

Abstract
Midbrain periaqueductal gray (PAG) and spinal cord dorsal horn are major action sites of opioid analgesics in the pain pathway. Our previous study has shown that opioid antagonists activate MORS196A-CSTA (a mutant of mu-opioid receptor) as full agonists in vitro cell models and naloxone showed antinociceptive effects after the expression of MORS196A-CSTA in the spinal cord in mice. The purpose of this study is to investigate the site directed antinociceptive effects of naloxone in mice injected with dsAAV-MORS196A-CSTA-EGFP at spinal cord or at periaqueductal gray. MORS196A-CSTA-EGFP was administered to ICR mice using dsAAV as vector. We measured MORS196A-CSTA-EGFP expression by detecting the EGFP visualization with a fluorescence microscope. The antinociceptive effect of naloxone was determined by tail-flick test and hot plate test. Drug rewarding effect was evaluated by the conditioned place preference test. Naloxone (10 mg/kg, s.c.) elicited both supraspinal and spinal antinociceptive responses in mice injected with the virus at PAG while only spinal antinociceptive response was observed in mice injected with virus at dorsal horn region. Chronic naloxone treatment did not induce physical dependence or rewarding effect in mice injected with MORS196A-CSTA-EGFP in spinal cord or PAG.
These data suggest that the observed naloxone-induced antinociceptive response is the consequence of the local expression of MORS196A-CSTA at specific sites of pain pathway. Injection of such MOR mutant and the systemic administration of naloxone can be a new strategy in the management of chronic pain without the various side effects associated with the use of morphine.
INTRODUCTION
Although opioids are used successfully for the treatment of moderate to severe pain, chronic uses of the drugs have led to tolerance and addiction to the drugs, thus limiting their uses. In the past, several approaches have been used to address the problems of tolerance and addiction to the drug during long-term treatment. Clinical studies indicated that the mixed agonist-antagonists, especially those that could activate kappa-opioid receptor (KOR) and are antagonists in mu-opioid receptor (MOR), have lower addictive liability (Rosow, 1987). However, with their dysphoric properties, KOR agonists have lower patient compliance and therefore they are not ideal analgesic agents. Another approach takes advantage of the antagonistic properties of dextromethorphan on the NMDA receptor, which has been implicated in tolerance mechanism. Although in animal studies dextromethorphan has been shown to block or reverse tolerance (Elliott et al., 1995a; Elliott et al., 1995b; Huang et al., 2003; Wong et al., 1999), high incidents of adolescent abuse due to its sigma and serotonergic receptors activities (Miller, 2005; Schwartz, 2005) has limited this approach to clinical use. Another approach is the use of bivalent ligands by “bridging” of neighboring pharmacophores with optimal spacer length (Portoghese et al., 1986a; Portoghese et al., 1985; Portoghese et al., 1986b). Such bivalent ligands with pharmacophores that select for mu and delta receptors were shown to exhibit minimal tolerance or dependence responses in mice chronically treated with these ligands (Daniels et al., 2005).
Instead of the above approaches, we have decided to develop the approach based on receptor activated solely by synthetic ligands (RASSL) (Coward et al., 1998). If a ligand that could activate RASSL but not the endogenous receptor can be identified, then RASSL can be used as probable therapeutic agents without side effects. Our unique MOR mutant, MORS196A is such a RASSL, in which opioid antagonists naloxone or naltrexone inhibited forskolin-stimulated adenylyl cyclase activity or activation of Kir3.1 channels (Claude et al., 1996). The in vivo activity of this mutant MOR was demonstrated by the S196A knock-in mice or by dsAAV2-mediated delivery of the mutant MOR at the dorsal horn area of ICR mouse, where naloxone or naltrexone produced antinociceptive effects without tolerance and dependence development (Chen et al., 2007; Yang et al., 2003).
However, whether the expression of such RASSL at the supraspinal sites within the pain pathway will result in responses to the activation of the receptor without tolerance and dependence is unknown. Therefore, in the present study, dsAVV2 was used to deliver a mutant MOR, MORS196A-CSTA into the periaqueductal grey area (PAG). This mutant in which S196A mutation was combined with the C330S and the T327A mutation has been shown to result in that antagonists exhibit full agonistic properties in vitro cell models (Claude-Geppert et al., 2005). Whether naloxone exhibited supraspinal/spinal acute and chronic antinociceptive effects without the development of tolerance and dependence in the mice injected with MORS196A-CSTA at the PAG area was compared to mice injected with the virus at the dorsal horn of S2/S3 region of the ICR mice.
MATERIALS AND METHODS
dsAAV microinjection
The virus dsAAV serotype 2 containing the MORS196A-CSTA-EGFP sequence was constructed as previously described (Chen et al., 2007). The virus titer of the dsAAV2 was determined to be 1013 vp/ml. Male ICR wild type mice that weighed 30~40 g were used in this study. All mice were kept under a 12 h light/dark cycle, at a temperature of 25 ± 2°C and humidity of 55% with food and water provided ad libitum. NIH guidelines for the care for the animals were followed and the protocol was approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center, Taiwan, R. O. C.
For both spinal cord and PAG injection, mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and placed under a dissecting microscope. For dorsal horn injection, a partial dorsal laminectomy was performed. Lumbar process at L2 was carefully removed to expose a segment of spinal cord. Mice were then placed in a spinal frame holder and mounted under a stereotaxic frame with a microinjector attachment that includes a 10-μl Hamilton syringe with a micro-tipped glass pipette. Six 0.5 μl injections of dsAAV2-MORS196A-CSTA-EGFP (1013 vp/ml) were injected at a depth of ~0.3 mm bilaterally. After surgery, muscle and skin around the wound were sutured and three micro surgical wound clips were applied. For the vlPAG injection, under pentobarbital anesthesia, a small incision was made on the skull to access the bregma. After a hole at the top of the skull was made with a drilling machine, the dsAAV2-MORS196A-CSTA-EGFP was stereotaxically administered through puncture points (A: −3.28 mm, L: ±0.3 mm, V: −3.0 mm to the bregma) using microinjection pump with a truncated 26 gauge needle at the rate of 0.2 μl/min.
