

Background: CaMKII is up-regulated in heart failure and modulates Na+ current (INa), yet the mechanism is unclear.
Result: CaMKII phosphorylates several sites in the first intracellular loop of NaV1.5, thereby altering INa gating properties.
Conclusion: This multisite phosphorylation may contribute to acquired arrhythmogenesis.
Significance: Identification of these regulatory sites is critical for potential therapeutic targeting of CaMKII and NaV1.5 in failing hearts.
Keywords: Calcium Signaling, CaMKII, Ion Channels, Protein Phosphorylation, Sodium Channels, Arrhythmia, Na+ Current, Ca2+/Calmodulin-dependent Protein Kinase
Abstract
The cardiac Na+ channel NaV1.5 current (INa) is critical to cardiac excitability, and altered INa gating has been implicated in genetic and acquired arrhythmias. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is up-regulated in heart failure and has been shown to cause INa gating changes that mimic those induced by a point mutation in humans that is associated with combined long QT and Brugada syndromes. We sought to identify the site(s) on NaV1.5 that mediate(s) the CaMKII-induced alterations in INa gating. We analyzed both CaMKII binding and CaMKII-dependent phosphorylation of the intracellularly accessible regions of NaV1.5 using a series of GST fusion constructs, immobilized peptide arrays, and soluble peptides. A stable interaction between δC-CaMKII and the intracellular loop between domains 1 and 2 of NaV1.5 was observed. This region was also phosphorylated by δC-CaMKII, specifically at the Ser-516 and Thr-594 sites. Wild-type (WT) and phosphomutant hNaV1.5 were co-expressed with GFP-δC-CaMKII in HEK293 cells, and INa was recorded. As observed in myocytes, CaMKII shifted WT INa availability to a more negative membrane potential and enhanced accumulation of INa into an intermediate inactivated state, but these effects were abolished by mutating either of these sites to non-phosphorylatable Ala residues. Mutation of these sites to phosphomimetic Glu residues negatively shifted INa availability without the need for CaMKII. CaMKII-dependent phosphorylation of NaV1.5 at multiple sites (including Thr-594 and Ser-516) appears to be required to evoke loss-of-function changes in gating that could contribute to acquired Brugada syndrome-like effects in heart failure.
Introduction
Inward Na+ current (INa)4 in the heart is produced primarily by the NaV1.5 channel. INa is responsible for the rapid upstroke of the cardiac myocyte action potential and for rapid propagation of depolarization throughout the heart. INa normally activates and inactivates very rapidly, but genetic or acquired alterations in cardiac sodium channel gating can lead to long QT syndrome (where INa fails to completely inactivate) and Brugada syndrome (where INa availability is reduced), which can predispose patients to ventricular arrhythmias (1). Moreover, one human NaV1.5 mutation (1795InsD) is associated with both long QT syndrome at low heart rates and Brugada syndrome at high heart rates (2). These genetic channelopathies, although relatively rare, have been extremely important in understanding the arrhythmogenic basis of channel gating alterations. Genetically normal channels may exhibit altered channel gating due to acquired post-translational modifications that may accompany common diseases, such as ischemic heart disease and heart failure (3).
Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) expression and activity levels are increased in both animal models of heart failure and in failing human hearts (4–7). CaMKII also co-immunoprecipitates with and phosphorylates NaV1.5 in cardiomyocytes (8). Overexpression of δC-CaMKII, the predominant cardiac myocyte isoform, either acutely in rabbit myocytes or transgenically in mouse myocytes, produces complex effects on INa gating (8). Notably, these effects phenocopy the human NaV1.5 mutation (1795InsD), which causes combined long QT syndrome and Brugada syndrome (2). These include a hyperpolarizing shift in voltage dependence of availability, enhanced intermediate inactivation, and slowed recovery from intermediate inactivation, all of which are loss-of-function effects, consistent with Brugada syndrome (8). In addition, CaMKII increases late or persistent INa, which has also been reported in stressed myocytes and in heart failure (9) and resembles long QT syndrome. These effects were blocked by acute CaMKII inhibition, confirming that CaMKII-dependent phosphorylation is involved. Because CaMKII is up-regulated in heart failure and can modulate NaV1.5, understanding its mechanism of action is essential to understanding the physiology and pathophysiology of INa gating.
Here, we sought to identify the molecular target of CaMKII-dependent regulation of INa. Biochemical approaches were used to identify sites at which CaMKII phosphorylates NaV1.5, and whole cell patch clamp recordings of wild-type (WT), non-phosphorylatable, and phosphomimetic mutant NaV1.5 were used to test for CaMKII-induced INa gating changes.
EXPERIMENTAL PROCEDURES
Recombinant CaMKII
α-CaMKII was expressed using a baculoviral expression system in insect cells and purified as described previously (10, 11). δC-CaMKII (expressed with a His6 tag in the baculoviral expression system) was applied to Ni+-NTA-agarose and eluted using an imidazole gradient (0–1 m). Fractions containing the kinase were then added to a CaM-Sepharose column in the presence of 50 mm HEPES, pH 7.4, 0.1 mm EDTA, and 2 mm CaCl2, washed, and eluted in 50 mm HEPES, pH 7.4, 400 mm NaCl, 0.1 mm EDTA, and 10 mm EGTA. A final gel filtration column purification step was performed as described previously (10, 11). Monomeric δC-CaMKII (δC-CaMKIImono) with an intein fusion tag was expressed in Rosetta2 DE3 bacteria, lysed, and enriched using a chitin affinity column. The kinase was eluted in 50 mm HEPES, pH 7.4, 0.1 mm EDTA, and 50 mm DTT and subsequently applied to a CaM-Sepharose column/gel filtration column as described above for δC-CaMKII.
CaMKII Phosphorylation of NaV1.5
For NaV1.5-stably expressing HEK293 cell lysates, 10-cm dishes were lysed in 20 mm Tris, pH 7.4, 200 mm NaCl, 0.1 mm EDTA, and 1% Triton X-100, and NaV1.5 was immunoprecipitated using polyclonal NaV1.5 antibody (Alomone, catalog no. ASC-005) and protein A/G-agarose. The immunoprecipitated proteins were extensively washed in lysis buffer, and the immunoprecipitates were phosphorylated with 1 μg of α-CaMKII in reaction mix (50 mm HEPES, pH 7.4, 100 mm NaCl, 10 mm MgCl2, 100 μm ATP, 2 mm CaCl2, 5 μm CaM, and [γ-32P]ATP (10 μCi/reaction) (12). After a 4-min incubation at 30 °C, the reaction was quenched with 50 mm Tris, pH 7.4, and 50 mm EDTA in phosphate-buffered saline. Immobilized proteins were collected by centrifugation, and the reaction mix was removed and extensively washed in quenching buffer. Immobilized proteins were solubilized in 4× LDS sample buffer and β-mercaptoethanol. Following gel electrophoresis, Western blotting was performed using monoclonal pan-NaV antibody (Sigma, catalog no. S8809) and Alexa680 goat anti-mouse secondary. The immunoprecipitated NaV1.5 protein was visualized using a LI-COR/Odyssey version 3.0 imaging station, and the phosphorylated proteins were visualized using autoradiography.
