sp1 binding elements, GGCGGG (-73 to -68; site A) and GGGCGG (-40 to -45; site B), which play a major role in the regulation of CD1d promoter activity.9 Consistently, we found that mutation of either Sp1 site will reduce the affinity of Sp1 to DNA. It has been reported that Sp1 plays a critical role in HDAC inhibitor-mediated gene expression, such as p21, CYP46A1 and 5-lipoxygenase.26-29 Sp1 can be linked to chromatin remodeling through interactions with chromatin-modifying factors such as CBP/p300 and HDACs. HDAC inhibition relieves the repression of HDACs from Sp1 sites and recruits CBP/p300 that possess intrinsic HAT activity, leading to the acetylation of surrounding histones and the enrichment of RNA polymerase II to initiate gene transcription.27-30 Our previous study also demonstrates that abrogation of HDAC activity by statins induces p21 expression through dissociation of HDAC1/2 and association of CBP, leading to histone acetylation on the Sp1 sites of p21 promoter.31
HDACs are divided into four classes: class I (HDAC1, 2, 3, and 8), class II (class IIa: HDAC4, 5, 7, and 9; class IIb: HDAC6 and 10), class III (SIRT1–7), and class IV (HDAC11).32 Several HDAC inhibitors are currently being developed and show different specificities.33 Among HDAC inhibitors used in this study, both TSA and SAHA are pan-HDAC inhibitors that significantly induced CD1d gene expression. Recently, a chemical phylogenetic analysis indicates that class I and class IIb are the main targets for the current HDAC inhibitors.34 By isoform-specific inhibitors and siRNA knockdown analysis, we found that inhibition of HDAC1/2 was responsible for the induction of CD1d expression. We also found that HDAC1/2/3-selective inhibitor MS-275 induced CD1d mRNA expression. The HDAC2/3-selective inhibitor apicidin also induced CD1d mRNA expression but not enhanced by combination with 1 μM MS-275 in which mimicked the effect of TSA and SAHA (Fig. S2). To get more insights of these HDAC inhibitors, the IC50 values to inhibit HDAC1–3 were compared according to published reports (Table S1).17,35-39 The effect of MS-275 and apicidin to inhibit HDAC1 activity is still controversial, and the induction of CD1d expression by apicidin was probably due to its inhibition on HDAC1/2.
Downregulation of CD1d expression is correlated with increasing metastasis in human breast cancer cells and disease progression in multiple myeloma.40,41 Engagement of CD1d by anti-CD1d monoclonal antibodies (mAbs) induces cell death of CD1d-transfected myeloma cell lines, suggesting the role of CD1d in the regulation of cell death.41 In addition, ATRA induces CD1d expression in human leukemia HL-60 cells and ligation with anti-CD1d mAbs promotes ATRA-induced apoptosis.42 We have recently shown that HDAC inhibitors induces apoptosis and autophagic cell death in hepatocellular carcinoma.43 It is possible that combination of anti-CD1d mAbs could enhance the anticancer effects of HDAC inhibitors.
NKT cells play pivotal roles in many immune responses including antitumor immunity. They specifically recognize exogenous ligand α-GalCer in conjunction with CD1d. After activation, NKT cells exert antitumor activity directly through death receptor-induced tumor cell apoptosis or indirectly through secretion of interferon-γ to activate NK and CD8+ T cells.44 In addition, activated NKT cells have been shown to directly kill CD1d-bearing tumor cells in a CD1d-dependnent manner in vitro. The expression levels of CD1d in tumors are correlated with their sensitivity to NKT-mediated antitumor immunity in vivo.45,46 Tumor cells transfected with CD1d can present α-GalCer to NKT cells and lead to NKT activation.47 Our results demonstrated that HDAC inhibitors induced CD1d expression in tumor cells, which might favor the presentation of glycolipids to NKT cells.
In the present study, we provide evidence that HDAC inhibition by TSA and SAHA induced CD1d expression in tumor cells through inhibition of HDAC1/2 and activation of transcription factor Sp1. The histone H3 acetylation on Sp1-responsive elements within CD1d promoter was increased, then leading to the increase of Sp1 transactivation and transcriptional activities. Our results provide a molecular basis for the application of HDAC inhibitors in cancer immunotherapy.