Antinociceptive testing
Tail-flick test
Drug-induced antinociceptive effect was evaluated by using the tail-flick test (D’Amour and Smith, 1941). Using a tail-flick apparatus (Ugo Basile, Italy), the basal tail-flick latency was controlled between 2.5 to 4 s for all animals. A cut-off time of 10 s was applied to avoid tail injury. The antinociceptive effects of the drugs are reported as time-response curves (area-under curve; AUC) or ED50 values. After 30, 60, 90, 120, 180 min of drug administration (s.c.) or 10, 20, 30, 60, and 90 min of drug administration (i.t. or i.c.v.), the tail-flick latency was recorded. For i.t. and i.c.v. injections, drugs were delivered in 5 μl and 1 μl respectively as previously described (Chen et al., 2007). The AUC was calculated using the Trapezoidal and Simpson’s rules. The ED50 was determined by up and down method (Dixon, 1965). The antinociceptive effect of saline, morphine (10 mg/kg, s.c.) or naloxone (10 mg/kg, s.c.) was used as the pre-test data before microinjection of dsAAV2 vector. After gene transfection for two weeks, the antinociceptive effect of saline, morphine (10 mg/kg, s.c.) or naloxone (10 mg/kg, s.c.) was tested again.
Hot plate assay
In the hot plate test, mice were placed on a 25 × 25-cm2 metal surface surrounded by a 30-cm-high Plexiglas wall with the temperature controlled at 50 ± 0.5°C (Digital DS-37 Socrel model). Latency (in seconds) required for mice to either lick or shake its hind paw or jump was recorded as the pain threshold. The baseline pain threshold was obtained by the mean values of 3 measurements before drug administration. Following drug administration, test latency was determined at 30, 60, and 90 min and a cut-off time of 30 s in mice was used in order to minimize injury to the animals. The AUC values of the drug(s) were calculated.
VonFrey test for mechanical allodynia
In a quiet room, mice were placed in acrylic cages (9×9×16 cm) with wire grid floors, 60–90 min before the start of testing. The test was consist of evoking a hind paw flexion reflex with a hand-held force transducer (electronic von Frey anesthesiometer; IITC Life Science, Woodland Hills, CA) adapted with a polypropylene tip. The investigator applies the tip perpendicularly to the central area of the hind paw with a gradual increase in pressure. The end point is characterized by the removal of the paw followed by clear flinching movements. After the paw withdrawal, the intensity of the pressure was recorded automatically. The value for the response is an averaging of 3~5 measurements.
Chronic morphine/naloxone treatment
To examine whether the drugs would induce tolerance after chronic treatment, the mice were injected with morphine (10 mg/kg, s.c.) or naloxone (10 mg/kg, s.c.) b.i.d. for 6 days. The ED50 (tested in the seventh day) and AUC (tested in the eighth day) values of the drugs were tested again after 6 days of treatment.
To determine the development of physical dependence, natural withdrawal symptoms were measured. Two weeks after gene transfer, the mice were treated daily with increasing doses of morphine or naloxone (10, 15, 20, 30, 40, and 50 mg/kg, s.c.) twice a day for 6 days. The mice were put into test chambers consisting of transparent round plastic boxes (20 cm diameter, 33 cm height). The somatic signs of opiate withdrawal (jump, paw tremor, wet dog shaking, teeth chattering, diarrhea, ptosis) were counted for 30 min at 14, 38 and 62 h after the last dose of the drug was injected. Jump and paw tremor frequencies were recorded during the test time, and a score of 1 was assigned to every three jumps or five paw tremors. Diarrhea and ptosis events were recorded for every 5-min interval in which they occurred (maximal score = 6). A total opiate natural withdrawal score was calculated by summing the values for each sign (Papaleo and Contarino, 2006).
Conditioned Place Preference (CPP) test
Drug rewarding effect was measured by the CPP test which was similar to that described previously (Huang et al., 2003), except the size of the apparatus was adjusted for mice. The CPP test apparatus, made from an acrylic plastic box (33 × 15 × 15 cm), was divided into three compartments. Two identically sized compartments (15 × 15 × 15 cm) were constructed at both sides, separated by a narrower compartment (3 × 15 × 15 cm). The compartments were connected by guillotine doors (7 × 3.5 cm) in the central unit. One of the large sides had a distinct environment (covered by mosaic-type paper with blue light) associated is repeatedly paired with the effects of a drug, the other side had a different environment (covered by white paper with red light) associated with non-drug treated state. During the experiments, the CPP apparatus was kept in an isolated room away from noise and light. For CPP conditionings, the mice were given saline in the morning and morphine (10 mg/kg, i.p.) or naloxone (10 mg/kg, i.p.) in the afternoon for 6 days. By repeated association of a distinct environment with the effects of the drug, mice were kept for 40 min in the corresponding compartment with the guillotine doors closed. CPP tests were performed before conditioning and after conditioning (day 7). We determined the place preference by placing the mice into the central compartment of the apparatus with the guillotine doors opened for 15 min. The time that the mice stayed in each compartment was recorded to determine the place preference. The measurement of drug rewarding effect was determined by an increase in the time spent in the compartment previously paired with drug injection than the time spent in the saline-paired compartment.
Immunohistochemistry
Mice were anesthetized with chlorohydrade (400 mg/kg, i.p.) and subjected to transcardial perfusion with Tyrode calcium free buffer (NaCl 116 mM, KCl 5.36 mM, MgCl2·6H2O 1.57 mM, MgSO4 0.405 mM, NaH2PO4 1.23 mM, Glucose 5.55 mM, NaHCO3 26.2 mM, pH 7.4), followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (KH2PO4 187.5 mM, Na2HPO4·12H2O 212.5 mM). The lumber spinal cord, dorsal root ganglion (DRG) and whole brain were dissected and then cryoprotected in 20% sucrose solution overnight at 4°C. Tissue was then embedded in OCT compound and immediately frozen in a −80°C freezer. Tissue was sectioned by a cryostat at a thickness of 10 μm at −20°C. The slices were mounted on SuperFrost Plus slides (Menzel-Glaser, Germany) and the EGFP expression was visualized with a fluorescence microscope. For immunohistochemistry, antibodies were diluted in blocking buffer (0.1 M PBS containing 1% goat serum and 0.1% Triton X-100). Anti-NeuN (MAB377; Chemicon, Temecula, CA, USA) antibody was used at a dilution of 1:500. The antibodies were visualized using goat anti rabbit or goat anti mouse IgG at a dilution of 1:200 (Jackson, USA) which was coupled with rhodamine for immunofluorescence detection. Sections were then washed with PBS, cleared and coverslip was mounted with mounting medium (Serotec, HIS002B). Fluorescence was detected using an Olympus fluorescence instruments (model: BH2-RFL-T3, Japan) on an upright microscope and an Olympus (Tokyo, Japan) BX50 camera with SPOT software (version 4.6; Diagnostic Instruments, Inc. U.S.A).