CaMKII Phosphorylation of GST-Proteins and Soluble Peptides
GST-tagged intracellular regions of NaV1.5 (N terminus, L1–L3, and C terminus) as well as GST-tagged mutants of the L1 were expressed in Bl-21 cells overnight at 16 °C using 0.1 mm isopropyl 1-thio-β-D-galactopyranoside. Cell pellets were lysed in lysis buffer (20 mm Tris, pH 7.4, 200 mm NaCl, 0.1 mm EDTA, and 2× protease inhibitors (Calbiochem, catalog no. 539137)), and GST fusion proteins were bound to glutathione beads in binding buffer (20 mm Tris, pH 7.4, 200 mm NaCl, 1 mm EDTA, and 0.1% Tween 20) for 2 h at 4 °C (12, 13). Following binding, beads were extensively washed (in binding buffer) to remove any unbound lysate. GST-bound fragments were then phosphorylated with 25 ng of δC-CaMKII in reaction mix (see above). δC-CaMKIImono was substituted for δC-CaMKII in Fig. 3, E and F, to avoid potential contaminating quantification of L1 phosphorylation with autophosphorylated δC-CaMKII. After incubation (1 min to 3 h), the reaction was quenched with 50 mm Tris, pH 7.4, and 50 mm EDTA in PBS, beads were extensively washed, and protein fragments were solubilized and denatured using 2× LDS sample buffer with β-mercaptoethanol at 70 °C. Radioactivity of phosphorylated proteins was detected directly on the beads using scintillation counting as described previously (13). Gel electrophoresis followed by Coomassie staining allowed for the visualization of GST fusions. CaMKII phosphorylation of GST fusions was visualized in the gel with autoradiography or phosphorimaging using MultiGauge version 3.0. Soluble peptides from different regions of the intracellular loop 1 (control, EEQNQATIAETEEKE; 483/484, KRRKRMSSGTEECGE; 516, NHLSLTRGLSRTSMK; 571, LRRTSAQGQPSPGTS; 593/594, LHGKKNSTVDCNGVV; and 571*, LLVPWPLRRTSAQGQ) were purchased from New England Peptide (Gardner, MA) or synthesized/HPLC-purified and reconstituted in dimethyl sulfoxide. For soluble peptide experiments, δC-CaMKII (25 ng) was incubated with the same reaction mix listed above and 100 μm (or 500 μm) peptide of interest for 1 min to 1 h at 30 °C. The solution was then transferred to P-81 filter papers (Whatman, GE Healthcare), washed with 75 mm phosphoric acid, and counted in a Beckman β-counter. The purified α-CaMKII, δC-CaMKII, and δC-CaMKIImono all had specific activity of 4–15 μmol/min/mg, as determined using the peptide AC-2 as a substrate. Statistical analysis of phosphorylation was performed using a one-way ANOVA with post hoc Dunnett's test with a statistical significance accepted at p < 0.05. To determine the stoichiometry of peptide phosphorylation, 100 μm peptide (483/484, 516, 571, and 593/594) was phosphorylated in vitro by 25 ng of δC-CaMKII as described above for 3 h. Using the ATP concentration, specific activity of the in vitro kinase assay, and the known peptide concentrations, the moles of phosphate/mole of peptide was calculated. To determine the stoichiometry of GST-L1 fragment phosphorylation, protein levels in the Coomassie-stained ∼58 kDa band corresponding to full-length GST-L1 were quantified using LI-COR/Odyssey version 3.0. The concentration of GST-L1 protein was quantified against a standard curve of BSA (60 kDa; linear from 0.5 to 10 μg). The moles of GST-L1 were then calculated using the determined concentration and the expected molecular mass of full-length GST-L1 (58.8 kDa). The full-length GST-L1 band was then cut out, and 32P incorporation in the band was quantified using Cerenkov counting in a scintillation counter. Using the ATP concentration and specific activity of the reaction, the moles of ATP/GST-L1 band were calculated and compared with the moles of protein.
FIGURE 3.

CaMKII phosphorylates Ser-516 and Ser-593/Thr-594 in L1. A, phosphor image of immobilized tiled peptide array of L1 after δC-CaMKII phosphorylation with [γ-32P]ATP (darkness indicates 32P incorporation on that peptide). Single-letter amino acid codes are shown. B, phosphorylation intensity of each peptide spot in A, detected using MultiGauge version 3.0. C, soluble peptide (50 μm) phosphorylation by monomeric δC-CaMKII (n = 3, ±S.D. (error bars); *, p < 0.05 versus control peptide). 15-mers were centered near the indicated sites. D, time course of monomeric δC-CaMKII phosphorylation of 500 μm control peptide, original unbiased Ser-571 peptide, the biased 571* peptide (see “Experimental Procedures” for sequence), and 50 μm AC2, a known CaMKII substrate. E, average phosphorylation (n = 3, ±S.D.) of soluble peptides in D at the 1 min time point. F, average phosphorylation (n = 3, ±S.D.) of immobilized peptides with wild-type control, as well as Ser-593/Thr-594 single point Ala mutations and a double-point Ala mutation. S593A/T594A and T594A exhibit decreased phosphorylation compared with wild-type control (n = 3, ±S.D.; *, p < 0.05 versus control Ser-493/Thr-494; one-way ANOVA, post hoc Dunnett's test).
CaMKII Co-immunoprecipitation with NaV1.5
HEK293 cells were transfected with hNaV1.5 and GFP-δC-CaMKII using Lipofectamine 2000. Following a 48-h incubation, the transfected cells were lysed in lysis buffer (see above), centrifuged (14,000 × g at 4 °C for 30 min), and precleared with Protein A- and G-agarose. GFP-CaMKII was immunoprecipitated with monoclonal GFP antibody (Clontech, catalog no. 632375). Mouse IgG was used as a control. Primary antibodies were pulled down using Protein G-agarose. The agarose was washed repeatedly using lysis buffer, and the agarose-bound proteins were solubilized in 4× LDS sample buffer and β-mercaptoethanol. Following gel electrophoresis and transfer, parallel Western blotting was performed using monoclonal GFP and monoclonal pan-NaV antibodies. The proteins were detected using a DyLight800 goat anti-mouse secondary antibody and visualized using a LI-COR/Odyssey version 3.0 imaging station.
CaMKII Targeting to L1 Domain of NaV1.5
GST-tagged intracellular regions of NaV1.5 (N terminus, L1–L3, and C terminus) were bound to glutathione-Sepharose as described (12, 13). Prior to the application of CaMKII, the immobilized proteins on the Sepharose beads were blocked in binding buffer with the addition of 5% BSA. Purified δC-CaMKII (1 μg total (unlabeled and DyLight800-labeled; 3:1 ratio)) was then bound to the beads for 1 h at 4 °C. To ensure that CaMKII concentration was not limited in assays determining the stoichiometry of binding to GST-L1, 20 μg of unlabeled kinase was added to the reaction. For treatment groups with naive CaMKII, δC-CaMKII was diluted in 50 mm HEPES, pH 7.4, and 1 mm EGTA for 2 min on ice prior to the addition to the binding reaction. For treatment groups with autophosphorylated CaMKII, δC-CaMKII was incubated in 50 mm HEPES, pH 7.4, 0.5 mm CaCl2, 5 μm CaM, 5 mm MgCl2, and 1 mm ATP for 2 min on ice and was subsequently added to the binding reactions. Following incubation with δC-CaMKII, the beads were then extensively washed. The beads were then applied to an in vitro CaMKII assay with 20 mm HEPES, pH 7.4, 100 mm NaCl, 2 mm CaCl2, 5 μm CaM, 10 mm MgCl2, 100 μm ATP, 50 μm syntide-2, and 6 μCi/reaction [γ-32P]ATP for 3 min at 30 °C (12). 32P incorporation on syntide-2 was assessed using a Beckman β-counter after transferring the solution to P-81 filter papers and washing unincorporated 32P with 75 mm phosphoric acid. CaMKII binding was also visualized using a LI-COR imaging station to detect DyLight800-labeled CaMKII in the Coomassie-stained electrophoretic gel. Student's t test was performed for statistical comparison with significance accepted at p < 0.05. Stoichiometry of binding was assessed by quantifying protein levels of GST-L1 and δC-CaMKII in the Coomassie-stained gel using LI-COR/Odyssey version 3.0 analysis software. The protein bands at 52 kDa (CaMKII subunit) and 58 kDa (GST-L1) were compared with a standard curve of increasing amounts of δC-CaMKII (linear from 0.5 to 10 μg). This allowed for the determination of each protein concentration. Using this concentration and the predicted molecular weight of each protein, the moles of CaMKII and GST-L1 were calculated.
Peptide SPOTS Arrays
Peptide arrays were constructed using the SPOTS synthesis method (11). Following synthesis, the peptide membrane is blocked at 4 °C overnight in binding buffer plus 5% BSA. δC-CaMKII (∼34 nm) was added to 50 mm HEPES, pH 7.4, 100 mm NaCl, 10 mm MgCl2, 100 μm ATP, 2 mm CaCl2, 5 μm CaM, 3 μCi/ml [γ-32P]ATP. The reaction was incubated for 5 min at room temperature, washed in 75 mm phosphoric acid three times, and visualized using phosphorimaging (Fuji phosphor imager). Phosphorylation of each peptide spot was detected and quantified using MultiGauge version 3.0. Statistical comparison was by one-way ANOVA with post hoc Dunnett's test.
