

Abstract
Entamoeba histolytica is an intestinal parasite that causes dysentery and liver abscess. Parasite cell surface receptors, such as the Gal/GalNAc lectin, facilitate attachment to host cells and extracellular matrix. The Gal/GalNAc lectin binds to galactose or N-acetylgalactosamine residues on host components and is composed of heavy (Hgl), intermediate (Igl), and light (Lgl) subunits. Although Igl is constitutively localized to lipid rafts (cholesterol-rich membrane domains), Hgl and Lgl transiently associate with this compartment in a cholesterol-dependent fashion. In this study, trophozoites were exposed to biologically relevant ligands to determine if ligand binding influences the submembrane distribution of the subunits. Exposure to human red blood cells (hRBCs) or collagen, which are bona fide Gal/GalNAc lectin ligands, was correlated with enrichment of Hgl and Lgl in rafts. This enrichment was abrogated in the presence of galactose, suggesting that direct lectin-ligand interactions are necessary to influence subunit location. Using a cell line that is able to attach to, but not phagocytose, hRBCs, it was shown that physical attachment to ligands was not sufficient to induce the enrichment of lectin subunits in rafts. Additionally, the mutant had lower levels of phosphatidylinositol (4,5)-bisphosphate (PIP2); PIP2 loading restored the ability of this mutant to respond to ligands with enrichment of subunits in rafts. Finally, intracellular calcium levels increased upon attachment to collagen; this increase was essential for the enrichment of lectin subunits in rafts. Together, these data provide evidence that ligand-induced enrichment of lectin subunits in rafts may be the first step in a signaling pathway that involves both PIP2 and calcium signaling.
INTRODUCTION
Entamoeba histolytica is an intestinal parasite responsible for dysentery and amoebic liver abscess (22). Amoebiasis is a food- and waterborne illness and is prevalent in underdeveloped countries lacking proper sanitation practices. As of 2010, it is estimated that 2.6 billion people worldwide do not use modern sanitation practices, and 886 million do not have access to clean drinking water sources (54). Thus, there is considerable global risk for acquiring E. histolytica infection.
Amoebiasis occurs when food or water contaminated with the environmentally resistant cyst form of the parasite is ingested; excystation leads to the release of amoeboid trophozoites in the small intestine. Trophozoites then move to and colonize the large intestine. Serious complications arise when trophozoites invade the colonic epithelium, enter the bloodstream, and travel to extraintestinal sites such as the liver, lungs, and brain. During colonization of the host, trophozoites attach to numerous ligands, including red blood cells (RBCs), extracellular matrix (ECM) components (e.g., collagen and fibronectin), intestinal flora, colonic mucins, and leukocytes (6, 15, 39). Therefore, adhesion is an important virulence function for the parasite.
In mammalian cells, integrins are dimeric transmembrane receptors that are responsible for cell-cell and cell-ECM adhesion and signal transduction. Although no integrin homologs have been identified in the E. histolytica genome (27), attachment to ligands in the host can occur through cell surface receptors, which share sequence homology with integrins. One such receptor is the heterotrimeric protein complex galactose/N-acetylgalactosamine lectin (Gal/GalNAc lectin). This adhesin binds to galactose and N-acetylgalactosamine residues on host cells and is composed of heavy (Hgl), light (Lgl), and intermediate (Igl) subunits. Hgl is a transmembrane protein that is disulfide linked to a glycophosphatidylinositol (GPI)-anchored Lgl. The heterodimer noncovalently associates with a GPI-anchored Igl. Both Hgl and Igl share sequence homology with β integrins (12, 46–48, 51), suggesting that they may also play a role in signaling.
Attachment of E. histolytica to human red blood cells (hRBCs) or collagen is inhibited in the presence of galactose, suggesting that the Gal/GalNAc lectin is an important receptor for these ligands (2, 33). On the other hand, binding of amoebae to fibronectin is not significantly inhibited by galactose, suggesting that the Gal/GalNAc lectin may not be the major receptor for this ligand (33). The functional regulation of the Gal/GalNAc lectin is not well understood.