Data analysis
Data are shown as means ± SEM. One-way ANOVA and Newman–Keuls test were used for data analysis. A difference was considered to be significant when p ≤ 0.05.
Materials
Morphine hydrochloride was purchased from TFDA (Taipei, Taiwan, R.O.C.). Naloxone, naltrexone and CTOP were supplied by Sigma Co. (U.S.A.). Buffer salts were purchased from Nacalai Tesque (Japan) or J. T. Baker (U.S.A.).
RESULTS
In vivo dsAAV2-MORS196A-CSTA-EGFP gene transfer and expression
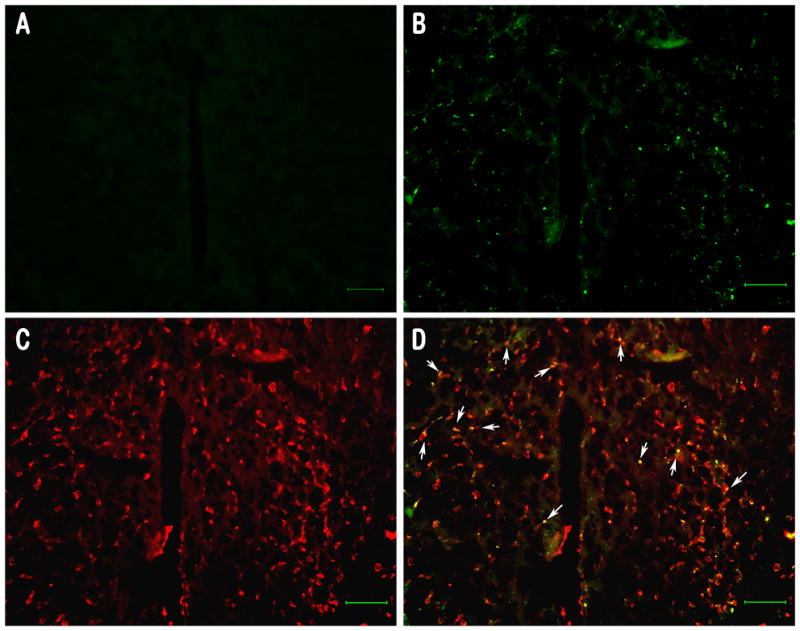
To confirm the expression of MORS196A-CSTA, we constructed the dsAAV2 vector containing EGFP protein in MORS196A-CSTA C-terminus as described previously (Chen et al., 2007). The expression of MORS196A-CSTA in mice injected with dsAAV2-MORS196A-CSTA-EGFP into the spinal cord dorsal horn was similar to the dsAAV2-MORS196A-EGFP expression reported previously (Chen et al., 2007). The expression of the mutant receptor as indicated by the ability of naloxone to induce antinociceptive responses could be detected 2 weeks after dsAAV2 injection and remained stable for at least 6 months [AUC = 240.9 ± 13.4 min × s (6 months) v.s. 35.3 ± 2.8 min × s (pre-injection)]. Similarly, when dsAAV2-MORS196A-CSTA-EGFP was injected into vlPAG, naloxone-induced antinociceptive activity could also be observed 2 weeks after injection and lasted for at least 6 months [AUC =189.9 ± 14.3 min × s (6 months) v.s. 26.9 ± 5.1 min × s (pre-injection)]. Expression of the MORS196A-CSTA at the injection PAG sites can be detected with the EGFP fluorescence at PAG 2 weeks after gene transferred (Fig. 1B). Similar PAG area from mice injected with saline did not reveal any EGFP fluorescence (Fig. 1A). It can be shown that the MOR mutant was expressed in neurons at PAG by the colocalization of the immunofluorescence of EGFP and NeuN, a neuronal marker commonly used to identify neurons (Figs. 1C and D). The expression of the EGFP fluorescence in neurons could be detected for at least 6 months in PAG and 12 months in the dorsal horn area of the spinal cord.
Figure 1.
Transfection of MORS196A-CSTA into the spinal cord or PAG did not alter the acute or chronic antinociceptive effects of morphine
Since the injection procedure may cause damage to the nociceptive neurons at the spinal cord or PAG area, or the over-expression of mutated MOR could cause an increase in morphine efficacy (Xu et al., 2003), we decided to determine the morphine in vivo responses in mice injected with the dsAAV2. To rule out the possible neuronal injury and changing mechanical nociception after operation and injection, basal tail-flick latencies and paw withdrawal threshold (von-Frey test) were determined. Only mice responded similarly in these two tests before and after the dsAAV2 injection were used in following studies. We found there was no difference in basal tail-flick latency or paw withdrawal threshold between the day before surgery and 2 weeks after surgery. To evaluate the drug-induced antinociception, tail-flick test was used, and the ED50 values were determined. As summarized in Table 1, injection of virus either at the dorsal horn of the spinal cord or at the vlPAG did not alter the potency of morphine (s.c.) to inhibit the tail-flick response. Morphine exhibited similar antinociceptive effects before and after MORS196A-CSTA-EGFP injection into these two sites. As expected, the antinociceptive effect of morphine was dramatically reduced after chronically injected with morphine for 6 days. The ED50 value of morphine increased by 4.8 fold when fixed doses of morphine (10 mg/kg, s.c.) were given b.i.d. for 6 days (Table 1). Such increase in the ED50 value and thus decrease in morphine potency was observed independent of the site of dsAAV2 injection.
Table 1.