Materials and Methods
Materials
Acetyl histone H3 (Ac-H3; 06–599) and Sp1 (07–645) antibodies for chromatin immunoprecipitation (ChIP) were from Millipore. Sp1 (sc-17824), CD1d (sc-19632) and Actin antibodies were from Santa Cruz Biotechnology. Zebularine (Z4775), 5-aza-2’-deoxycytidine (A3635), trichostatin A (TSA; T8552), MS-275 (M5568), sodium butyrate (B5887), oxaflatin (O3139) and valproic acid (VPA; P4543) were from Sigma. Suberoylanilide hydroxamic acid (SAHA) was provided by MSD Taiwan (Merck). Tubacin was provided by Dr. Ralph Mazitschek (Broad Institute and Chemical Biology Program, Harvard Univeristy and Massachusetts Institute of Technology). PCI-34051 was provided by Dr. Wei-Jan Huang (Graduate Institute of Pharmacognosy, College of Medicine, Taipei Medical University). 3xRARE-tk luciferase reporter construct was provided by Dr. Rene Bernards (The Netherlands Cancer Institute). Sp1-luciferase reporter, GAL4-Sp1 and Fc-luciferase repoter constructs were provided from Dr. Zee-Fen Chang (College of Medicine, National Taiwan University).
Cell culture
The human alveolar epithelial A549 cells and mouse melanoma cells B16/F0 were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; GIBCO, 12800–017). Human lung adenocarcinoma epithelial NCI-H292 cells and mouse lung carcinoma TC-1 cells that are kindly provided by T-C Wu (Department of Pathology, Johns Hopkins Medical Institutions) were cultured in RPMI Medium 1640 (GIBCO, 31800–022). All medium were supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, 10437), 1% L-glutamine (GIBCO, 25030), 1% Antibiotic:Antimycotic Solution (GEMINI Bio Products, 400–101), and incubated at 37°C in a humidified incubator containing 5% CO2.
Western blot analysis
Proteins in the cell lysate (50 μg) are separated on a 10% SDS-polyacrylamide gel, then transferred electrophoretically onto a nylon cellulose membrane (Amersham, RPN303E). The membrane is pre-hybridized in 20 mM TRIS-HCl, pH 7.5, 1.5 M NaCl, 0.05% Tween-20 (TBST buffer) and 5% skim milk for 1 h, then transferred to a solution containing 1% BSA/TBST and primary antibody and incubated overnight at 4°C. After washing with the TBST buffer, the membrane is submerged in 1% BSA/TBST containing the horseradish peroxidase conjugated secondary antibody for 1 h. The membrane is washed with TBST buffer, then developed by WESTERN LIGHTNING Plus-ECL (PerkinElmer, NEL105).
Semi-quantitative RT-PCR and real time PCR
Total RNA is isolated from A549 cells using TRIZOL reagent (Invitrogen, 15596–018). Reverse transcription reaction is performed using 2 μg of total RNA and reverse transcribed into cDNA using oligo dT primer. For RT-PCR, cDNA was amplified using primers specific for CD1d (5′-CAGATCTCGTCCTTCGCCAA-3′ and 5′-GCTCGGAGATACCATGACTC) and β-actin (5′-TGACGGGGTCACCCAC ACTGTGCCCATCTA-3′ and 5′-CTAGAAGCATTTGCGGGGACGATGGAGGG- 3′). The PCR products are subjected to 1% agarose gel electrophoresis. Real time PCR was performed using KAPA SYBR FAST qPCR Kit (KAPA Biosystems, KK4603) in ABI PRISMTM 7900 Sequence Detection System (Applied Biosystems). The primer sequences for CD1d and GAPDH were (CD1d, 5′-GAGCAACCCTGGATGTGGT-3′ and 5′-TCAAGCCCATGGAGGTGTA-3′) and (GAPDH, 5′-AGCCACATCGCTCA GACAC-3′ and 5′-GCCCAATACGACCAAATCC-3′). All samples were read in triplicate, and values were normalized to GAPDH expression.