Transfection
Electrophysiological recordings were performed in HEK293 cells (Invitrogen, catalog no. 70507), which provide a well controlled environment to study the direct influence CaMKII activity has on channel function. The HEK293 cells were cultured in DMEM supplemented with 10% FBS and 5% penicillin/streptomycin and passaged to maintain them subconfluent (maximum of 20 passages). One day before transfection, cells were plated on 35 × 50-mm coverslips adapted for electrophysiology. They were transfected using Lipofectamine 2000 (0.5 μl/cm2) in Opti-MEM (Invitrogen) per the manufacturer's instructions, with plasmids (hNaV1.5 WT or mutant, in all cases with the C373S mutation to reduce tetrodotoxin sensitivity) and, where indicated, GFP-δC-CaMKII, all at 0.5 μg/cm2) for 5–6 h and then washed and incubated for at least 24–36 h before experiments. This overexpression within our reduced system mimics physiological disease states, such as heart failure, which exhibit increased CaMKII expression and activity (4–7). Cells that were visibly fluorescent were identified as expressing δC-CaMKII. For all experiments, single cells expressing NaV1.5 were selected based on their appearance and on the presence of INa without secondary artifacts.
Solutions for Electrophysiology
Pipette solutions and recording protocols were standard for Na+ channel electrophysiology as described previously (8). Cells were bathed in Tyrode solution consisting of 140 mm NaCl, 1 mm MgCl2, and 0.5 mm CaCl2 (to help maintain stable pipette seals), with 4 mm CsCl substituted for KCl. The base pipette solution contained 10 mm NaCl, 50 mm CsCl, 90 mm cesium glutamate, 5 mm Tris-ATP, 0.3 mm Li-GTP, 5 mm Cs4-1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, and 10 mm HEPES, titrated to pH 7.2 with CsOH at room temperature. All pipette solutions contained 1 μm calmodulin (Calbiochem/EMD, catalog no. 208690). CaCl2 and MgCl2 were added to yield 1 mm free [Mg2+] and 1 μm free [Ca2+] (high Ca2+ condition in all experiments here) or, in preliminary studies, <100 nm free [Ca2+] (low Ca2+; calculated by MaxChelator). To inhibit CaMKII in select experiments, 1 μm AIP was added to pipette solution. Notably, AIP substantially lower than 1 μm did not unambiguously block δC-CaMKII responses. Pipette and bath solutions were designed with closely matching osmolarity, which stabilized pipette access and improved longevity of recording. All pipette solutions were stored frozen at −80 °C in 500-μl single-use aliquots. Fluoride was not used in the solutions.
Electrophysiological Recording
Whole cell INa was recorded using ruptured patch voltage clamp at room temperature, with an Axopatch 200B amplifier, 1322A interface, and pClamp 8.2 software (Molecular Devices). Seal resistance for successfully recorded cells was typically >5 gigaohms, and uncompensated on-cell access resistance ranged from 1 to 4 megaohms. Pipettes were made from either type 7052 or 8250 glass, were thin-walled and broadly tapered to promote diffusion, and were heat-polished. After patch rupture, 2 min was allowed for diffusion, at which time each cell was checked for expression of INa. Records used experimentally were recorded ≥4 min postrupture, and where possible the order of protocols was standardized to limit possible implicit time dependence. A liquid junction potential between pipette and bath solutions, −9 mV, pipette-negative, originated due to substitution of glutamate for most of the chloride in the pipette solution, and was corrected during recording. Capacitance transients due to pipette and cell membrane were cancelled, and series (access) resistance was compensated to >70%. The quality of transient cancellation and clamp compensation was continuously monitored except during the actual acquisition of data.
Electrophysiological Protocols
At the start of each protocol as well as between episodes, Na+ channels were held at −140 mV, promoting initial rest and subsequent return to the fully available closed state. During intervals between protocols, cells were held at −110 mV to promote longer experimental life.
To establish activation voltage dependence, the current-voltage relation was recorded. Cells were step-depolarized for 50 ms to test potentials between −80 and +30 mV. To measure steady state inactivation voltage dependence, cells were predepolarized for 500 ms to potentials between −140 and −20 mV. The extent of inactivation was measured immediately after the prepulse by applying a −20 mV depolarization for 50 ms. After each test pulse, 140 ms was allowed at −140 mV for recovery, followed by a second −20 mV pulse, which provided a full availability reference current for the particular trace.
Time-dependent properties were measured as follows. Decay of current after its peak at −30 mV during the above current-voltage protocol was used to measure the rate of fast inactivation. Recovery from fast inactivation was studied by applying a 50-ms pulse to −20 mV, which initialized maximal fast inactivation. Recovery after increasing intervals at −140 mV (duration range 0.5–128 ms) was measured by a second pulse to −20 mV. Peak INa recorded during the initial pulse served as a reference for fractional recovery in each episode. INa decay was fit well by a biexponential function (τ1 0.736 ± 0.16 and τ2 2.3 ± 0.16 ms (S.D.) with 89% in the faster component; n = 28) and did not differ among NaV1.5 mutants or with/without AIP. Recovery from fast inactivation was also biexponential (τ1 = 2.98 ± 1.04, τ2 = 149 ± 182 ms (S.D.), with 89% in the faster component; n = 8) and did not differ among NaV1.5 mutants or with/without AIP.
To measure the rate and extent of entry into intermediate and slow inactivated states, pulses at −20 mV were applied for 5 ms to 4.75 s, and recovery at −140 mV was allowed for 20 ms to allow full recovery from rapid inactivation (see “Results”), after which a test pulse at −20 mV was applied. In this protocol, the peak current during the initial inactivating pulse provided a reference against which the ensuing inactivation was measured. Corresponding with the recovery from fast inactivation protocol, recovery from intermediate and slow inactivation was measured using a 3-s inactivating prepulse at −20 mV (shown to be effective at inducing slow inactivation; see “Results”), after which recovery at −140 mV was allowed for durations from 0.5 ms to 3 s. This protocol was preceded and followed by separate reference depolarizations to −20 mV applied under full availability conditions.
Voltage step and time interval sequences in the above protocols were chosen for efficiency. Increments were adjusted to concentrate data in the voltage or time ranges expected to show the most variation, and the total number of episodes was chosen sufficient for robust curve fitting without requiring undue experimental time.
Electrophysiological Analysis
All records were corrected for passive leak. Average current measured during a −140 mV interval (usually the beginning) within each episode when INa was assuredly not active was expressed as a point estimate of conductance (Gleak = Ileak × Vhold) which was then used to infer and subtract leak current at each time and voltage during the rest of the episode.
Activation voltage dependence was used to screen cells for technical errors that would preclude further analysis. In records with good voltage control (based on the slope of the activation curve), we found that half-activation voltage did not differ significantly among transfects or treatments (see “Results”). Leak-corrected peak current versus voltage data were parameterized using the Boltzmann equation,
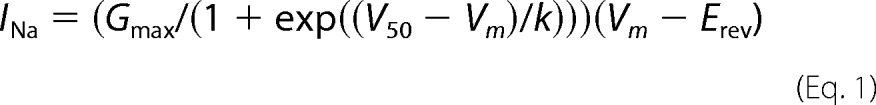 |
in which Gmax is the maximum conductance, V50 is the half-activation voltage, Vm is the test potential, k is the slope of voltage dependence, and Erev is the current reversal potential. Similarly, the steady state inactivation voltage dependence (availability curve) was found by fitting the following,
where Imax is a surrogate for Gmax because test Vm is constant in the inactivation protocol. Time dependences were fit by either decreasing the following,
 |
or increasing the following,
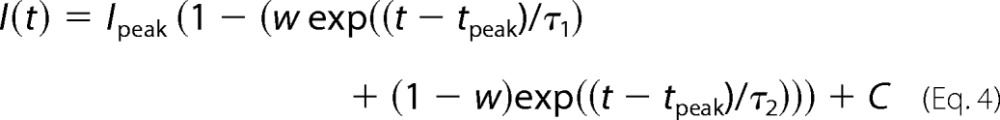 |
biexponential functions, where Ipeak and tpeak were measured directly from records, and w (weight; 0 < w <1), τ1 and τ2 (time constants), and C (asymptotic value) were fit. In some analyses, C or w was constrained to 0, or a plateau was included before a single decreasing exponential.
Preliminary INa experiments under several conditions (pipette solutions with and without AIP and low versus high Ca2+, transfection of hNaV1.5 ± GFP-δC-CaMKII) were used to optimize detection of CaMKII effects (supplemental Fig. S3). All data reported here were recorded with GFP-δC-CaMKII and hNaV1.5 co-transfected and with pipette solutions containing 1 μm CaM and 1 μm free Ca2+ ± 1 μm AIP unless noted otherwise. All data were processed and parameterized for statistical treatment using home-written Microsoft Visual Basic scripts run with Microsoft Excel. We analyzed parameterized data when appropriate with unpaired Student's t tests and one-way or two-way ANOVA followed by post hoc Tukey test for all pairwise comparisons, using the NaV1.5 isoform and the presence of AIP as factors, using GraphPad Prism software, version 5.02 for Windows (GraphPad Software, San Diego, CA).