Tolerance induced by morphine but not naloxone after MORS196A-CSTA gene transferred locally into spinal cord or vlPAG.
| Gene transfer site | Spinal cord | vlPAG | ||
|---|---|---|---|---|
| morphine | naloxone | morphine | naloxone | |
| ED50, 2 weeks after gene transfection | 1.55 ± 0.45 | 8.42 ± 1.23 | 1.30 ± 0.24 | 7.49 ± 0.84 |
| ED50, after chronic drug (10 mg/kg, s.c., b.i.d. for 6 days) | 7.49 ± 1.16 (4.8 fold) | 8.42 ± 1.23 (1.0 fold) | 5.96 ± 0.87 (4.6 fold) | 8.42 ± 1.23 (1.1 fold) |
The ED50 (mg/kg) of drugs to inhibit the tail-flick responses were determined 2 weeks after gene transfer into spinal cord or vlPAG by the up-down method. After chronically treated with drugs (10 mg/kg, s.c., b.i.d. for 6 days), animals were tested for the ED50 of drugs again. The values represent the mean ± S.E.M. (n= 6–8)
Naloxone elicited spinal antinociception without tolerance development in mice injected with MORS196A-CSTA-EGFP at the spinal cord
Naloxone, being an opioid antagonist, did not produce any antinociceptive effect before the transfer of MORS196A-CSTA-EGFP gene into the spinal cord (Fig. 2A). However 2 weeks after the gene transfer, ~90 % mice exhibited responses to systemic injection of naloxone (10, 20 or 30 mg/kg, s.c.) and significant antinociceptive effects induced by naloxone were observed (Fig. 2A). At 10 mg/kg of naloxone, maximal antinociceptive response was achieved. However, the maximal antinociceptive activity produced by naloxone was much less than that elicited by morphine at the same dose (Fig. 2C). Similar to the observations with spinal cord injection of the dsAAV2-MORS196A (Chen et al., 2007), when hot plate test was used to monitor the supraspinal antinociceptive effect, naloxone (30 mg/kg, s.c.) did not induce any significant antinociceptive effect (AUC = 58.1 ± 8.8 min × s) in mice injected with the dsAAV2-MORS196A-CSTA at the dorsal horn of the spinal cord (Fig. 2C). On the other hand, morphine (10 mg/kg, s.c.) produced a significant antinociceptive effect (AUC = 813.1 ± 39.9 min × s) by the same measurement in mice injected with dsAAV2 (Fig. 2C).
Figure 2.
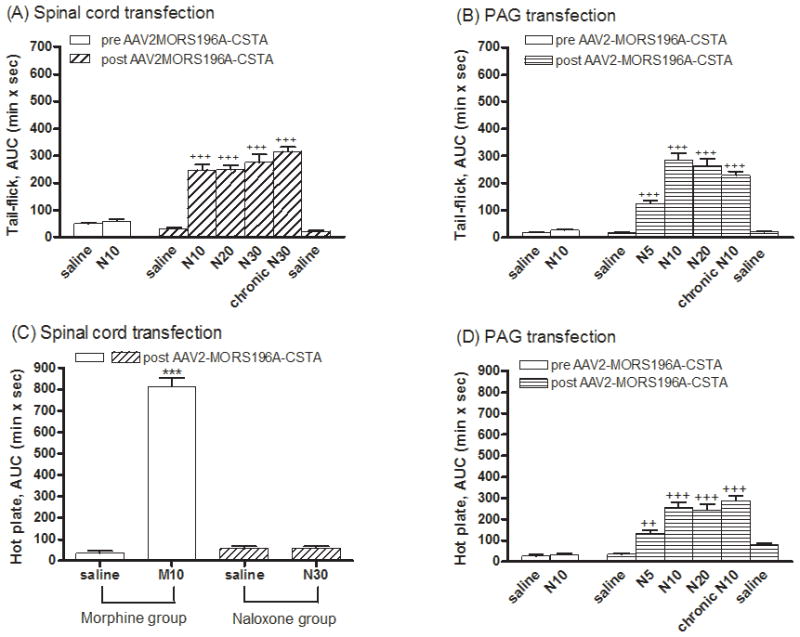
Chronic administration of morphine results in tolerance development. Injection of dsAAV2-MORS196A-CSTA into the dorsal horn area of the spinal cord did not alter chronic morphine induced tolerance development. The degree of tolerance induced by injecting a fixed dose (10 mg/kg, s.c., b.i.d.) of morphine for 6 days is about 5 fold (4.8 fold increase in the ED50 value) (Table 1). In contrast, when mice injected with the dsAAV2 at spinal cord were treated with naloxone (10–30 mg/kg, s.c., b.i.d.) for 6 days, neither the magnitude (AUC = 314.5 ± 17.6 min × s) nor the potency (ED50) of naloxone was different from the acute antinociceptive effect as measured by the tail-flick test (Table 1).
Naloxone elicited supra-spinal antinociceptive effect without tolerance development in mice injected with MORS196A-CSTA-EGFP at vlPAG
Similar to the injection of dsAAV2 at the dorsal horn area of spinal cord, injection of the mutant MOR containing dsAAV2 into the vlPAG area resulted in the antinociceptive response to naloxone as measured by tail-flick latency. As shown in Figs. 2B and 2D, naloxone (5–20 mg/kg, s.c.) elicited antinociceptive effects 2 weeks after gene transfer into the vlPAG area as determined by both tail-flick (Fig. 2B) and hot plate (Fig. 2D) tests. Again, the naloxone-induced antinociceptive effect in mice injected with dsAAV2 at vlPAG reached maximal response about 10 mg/kg. The naloxone-induced maximal antinociceptive response was again smaller than that observed with morphine. When morphine (10 mg/kg, s.c., b.i.d.) was given sub-chronically for 6 days, it induced tolerance development (Table 1). However, when the mice injected with dsAAV2-MORS196A-CSTA were sub-chronically treated with naloxone (10 mg/kg, s.c., b.i.d.) for 6 days, neither the magnitude (229.3 ± 13.6 min × s) nor the potency of naloxone differed greatly from the acute magnitude and potency values as measured by tail-flick test (Table 1), or by the hot plate test (Fig. 2D). These results indicated that chronic treatment with naloxone in mice injected with the dsAAV2 virus at vlPAG did not result in tolerance development to both spinal and supraspinal antinociceptive responses.
