

Abstract
While numerous studies have examined gene expression changes from homogenates of heart tissue, this prevents studying the inherent stochastic variation between cells within a tissue. Isolation of pure cardiomyocyte populations through a collagenase perfusion of mouse hearts facilitates the generation of single cell microarrays for whole transcriptome gene expression, or qPCR of specific targets using nanofluidic arrays. We describe here a procedure to examine single cell gene expression profiles of cardiomyocytes isolated from the heart. This paradigm allows for the evaluation of metrics of interest which are not reliant on the mean (for example variance between cells within a tissue) which is not possible when using conventional whole tissue workflows for the evaluation of gene expression (Figure 1). We have achieved robust amplification of the single cell transcriptome yielding micrograms of double stranded cDNA that facilitates the use of microarrays on individual cells. In the procedure we describe the use of NimbleGen arrays which were selected for their ease of use and ability to customize their design. Alternatively, a reverse transcriptase - specific target amplification (RT-STA) reaction, allows for qPCR of hundreds of targets by nanofluidic PCR. Using either of these approaches, it is possible to examine the variability of expression between cells, as well as examining expression profiles of rare cell types from within a tissue. Overall, the single cell gene expression approach allows for the generation of data that can potentially identify idiosyncratic expression profiles that are typically averaged out when examining expression of millions of cells from typical homogenates generated from whole tissues.
Keywords: Molecular Biology, Issue 58, Single cell analysis, Microarray, Gene expression, Cardiomyocyte, Mouse heart perfusion, mice, qPCR
Protocol
1. Step 1 - Cardiomyocyte isolation
- Prepare the digestion buffer as follows: 10 x Digestion Buffer (made in ultra-pure H2O)
Final Conc. g/L g/500 mL 10x solution g/L NaCl 130 mM 7.597 3.3 75.97 KCl 5 mM 0.4 0.2 4.0 Pyruvic acid 3 nM 0.33 0.165 3.30 HEPES 25 mM 5.96 2.85 59.6 MgCl2 0.5 mM 0.101 0.05 1.01 NaH2PO4monobasic 0.33 mM 0.04 0.02 0.40 Dextrose 22 mM 3.96 1.98 39.6 Using the 10x Digestion Buffer, make the following four solutions for each heart that will be perfused (made in ultra-pure H2O):
Digestion Buffer A (Make three aliquots of this solutions)
- 50 mL - 1x Digestion Buffer containing:
- 75 μL - EGTA solution (100 mM EGTA)
Digestion Buffer B (Make one of these)
- 25 mL - 1x Digestion Buffer containing:
- 1.25 μL - CaCl2 Solution (1 M)
- 1 mg - Protease
- 25 mg - Collagenase (can also use 75% of this amount = 18.75 mg)**
Collection Buffer (Make one of these)
- 2.5 mL - 1x Digestion Buffer containing:
- 1.25 μL - CaCl2 Solution (1 M)
- 0.5 mg - Protease
- 15 mg - Collagenase (75% = 11.25 mg)**
- 125 mg - BSA
Neutralization Wash Buffer (Make one of these)
- 25 mL - 1x Digestion Buffer containing:
- 6.25 μL - CaCl2 Solution (1 M)
- 250 mg BSA
** Depending on the source and batch of collagenase, the enzyme activity may vary. It may be necessary to optimize the amount of enzyme added to this buffer to prevent over-digestion.
Set up the isolation system as previously described in detail 1,2 with three 15 mL beakers of Digestion Buffer A on ice, one 50 mL tube of Digestion Buffer A and the 25 mL Digestion Buffer B ready for perfusion (Digestion Buffer A should be flushed through the line, at minimum of 5 mL run through the system), Collection Buffer warmed in a water bath to raise the temperature to 37°C, and the Neutralization Wash Buffer at RT.
Fill the perfusion cannula and the dissection dish where the heart will be cleaned and tied onto the perfusion tip with cold Digestion Buffer A. Loosely knot some suture (4-O to 6-O size) around the upper portion of the cannula. This will later be used to hold the aorta to prevent the heart slipping off the cannula.
Terminally sedate the mouse with an I.P. injection of anesthetic agent (0.25 mL Sodium Pentobarbital solution) and ensure the mouse is unconscious and unresponsive to pain stimuli. Cut open the thoracic cavity carefully along the thoracic margin by cutting the diaphragm (Figure 2). Cut superiorly upwards and reflect the anterior ribcage (these incisions are designated B and A, respectively in Figure 2). Critical - the mouse must still be breathing prior to these incisions.