In other systems, lipid rafts play a role in regulating the function of cell surface receptors, including integrins (24). Lipid rafts are tightly packed cholesterol- and sphingolipid-rich membrane microdomains. Lipid rafts are thought to serve as platforms within which signaling proteins interact. The removal of cholesterol, resulting in the disruption of lipid rafts, significantly inhibits the adhesion of E. histolytica trophozoites to host cells (23) and collagen (33) but only slightly inhibits the adhesion of trophozoites to fibronectin (33). This suggests that E. histolytica lipid rafts play a significant role in binding to host cells and collagen and a lesser role in binding to host fibronectin. The parallel roles of the Gal/GalNAc lectin and lipid rafts in binding to collagen, but not fibronectin, suggest that these membrane domains regulate the function of the lectin.
In addition to protein receptors, lipids can also participate in signaling pathways that emanate from lipid rafts. One such family of signaling lipids are the phosphoinositides. Two phosphorylated members of the phosphoinositide family are phosphatidylinositol (4,5)-bisphosphate (PIP2) and phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Both of these lipids play important roles in cellular processes such as phagocytosis, protein kinase activation, and actin polymerization (9, 18). PIP2 also regulates calcium signaling (8, 19, 28). For example, signal transduction can lead to hydrolysis of PIP2, resulting in the production of second-messenger molecules, inositol trisphosphate (IP3) and diacylglycerol (DAG) (20). These, in turn, facilitate the release of calcium into the cytoplasm from intracellular calcium stores and from the extracellular space through channels in the plasma membrane (16, 40).
Phosphoinositides can also facilitate signaling by recruiting downstream proteins that have specific phosphoinositide binding domains. For example, FYVE-finger domains, which were originally observed in Fab1p, YOTB, Vac1p, and EEA1 proteins, bind specifically to phosphatidylinositol 3-phosphate (44). Additionally, certain pleckstrin homology (PH) domains, such as that from Bruton's tyrosine kinase (PHBTK), have been shown to specifically bind PIP3 (42). Overexpression of green fluorescent protein (GFP)–FYVE-finger domains or GFP-PHBTK domains has been used to localize phosphoinositides in real time in E. histolytica (4, 38).
Previously, we demonstrated that cholesterol loading of parasite membranes induced the enrichment of the Gal/GalNAc lectin subunits in lipid rafts, which in turn increased the activity of the Gal/GalNAc lectin (53). In this study, we have examined the localization of Gal/GalNAc lectin subunits after attachment to biologically relevant extracellular ligands. We show that binding to human red blood cells (hRBCs) and collagen results in the enrichment of Hgl and Lgl in lipid rafts, while attachment to fibronectin does not change the localization of the subunits. We also demonstrate that cells expressing GFP-PHBTK exhibit reduced PIP2 levels. In these cells, attachment to ligand is not correlated with enrichment of Hgl and Lgl in lipid rafts; the phenotype is reversible upon the addition of exogenous PIP2, indicating a role for PIP2 in regulating the submembrane position of the Gal/GalNAc lectin. Finally, intracellular calcium levels increase upon attachment to collagen; increased intracellular calcium levels appear to be essential for the enrichment of lectin subunits in rafts. Together, our data suggest that colocalization of Gal/GalNAc lectin subunits in rafts may be the first step in the activation of a signaling pathway and that PIP2 and calcium may be involved in this pathway.
MATERIALS AND METHODS
Strains and culture conditions.
E. histolytica trophozoites (strain HM1:IMSS) were grown axenically in TYI-S-33 medium (11) in 15-ml glass screw-cap tubes or T25 cell culture flasks (Sarstedt, Newton, NC) at 37°C. The construction of a cell line conditionally expressing GFP-PHBTK (tetracycline inducible) is described elsewhere (4). GFP-PHBTK-expressing trophozoites were maintained in TYI-S-33 medium supplemented with 6 μg/ml G418 and 15 μg/ml hygromycin. The expression of GFP-PHBTK was induced with 5 μg/ml tetracycline for 24 h prior to use in assays. Prior to performance of assays, cells were incubated on ice for 10 or 20 min in order to release them from tube or flask surfaces, respectively.