RESULTS
Identification of CaMKII Phosphorylation Sites on NaV1.5
Sodium channels are multiprotein complexes. The α-subunit (e.g. NaV1.5) contains the channel pore and major determinants of gating and drug binding. These large transmembrane proteins (>220 kDa) contain four domains (I–IV in Fig. 1A), each containing six α-helical transmembrane segments, with a voltage sensor and a pore loop. The NaV1.5 α-subunit has intracellular N and C termini, plus three major intracellular loops connecting domains I–IV, termed L1–L3, respectively (Fig. 1A). When we express human NaV1.5 in HEK293 cells, it is a substrate for recombinant rat α-CaMKII, based on 32P incorporation (Fig. 1B), similar to rodent NaV1.5 immunoprecipitated from heart (8). No CaMKII phosphorylation is observed in untransfected HEK293 cells at the molecular weight for NaV1.5. To narrow down the CaMKII phosphorylation site(s), we constructed GST fusions of the major cytoplasmic regions of hNaV1.5 (L1–L3 and the N and C termini). Bacterially expressed GST fusion proteins were purified and phosphorylated directly on glutathione-Sepharose by recombinant human δC-CaMKII (the predominant cytoplasmic cardiac isoform). Relative expression patterns for these GST fusion proteins are shown in the Coomassie-stained gel in supplemental Fig. S1A (which also shows the predicted molecular weight for each fusion protein). CaMKII phosphorylation assessed by 32P incorporation and autoradiography was limited primarily to the L1 region of NaV1.5, as illustrated by the autoradiographic image (Fig. 1C) and quantified data (Fig. 1D); the L1 domain is reproducibly the best CaMKII substrate tested under these conditions. When phosphorylation of the GST-tagged intracellular regions is normalized to the protein expression level of the predicted molecular weight for each fusion protein, the L1 is still the predominant CaMKII substrate (supplemental Fig. S1B). An estimated 1.59 ± 0.10 mol of phosphate were incorporated/mol of GST-L1, suggesting that there is more than one phosphoacceptor site per full-length L1. These data are in good agreement with recent reports using a similar strategy to confirm L1 as a CaMKII phosphorylation hot spot (14, 15).
FIGURE 1.

CaMKII phosphorylates L1 of NaV1.5. A, human NaV1.5 has five major intracellular regions (N and C termini plus loops 1–3 (L1–L3). Potential CaMKII phosphorylation sites in the L1 are highlighted. Single-letter amino acid codes are shown. B, hNaV1.5 immunoprecipitated from HEK293 cells was exposed to activated α-CaMKII with [γ-32P]ATP. Western blot (left) indicates hNaV1.5 in stably expressing HEK293 cells, and the autoradiogram (right) indicates phosphorylation at NaV1.5 molecular weight. C and D, bacterially expressed GST fusion proteins of intracellular hNaV1.5 regions were exposed to activated δC-CaMKII with [γ-32P]ATP. C, autoradiograph indicates phosphorylation of the L1 after δC-CaMKII phosphorylation. D, average 32P incorporation (n = 3, ±S.D. (error bars)) of GST fusion proteins detected following Cerenkov counting.
CaMKII Binding to L1
The proximity of protein kinases to their substrates is important for the timing and specificity of signal transduction (16). CaMKII was previously shown to coimmunoprecipitate with cardiac NaV1.5 (8). We first verified these findings in HEK293 cells expressing hNaV1.5 and GFP-δC-CaMKII. As expected, δC-CaMKII co-immunoprecipitated with hNaV1.5 (Fig. 2A). No GFP-δC-CaMKII or hNaV1.5 were detected when pulled down with an IgG control (Fig. 2A). Next, we aimed to identify which intracellular domain of NaV1.5 (N terminus, L1–L3, and C terminus) stably interacted with CaMKII. For this, δC-CaMKII (naive, in the presence of Ca2+/CaM, and autophosphorylated) was applied to beads bound with GST-tagged intracellular fragments or GST alone as described previously (11, 12). Gel electrophoresis with Western blot detection of CaMKII indicated that autophosphorylated CaMKII preferentially targets to GST-L1 (Fig. 2B). This finding was verified using DyLight800-labeled δC-CaMKII. Fluorescent detection of CaMKII indicated that autophosphorylated CaMKII forms a stable interaction with the first major intracellular loop of NaV1.5 (Fig. 2C; for full Coomassie-stained gel, see supplemental Fig. S2A). There was appreciably less binding when naive CaMKII (non-activated) was incubated with either GST or GST-L1, suggesting that, analogous to other ion channels and accessory proteins (12, 17–19), CaMKII activation exposes a targeting site for interacting with the L1 domain of hNaV1.5. The stoichiometry of CaMKII binding was 0.92 ± 0.11 mol of kinase subunit (52 kDa)/mol of full-length GST-L1. The binding of CaMKII to L1 was also quantified using an in vitro kinase assay (12, 13). In this assay, an increase in syntide-2 phosphorylation is associated with CaMKII binding to a target protein. A significant increase in syntide-2 phosphorylation was only observed when autophosphorylated CaMKII was incubated with and allowed to bind GST-L1 (Fig. 2D). These data demonstrate for the first time that activated CaMKII forms a stable interaction with the L1 domain of NaV1.5.
FIGURE 2.

Autophosphorylated CaMKII tethers to L1 of NaV1.5. A, HEK293 cells expressing hNaV1.5 and GFP-δC-CaMKII were lysed, pulled down with monoclonal GFP antibody, and subsequently blotted for GFP-δC-CaMKII (right) and hNaV1.5 (left). Full-length hNaV1.5 co-immunoprecipitated (IP) with GFP-δC-CaMKII. B, Western blot detection of δC-CaMKII bound to GST-tagged fragments of the major intracellular loops of hNaV1.5 under naive conditions (top), in the presence of Ca2+/CaM (middle), and when δC-CaMKII was preautophosphorylated (bottom). C, image of 1 μg of Ca2+/CaM/autophosphorylated DyLight800-labeled δC-CaMKII bound to GST-L1 and not GST alone (right lanes). Naive δC-CaMKII (left lanes) did not bind GST-L1. D, average phosphorylation (n = 3, ± S.D. (error bars)) of syntide-2 with GST-L1 pull-down fragments bound to autophosphorylated δC-CaMKII (right bars) and not naive CaMKII (left bars). Data are represented as specific activity with 1 μg of multimeric human δC-CaMKII. *, significant difference compared with GST alone (p < 0.05; t test).
Identification of CaMKII Phosphorylation Sites in L1
There are seven consensus CaMKII phosphorylation sites in five regions of L1 conforming to Arg/Lys-X-X-Ser/Thr (20) (Fig. 1A). However, CaMKII can also phosphorylate non-canonical phosphorylation motifs (21). Therefore, we experimentally scanned the entire L1 domain for potential CaMKII phosphorylation sites using a non-biased overlapping peptide array. A total of 141 peptides were constructed using a robotic peptide synthesizer (Intavis, MultiPep). Each peptide is 15 amino acids in length, and tiled peptides are shifted by two amino acids (13 overlapping amino acids per peptide). Thus, each potential CaMKII phosphorylation motif is represented within multiple peptides, creating several opportunities for the Ser/Thr residues to be phosphorylated as they move in and out of a family of overlapping peptides. These immobilized peptides are synthesized on a modified cellulose membrane using routine Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry (22, 23). Activated δC-CaMKII was applied to the immobilized peptides (11) in the presence of [γ-32P]ATP. Phosphorylated peptides were imaged on autoradiographic film (Fig. 3A) and quantified using phosphorimaging (Fig. 3B). In this non-biased strategy, several overlapping peptide motifs were phosphorylated, including Ser-483/Ser-484, Ser-516, and Ser-593/Thr-594 (Fig. 3, A and B). Our in vitro phosphorylation approach did not recognize Ser-571, a site recently reported to be a CaMKII substrate on NaV1.5 (15).