Using a plastic pipette cut to size, gently suck the heart into the pipette. Cut the heart out of the thoracic cavity being careful to leave enough of the aorta on the heart to clearly identify the aorta branch (Figure 3). A pipette is used since it causes minimal damage to the heart.
Drop the heart into one beaker of ice cold Digestion Buffer A and rinse the blood away. After 15 - 45 sec place the heart into the second beaker of ice cold Digestion Buffer A to briefly rinse again. Transfer the heart into the petri dish also filled with cold Digestion Buffer A.
View the heart in the dish of Digestion Buffer A under the dissection scope and carefully isolate the aorta and its branches. Carefully trim just inferior to the last branch (Figure 4).
Fit the cannula tip into the aorta and ligate in place with the 4-O to 6-O suture using the stage setup under a dissection microscope. Ensure that the cannula does not sit too low within the aorta and remains above the coronary arteries (Figure 4).
With Digestion Buffer A flowing through the perfusion setup, place the perfusion tip on to the setup, and begin to perfuse the heart until the majority of blood is washed from the heart and fluid leaving the heart is clear. Critical - the temperature of the perfusion fluid must be as close as possible to 37°C when it comes out of the catheter. If the solution is too hot or too cold then the perfusion will kill the tissue. The time until perfusion of the heart is also critical. The time window must be less than 5-10 min from removal of the heart until perfusion begins. The heart must be removed under anesthetic, not CO2 affixation or cervical dislocation.
Once the blood is washed from the heart, switch to the Digestion Buffer B from the Digestion Buffer A. Ensure that the Digestion Buffer B is now flowing through the heart. The solution will begin to have a yellow tint. Wait 2-3 min until the heart shows signs of losing its shape and starts to look rounded which is an indication that the digestion is working. The myocardium should appear lighter as the perfusion proceeds.
Cut the ventricles off with scissors and place into a 50 mL conical tube containing 15 mL of Collection Buffer at 37°C. Gently cut the ventricle into small pieces while placing the tissue into the Collection Buffer. Mix gently with a plastic pipette to dissolve the cell-cell adhesions and make a single cell cardiomyocyte mixture.
When most of the tissue is dissolved, (less than 1 min) let the larger tissue pieces sink to the bottom. Remove and save the supernatant to a 15 mL conical tube. Let gravity pull a pellet from the supernatant over 10 to 20 min (15 min preferred) at room temperature.
Remove and discard the supernatant. Wash pellet by adding 5 mL of the Neutralization Wash Buffer to the pellet and gently mix the cells to resuspend them. At this point the cells are immediately ready to be selected for single cell analysis (proceed to step 2).
2. Step 2 - Isolation of single cells
Prepare a micromanipulator using a flame and a glass capillary tube which will be used to manipulate cells. The micromanipulator is simply a very fine capillary tube which is then attached to a long piece of flexible tubing which can move or pick up cells when suction is applied.
Immediately proceed to selection of the individual cells under a microscope (Nikon Eclipse TS100, 20x) using a micromanipulator which was created with a pulled pipette and an airlock to prevent material from being transferred to the single cell samples sample. Ensure that the cell population is healthy (70% viable) and not damaged from the isolation process.
Dilute the cells down in a small Petri dish so that they may be selected individually. When picking cells be sure to capture the cells in a small volume (less than 2 μL). Select only healthy cells that have the distinctive normal cardiomyocyte morphology (Figure 5).
Place individual cells into the bottom of individual PCR tubes and freeze them immediately on dry ice. These cells are then stored at -80°C until they are to be used for gene expression analysis.
3. Step 3A - Single cell qPCR gene expression
To perform qPCR on single cells employ a protocol adapted from one developed for single cell gene expression by Fluidigm Corporation for their Biomark Nanofluidic qPCR arrays (Fluidigm - Advance Development Protocol #30)3,4.
Preparing the Reverse transcription - Specific Target Amplification (RT-STA). To begin, resuspend all primer pairs (we have used up to 125 primer pairs successfully) for the genes of interest at 100 μM in 1x DNA suspension buffer (10 mM Tris, pH 8.0, 0.1 mM EDTA). Combine and further dilute the forward and reverse primer pairs to 20 μM for each assay. Lastly, dilute all primer pairs into one solution with a final concentration of 200 nM for each assay that will be amplified in the RT-STA reaction. To do this, take each primer pair and dilute them 1:100. For example, if you have 30 assay primer pairs at 20 μM, you will need to add 1 μL of each pair (30 μL total), and then make up the remainder of the volume with 70 μL of DNA suspension buffer to reach the final dilution volume of 100 μL.