Naloxone-mediated antinociceptive responses correlated with the expression sites of MORS196A-CSTA
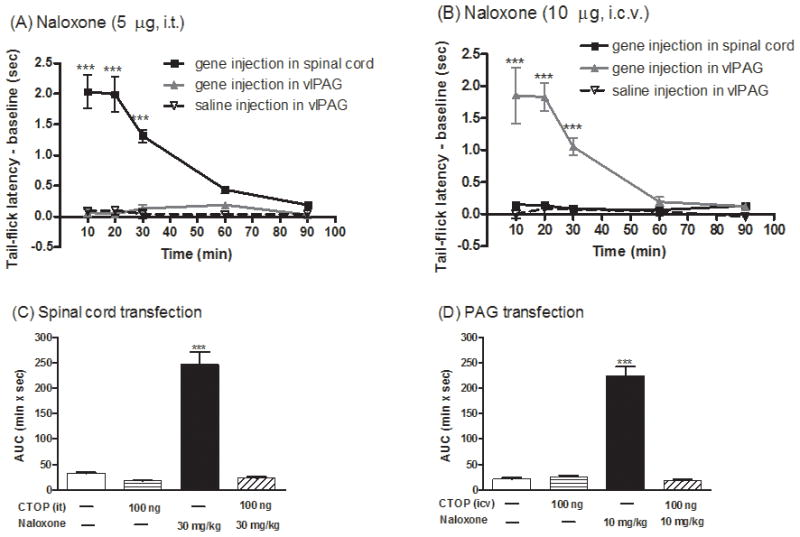
Low doses of naloxone have been reported to produce analgesia (Ueda et al., 1986). Further, in a transgenic mouse model for sickle cell anemia, naloxone was reported to elicit antinociceptive response involving the CCR5/CCR1 chemokine receptor (Lunzer et al., 2007). Thus, the observed naloxone-induced antinociceptive responses after dsAAV2-MORS196A-CSTA-EFP injection could be independent of the expression of mutant MOR. In order to demonstrate the direct relationship between the site of mutant MOR injection and the antinociceptive responses, naloxone was injected intrathecally (i.t., 5 μg) or intracerebroventricularly (i.c.v., 10 μg) respectively in mice injected with the virus either at the dorsal horn of spinal cord or at vlPAG area. In mice injected with the dsAAV2-MORS196A-CSTA-EGFP into S2/S3 spinal cord dorsal horn, naloxone elicited antinociceptive effects only when it was given by i.t. (Fig. 3A) but not by i.c.v. route (Fig. 3B). On the contrary, in mice injected with dsAAV2-MORS196A-CSTA-EGFP in vlPAG, naloxone elicited antinociceptive effects only when it was given by i.c.v. (Fig. 3B) but not by the i.t. route (Fig. 3A). In order to demonstrate the observed naloxone antinociceptive effect was indeed due to the activation of the mutant MOR, the mice were pretreated with a MOR-selective peptide antagonist, CTOP, prior to the administration of naloxone. CTOP by itself (100 ng) could not elicit the antinociceptive effect in mice transfected with MORS196A-CSTA-EGFP into the spinal cord (AUC = 17.1 ± 2.1 min × s, Fig. 3C) or into the vlPAG area (AUC = 25.2 ± 3.5 min × s, Fig. 3D). When 100 ng of CTOP was injected i.t. 5 min before naloxone (30 mg/kg, s.c.), this peptide antagonist blocked the naloxone-induced antinociceptive response from the calculated AUC = 246.8 ± 24.9 min × s to the control AUC = 23.0 ± 2.6 min × s in mice injected with MORS196A-CSTA-EGFP at the spinal cord (Fig. 3C). Similarly, the antinociceptive effect of naloxone (10 mg/kg, s.c.) was reduced from the AUC = 224.1 ± 18.5 min × s to AUC = 17.3 ± 3.3 min × s when CTOP (100 ng) was injected i.c.v. 5 min before naloxone treatment (Fig. 3D). These data indicated that naloxone-mediated antinociceptive responses were the results of the opioid antagonist activating the mutant MOR at the site of virus injection.
Figure 3.
Naloxone did not induce physical dependence or rewarding effect in mice injected with MORS196A-CSTA-EGFP
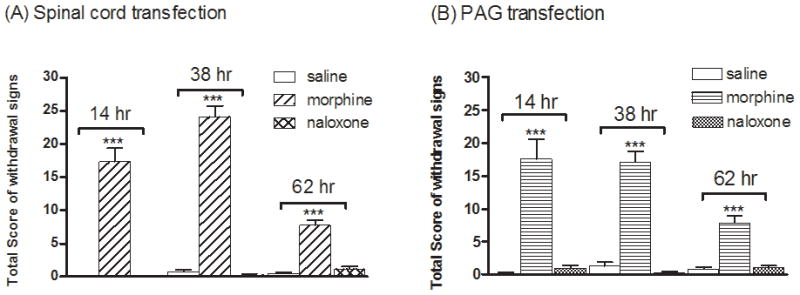
In previous studies, naloxone-induced activation of the mutant MOR (MORS196A) after injection of dsAAV2-MORS196A at dorsal horn of spinal cord did not result in development of physical dependence or rewarding effect in the mice. Whether the injection of the mutant MOR containing virus at the vlPAG area will result in the development of physical dependence or reward effect is uncertain. Thus, the natural withdrawal signs after chronic drug treatment were measured and the CPP test was carried out to monitor the physical dependence and rewarding effect in mice. As expected, chronic treatment with morphine (10–50 mg/kg, s.c., b.i.d. for 6 days) in mice injected with MORS196A-CSTA either at spinal cord or at vlPAG resulted in significant involuntary withdrawal behaviors 14, 38 or 62 h after the withdrawal of morphine (Fig. 4). However, chronic treatment with naloxone in these mice did not elicit any withdrawal signs (Fig. 4).
Figure 4.
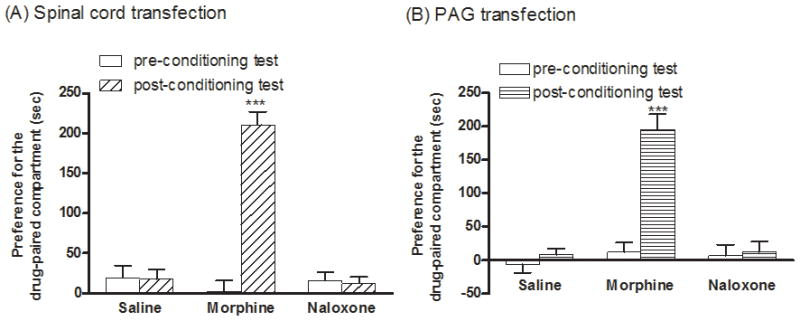
For the rewarding effect determined by CPP test, no preference in the control group conditioned with saline was observed indicating that the current CPP apparatus provided an unbiased design. Mice injected with the dsAAV2-MORS196A-CSTA at spinal cord treated with morphine (10 mg/kg, i.p. per day) and were conditioned for 6 days exhibited significantly increase the time spent in the morphine-paired compartment (210.6 ± 16.0 s, P < 0.001, Fig. 5A). Such observations are in accord with those reported on chronic morphine treatment induced conditioned place preference (Dallimore et al., 2006; Guo et al., 2008). In contrast, though naloxone could induce antinociceptive response in mice injected with the dsAAV2-MORS196A-CSTA at spinal cord, chronic naloxone treatment (10 mg/kg, i.p. per day) did not produce any rewarding effect as measured by the CPP test (Fig. 5A). When parallel CPP tests were carried out with mice injected with the dsAAV2-MORS196A-CSTA into the vlPAG, again, morphine treatment produced CPP response while chronic naloxone treatment did not (Fig. 5B). Thus, similar to the local injection of the dsAAV2 in spinal cord, expression of the mutant MOR in the vlPAG and activation of the receptor by naloxone result in the supraspinal and spinal antinociceptive responses without the development of tolerance and dependence or the rewarding effect associated with chronic morphine treatment.