Bisulfite DNA sequencing
Extracted DNA is bisulfite-modified by using the EZ DNA Methylation-Gold™ Kit (Zymo Research, D5005). Modified DNA samples are amplified by PCR and then subcloned into pGEM-T Easy vectors (Promega, A1360). These DNA clones are directly sequenced. PCR primers for generating the CD1d fragment are 5′-TTGTGAAATTTATTGAAGTGAG-3′ and 5′-AAACAACAAAAACAACAAAC AC-3′. Conditions for PCR are as follows: 1 cycle at 95°C for 5 min; 35 cycles at 95°C for 30 sec, 54°C for 30 sec, and 72°C for 1 min; and 1 cycle at 72°C for 5 min.
siRNA knockdown analysis
The siGENOMIC SMARTpool HDAC1 (Dharmacon, M-003943–02), HDAC2 (M-003495–02) and HDAC3 (M-003496–02) siRNAs were purchased from Dharmacon RNAi Technologies. These siRNAs were transiently transfected into cells with DharmaFECT 4 siRNA Transfection Reagent (T-2004). After 48 h, the cells were harvested and subjected to RT-PCR or western blotting assay.
Luciferase reporter assay
A549 cells, grown to 50% confluent in 12-well plates, were transfected with the luciferase reporter DNA and β-galactosidase DNA using Arrest-In transfection reagent (Open Bioystems, ATR1740) according to the manufacturer's recommendations. Briefly, reporter DNA (0.4 μg) and β-galactosidase DNA (0.2 μg; plasmid pRK containing the β-galactosidase gene driven by the constitutively active SV40 promoter, used to normalize the transfection efficiency) were mixed with 0.6 μL of Tfx-50 in 1 mL of serum-free DMEM. After 10–15 min incubation at room temperature, the mixture was applied to the cells, then, 1 h later, 1 mL of complete growth medium was added. On the following day, the medium was replaced with fresh medium. Forty-eight hours after transfection, the cells were treated with HDAC inhibitors for 16 h. Cell extracts were then prepared and luciferase and β-galactosidase activities measured, the luciferase activity being normalized to the β-galactosidase activity.
Chromatin immunoprecipitation (ChIP) assay
A549 cells were incubated with HDAC inhibitors. 1% formaldehyde was added to the culture medium and after incubation for 20 min at 37°C, cells were washed twice in PBS, scraped and lysed in lysis buffer (1% SDS, 10 mM TRIS-HCl pH 8.0, with 1 mM PMSF, pepstatin A and aprotinin) for 10 min at 4°C. Lysates were sonicated 5 times for 10 sec each and the debris was removed by centrifugation. One third of the lysates was used as DNA input control. The remaining two thirds of the lysates were diluted 10-fold with a dilution buffer (0.01% SDS, 1% Triton X 100, 1 mM EDTA, 10 mM TRIS-HCl pH 8.0, and 150 mM NaCl) followed by incubation with an anti-Sp1, Ac-H3 antibody or a non-immune rabbit IgG overnight at 4°C. Immunoprecipitated complexes were collected by using protein A-sepharose beads. The precipitates were extensively washed and incubated in the elution buffer (1% SDS and 0.1 M NaHCO3) at room temperature for 20 min. Cross-linking of protein-DNA complexes was reversed at 65°C for 5 h, followed by treatment with 100 μg/mL protease K for 3 h at 50°C. DNA was extracted 3 times with phenol/chloroform and precipitated with ethanol. Pellets were resuspended in TE buffer and subjected to PCR amplification using specific primer: 5′-CAACCTTATGTCCTGCTTCCAA-3′ and 5′-CAGAAACAGCAGGCACCCCAT A-3′. PCR products are then resolved by 1.5% agarose gel electrophoresis.