Exposure to ligands.
Wild-type or GFP-PHBTK-expressing cells (3.5 × 106) were incubated in serum-free medium for 30 min and then exposed to various ligands prior to lipid raft extraction. For hRBC exposure, trophozoites were incubated in the presence of 3.5 × 108 hRBCs (U.S. Biological, Swampscott, MA) for 5 min at 37°C. For exposure to collagen and fibronectin, cells were incubated on ECM-coated flasks (BD Biosciences, Bedford, MA) or uncoated flasks (Sarstedt) for 15 min at 37°C.
Lipid raft extraction.
After exposure to ligands, isolation and characterization of lipid rafts were carried out as previously described (23). Extracted raft-associated proteins were characterized by SDS-PAGE and Western blotting as described previously (23). Primary antibodies included a mixture of monoclonal anti-Lgl antibodies (3C2, IC8, IA9, and ID4) (1:4,000 dilution), polyclonal anti-Hgl antibodies (1:5,000 dilution), monoclonal anti-Hgl antibodies (1G7) (1:1,000 dilution), or a mixture of monoclonal anti-Igl antibodies (3G5-A3-G3, 5H1-F11-D11, and 4G2-D8-H1) (1:4,000 dilution) (antibodies were kind gifts from William A. Petri, Jr., University of Virginia School of Medicine, Charlottesville, VA). Western blots were analyzed by densitometry using ImageJ software (version 1.42q; U.S. National Institutes of Health, Bethesda, MD).
Whole-cell PIP2 extraction and lipid dot blots.
Total lipid was extracted from wild-type and GFP-PHBTK-expressing trophozoites according to the methods of Gray et al. (14). Briefly, 1 × 106 cells were washed twice with phosphate-buffered saline (PBS). Lipids were precipitated by the addition of 5 ml of 0.5 M trichloroacetic acid (TCA) and centrifuged at 500 × g for 5 min at 4°C. The pellets were washed with 3 ml of 5% (wt/vol) TCA–1 mM EDTA and centrifuged at 500 × g for 5 min. To the pellets, 3 ml of methanol-chloroform (2:1) was added, and the mixture was vortexed 3 times over a period of 10 min at room temperature to facilitate neutral lipid extraction. The extracted lipids were centrifuged at 500 × g for 5 min at 4°C. To the pellet, 2.25 ml methanol-chloroform-12.1 N HCl (80:40:1) was added, and the mixture was vortexed 4 times over 15 min at room temperature and centrifuged at 500 × g. The resulting supernatant was subjected to phase split by the addition of 750 μl chloroform and 1.35 ml 0.1N HCl. The solution was centrifuged at 500 × g for 5 min at 4°C. After centrifugation, the organic phase was collected and dried using a MiVac Duo Sample Concentrator Speed Vac centrifuge (GeneVac, Gardiner, NY).
The vacuum-dried lipid pellets were resuspended in a methanol-chloroform-water mixture (2:1:0.8) and vortexed for 30 s, followed by sonication in a cold water bath for 10 min. The lipids were then spotted onto a nitrocellulose membrane. The membrane was blocked with 1.5% fatty acid-free bovine serum albumin (BSA) for 1 h at room temperature and probed with mouse anti-PIP2 (Abcam, Cambridge, MA) or mouse anti-PIP3 (Echelon Biosciences, Salt Lake City, UT) antibodies. Densitometric analysis was performed using Image J software.
PIP2 loading.
GFP-PHBTK-expressing cells were loaded with PIP2 using a shuttle PIP2 kit (Echelon Biosciences, Salt Lake City, UT) according to the manufacturer's guidelines. Concentrations of 25 μM PIP2 and 12.5 μM PIP2 carrier histone (H1) were used. Loading was carried out for 30 min at 37°C. PIP2 loading was confirmed using fluorescence microscopy of a BODIPY-labeled PIP2 (Nikon Eclipse TI-E spectral confocal microscope; Nikon Instruments Inc., Lewisville, TX). After PIP2 loading, cells were exposed to hRBCs and lipid rafts were extracted as described above.
Calcium assay.