Figure 5.
DISCUSSION AND CONCLUSIONS
Despite decades of study, the development of tolerance and dependence still present serious difficulties for the use of opioids for pain control. In our previous study, we have demonstrated the ability of naloxone to elicit antinociceptive response without tolerance and dependence in mice locally injected MORS196A in S2/S3 region of spinal cord dorsal horn. A fact worth noticed was the antinociceptive response only showed at spinal level, but not at supraspinal level.
In this study, we used dsAAV again to carry the gene of MORS196A-CSTA mutant instead of MORS196A as the delivered gene. The reason we chose to deliver the MORS196A-CSTA instead of MORS196A was based on the observed higher efficacy exhibited by naloxone and naltrexone in the in vitro cell models. The effect of naloxone has been studied in mice transfected with MORS196A into spinal cord dorsal horn (Chen et al., 2007). It is highly conceivable that naloxone would also have antinociceptive effects in mice transfected with MORS196A-CSTA into the spinal cord and even have better effect because naloxone acted as a full agonist at this mutated receptor. However, the efficacy of naloxone in mice transfected with MORS196A-CSTA was in line with mice transfected with MORS196A in spinal cord dorsal horn region. The lower efficacy exhibited by naloxone as compared to morphine may be due to the low level of the opioid receptor mutant expressed at the injection sites as compared to endogenous opioid receptors. To confirm that naloxone would act as a full agonist at MORS196A-CSTA mutant receptor in vivo, generation of the MORS196A-CTSA knock-in mice or locally injection of the MORS196A-CSTA virus into mu-opioid receptor knockout mice should be carried out to test the hypothesis.
Naloxone did not elicit any antinociceptive effect at supraspinal level in mice transfected with MORS196A-CSTA at spinal cord (Fig. 3B). This is consistent with our previous study. The midbrain periaqueductal gray especially the ventral lateral part (vlPAG) is thought to play a key role in the descending modulation of nociception. Microinjection of morphine into the vlPAG produces antinociception through the activation of the mu-opioid receptors (Jacquet and Lajtha, 1974). In order to investigate whether naloxone has supraspinal/spinal antinociceptive effect, we also transfer the gene of MORS196A-CSTA into vlPAG. By slicing the PAG region of the mice, we found that almost every mouse showed significant MORS196A-CSTA-EGFP expression in PAG (Fig. 1). Nearly 90% (36 of 42 mice) of mice transfected with MORS196A-CSTA into spinal cord dorsal horn and 100% (16 of 16 mice) of mice transfected with MORS196A-CSTA into vlPAG responded to naloxone and exhibited significant antinociceptive effect measured by tail-flick test. However, the proportion of EGFP expression was relatively low. We could not control the expression of mutant receptor in the same neurons as endogenous one due to the use of CMV promoter in the viral constructs. Although AAV5-based vectors have been shown to mediate higher levels of transgene expression in neuron (Tenenbaum et al., 2004), more neuron-specific promoters could be incorporated into future designs of targeting vectors in the delivery of MORS196A-CSTA so as to improve the agonistic activity of naloxone.
Except tail-flick test which measures pain depending on a spinal reflex, we also used hot plate test which measures a reflex that requires circuitry in the brain as well as in the spinal cord in the present study. We found that the antinociceptive effect of naloxone could only be shown by tail-flick test but not hot plate test if the gene of MORS196A-CSTA was expressed in the spinal cord. On the other hand, the antinociceptive effect of naloxone could be shown both by tail-flick and hot plate tests if the gene of MORS196A-CSTA was expressed in PAG (Fig. 2). These results indicated that naloxone elicited both supraspinal and spinal antinociceptive responses in mice injected with the virus at PAG while only induced spinal antinociceptive response in mice injected with virus at spinal dorsal horn region.
We also investigated the tolerance, dependence and rewarding effects after chronic morphine/naloxone treatment in normal or locally gene transfected mice at spinal cord or vlPAG. The results were in line with our prediction that chronic naloxone treatment did not induce significant tolerance. Roy et al. (2005) has provided evidences that simultaneous activation of both mu- and delta-opioid receptors results in tolerance development, mu-opioid receptor activation in conjunction with delta-opioid receptor blockade significantly attenuates the development of tolerance (Roy et al., 2005). The absolute dependence on delta-opioid receptor activity for tolerance development duringchronic morphine treatment was demonstrated unequivocally in the DOR−/− null mouse (Zhu et al., 1999). Since naloxone acted as an agonist in MORS196A-CSTA receptor locally expressed at spinal cord or PAG but still acted as an antagonist in endogenous mu- and delta-opioid receptors, it explains why naloxone did not induce tolerance in these mice. Furthermore, chronic naloxone treatment did not induce significant dependence in mice injected with these genes either. Mesolimbic areas were the major sites related to the rewarding effects produced by abused drugs (Wise, 1989). Among these areas, the dopamine projection from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and medial prefrontal cortex (mPFC) are thought to be primarily involved in the rewarding effects (Simon et al., 1980). Further, a number of studies have indicated that the central extended amygdala and nucleus accumbens (NAc) could mediate opioid withdrawal (Gracy et al., 2001; Le Guen et al., 2001; Walters et al., 2000). Since the site(s) in which mutated opioid receptors expressed in our study were spinal cord or PAG which were not regions related to the withdrawal or rewarding effects of opioids, it is reasonable to expect that naloxone would not induce withdrawal or rewarding effects.