We further investigated the potential CaMKII phosphorylation sites identified in the immobilized peptide array using soluble peptides (Fig. 3C). For comparison, a peptide containing Ser-571 was also included. Using this assay, only soluble peptides encompassing the region around Ser-516 and Ser-593/Thr-594 exhibited significantly greater 32P incorporation than a control peptide (encompassing residues 417–431 of the L1 domain) (Fig. 3C). The stoichiometry of this phosphorylation following a 3-h incubation was 1.18 ± 0.12 mol of phosphate/mol of Ser-516 peptide and 0.44 ± 0.01 mol of phosphate/mol of Ser-593/Thr-594 peptide. These data suggest that the Ser-516 peptide is a better CaMKII substrate; however, it is also conceivable that the reduced stoichiometry displayed by the 593/594 peptide is due to a Cys residue forming disulfide bonds between peptides. We did not observe significant phosphorylation over background for a soluble peptide harboring the Ser-483/Ser-484 motif (0.01 mol of phosphate/mol of peptide), consistent with previous work showing that this region was not important for CaMKII regulation of NaV1.5 (15). Although we do not fully understand this discrepancy, it is conceivable that the Ser-483/Ser-484 motif is picked up as a substrate using the immobilized assay because multivalent proteins bind to immobilized peptides with a much higher affinity on a two-dimensional surface (24). As observed with immobilized peptides, a soluble peptide containing the Ser-571 motif was again not phosphorylated above background (Fig. 3C), with only 0.02 mol of phosphate incorporated/mol of peptide. However, in this Ser-571 peptide, the phosphorylation site was located toward the N-terminal end of the peptide (as derived from the unbiased immobilized array; see “Experimental Procedures” for sequences). Because it was possible that this unbiased approach resulted in a peptide that was missing a component of the CaMKII targeting sequence, another peptide with the Ser-571 phosphorylation site at the center of the sequence was generated, termed 571*. Similar to the original Ser-571 peptide, 571* was not a CaMKII substrate in vitro even when incubation was extended from 1 to 60 min or when the working peptide concentration was increased to 500 μm (Fig. 3, D and E). Thus, Ser-571 is not phosphorylated by CaMKII in vitro.
To identify the contribution of Ser-593 versus Thr-594 as the CaMKII phosphoacceptor site, immobilized peptides with point mutations of each site individually as well as in combination were generated and phosphorylated with δ-CaMKII. This revealed that Thr-594 is the preferential CaMKII target in peptides containing both Ser-593 and Thr-594 (Fig. 3F).
To examine these CaMKII substrate sequences in the context of the L1 domain, we constructed GST fusions of the full L1 domain harboring mutations of each phosphoacceptor Ser/Thr amino acid to Ala (Fig. 4, A and B). We included Ala substitutions for Ser-571 and Ser-593. The extent of phosphorylation following a 1-h incubation with δC-CaMKIImono was compared between WT L1 and each point mutation in L1 along with GST alone. Monomeric δC-CaMKII was used to minimize potential contamination of the 32P signal in GST-L1 and its mutants with autophosphorylated CaMKII. Protein levels for each L1 fusion protein (including GST) are shown in the upper panel of Fig. 4A. Because GST is a much smaller protein (28 kDa versus 60 kDa for L1), a Coomassie-stained cutout is shown for this protein (the full Coomassie-stained gel is shown in supplemental Fig. S2B). An autoradiograph is shown in Fig. 4A, but CaMKII phosphorylation was quantified using phosphorimaging. To compensate for differences in protein expression levels and GST pull-down, each GST fusion was normalized for protein levels using LI-COR imaging of the Coomassie-stained bands. The relative phosphorylation for each mutation normalized to WT L1 is shown in Fig. 4B. Compared with unconjugated GST, WT L1-GST exhibited significant phosphorylation as expected. Phosphorylation of the L1 was significantly reduced when either Ser-516 or Ser-594 was mutated to Ala (Fig. 4, A and B). The greatest reduction in phosphorylation was observed when both Ser-516 and Ser-594 were mutated in combination (S516A/S594A) (Fig. 4, A and B). Neither the S593A nor the S571A mutation reduced L1 phosphorylation significantly (Fig. 4, A and B). A full time course of GST phosphorylation up to 3 h indicated that S516A and T594A but not Ser-571 reduced phosphate incorporation in the L1 fragment (Fig. 4C), again indicating that Ser-516 and Thr-594, but not Ser-571, are CaMKII phosphoacceptor sites in L1. Phosphorylation of L1 with phosphomimetic (Glu) substitutions was also examined. The full time course (5 min to 3 h) of the phosphomimetic phosphorylation directly mimicked the Ala mutants shown in Fig. 4C (see supplemental Fig. S2C). As with the Ala mutants, S516E and T594E exhibited a significant reduction in phosphorylation at the 3 h time point (the point of saturation; Fig. 4D). This decrease was not observed with S571A or S571E. These data and the fact that the Ser-571 putative phosphorylation was not identified by the Scansite 2.0 prediction tool, even on lowest stringency (25), suggest that this sequence is not a phosphoacceptor site for CaMKII.
FIGURE 4.

CaMKII phosphorylation of Nav1.5 L1-GST fragments. A, Coomassie stain (top) and phosphor image (bottom) of GST-tagged wild-type L1 and various L1 mutants. B, average phosphorylation (n = 3, ±S.D. (error bars)) of L1 constructs shown in A. S516A, T594A, and S516A/T594A are significantly decreased compared with wild-type GST-L1, as indicated (*, p < 0.05, one-way ANOVA, post hoc Dunnett's test). C, phosphorylation time course of GST-L1 wild-type and mutant fragments. D, stoichiometry of phosphorylation of GST-L1 wild-type and mutant fragments following 180-min phosphorylation by monomeric δC-CaMKII (time of saturation in C). Ala and Glu substitutions at Ser-516 and Thr-594 result in significantly decreased phosphorylation compared with wild-type and Ser-571 mutants (n = 3, ±S.D.; *, p < 0.05, one-way ANOVA, post hoc Dunnett's test).
Electrophysiological Analysis of CaMKII Effects on WT and Mutant NaV1.5 Activation
To examine functional effects of these phosphorylation sites on INa, we expressed full-length WT and mutant hNaV1.5 in HEK293 cells and analyzed INa using voltage clamp. In vitro phosphoacceptor sites, Ser-516 and Thr-594, were mutated to non-phosphorylatable Ala or phosphomimetic Glu residues. We also tested S571A and S571E for comparison with the results of Hund et al. (15) and S593A. WT and Ser to Ala mutant channels were co-transfected with GFP-δC-CaMKII. Based on extensive preliminary studies (e.g. see supplemental Fig. S3), we included 1 μm free [Ca2+] and 1 μm CaM (to activate CaMKII) in all pipette solutions. To assess CaMKII-specific effects, we compared INa with and without the selective CaMKII peptide inhibitor AIP (1 μm) in the pipette. The presence of AIP controls for potential Ca2+ and CaM effects on INa and ensures that the effects measured are via CaMKII.
Representative INa traces from a holding potential (VH) of −140 mV and INa-voltage relationship for WT, S516A, S593A/T594A, T594A, S571A, and S593A (with and without AIP) are shown in Fig. 5A. There was no difference in either INa amplitude or the shape of the I-V curves. Fig. 5, B and C, shows that CaMKII had no effect on the V50 of activation for any channel; nor was there any significant difference between mutant and WT channels. This agrees with results in cardiomyocytes in which CaMKII did not alter INa activation (8) and also shows that these mutant channels have normal WT activation.
FIGURE 5.

CaMKII shifts INa availability but not activation. INa was measured in HEK cells expressing WT, S516A, S593A/T594A, T594A, S571A, or S593A mutant hNaV1.5 plus GFP-δC-CaMKII (pipette containing 1 μm Ca2+ plus 1 μm CaM with or without AIP). A, representative raw INa and mean current-voltage relationships. Protocols for I-V (top) and steady state inactivation (SSI; bottom) for D and E are shown. B and C, voltage dependence and V50 of activation were not different among groups or with/without AIP. D and E, INa availability was shifted to more negative Vm by δC-CaMKII in WT and S593A control, which was prevented by AIP. S516A, S593A/T594A, T594A, and S571A did not show this effect (n as indicated, ±S.E. (error bars); *, p < 0.05 by t test versus with AIP and two-way ANOVA but no difference among the indicated groups).
Electrophysiological Analysis of CaMKII Effects on WT and Mutant NaV1.5 Inactivation
Steady state inactivation (or availability) has been reported to be negatively shifted by CaMKII in cardiac myocytes (8). The availability of INa was measured after a 500-ms pulse to potentials between −140 and −20 mV (Fig. 5, D and E). When CaMKII was activated, there was a hyperpolarzing shift in the V50 for availability in both WT NaV1.5 and the S593A channel (versus when AIP was present). Furthermore, the S593A control was not significantly different from WT under CaMKII-activating conditions. Unlike WT and S593A, the channels with Ala substitution at Ser-516, Ser-571, and Thr-594 did not display a hyperpolarizing shift upon CaMKII activation. The V50 of availability for phosphomutant channels was similar to that in WT channels with CaMKII blocked by AIP. Thus, mutating any one of Ser-516, Ser-571, or Thr-594 to Ala can prevent the CaMKII-dependent shift in Na+ channel availability. The implication is that all three sites are involved in the CaMKII-dependent shift in INa availability. Preliminary studies with S483A/S484A exhibited behavior that was inconsistent with block of CaMKII-induced negative shifts in availability and was not used for further analysis.