Relative intracellular calcium levels were assessed using the calcium indicator fluo-4/AM according to the manufacturer's instructions. Fluo-4/AM is fluorescent when bound to calcium. Wild-type cells were washed twice with calcium stain loading buffer (CSB) (50) and then incubated in CSB supplemented with 5 μM fluo-4/AM (Invitrogen, Carlsbad, CA) or an equivalent volume of dimethyl sulfoxide (DMSO) (diluent control) for 30 min at 37°C. After staining, cells were washed twice with CSB, and 1 × 105 cells (stained or control) were added to the wells of a 12-well plate which contained 1 mM CaCl2 (5) and a glass coverslip coated with collagen or fibronectin (BD Biosciences). After 3 min, plates were transferred to a BioTek Flx800-I microplate reader (BioTek, Winooski, VT) and incubated at 37°C, and fluorescence (excitation, 485 nM; emission, 525 nM) was monitored at 5-min intervals for 10 min. To account for background fluorescence, the fluorescence value of control cells (DMSO) was subtracted from the fluorescence value of fluo-4/AM-stained cells.
Calcium chelation.
To chelate intracellular calcium, cells were incubated in the presence of 50 μM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA/AM) (EMD Chemicals Group, Darmstadt, Germany) in serum-free medium for 30 min at 37°C. Cells were then exposed to collagen-coated coverslips, and the calcium assay was performed as described above. Cells were also exposed to collagen-coated flasks, and lipid rafts were isolated and characterized as described above.
Adhesion assay.
To determine the effect of intracellular calcium chelation on adhesion, we used a previously described adhesion assay (33, 38). Cells were preexposed to serum-free medium with or without 50 μM BAPTA/AM for 30 min at 37°C in the presence of the fluorescent vital stain calcein-AM (5 μg/ml). Cells (3 × 104) were seeded in the wells of a 96-well collagen-coated plate (BD Biosciences) (in triplicate) for each condition and incubated at 37°C for 15 min. The wells were then washed with warm PBS to remove nonadherent cells. Fluorescence was measured using a BioTek Flx800-I microplate reader (excitation, 485 nM; emission, 525 nM). Values were reported as percentages of the control value, which was arbitrarily set to 100%.
Statistical analysis.
All data are reported as a mean ± standard deviation (SD). Statistical analyses were carried out using GraphPad Instat V.3. Comparisons were carried out using a one-way analysis of variance (ANOVA) with posttest. P values of less than 0.05 (*) were considered statistically significant, and values of less than 0.01 (**) or 0.001 (***) were considered highly statistically significant.
RESULTS
Exposure to hRBCs correlates with enrichment of Hgl and Lgl subunits in lipid raft fractions.
In mammalian cells, binding to ligand induces clustering of integrins in lipid raft domains (17). To determine if ligand engagement also influences the submembrane distribution of the subunits of the Gal/GalNAc lectin, we exposed trophozoites to hRBCs and isolated and characterized lipid rafts as described previously (23). The composition of lipid rafts confers detergent resistance to these membrane domains. Therefore, purification of lipid rafts was initiated by extraction with cold Triton X-100. This resulted in the isolation of detergent-resistant membrane (DRM), which consists of both lipid raft and actin-rich membrane. Since the buoyant density of lipid rafts is less than that of actin-rich membrane, these two membrane domains were further separated by sucrose density gradient centrifugation. To address possible contamination of DRM from hRBCs, whole-cell lysates from hRBCs were tested by Western blotting with antibodies for Hgl, Lgl, and Igl and were shown to have no cross-reacting proteins (data not shown).
Western blot analysis of gradient fractions revealed that the majority of Igl was found in a low-density region (fractions 9 to 14) (Fig. 1). Previously, these fractions were shown to possess the highest levels of cholesterol among other detergent-resistant fractions (23). Thus, these fractions are identified as lipid rafts. The localization of Igl to these low-density rafts was consistent with previous reports (23, 53). In control cells, the majority of Hgl and Lgl was associated with less buoyant, actin-rich fractions (fractions 17 to 20) (Fig. 1). However, after exposure to hRBCs, there was an increase in the proportion of Hgl and Lgl that was localized to lipid raft fractions (fractions 9 to 14), whereas the submembrane distribution of Igl remained unchanged (Fig. 1). This observation suggests that binding to at least one ligand, hRBCs, can induce the enrichment of Hgl and Lgl in lipid rafts.