DNA affinity precipitation assay (DAPA)
The DAPA probes were prepared by annealing the biotinated sense oligonucleotides and non-biotinated antisense oligonucleotides: 5′-AGCTGAGCGGC GGGGGAGAAGAGTGCGCAGGTCAGAGGGCGGCGCGCAGC-3′ and 5′-GCTG CGCGCCGCCCTCTGACCTGCGCACTCTTCTCCCCCGCCGCTCAGCT-3′ (WT), 5′-AGCTGAGCGTTTTGGGAGAAGAGTGCGCAGGTCAGAGGGCGGCGCGCAGC-3′ and 5′-GCTGCGCGCCGCCCTCTGACCTGCGCACTCTTCTCCCAAAAC GCTCAGCT-3′ (mutA), and 5′-AGCTGAGCGGCGGGGGAGAAGAGTGCGCAG GTCAGAGTTTTGCGCGCAGC-3′ and 5′-GCTGCGCGCAAAACTCTGACCTGC GCACTCTTCTCCCCCGCCGCTCAGCT-3′ (mutB), 5′-AGCTGAGCGTTTTGGG AGAAGAGTGCGCAGGTCAGAGTTTTGCGCGCAGC-3′ and 5′-GCTGCGCGCA AAACTCTGACCTGCGCACTCTTCTCCCAAAACGCTCAGCT-3′ (mutAB). The nuclear extract (200 µg) was precleared at room temperature for 1 h with 10 µL of the 4% streptavidin-coated beads mixed with 50% slurry to reduce nonspecific binding. Precleared nuclear extract was incubated with 2 µg of biotinylated DNA oligonucleotides and 20 µL of 4% streptavidin-agarose beads with 50% slurry in 400 µL of PBS at room temperature for 1 h with shaking. Beads were then collected by centrifugation at 2000 rpm for 2 min and washed with cold PBS three times. DNA-protein complexes bound to the beads were eluted with 30 µL of SDS sample buffer. Nuclear proteins were denatured by putting on dry bath at 95°C for 5 min and subjected to western blot analysis probed with anti-Sp1 antibody.
Statistical analysis
Means and standard deviations of samples (performed in triplicate) were calculated from the numerical data generated in this study. Significant differences between treatments were determined by Student’s t-test.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by research grants from National Health Research Institutes (NHRI-EX100–9830BI) and National Science Council (NSC 99–2321-B-002–016-MY2), Taiwan.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/19373
References
- 1.Hansen TH, Huang S, Arnold PL, Fremont DH. Patterns of nonclassical MHC antigen presentation. Nat Immunol. 2007;8:563–8. doi: 10.1038/ni1475. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004;114:1379–88. doi: 10.1172/JCI23594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Exley MA, Lynch L, Varghese B, Nowak M, Alatrakchi N, Balk SP. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: reversing tumor-induced defects. Clin Immunol. 2011;140:184–95. doi: 10.1016/j.clim.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bleicher PA, Balk SP, Hagen SJ, Blumberg RS, Flotte TJ, Terhorst C. Expression of murine CD1 on gastrointestinal epithelium. Science. 1990;250:679–82. doi: 10.1126/science.1700477. [DOI] [PubMed] [Google Scholar]
- 5.Brossay L, Jullien D, Cardell S, Sydora BC, Burdin N, Modlin RL, et al. Mouse CD1 is mainly expressed on hemopoietic-derived cells. J Immunol. 1997;159:1216–24. [PubMed] [Google Scholar]
- 6.Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SM, et al. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology. 2000;100:37–47. doi: 10.1046/j.1365-2567.2000.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandal M, Chen XR, Alegre ML, Chiu NM, Chen YH, Castaño AR, et al. Tissue distribution, regulation and intracellular localization of murine CD1 molecules. Mol Immunol. 1998;35:525–36. doi: 10.1016/S0161-5890(98)00055-8. [DOI] [PubMed] [Google Scholar]
- 8.Roark JH, Park SH, Jayawardena J, Kavita U, Shannon M, Bendelac A. CD1.1 expression by mouse antigen-presenting cells and marginal zone B cells. J Immunol. 1998;160:3121–7. [PubMed] [Google Scholar]
- 9.Chen QY, Jackson N. Human CD1D gene has TATA boxless dual promoters: an SP1-binding element determines the function of the proximal promoter. J Immunol. 2004;172:5512–21. doi: 10.4049/jimmunol.172.9.5512. [DOI] [PubMed] [Google Scholar]
- 10.Chen QY, Zhang T, Pincus SH, Wu S, Ricks D, Liu D, et al. Human CD1D gene expression is regulated by LEF-1 through distal promoter regulatory elements. J Immunol. 2010;184:5047–54. doi: 10.4049/jimmunol.0901912. [DOI] [PubMed] [Google Scholar]
- 11.Geng Y, Laslo P, Barton K, Wang CR. Transcriptional regulation of CD1D1 by Ets family transcription factors. J Immunol. 2005;175:1022–9. doi: 10.4049/jimmunol.175.2.1022. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Ross AC. Retinoic acid regulates CD1d gene expression at the transcriptional level in human and rodent monocytic cells. Exp Biol Med (Maywood) 2007;232:488–94. [PMC free article] [PubMed] [Google Scholar]
- 13.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 14.Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, et al. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. 1999;19:6415–26. doi: 10.1128/mcb.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 16.Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet. 2003;34:187–92. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- 17.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 18.Pan LN, Lu J, Huang B. HDAC inhibitors: a potential new category of anti-tumor agents. Cell Mol Immunol. 2007;4:337–43. [PubMed] [Google Scholar]