We also tested for enhanced INa entry into intermediate inactivation (another reported effect of CaMKII on INa in cardiac myocytes) (8). Cells were depolarized to −20 mV for various times, with a subsequent 20-ms repolarization to allow recovery from fast inactivation before a test pulse (Fig. 6A). Fig. 6, B and C, shows that CaMKII activation in WT NaV1.5 exhibits significant accumulation into intermediate inactivation, and this was inhibited by the inclusion of AIP. Like the availability shift, these changes resemble measurements in adult ventricular myocytes, where CaMKII enhanced intermediate inactivation (8). Neither S516A nor T594A with CaMKII activation exhibited as much intermediate inactivation as WT (Fig. 6B). Ala substitution at Ser-516, Thr-594, and Ser-571 (and double mutant at both Ser-516 and Thr-594) all prevent the CaMKII-dependent accumulation of intermediate inactivation, and with CaMKII, all are similar to WT plus AIP (Fig. 6C).
FIGURE 6.

CaMKII enhances INa entry into intermediate inactivation. A, representative raw WT INa trace of enhanced entry into intermediate inactivation and two-pulse voltage protocol. B, time dependence of accumulation into intermediate inactivation during pulses to −20 mV was significantly greater in WT under CaMKII-activating conditions and 516E/594E phosphomimetic (+AIP) versus WT + AIP and phosphomutants. C, average accumulation of intermediate inactivation at 2.5 s from the bounding box in B (n as indicated, ±S.E. (error bars); *, p < 0.05 by ANOVA or t test versus with AIP; #, p < 0.05 by t test versus with AIP; p = not significant versus WT-AIP).
Phosphomimetic mutant NaV1.5 channels with Glu replacing Ser-516, Thr-594, or Ser-571 or both Ser-516 and Thr-594 were used to test whether those mimic CaMKII phosphorylation effects on INa. Phosphomimetic mutants were studied without CaMKII co-transfection and with AIP in the pipette to eliminate potentially confounding effects of endogenous CaMKII phosphorylation at other sites.
With respect to enhanced accumulation into intermediate inactivation observed for WT upon CaMKII activation, we tested the phosphomimetic channels with AIP in the pipette. No single phosphomimetic (S516E, S571E, or T594E) was able to recapitulate WT accumulation into intermediate inactivation (fraction INa inactivated at 2.5 s of 0.10 ± 0.005 (n = 3), 0.093 ±.04 (n = 4), and 0.11 ± 0.012 (n = 3) respectively). However, the S516E/T594E double phosphomimetic (+AIP) enhanced accumulation into intermediate inactivation (versus WT + AIP) and was not significantly different from WT with activated CaMKII. These results are consistent with multiple sites (Ser-516 and Thr-594) being involved in the CaMKII-dependent enhancement of intermediate inactivation.
We also repeated INa activation and availability measurements for S516E, T594E, S571E, and S516E/T594E channels (Fig. 7). The V50 of activation was again unaltered from WT in any of these mutants (Fig. 7A). However, the T594E and S516E mutants exhibit a strong negative shift of availability compared with WT + AIP and are not different from that seen in WT with activated CaMKII (Fig. 7B).
FIGURE 7.

S516E and T594E phosphomimetics shift INa availability independent of CaMKII. A, V50 of activation was not different among phosphomimetics or compared with WT. B, T594E and S516E INa V50 of availability was shifted to negative Vm in the presence of AIP and without δC-CaMKII co-expression (indicated by striping). V50 of availability for S571E and S516E/T594E was not significantly different from WT with AIP (*, p < 0.05 by ANOVA versus WT with AIP; bracket indicates no difference by t test for S516E and T594E versus WT with δC-CaMKII). C, expanded version of B, for comparing the effect of AIP omission (versus with AIP) in the pipette for different NaV1.5. For the T594E phosphomimetic, availability was not altered when endogenous CaMKII was allowed to function (versus with AIP). For S516E, endogenous CaMKII partly reversed the effect of the phosphomimetic. S571E, which did not mimic the CaMKII-induced shift in V50 when AIP was present (in contrast to S516E and T594E) could still shift V50 negative by endogenous CaMKII activity. (n as indicated, ±S.E. (error bars); AIP and exogenous CaMKII present only where indicated). #, p < 0.05 by t test versus with AIP for the same NaV1.5 type.
We also measured availability in phosphomimetic channels when AIP was omitted, a case where endogenous CaMKII would be active and could phosphorylate other sites (Fig. 7C). When AIP was omitted, T594E was not altered versus the AIP condition. This suggests that there is little additional CaMKII effect once Thr-594 phosphorylation is prevented. The S571E phosphomimetic did not shift with AIP present but did when CaMKII activity was allowed. That indicates that S571E could not mimic the CaMKII effect but that another available CaMKII target site (e.g. Thr-594) could. S516E mimics CaMKII effects in the presence of AIP, but surprisingly when CaMKII was allowed, the availability moved back toward WT + AIP. A similar availability result was seen with the S516E/T594E (with and without AIP) or the triple mutant (including S571E; Fig. 7C). It is possible that when Ser-516 is a Glu, either a Glu or a phosphate at Thr-594 antagonizes the functional mimic of CaMKII effects on INa availability seen with either site mutant. This indicates a complex functional interplay between these phosphorylation sites that will merit further study.
In the above studies, we also tested for the appearance of sustained or late INa, another characteristic effect of CaMKII on native cardiac INa (8, 14). We saw no evidence of late INa in any of the mutant NaV1.5 channels studied; nor were the kinetics of fast inactivation altered (not shown). This is consistent with the notion that an additional protein(s) indigenous to myocytes but not present in HEK293 cells (e.g. β-subunit) is required for the appearance of late INa (26–30).
These data also suggest that the phosphorylation status of Ser-516 and Thr-594 on NaV1.5, even in the absence of the β-subunit, is an important mediator of the CaMKII-dependent negative shift in NaV1.5 INa availability and accumulation of intermediate inactivation.
DISCUSSION
The α-subunit of the human cardiac voltage-gated sodium channel is phosphorylated by CaMKII in the L1 loop. These data are consistent with the L1 region as a hot spot for CaMKII phosphorylation as noted before (14, 15). We show for the first time that activated CaMKII forms a stable interaction with the L1 loop, a targeting mechanism that may contribute to co-localization of CaMKII with NaV1.5 previously observed in myocytes by immunohistochemistry and heart tissue by immunoprecipitation (8, 31). Recently, Ser-571 was identified as an important CaMKII phosphorylation site in L1 (15), a site implicated in regulating NaV1.5 INa. However, using multiple biochemical assays, we did not observe Ser-571 to be a CaMKII phosphorylation target in our study. Rather, our unbiased measurement of CaMKII phosphorylation sites in L1 identified three phosphorylation motifs for CaMKII at positions Ser-483/Ser-484, Ser-516, and Thr-594. Unlike Ser-483/Ser-484, the Ser-516 and Thr-594 sites were verified as CaMKII substrates using multiple approaches, including GST fusion mutations of L1 directly, as well as using soluble and immobilized peptides. Thus, these phosphorylation sites were the best candidates for further characterization using functional studies.
To understand the influence of CaMKII phosphorylation of these sites, we co-transfected GFP-δC-CaMKII, the predominant cytosolic CaMKII isoform found in the heart, along with WT and mutant forms of hNaV1.5, into HEK293 cells and measured INa using intracellular pipette conditions supporting acute CaMKII activation (1 μm free Ca2+ plus CaM). Interestingly, high Ca2+ in the pipette without CaMKII overexpression was sufficient to produce a small but significant hyperpolarizing shift in WT NaV1.5 availability (supplemental Fig. S3). Thus, endogenous CaMKII may be activated by 1 μm pipette Ca2+ (32), but higher levels of CaMKII were necessary for maximal functional effects. The potent peptide CaMKII inhibitor AIP was included to verify that changes in INa were due to CaMKII and not direct effects of Ca2+ (33–36) and/or Ca2+/CaM (34, 35, 37–41). We found that Ala point mutations at Ser-516 and Thr-594 identified in our biochemical phosphorylation assays disrupted CaMKII-induced alterations in INa gating. In further support of Ser-516 and Thr-594 being functionally relevant CaMKII targets, we observed that phosphomimetic mutations (S516E and T594E) functioned to recapitulate CaMKII effects on Na+ channel availability (in the absence of CaMKII overexpression and presence of AIP).