Fig 1.

Exposure to hRBCs is correlated with enrichment of Hgl and Lgl in lipid rafts. Trophozoites (3.5 × 106) were serum starved and exposed to hRBCs. Detergent-resistant membrane (DRM) was extracted and fractionated using sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to SDS-PAGE and Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. In both control trophozoites and trophozoites exposed to hRBCs, Igl is localized predominantly to fractions 9 to 14, previously identified as lipid rafts. Hgl and Lgl, which are localized to dense, actin-rich fractions 17 to 20 in control cells, are enriched in lipid rafts (fractions 9 to 14) upon hRBC exposure.
To determine if enrichment of Hgl and Lgl in lipid rafts was dependent on a physical interaction between the Gal/GalNAc lectin and its ligand, cells were pretreated with galactose, a competitive inhibitor of lectin-ligand binding, or mannose (a control sugar) prior to hRBC exposure. Incubation with galactose prevented the enrichment of Hgl and Lgl in lipid raft fractions after hRBC exposure, while incubation with mannose did not inhibit the enrichment of Hgl and Lgl in lipid raft fractions after hRBC exposure (Fig. 2). The localization of Igl in lipid raft domains was unaffected in the presence of galactose or mannose. These data suggest that physical interaction between the Gal/GalNAc lectin and its ligand is necessary for raft enrichment of Hgl and Lgl.
Fig 2.

Enrichment of Hgl and Lgl in lipid rafts upon exposure to hRBCs is inhibited in the presence of galactose. Trophozoites (3.5 × 106) were serum starved and exposed to 10 mM galactose (gal) or 10 mM mannose (man) prior to exposure to hRBCs. DRM was isolated and fractionated using sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. The localization of Igl remained unchanged after exposure to galactose or mannose followed by hRBCs. The enrichment of Hgl and Lgl in lipid rafts after exposure to hRBCs was inhibited in the presence of galactose but not mannose.
Exposure to collagen type I correlates with galactose-sensitive enrichment of Hgl and Lgl subunits in lipid rafts.
To determine if another ligand also induces the enrichment of Gal/GalNAc lectin subunits in lipid rafts, we exposed trophozoites to collagen type I, which has been shown to initiate signaling in E. histolytica (7, 10, 35). Trophozoites were incubated on collagen-coated flasks or uncoated control flasks. Lipid rafts were extracted and characterized. Similar to the case for incubation with hRBCs, incubation on collagen was accompanied by an increase in the levels of Hgl and Lgl subunits in high-buoyancy lipid raft fractions (Fig. 3). Interestingly, the fractions with the highest levels of Hgl and Lgl (fractions 13 to 16) (Fig. 3) differed from those with the highest levels of Hgl and Lgl after exposure to hRBCs (fractions 9 to 14) (Fig. 2). This suggests that the molecular mechanism governing the submembrane distribution of the Gal/GalNAc lectin subunits differs in a ligand-specific manner. This enrichment was prevented by the addition of galactose but not by the addition of mannose (Fig. 4). Therefore, physical interaction of trophozoites with collagen also appears to be necessary for enrichment of Hgl and Lgl in lipid rafts.
Fig 3.

Exposure to collagen is correlated with a calcium-dependent enrichment of Hgl and Lgl in lipid rafts. Trophozoites (3.5 × 106) were serum starved or serum starved and incubated in the presence of BAPTA/AM and incubated on collagen-coated flasks (coll) or uncoated control flasks. DRM was isolated and fractionated by sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. In cells treated with collagen, the distribution of Igl was not different from that in control cells. Hgl and Lgl subunits were enriched in fractions 13 to 16 upon exposure to collagen. Enrichment of Hgl and Lgl in lipid rafts was inhibited in the presence of BAPTA/AM.
Fig 4.