- Now that the primer solutions are prepared, assemble the RT-STA reaction master mix. Prepare enough master mix for each of the individual cells you wish to amplify.
Reagent Volume in μL (per reaction) CellsDirect 2x Reaction mix 5.0 SuperScript III RT Plantinum Taq mix 0.2 RT-STA Primer mix (200 nM each assay) 2.5 Nuclease Free H2O (or 1x DNA suspension buffer) 1.3 Total 9.0 - Immediately remove frozen cells and centrifuge at maximum RCF for 30 seconds to ensure that the cells are positioned at the bottom of the tube. Add 9 μL of RT-STA mix to the tube and proceed to the reaction in a thermocycler.
- 50°C - 15 minutes
- 95°C - 2 minutes
- 18 cycles of
- 95°C for 15 seconds
- 60°C for 4 minutes
- Hold at 4°C
- Remove the tubes from the PCR machine and add 4 μL of ExoSAP-IT to the reaction. ExoSAP-IT is a PCR cleanup reagent which will remove excess primers and enzyme from the RT-STA reaction. Run the tubes through the following reaction in a PCR machine.
- 37°C - 15 minutes
- 80°C - 15 minutes
- Hold at 4°C
When the ExoSAP-IT treatment is complete; dilute the samples 1:5 with DNA suspension buffer. The samples are now prepared for qPCR analysis. We use this material in conjunction with Fluidigm's Gene expression qPCR Arrays (48.48, or 96.96 plates). It is also possible to use this approach with more traditional qPCR instrumentation (96 well or 384 well formats). However, these instruments typically require much more sample input which restricts the number of assays that can be performed from a single cell.
3. Step 3B - Whole transcriptome amplification and microarray of single cells
Extraction and Amplification
Use Sigma's WTA2 kit (Cat# WTA2) to amplify double stranded cDNA from both pellets and single cells. Single cells are "extracted" via the first step of Sigma's WTA2 protocol according to manufacturer's instructions. During this first step the incubation with library synthesis buffer at 37°C for 5 minutes disrupts the membrane of the single cells and "extracts" the message for amplification.
Amplify single cells in two rounds, using the directions from the Sigma-Aldrich's WTA2 kit. Perform the library prep step according to the manufacturer's protocol; then add 1/5 of the library sample to 70 μL of the WTA2 amplification Master Mix. Amplify the samples for 25 cycles using the recommended cycling parameters.
Purify the samples using Qiagen's QIAquick PCR Purification Kit (Cat# 28104) and process on Qiagen's Qiacube using the "Cleanup QIAquick PCR Amplification Reactions Standard V4" protocol.
Concentrate the purified samples to 5 μL, using a speed vacuum, and put through a second round of amplification for 17 cycles (WTA2 amplification protocol and cycling parameters repeated).
Purify samples using the QIAquick PCR Purification Kit and check quality using a NanoDrop spectrophotometer and Agilent's BioAnalyzer DNA 7500 kit according to manufacturers' protocol.
Labeling and NimbleGen Gene Expression Arrays
Label 2 μg of double stranded cDNA from each amplified cell sample using NimbleGen's One Color Labeling Kit (Cat# 05223555001) according to the manufacturer's protocol.
Hybridize 5 μg of Cy3 labeled sample to Mus musculus 12x135k, NimbleGen Gene Expression Arrays (Cat# 05543797001) according to manufacturer's protocol. After hybridization of the samples to the arrays, the slides should then be washed and dried before they can be imaged on a laser scanner.
Scan Gene Expression Arrays using a laser scanner. Here, we use a Molecular Devices GenePix 4200A Scanner with settings of 100POW, and 300-350PMT. Then generate the pair files for each array using NimbleGen's NimbleScan software and subsequently analyze for whole transcriptome expression from each single cell array (a table of typical stably expressed cardiomyocyte transcripts is included as a supplement; Table S1).