- 19.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 20.Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–42. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 21.Chou CW, Chen CC. HDAC inhibition upregulates the expression of angiostatic ADAMTS1. FEBS Lett. 2008;582:4059–65. doi: 10.1016/j.febslet.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 22.Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000;96:3847–56. [PubMed] [Google Scholar]
- 23.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–24. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 24.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- 25.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–45. doi: 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- 26.Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, et al. Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites. Biochem Biophys Res Commun. 1997;241:142–50. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- 27.Schnur N, Seuter S, Katryniok C, Rådmark O, Steinhilber D. The histone deacetylase inhibitor trichostatin A mediates upregulation of 5-lipoxygenase promoter activity by recruitment of Sp1 to distinct GC-boxes. Biochim Biophys Acta. 2007;1771:1271–82. doi: 10.1016/j.bbalip.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Nunes MJ, Milagre I, Schnekenburger M, Gama MJ, Diederich M, Rodrigues E. Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J Neurochem. 2010;113:418–31. doi: 10.1111/j.1471-4159.2010.06612.x. [DOI] [PubMed] [Google Scholar]
- 29.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–74. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Pufahl L, Katryniok C, Schnur N, Sorg BL, Metzner J, Grez M, et al. Trichostatin A induces 5-lipoxygenase promoter activity and mRNA expression via inhibition of histone deacetylase 2 and 3. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01420.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH, Chen CC. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008;68:2375–83. doi: 10.1158/0008-5472.CAN-07-5807. [DOI] [PubMed] [Google Scholar]
- 32.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 33.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 34.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–43. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, et al. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer. 2007;121:1138–48. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- 36.Blackwell L, Norris J, Suto CM, Janzen WP. The use of diversity profiling to characterize chemical modulators of the histone deacetylases. Life Sci. 2008;82:1050–8. doi: 10.1016/j.lfs.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–8. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 38.Li J, Staver MJ, Curtin ML, Holms JH, Frey RR, Edalji R, et al. Expression and functional characterization of recombinant human HDAC1 and HDAC3. Life Sci. 2004;74:2693–705. doi: 10.1016/j.lfs.2003.09.070. [DOI] [PubMed] [Google Scholar]
- 39.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hix LM, Shi YH, Brutkiewicz RR, Stein PL, Wang CR, Zhang M. CD1d-expressing breast cancer cells modulate NKT cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PLoS One. 2011;6:e20702. doi: 10.1371/journal.pone.0020702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spanoudakis E, Hu M, Naresh K, Terpos E, Melo V, Reid A, et al. Regulation of multiple myeloma survival and progression by CD1d. Blood. 2009;113:2498–507. doi: 10.1182/blood-2008-06-161281. [DOI] [PubMed] [Google Scholar]
- 42.Ozeki M, Shively JE. Differential cell fates induced by all-trans retinoic acid-treated HL-60 human leukemia cells. J Leukoc Biol. 2008;84:769–79. doi: 10.1189/jlb.1207817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy. 2010;6:1057–65. doi: 10.4161/auto.6.8.13365. [DOI] [PubMed] [Google Scholar]
- 44.Motohashi S, Nakayama T. Clinical applications of natural killer T cell-based immunotherapy for cancer. Cancer Sci. 2008;99:638–45. doi: 10.1111/j.1349-7006.2008.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haraguchi K, Takahashi T, Nakahara F, Matsumoto A, Kurokawa M, Ogawa S, et al. CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk Lymphoma. 2006;47:2218–23. doi: 10.1080/10428190600682688. [DOI] [PubMed] [Google Scholar]
- 46.Metelitsa LS, Weinberg KI, Emanuel PD, Seeger RC. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia. 2003;17:1068–77. doi: 10.1038/sj.leu.2402943. [DOI] [PubMed] [Google Scholar]
- 47.Shimizu K, Goto A, Fukui M, Taniguchi M, Fujii S. Tumor cells loaded with alpha-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol. 2007;178:2853–61. doi: 10.4049/jimmunol.178.5.2853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.