Enrichment of Hgl and Lgl in lipid rafts after exposure to collagen is inhibited by the presence of galactose but not mannose. Trophozoites (3.5 × 106) were serum starved and pretreated with 10 mM galactose (gal) or 10 mM mannose (man). Cells were then incubated on collagen-coated flasks (coll) for 15 min at 37°C. DRM was isolated and subjected to sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. The localization of Igl to lipid rafts (fractions 9 to 14) remained unchanged in the presence of galactose or mannose. The enrichment of Hgl and Lgl in lipid rafts after exposure to collagen was inhibited in the presence of galactose but not mannose.
Exposure to fibronectin does not correlate with an enrichment of Hgl and Lgl subunits in lipid rafts.
Because galactose and raft-disrupting agents have little effect on trophozoite-fibronectin interaction (33), it is likely that neither the Gal/GalNAc lectin nor lipid rafts play a primary role in the interaction between the parasite and this ECM component. Therefore, as a control, we incubated trophozoites on fibronectin-coated flasks and isolated and characterized lipid rafts. In both control cells and cells exposed to fibronectin, Hgl and Lgl were concentrated in the actin-rich fractions (fractions 17 to 20), while Igl was concentrated in lipid raft fractions (fractions 9 to 14) (Fig. 5). Therefore, exposure to fibronectin did not affect the localization of any of the Gal/GalNAc lectin subunits and in particular did not induce the enrichment of Hgl and Lgl in lipid raft domains of E. histolytica. This supports the authenticity of our finding that binding to a bona fide ligand of the Gal/GalNAc lectin (e.g., hRBCs and collagen) can influence the submembrane localization of this adhesin.
Fig 5.

Exposure to fibronectin is not associated with enrichment of Gal/GalNAc lectin subunits. Trophozoites (3.5 × 106) were serum starved and incubated on fibronectin-coated flasks or uncoated control flasks. DRM was isolated and fractionated using sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. In both control and fibronectin exposed cells, Igl was localized to fractions 9 to 14, previously identified as lipid rafts. Hgl and Lgl were localized primarily to fractions 17 to 20 in both fibronectin-exposed and control cells.
Attachment to hRBCs is not sufficient for enrichment of Hgl and Lgl in lipid rafts.
Previously, an E. histolytica cell line expressing GFP-labeled PH domain derived from Bruton's tyrosine kinase (GFP-PHBTK) was developed (4). The GFP-PHBTK-expressing cell line exhibited interesting phenotypes, including enhanced motility and a phagocytic defect characterized by the ability to bind to, but not internalize, hRBCs (4). The latter characteristic provided the opportunity to test the sufficiency of ligand binding in the regulation of Gal/GalNAc localization. GFP-PHBTK-expressing cells were exposed to hRBCs, and lipid rafts were purified and characterized. In this cell line, attachment to hRBCs was not correlated with the enrichment of Hgl and Lgl in lipid rafts (Fig. 6), suggesting that while necessary (Fig. 2 and 4), ligand binding is not sufficient to induce enrichment of Hgl and Lgl in lipid rafts.
Fig 6.

PIP2 plays a role in Hgl and Lgl enrichment in lipid rafts. GFP-PHBTK-expressing trophozoites (3.5 × 106) were serum starved and exposed to hRBCs. DRM was isolated and fractionated using sucrose gradient density centrifugation. Nineteen fractions and a pellet (20P) were collected and subjected to Western blot analysis using antibodies specific for Hgl (A), Lgl (B), or Igl (C). Average values and standard deviations for densitometric scans (n = 2) are reported as the percentage of total detergent-resistant protein for each subunit. In the mutant, the submembrane distribution of the three subunits remained unchanged upon exposure to hRBCs. PIP2 loading restored the enrichment of Hgl and Lgl in lipid raft fractions.
PIP2 regulates the submembrane distribution of Hgl and Lgl.