If the heart perfusion worked well there should be a high percentage of healthy heart cells that retain their typical rectangular morphology upon isolation. If the perfusion did not proceed well then there will be a large percentage of dead cells (see images in Figure 5). If the cells are correctly amplified using either the WTA2 or RT-STA methods then the end products should pass subsequent quality control tests for quality of mRNA samples by NanoDrop and BioAnalyser (Figure 6). For the microarray workflow, this is completed after WTA amplification where the mRNA is tested by both NanoDrop and Bioanalyzer. Representative positive results for this analysis are shown in Figure 6. The WTA2 samples should show a robust amplification with both the NanoDrop spectrophotometer reading, as well as the electropherogram readout from the Bioanalyzer (BA) chip (Figure 6). A table of genes which were stably detected via microarray of single cells is included as an example (Table S1). For the qPCR process, the quality of the data can be assessed by performing a melt step at the end of the PCR reaction to ensure that the primer sets are amplifying the desired product (Figure 7). If correct, this melt step should generate one specific melt peak. To illustrate this point, a subset of nanofluidic qPCR data is shown in a heatmap of peak melt temperature (Figure 8) 8. It is possible to assess the quality of a PCR product by its melt temperature to ensure the absence of non-specific products 5. Each row of this map represents one cell sample (S1-S40) tested in 43 separate qPCR assays (A1-A43). In this figure, it is clear that some assays are quite variable and are probably less reliable than the more stable assays with higher PCR specificity.
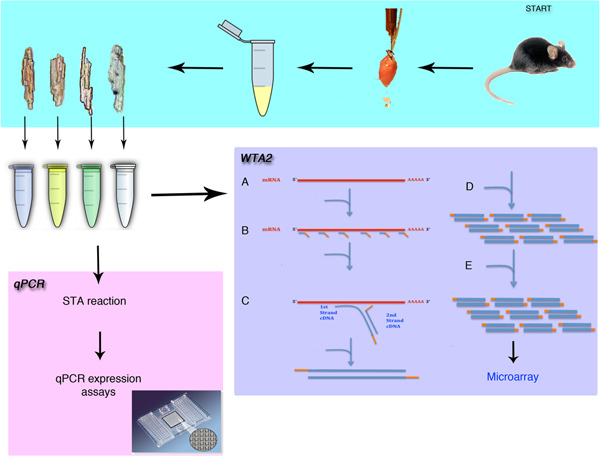 Figure 1. Overview of the single cell isolation and gene expression analysis. The procedure will demonstrate the surgical removal of the mouse heart and the isolation of individual cardiomyocytes. The methods discussed in this procedure include the methods for either qPCR or microarray analysis after whole transcriptome amplification (WTA). The WTA procedure begins with the lysis (A) then the binding of Sigma's universal primers (B) to the mRNA pool. The extension of these primers (C) and the amplification (D) phase create micrograms of amplified material. This amplification is then repeated (E) to generate enough material for the microarray procedure.
Figure 1. Overview of the single cell isolation and gene expression analysis. The procedure will demonstrate the surgical removal of the mouse heart and the isolation of individual cardiomyocytes. The methods discussed in this procedure include the methods for either qPCR or microarray analysis after whole transcriptome amplification (WTA). The WTA procedure begins with the lysis (A) then the binding of Sigma's universal primers (B) to the mRNA pool. The extension of these primers (C) and the amplification (D) phase create micrograms of amplified material. This amplification is then repeated (E) to generate enough material for the microarray procedure.
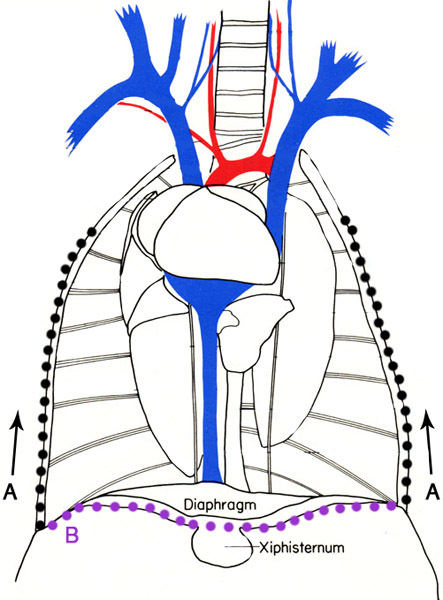 Figure 2. Representation of the location of the incisions to remove the heart from the mouse. Cut along the lateral wall of the rib cage (A), then cut along the inferior margin of the rib cage (B). Cutting the vessels above and below the heart allow for removal while still leaving enough of the aorta to cannulate the heart. Images adapted from M.J. Cook 6.