Important progress in our study is that the dsAAV2-MORS196A-CSTA-EGFP could be delivered and be activated in different parts of the pain pathway. Further, the observed naloxone antinociceptive activities in hot plate and tail-flick assays are dependent on the site of virus injection clearly demonstrated the observed naloxone effect was not due to the activation of endogenous opioid receptors or other receptors. Our current studies also indicate to us that the mutant opioid receptor gene can be delivered at any part of the pain pathway and will result in naloxone antinociceptive activities. Although in vivo injection of the MORS196A-CSTA receptor mutant and systemic administration of naloxone constitutes a promising therapeutic approach, the gene delivery methods are too invasive to be applied for human use. Maybe the dsAAV2 can be delivered in other ways such as i.t. injection in order to alleviate the invasive method of vlPAG injection of the virus. Recently we have carried out this experiment in which the dsAAV2 was locally injected into the subarachnoid space of the spinal cord by intrathecal administration and found out that naloxone (10 mg/kg, s.c., b.i.d. for 6 days) elicited antinociceptive effect without tolerance and significant withdrawal symptoms or rewarding effect (Kao et al., 2010). Lumbar puncture has been used widely for spinal anesthesia with minimal danger in injuring the spinal cord and nerve roots. Thus, i.t. injection of the virus should be an optimal route in the delivery of MORS196A-CSTA mutant receptor.
With the absence of tolerance, dependence or addiction to the systemic administration of the antagonist, our approach of using the mutant opioid receptor in combination with the antagonist could present an alternative path for the chronic pain management. However the use of viral vector for gene delivery has to consider the possibility that any integration of the transgenes into sites within the transcription active regions could result in disrupting the expression of tumor suppressor gene or activating an oncogene resulting in the malignant transformation of cells (Schroder et al., 2002; Woods et al., 2003). Even with the advantage that AAV vector possesses, i.e., only known mammalian virus that is capable of site-specific integration into human cells and can achieve sustained and high-level of gene delivery without immune complications and vector associated toxicity (Song et al., 2001; Xiao et al., 1996; Xiao et al., 2000), The use of dsAAV for the delivery of mutant opioid receptor as a therapeutic treatment of chronic pain must consider probable transformation of the neurons that the virus infected.
Acknowledgments
This study was supported by grants from National Science Council (NSC97-2320-B-016-003), Taiwan, Republic of China and National Institutes of Health, USA [Grant R01 DA023905, R01 DA011806, K05 DA000513]. PY Law is the recipient of K05 DA000513.
Abbreviations
- dsAAV
double-stranded adenoassociated virus
- MOR
mu-opioid receptor
- KOR
kappa-opioid receptor
- DOR
delta-opioid receptor
- RASSL
receptor activated solely by synthetic ligands
- vlPAG
ventral lateral periaqueductal gray
- CPP
conditioned place preference
Contributor Information
Shu-Husan Chou, Beitou Armed Forces Hospital, Pharmacy.
Jen-Hsin Kao, National Defense Medical Center, Graduate Institute of Life Science.
Pao-Luh Tao, National Health Research Institutes, Division of Mental Health and Addiction Medicine.
Ping-Yee Law, University of Minnesota Medical School, Department of Pharmacology.
Horace H. Loh, University of Minnesota Medical School, Department of Pharmacology
References
- Chen SL, Ma HI, Han JM, Tao PL, Law PY, Loh HH. dsAAV type 2-mediated gene transfer of MORS196A-EGFP into spinal cord as a pain management paradigm. Proc Natl Acad Sci U S A. 2007;104(50):20096–20101. doi: 10.1073/pnas.0703409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude-Geppert PA, Liu J, Solberg J, Erickson-Herbrandson LJ, Loh HH, Law PY. Antagonist efficacy in MORS196L mutant is affected by the interaction between transmembrane domains of the opioid receptor. The Journal of pharmacology and experimental therapeutics. 2005;313(1):216–226. doi: 10.1124/jpet.104.076505. [DOI] [PubMed] [Google Scholar]
- Claude PA, Wotta DR, Zhang XH, Prather PL, McGinn TM, Erickson LJ, Loh HH, Law PY. Mutation of a conserved serine in TM4 of opioid receptors confers full agonistic properties to classical antagonists. Proc Natl Acad Sci U S A. 1996;93(12):5715–5719. doi: 10.1073/pnas.93.12.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Controlling signaling with a specifically designed Gi-coupled receptor. Proc Natl Acad Sci U S A. 1998;95(1):352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amour FE, Smith DL. A method for determining loss of pain sensation. The Journal of pharmacology and experimental therapeutics. 1941;72(1):74–79. [Google Scholar]
- Dallimore JE, Mickiewicz AL, Napier TC. Intra-ventral pallidal glutamate antagonists block expression of morphine-induced place preference. Behav Neurosci. 2006;120(5):1103–1114. doi: 10.1037/0735-7044.120.5.1103. [DOI] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102(52):19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. The up-and-down method for small sample. J Amer Stat Assoc. 1965;60:967–978. [Google Scholar]
- Elliott K, Kest B, Man A, Kao B, Inturrisi CE. N-methyl-D-aspartate (NMDA) receptors, mu and kappa opioid tolerance, and perspectives on new analgesic drug development. Neuropsychopharmacology. 1995a;13(4):347–356. doi: 10.1016/0893-133X(95)00083-P. [DOI] [PubMed] [Google Scholar]
- Elliott KJ, Brodsky M, Hyanansky A, Foley KM, Inturrisi CE. Dextromethorphan shows efficacy in experimental pain (nociception) and opioid tolerance. Neurology. 1995b;45(12 Suppl 8):S66–68. doi: 10.1212/wnl.45.12_suppl_8.s66. [DOI] [PubMed] [Google Scholar]
- Gracy KN, Dankiewicz LA, Koob GF. Opiate withdrawal-induced fos immunoreactivity in the rat extended amygdala parallels the development of conditioned place aversion. Neuropsychopharmacology. 2001;24(2):152–160. doi: 10.1016/S0893-133X(00)00186-X. [DOI] [PubMed] [Google Scholar]
- Guo N, Garcia MM, Taylor BK, Zadina JE, Harlan RE. Blockade of micro-opioid receptors in the medial thalamus inhibits acquisition, but not expression, of morphine-induced conditioned place preference. Neuroscience. 2008;151(4):948–954. doi: 10.1016/j.neuroscience.2007.10.058. [DOI] [PubMed] [Google Scholar]