Given the phenotype of the GFP-PHBTK-expressing cells (4), we hypothesized that phosphoinositide signaling was altered in the mutant. Therefore, we used lipid dot blots to determine the levels of PIP2 in the transgenic cell line. Compared to that in wild-type cells, the level of PIP2 in GFP-PHBTK-expressing cells was decreased approximately 77% (Fig. 7). Since the regulation of integrin function depends on PIP2 signaling (21, 25, 26), it is conceivable that alterations in the levels of this lipid could influence the enrichment of Hgl and Lgl in lipid rafts upon ligand binding. In other systems, it has been established that PIP2 resides in rafts (reviewed in references 29, 37, and 52). Since the GFP-PHBTK-expressing cell line had reduced levels of PIP2, we determined whether addition of exogenous PIP2 to this mutant could rescue the Hgl and Lgl raft enrichment defect. The mutant cell line was loaded with PIP2 using a Shuttle PIP2 kit (Echelon Biosciences), and the successful addition of PIP2 to cells was confirmed by fluorescence microscopy using BODIPY-labeled PIP2 (Fig. 7). Interestingly, loading of PIP2 resulted in restoration of the ability of this cell line to respond to hRBC exposure with enrichment of Hgl and Lgl in lipid raft domains (Fig. 6). However, PIP2 addition did not completely reverse the phenotype, since the percent enrichment of Hgl and Lgl in rafts was less in the PIP2-loaded mutant than in wild-type cells (Fig. 1). These data provide genetic evidence of a role for PIP2 in regulating the lectin subunit localization in lipid rafts.
Fig 7.

GFP-PHBTK-expressing cells have altered PIP2 levels, and can be loaded with PIP2. (A) Phosphoinositides were extracted from whole-cell lysates, and PIP2 levels were measured using dot blots with antibodies specific to PIP2. Levels were analyzed and assigned a value of arbitrary densitometric units. PIP2 levels were lower in GFP-PHBTK-expressing cells than in wild-type cells. (B) PIP2 loading in GFP-PHBTK-expressing cells was confirmed using a BODIPY-labeled PIP2. Both differential interference contrast (DIC) and fluorescence images are shown. Scale bars represent 25 μm.
Calcium signaling is necessary for the enrichment of Hgl and Lgl in rafts after ligand binding.
In other systems, PIP2 can be hydrolyzed into IP3 and DAG, which facilitates calcium signaling (8, 43). Given the importance of PIP2 in the localization of Hgl and Lgl to lipid rafts, we measured intracellular calcium levels after exposure to collagen and fibronectin using a fluorescence-based calcium assay. We observed a significant increase in intracellular calcium levels after exposure to collagen but not after exposure to fibronectin (Fig. 8).
Fig 8.
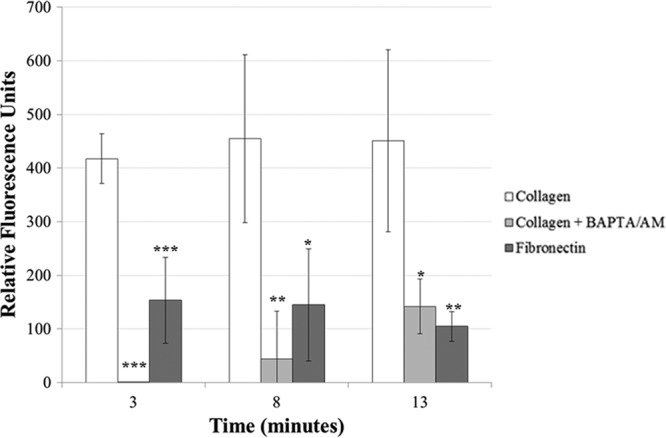
Intracellular calcium levels are significantly higher in collagen-exposed cells than in collagen/BAPTA/AM- or fibronectin-exposed cells. Intracellular calcium levels were measured for wild-type trophozoites that were exposed to collagen, with or without BAPTA/AM, or fibronectin. Compared to those in collagen-exposed cells (n = 3), calcium levels in BAPTA/AM-exposed or in fibronectin-exposed cells (n = 4) were significantly lower at all tested time points.