Figure 2. Representation of the location of the incisions to remove the heart from the mouse. Cut along the lateral wall of the rib cage (A), then cut along the inferior margin of the rib cage (B). Cutting the vessels above and below the heart allow for removal while still leaving enough of the aorta to cannulate the heart. Images adapted from M.J. Cook 6.
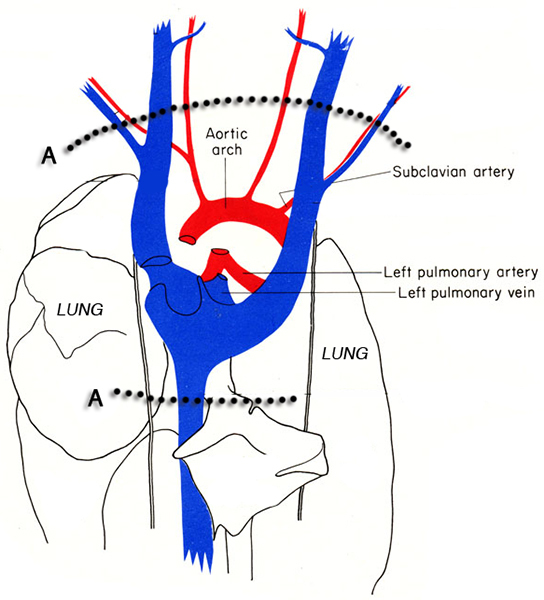 Figure 3. Diagram of the thorax of the mouse. The dotted lines indicate the locations of the incisions (A) for excising the heart from the thoracic cavity without damaging the heart tissue. Images adapted from MJ Cook 6.
Figure 3. Diagram of the thorax of the mouse. The dotted lines indicate the locations of the incisions (A) for excising the heart from the thoracic cavity without damaging the heart tissue. Images adapted from MJ Cook 6.
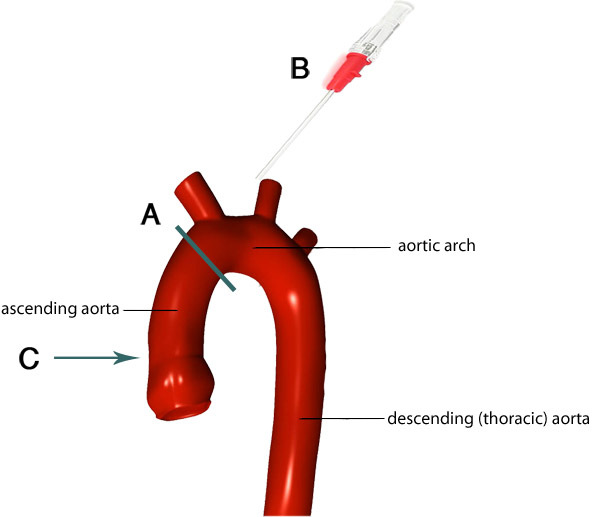 Figure 4. Image of the aortic arch and its branches. The gray line indicates the ideal location of where to trim the aorta (A). The remaining aorta attached to the heart is cannulated so that the tip of the cannula (B) goes to the appropriate level within the aorta (C). Images adapted from F. Gaillard 7.
Figure 4. Image of the aortic arch and its branches. The gray line indicates the ideal location of where to trim the aorta (A). The remaining aorta attached to the heart is cannulated so that the tip of the cannula (B) goes to the appropriate level within the aorta (C). Images adapted from F. Gaillard 7.
 Figure 5. Selection of single cells for gene expression analysis. Each panel shows cardiomyocytes imaged under the light microscope showing typical morphology of cardiomyocytes. Healthy cardiomyocytes are indicated by the green arrows. Cells which are dead or dying are shown with red arrows. These dead cells should not be used in the analysis.
Figure 5. Selection of single cells for gene expression analysis. Each panel shows cardiomyocytes imaged under the light microscope showing typical morphology of cardiomyocytes. Healthy cardiomyocytes are indicated by the green arrows. Cells which are dead or dying are shown with red arrows. These dead cells should not be used in the analysis.