It is unclear why the Ala mutagenesis data are consistent with both Ser-516 and Thr-594 phosphorylation and Ser-571 presence being required to shift INa availability, whereas Glu substitution suggests that either Ser-516 or Thr-594 phosphorylation may suffice. Of course, Glu substitution is an imperfect phosphomimetic and the Ser/Thr to Ala mutation might be less supportive of functional competency of phosphorylation at other sites.
Using a biased approach to screen CaMKII phosphorylation sites on NaV1.5 based on the Arg-X-X-Ser/Thr motif, Hund et al. (15) reported that of the mutations tested (including Ser-484, Ser-571, and Ser-664), only S571A blocked CaMKII-dependent regulation of NaV1.5 INa gating, whereas S571E was reported to mimic WT NaV1.5. We also observed that S571A disrupted CaMKII regulation of INa gating when CaMKII was co-expressed with the mutant channel. However, unlike S516E and T594E, the S571E mutant was not sufficient to recapitulate CaMKII functional changes in INa gating when CaMKII was inhibited by AIP. Moreover, S571E channel availability could only shift negative with active CaMKII, suggesting that phosphorylation at other CaMKII target sites (Ser-516 and/or Thr-594) might be required. Thus, although Ser-571 may be a site of phosphorylation, although not by CaMKII in our hands, we suspect that its role in CaMKII-dependent NaV1.5 gating might be less direct than that of Ser-516 and Thr-594. For example, Ser-571 might have an absolute requirement for βIV spectrin to allow CaMKII to gain access (15), or its direct phosphorylation might be mediated by another kinase. Notably, this site is conserved among animal species and other NaV1.X isoforms (supplemental Fig. S4). Moreover, bioinformatic screening indicates that Ser-571 could be a target for PKA, Akt, or PKG. Further work is required to clarify Ser-571 phosphorylation.
Whether the CaMKII regulatory sites identified in our study are simultaneously, sequentially, or individually phosphorylated in vivo by CaMKII is unclear at this point. The simplest interpretation of Fig. 5E would be that Ser-516, Ser-571, and Thr-594 must all be phosphorylated to produce the INa availability effects observed here. These data made it surprising that either T594E or S516E alone could recapitulate CaMKII-dependent INa availability shifts, whereas these sites appear additive in their effects on accumulation into intermediate inactivation. The ability of the S516E/T594E double phosphomimetic to fully recapitulate CaMKII effects of enhanced accumulation into intermediate inactivation in Fig. 6 but not the shift in INa availability (Fig. 7B) reinforces the complexity and dynamic nature of phosphorylative regulation at these sites. Additional work and new tools, including the use of myocytes, could further clarify this (e.g. kinetics and sequence of site phosphorylation, channel state dependence effects). An intriguing possibility is that, as seen for neuronal KV1.2 channel gating (42), graded regulation of NaV1.5 by phosphorylation may also regulate myocyte excitability via modulation of NaV1.5 gating. It is currently unclear whether the presence of multiple phosphorylatable sites in the L1 region of NaV1.5 represents redundancy or permits fine tuning and/or cross-talk between multiple protein kinases.
Notably, Thr-594 in L1 is perfectly conserved among the seven mammalian NaV1.5 channels (supplemental Fig. S4A). Among other NaV1.X channels (supplemental Fig. S4B), only SCN8A (NaV1.6) has the Thr-594 equivalent and also the basic Arg/Lys at the P−3 position (most others have Met at P−3 or Ala at the P0). Although Ser-516 is well conserved in NaV1.5 across species, all non-human mammalian NaV1.5 proteins possess a His rather than Arg at P−3, which could alter the ability of CaMKII to phosphorylate this site. Although ClustalW alignment of NaV1.X channels does not indicate conservation of Ser-516 in the other isoforms, neuronal voltage-gated sodium channels do contain Arg/Lys-X-X-Ser/Thr motifs in the region upstream from Ser-516 in NaV1.5. Thus, further exploration will be required to determine whether CaMKII also targets the sites studied here in other voltage-gated sodium channels.
We tested for, but did not detect, CaMKII-induced late INa (not shown). This indicates that NaV1.5 and δC-CaMKII alone are not sufficient to recapitulate CaMKII-induced late INa seen in cardiomyocytes (8, 9, 14). It is possible that CaMKII-induced late INa involves phosphorylation by CaMKII at the same sites on the sodium channel as studied here but that an additional protein (e.g. β-subunit) is required for translation into late INa (26–30). We also assayed for CaMKII-induced changes in recovery from inactivation but did not observe a significant difference across tested conditions (data not shown). Again, this may reflect the importance of regulatory β-subunits in modulating recovery from inactivation and other gating phenomenon (30, 43–46). Although the use of the heterologous expression system limits the number of functional effects observed, all of the CaMKII-induced functional changes in INa seen in HEK293 cells are consistent with those observed in myocytes under similar recording conditions (8, 15), indicating that this reduced system can be used to dissect components of CaMKII-dependent functional changes in NaV1.5, and suffice for the CaMKII-induced shifts in availability and intermediate inactivation.
The mechanism underlying the complex functional effects of CaMKII on cardiac sodium channel is of extreme interest because of the therapeutic potential of these targets in arrhythmias and heart failure (47–50). Indeed, the CaMKII-dependent effects on INa gating in cardiac myocytes (negative-shifted availability curve, accumulated intermediate inactivation, slowed recovery from inactivation, and enhanced late INa) directly phenocopy a human NaV1.5 arrhythmogenic mutant (Ins1795D), where individual patients exhibit both Brugada syndrome (due to loss of Na+ channel function at high heart rates and reduced cardiac conduction velocity) and long QT syndrome (where late INa causes action potential duration prolongation), and both can lead to life-threatening arrhythmias. Because CaMKII expression and activation is elevated in heart failure patients (and animal models of heart failure) (4–7), these CaMKII-dependent alterations in Na+ channel gating may cause arrhythmogenesis in many millions of people carrying the clinical diagnosis of heart failure. The altered sodium channel gating may also contribute to alterations in intracellular [Na+]i and [Ca2+]i regulation in this same large population, and this can contribute to both systolic and diastolic dysfunction in heart failure. Understanding the molecular and structural details of INa regulation by CaMKII-dependent phosphorylation will require further work, but our identification of Ser-516 and Thr-594 as functionally important CaMKII target sites on NaV1.5 is a major step forward.
Acknowledgment
We thank Dr. Laurie Parker for assistance with peptide synthesis.
This work was supported, in whole or in part, by National Institutes of Health Grants NS053422 (to T. R. C.), P01-HL080101 (to D. M. B.), and R37HL30077 (to D. M. B.). This work was also supported by American Heart Association National Scientist Development Grant 0930064N (to A. H.) and a grant from the Ralph W. and Grace M. Showalter foundation (to A. H.).

This article contains supplemental Figs. S1–S4.
- INa
- Na+ current
- CaM
- calmodulin
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- ANOVA
- analysis of variance
- AIP
- autoinhibitory peptide.