- Huang EY, Liu TC, Tao PL. Co-administration of dextromethorphan with morphine attenuates morphine rewarding effect and related dopamine releases at the nucleus accumbens. Naunyn Schmiedebergs Arch Pharmacol. 2003;368(5):386–392. doi: 10.1007/s00210-003-0803-7. [DOI] [PubMed] [Google Scholar]
- Jacquet YF, Lajtha A. Paradoxical effects after microinjection of morphine in the periaqueductal gray matter in the rat. Science. 1974;185(156):1055–1057. doi: 10.1126/science.185.4156.1055. [DOI] [PubMed] [Google Scholar]
- Kao JH, Chen SL, Ma HI, Law PY, Tao PL, Loh HH. Intrathecal delivery of a mutant micro-opioid receptor activated by naloxone as a possible antinociceptive paradigm. The Journal of pharmacology and experimental therapeutics. 2010;334(3):739–745. doi: 10.1124/jpet.109.165399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guen S, Gestreau C, Besson JM. Sensitivity to naloxone of the behavioral signs of morphine withdrawal and c-Fos expression in the rat CNS: a quantitative dose-response analysis. J Comp Neurol. 2001;433(2):272–296. doi: 10.1002/cne.1140. [DOI] [PubMed] [Google Scholar]
- Lunzer MM, Yekkirala A, Hebbel RP, Portoghese PS. Naloxone acts as a potent analgesic in transgenic mouse models of sickle cell anemia. Proc Natl Acad Sci U S A. 2007;104(14):6061–6065. doi: 10.1073/pnas.0700295104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SC. Dextromethorphan psychosis, dependence and physical withdrawal. Addict Biol. 2005;10(4):325–327. doi: 10.1080/13556210500352410. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Contarino A. Gender- and morphine dose-linked expression of spontaneous somatic opiate withdrawal in mice. Behav Brain Res. 2006;170(1):110–118. doi: 10.1016/j.bbr.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. Opioid agonist and antagonist bivalent ligands. The relationship between spacer length and selectivity at multiple opioid receptors. J Med Chem. 1986a;29(10):1855–1861. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Larson DL, Yim CB, Sayre LM, Ronsisvalle G, Lipkowski AW, Takemori AE, Rice KC, Tam SW. Stereostructure-activity relationship of opioid agonist and antagonist bivalent ligands. Evidence for bridging between vicinal opioid receptors. J Med Chem. 1985;28(9):1140–1141. doi: 10.1021/jm00147a002. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Ronsisvalle G, Larson DL, Takemori AE. Synthesis and opioid antagonist potencies of naltrexamine bivalent ligands with conformationally restricted spacers. J Med Chem. 1986b;29(9):1650–1653. doi: 10.1021/jm00159a014. [DOI] [PubMed] [Google Scholar]
- Rosow CE. The clinical usefulness of agonist-antagonist analgesics in acute pain. Drug Alcohol Depend. 1987;20(4):329–337. doi: 10.1016/0376-8716(87)90006-8. [DOI] [PubMed] [Google Scholar]
- Roy S, Guo X, Kelschenbach J, Liu Y, Loh HH. In vivo activation of a mutant mu-opioid receptor by naltrexone produces a potent analgesic effect but no tolerance: role of mu-receptor activation and delta-receptor blockade in morphine tolerance. J Neurosci. 2005;25(12):3229–3233. doi: 10.1523/JNEUROSCI.0332-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110(4):521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Schwartz RH. Adolescent abuse of dextromethorphan. Clin Pediatr (Phila) 2005;44(7):565–568. doi: 10.1177/000992280504400702. [DOI] [PubMed] [Google Scholar]
- Simon H, Scatton B, Moal ML. Dopaminergic A10 neurones are involved in cognitive functions. Nature. 1980;286(5769):150–151. doi: 10.1038/286150a0. [DOI] [PubMed] [Google Scholar]
- Song S, Laipis PJ, Berns KI, Flotte TR. Effect of DNA-dependent protein kinase on the molecular fate of the rAAV2 genome in skeletal muscle. Proc Natl Acad Sci U S A. 2001;98(7):4084–4088. doi: 10.1073/pnas.061014598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum L, Chtarto A, Lehtonen E, Velu T, Brotchi J, Levivier M. Recombinant AAV-mediated gene delivery to the central nervous system. J Gene Med 6 Suppl. 2004;1:S212–222. doi: 10.1002/jgm.506. [DOI] [PubMed] [Google Scholar]
- Ueda H, Fukushima N, Kitao T, Ge M, Takagi H. Low doses of naloxone produce analgesia in the mouse brain by blocking presynaptic autoinhibition of enkephalin release. Neurosci Lett. 1986;65(3):247–252. doi: 10.1016/0304-3940(86)90269-7. [DOI] [PubMed] [Google Scholar]
- Walters CL, Aston-Jones G, Druhan JP. Expression of fos-related antigens in the nucleus accumbens during opiate withdrawal and their attenuation by a D2 dopamine receptor agonist. Neuropsychopharmacology. 2000;23(3):307–315. doi: 10.1016/S0893-133X(00)00113-5. [DOI] [PubMed] [Google Scholar]
- Wise RA. Opiate reward: sites and substrates. Neurosci Biobehav Rev. 1989;13(2–3):129–133. doi: 10.1016/s0149-7634(89)80021-1. [DOI] [PubMed] [Google Scholar]
- Wong CS, Wu CT, Yu JC, Yeh CC, Lee MM, Tao PL. Preincisional dextromethorphan decreases postoperative pain and opioid requirement after modified radical mastectomy. Can J Anaesth. 1999;46(12):1122–1126. doi: 10.1007/BF03015519. [DOI] [PubMed] [Google Scholar]
- Woods NB, Muessig A, Schmidt M, Flygare J, Olsson K, Salmon P, Trono D, von Kalle C, Karlsson S. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101(4):1284–1289. doi: 10.1182/blood-2002-07-2238. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70(11):8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, Tsao YP, Dressman D, Hoffman EP, Watchko JF. Full functional rescue of a complete muscle (TA) in dystrophic hamsters by adeno-associated virus vector-directed gene therapy. J Virol. 2000;74(3):1436–1442. doi: 10.1128/jvi.74.3.1436-1442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Gu Y, Xu GY, Wu P, Li GW, Huang LY. Adeno-associated viral transfer of opioid receptor gene to primary sensory neurons: a strategy to increase opioid antinociception. Proc Natl Acad Sci U S A. 2003;100(10):6204–6209. doi: 10.1073/pnas.0930324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Law PY, Guo X, Loh HH. In vivo activation of a mutant mu-opioid receptor by antagonist: future direction for opiate pain treatment paradigm that lacks undesirable side effects. Proc Natl Acad Sci U S A. 2003;100(4):2117–2121. doi: 10.1073/pnas.0334906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24(1):243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]