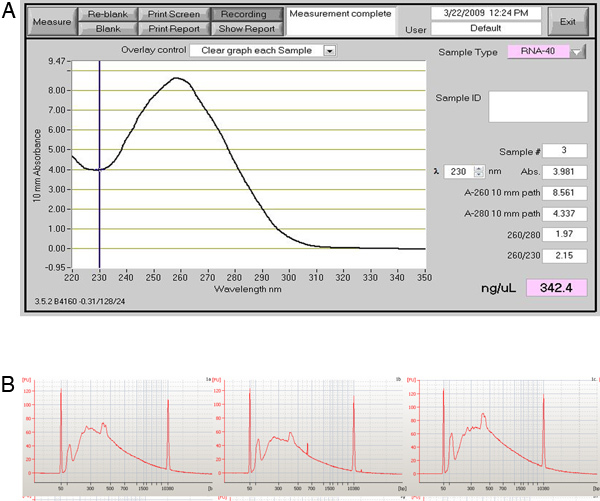 Figure 6. Quality control WTA results for the single cell amplification. (A) Image of a NanoDrop spectrophotometer readout that is appropriate for the amplified transcripts from the WTA reaction. (B) The readout from BioAnalyzer chip shows the results from three amplified cells.
Figure 6. Quality control WTA results for the single cell amplification. (A) Image of a NanoDrop spectrophotometer readout that is appropriate for the amplified transcripts from the WTA reaction. (B) The readout from BioAnalyzer chip shows the results from three amplified cells.
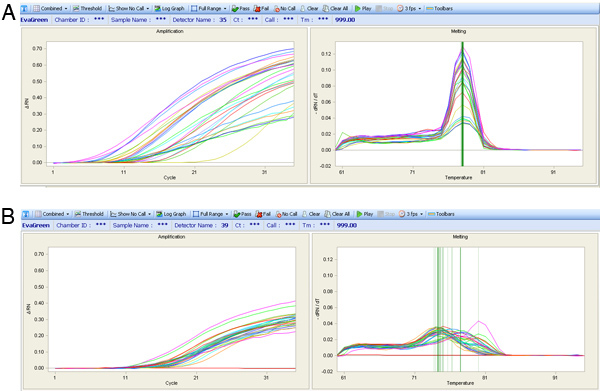 Figure 7. qPCR amplification curves (Left chart) and peak melt temperature curves (Right chart). (A) The expression results from a quality assay are shown with a single melt peak despite different amplification curves. (B) Melt curves for a poor primer set, which has highly variable melt peaks which is not ideal for expression analysis.
Figure 7. qPCR amplification curves (Left chart) and peak melt temperature curves (Right chart). (A) The expression results from a quality assay are shown with a single melt peak despite different amplification curves. (B) Melt curves for a poor primer set, which has highly variable melt peaks which is not ideal for expression analysis.
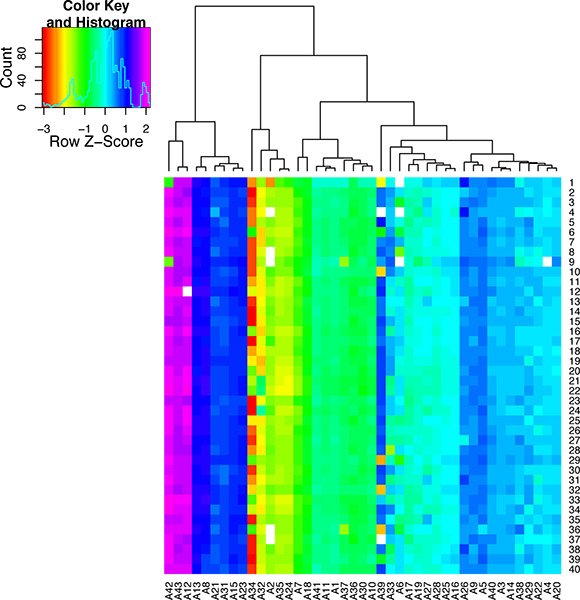 Figure 8. Quality control results for the single cell qPCR reactions. This heat map was created to display the melt temperatures as a color scale. The peak melting temperature for each qPCR assay (A1-A43) is shown for 40 individual cells (S1-40). The assays were clustered according their melt temperature. By looking down the column for each assay it is possible to see the variation melt across all the samples. This figure demonstrates that some qPCR assays are very specific while others are highly variable and thus are not suitable for single cell analysis.
Figure 8. Quality control results for the single cell qPCR reactions. This heat map was created to display the melt temperatures as a color scale. The peak melting temperature for each qPCR assay (A1-A43) is shown for 40 individual cells (S1-40). The assays were clustered according their melt temperature. By looking down the column for each assay it is possible to see the variation melt across all the samples. This figure demonstrates that some qPCR assays are very specific while others are highly variable and thus are not suitable for single cell analysis.