REFERENCES
- 1. Ruan Y., Liu N., Priori S. G. (2009) Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 6, 337–348 [DOI] [PubMed] [Google Scholar]
- 2. Veldkamp M. W., Viswanathan P. C., Bezzina C., Baartscheer A., Wilde A. A., Balser J. R. (2000) Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 86, E91–E97 [DOI] [PubMed] [Google Scholar]
- 3. Abriel H. (2007) Roles and regulation of the cardiac sodium channel NaV1.5. Recent insights from experimental studies. Cardiovasc. Res. 76, 381–389 [DOI] [PubMed] [Google Scholar]
- 4. Hoch B., Meyer R., Hetzer R., Krause E. G., Karczewski P. (1999) Identification and expression of δ-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721 [DOI] [PubMed] [Google Scholar]
- 5. Kirchhefer U., Schmitz W., Scholz H., Neumann J. (1999) Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42, 254–261 [DOI] [PubMed] [Google Scholar]
- 6. Zhang T., Maier L. S., Dalton N. D., Miyamoto S., Ross J., Jr., Bers D. M., Brown J. H. (2003) The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919 [DOI] [PubMed] [Google Scholar]
- 7. Ai X., Curran J. W., Shannon T. R., Bers D. M., Pogwizd S. M. (2005) Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322 [DOI] [PubMed] [Google Scholar]
- 8. Wagner S., Dybkova N., Rasenack E. C., Jacobshagen C., Fabritz L., Kirchhof P., Maier S. K., Zhang T., Hasenfuss G., Brown J. H., Bers D. M., Maier L. S. (2006) Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maltsev V. A., Reznikov V., Undrovinas N. A., Sabbah H. N., Undrovinas A. (2008) Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes. Similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 294, H1597–H1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bradshaw J. M., Hudmon A., Schulman H. (2002) Chemical quenched flow kinetic studies indicate an intraholoenzyme autophosphorylation mechanism for Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 277, 20991–20998 [DOI] [PubMed] [Google Scholar]
- 11. Ashpole N. M., Hudmon A. (2011) Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol. Cell Neurosci. 46, 720–730 [DOI] [PubMed] [Google Scholar]
- 12. Hudmon A., Schulman H., Kim J., Maltez J. M., Tsien R. W., Pitt G. S. (2005) CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hudmon A., Choi J. S., Tyrrell L., Black J. A., Rush A. M., Waxman S. G., Dib-Hajj S. D. (2008) Phosphorylation of sodium channel NaV1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J. Neurosci. 28, 3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aiba T., Hesketh G. G., Liu T., Carlisle R., Villa-Abrille M. C., O'Rourke B., Akar F. G., Tomaselli G. F. (2010) Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea pig ventricular myocytes. Cardiovasc. Res. 85, 454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hund T. J., Koval O. M., Li J., Wright P. J., Qian L., Snyder J. S., Gudmundsson H., Kline C. F., Davidson N. P., Cardona N., Rasband M. N., Anderson M. E., Mohler P. J. (2010) A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pawson T., Scott J. D. (1997) Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 [DOI] [PubMed] [Google Scholar]
- 17. Strack S., Colbran R. J. (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 273, 20689–20692 [DOI] [PubMed] [Google Scholar]
- 18. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 [DOI] [PubMed] [Google Scholar]
- 19. Grueter C. E., Abiria S. A., Wu Y., Anderson M. E., Colbran R. J. (2008) Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel β subunits. Biochemistry 47, 1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Songyang Z., Lu K. P., Kwon Y. T., Tsai L. H., Filhol O., Cochet C., Brickey D. A., Soderling T. R., Bartleson C., Graves D. J., DeMaggio A. J., Hoekstra M. F., Blenis J., Hunter T., Cantley L. C. (1996) A structural basis for substrate specificities of protein Ser/Thr kinases. Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell Biol. 16, 6486–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. White R. R., Kwon Y. G., Taing M., Lawrence D. S., Edelman A. M. (1998) Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shard and distinctive features. J. Biol. Chem. 273, 3166–3172 [DOI] [PubMed] [Google Scholar]
- 22. Frank R., Overwin H. (1996) SPOT synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol. Biol. 66, 149–169 [DOI] [PubMed] [Google Scholar]
- 23. Frank R. (2002) The SPOT synthesis technique. Synthetic peptide arrays on membrane supports. Principles and applications. J. Immunol. Methods 267, 13–26 [DOI] [PubMed] [Google Scholar]
- 24. Naffin J. L., Han Y., Olivos H. J., Reddy M. M., Sun T., Kodadek T. (2003) Immobilized peptides as high-affinity capture agents for self-associating proteins. Chem. Biol. 10, 251–259 [DOI] [PubMed] [Google Scholar]
- 25. Obenauer J. C., Cantley L. C., Yaffe M. B. (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aman T. K., Grieco-Calub T. M., Chen C., Rusconi R., Slat E. A., Isom L. L., Raman I. M. (2009) Regulation of persistent Na current by interactions between β subunits of voltage-gated Na channels. J. Neurosci. 29, 2027–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qu Y., Curtis R., Lawson D., Gilbride K., Ge P., DiStefano P. S., Silos-Santiago I., Catterall W. A., Scheuer T. (2001) Differential modulation of sodium channel gating and persistent sodium currents by the β1, β2, and β3 subunits. Mol. Cell Neurosci. 18, 570–580 [DOI] [PubMed] [Google Scholar]
- 28. Bant J. S., Raman I. M. (2010) Control of transient, resurgent, and persistent current by open-channel block by Na channel β4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 107, 12357–12362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maltsev V. A., Kyle J. W., Undrovinas A. (2009) Late Na+ current produced by human cardiac Na+ channel isoform NaV1.5 is modulated by its β1 subunit. J. Physiol. Sci. 59, 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mishra S., Undrovinas N. A., Maltsev V. A., Reznikov V., Sabbah H. N., Undrovinas A. (2011) Post-transcriptional silencing of SCN1B and SCN2B genes modulates late sodium current in cardiac myocytes from normal dogs and dogs with chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H1596–H1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoon J. Y., Ho W. K., Kim S. T., Cho H. (2009) Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J. Mol. Cell Cardiol. 47, 475–484 [DOI] [PubMed] [Google Scholar]
- 32. Tsui J., Inagaki M., Schulman H. (2005) Calcium/calmodulin-dependent protein kinase II (CaMKII) localization acts in concert with substrate targeting to create spatial restriction for phosphorylation. J. Biol. Chem. 280, 9210–9216 [DOI] [PubMed] [Google Scholar]
- 33. Wingo T. L., Shah V. N., Anderson M. E., Lybrand T. P., Chazin W. J., Balser J. R. (2004) An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 11, 219–225 [DOI] [PubMed] [Google Scholar]
- 34. Shah V. N., Wingo T. L., Weiss K. L., Williams C. K., Balser J. R., Chazin W. J. (2006) Calcium-dependent regulation of the voltage-gated sodium channel hH1. Intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. U.S.A. 103, 3592–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Young K. A., Caldwell J. H. (2005) Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J. Physiol. 565, 349–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chagot B., Potet F., Balser J. R., Chazin W. J. (2009) Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J. Biol. Chem. 284, 6436–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mori M., Konno T., Morii T., Nagayama K., Imoto K. (2003) Regulatory interaction of sodium channel IQ-motif with calmodulin C-terminal lobe. Biochem. Biophys. Res. Commun. 307, 290–296 [DOI] [PubMed] [Google Scholar]
- 38. Theoharis N. T., Sorensen B. R., Theisen-Toupal J., Shea M. A. (2008) The neuronal voltage-dependent sodium channel type II IQ motif lowers the calcium affinity of the C-domain of calmodulin. Biochemistry 47, 112–123 [DOI] [PubMed] [Google Scholar]
- 39. Kim J., Ghosh S., Liu H., Tateyama M., Kass R. S., Pitt G. S. (2004) Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279, 45004–45012 [DOI] [PubMed] [Google Scholar]
- 40. Tan H. L., Kupershmidt S., Zhang R., Stepanovic S., Roden D. M., Wilde A. A., Anderson M. E., Balser J. R. (2002) A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415, 442–447 [DOI] [PubMed] [Google Scholar]
- 41. Deschênes I., Neyroud N., DiSilvestre D., Marbán E., Yue D. T., Tomaselli G. F. (2002) Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ. Res. 90, E49–E57 [DOI] [PubMed] [Google Scholar]
- 42. Park K. S., Mohapatra D. P., Misonou H., Trimmer J. S. (2006) Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313, 976–979 [DOI] [PubMed] [Google Scholar]
- 43. Ko S. H., Lenkowski P. W., Lee H. C., Mounsey J. P., Patel M. K. (2005) Modulation of Na(v) 1.5 by β1- and β3-subunit co-expression in mammalian cells. Pflugers Arch. 449, 403–412 [DOI] [PubMed] [Google Scholar]
- 44. Fahmi A. I., Patel M., Stevens E. B., Fowden A. L., John J. E., 3rd, Lee K., Pinnock R., Morgan K., Jackson A. P., Vandenberg J. I. (2001) The sodium channel β-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J. Physiol. 537, 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meadows L. S., Chen Y. H., Powell A. J., Clare J. J., Ragsdale D. S. (2002) Functional modulation of human brain NaV1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2, and β3 subunits. Neuroscience 114, 745–753 [DOI] [PubMed] [Google Scholar]
- 46. Goldin A. L. (2003) Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol. 13, 284–290 [DOI] [PubMed] [Google Scholar]
- 47. Bers D. M., Grandi E. (2009) Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anderson M. E., Brown J. H., Bers D. M. (2011) CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell Cardiol. 51, 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grueter C. E., Colbran R. J., Anderson M. E. (2007) CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J. Mol. Med. 85, 5–14 [DOI] [PubMed] [Google Scholar]
- 50. Maier L. S. (2011) CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm 8, 474–477 [DOI] [PubMed] [Google Scholar]