Table S1. Supplemental table of microarray data. These genes are listed according to the robustness in which they are detected in single cardiomyocytes across a large dataset. This gene list indicates the sensitivity of detection for a number of genes that are typically expressed in single cardiomyocytes.
Discussion
This method has the possibility to generate cardiomyocytes for a number of functional as well as gene expression studies. The examination of single cells is a burgeoning area in gene expression analysis. The advantage of examining transcriptional levels at the level of the single cell is that it allows the examination of a pure cell population, which is not possible from whole tissue preparations. Additionally, single cell analysis allows for the examination of stochastic variation of mRNA levels in individual cells to define cell populations that were previously thought to be homogeneous 8,9. In addition to identifying genes which are potentially stochastic in their gene expression 10,11,12, the method also allows for the identification of rare cell populations defined by their gene expression profiles.
While this method provides a number of exciting potential uses in gene expression analysis, there are some caveats and considerations in the use of single cells. The primary limitation to single cell analysis, is the collection of enough cells in the experiment to achieve statistical significance with regards to the metric of interest, for example variance. In cases where the numbers of cells isolated from the tissue of interest is not a limitation, one can examine hundreds of cells per group, using approaches such as next-generation sequencing, or nanofluidic arrays. However, in some cases, there may be difficulty in obtaining sufficient cells from the tissue of interest. This can in part be caused by idiosyncratic time intensive procedures for the collection of the targeted cell type. However, careful planning and preparation prior to proceeding with single cell collection may still allow for rigorous single cell analysis with limited technical error. When examining the results it must be considered, that even though this procedure for isolating cells has been used in numerous studies (including microscopy, electrophysiology, etc.), the isolation itself could potentially effect gene expression. As with any method which manipulates biological samples, the results of your findings should be carefully validated to ensure the single cell expression is representative of the tissue itself and not technical bias. Methods such as in situ hybridization may prove useful to verify these results in the intact tissue. Lastly, it is critical to ensure that the data generated is carefully checked for quality control. The data shown in Figure 8 demonstrates that qPCR assays may be very robust in their specificity, such as assay #13 (A13) or have a high level of variability which can lead to technical variance such as assay #34 (A34).
Disclosures
Free access and production of this of this article is sponsored Sigma-Aldrich.
Acknowledgments
The authors would like to acknowledge the technical assistance of C. Zambataro during the filming of this protocol. We gratefully acknowledge the support of the Glenn Foundation for Medical Research (S.M.), The Hillblom foundation, and the National Institutes of Health for a Nathan Shock Center award (P30AG025708) and PO1AG025901. J.M.F. was supported by T32AG000266 awarded to the Buck Institute for Research on Aging.
References
- Wolska BM, Solaro RJ. Method for isolation of adult mouse cardiac myocytes for studies of contraction and microfluorimetry. Am. J. Physiol. 1996;271(3 Pt 2):H1250–H1255. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- Shioya T. A simple technique for isolating healthy heart cells from mouse models. J. Physiol. Sci. 2007;57(6):327–335. doi: 10.2170/physiolsci.RP010107. [DOI] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell. 2010;18(4):675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Narsinh KH, Sun N, Sanchez-Freire V, Lee AS, Almeida P, Hu S, Jan T, Wilson KD, Leong D, Rosenberg J, Yao M, Robbins RC, Wu JC. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J. Clin. Invest. 2011;121(3):1217–1221. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50(4):262–270. doi: 10.1016/j.ymeth.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Cook MJ. Informatics. Academic Press; 1965. The Anatomy of the Laboratory Mouse. [Google Scholar]
- Gaillard F. Aorta. Radiopaedia. 2008.
- Ståhlberg A, Andersson D, Aurelius J, Faiz M, Pekna M, Kubista M, Pekny M. Defining cell populations with single-cell gene expression profiling: correlations and identification of astrocyte subpopulations. Nucleic Acids Res. 2011;39(4):e24–e24. doi: 10.1093/nar/gkq1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong JF, Chen Y, Marcus JS, Scherer A, Quake SR, Taylor CR, Weiner LP. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab Chip. 2008;8(1):68–74. doi: 10.1039/b712116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441(7096):1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135(2):216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M, Levine A, Siggia E, Swain P. Stochastic gene expression in a single cell. Science. 2002;297(5584):1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]