To determine if the accumulation of intracellular calcium was essential for the localization of Hgl and Lgl in lipid rafts, we exposed trophozoites to BAPTA-AM, an intracellular calcium chelator, prior to exposure to collagen. Reduction of calcium by BAPTA-AM was confirmed using a fluorescence-based calcium assay (Fig. 8). Exposure to BAPTA/AM prior to exposure to collagen prevented the enrichment of Hgl and Lgl in lipid rafts (Fig. 3), suggesting that the accumulation of intracellular calcium is necessary for lipid raft association of Gal/GalNAc lectin subunits. It is possible that the failure of Hgl and Lgl to become enriched in lipid rafts after exposure to BAPTA/AM and collagen was due to decreased adhesion. We measured adhesion to collagen in the presence of BAPTA/AM. Adhesion to collagen was not significantly inhibited in the presence of 50 μM BAPTA/AM (Fig. 9). This suggests that any effects of BAPTA/AM exposure on intracellular calcium levels and the localization of Hgl and Lgl were not simply due to a decrease in adhesion to the collagen-coated surfaces.
Fig 9.
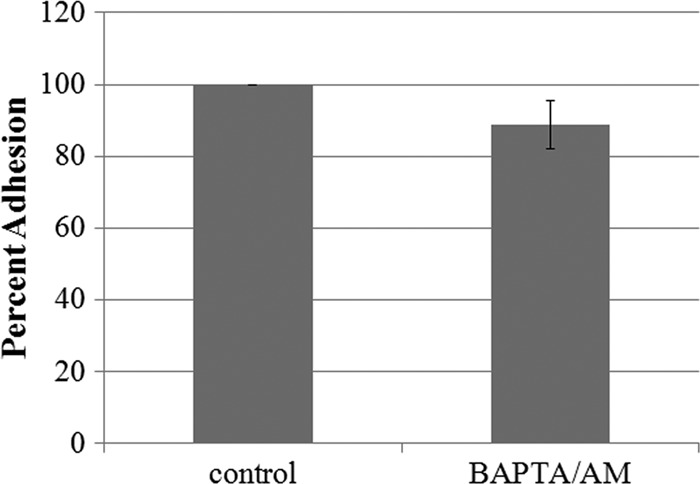
Adhesion to collagen is not significantly inhibited in the presence of BAPTA/AM. Adhesion to collagen was measured for wild-type cells that were exposed to serum-free medium with or without BAPTA/AM. Values were averaged, and adhesion is represented as a percentage of the control value, set to 100% (±SD; n = 3). Adhesion to collagen was not significantly inhibited in the presence of BAPTA/AM or control.
DISCUSSION
In this study, we have shown that exposure of E. histolytica to bona fide Gal/GalNAc lectin ligands (e.g.,. hRBCs or collagen) was accompanied by enrichment of the Gal/GalNAc lectin subunits, specifically Hgl and Lgl, in lipid raft domains. Previously, it was shown that cholesterol loading induced colocalization of Gal/GalNAc lectin subunits in rafts and increased activity of the Gal/GalNAc lectin (53). Here, we have provided evidence that another condition, namely, ligand binding, can also influence the submembrane localization of the Gal/GalNAc lectin subunits. We have also shown that binding to ligand was necessary, but not sufficient, to induce enrichment of Hgl and Lgl in lipid rafts after ligand binding. Our data also indicate that PIP2 and calcium participate in the enrichment of Gal/GalNAc lectin subunits in rafts.
Enrichment of Hgl and Lgl in high-buoyancy lipid raft domains after ligand binding is similar to the clustering and activation of mammalian integrins in lipid rafts. For example, in Jurkat T lymphocytes, attachment to collagen type IV or fibronectin induces lipid raft enrichment of α2β1 and α4β1 integrins, respectively (17). Furthermore, activation of another integrin in Jurkat T lymphocytes, lymphocyte function-associated antigen 1 (LFA-1), is correlated with its enrichment in lipid rafts (24). Although these signaling pathways are well understood in immune cells, the current study is an important first step toward the understanding of downstream signaling pathways that arise from lipid rafts in a parasite model.
The present study shows that attachment to ligand results in colocalization of the three lectin subunits in lipid raft fractions. Previously, it was shown by immunoprecipitation that Igl associates with Hgl (30). Importantly, we have not shown that Hgl and Lgl physically interact with Igl in lipid rafts. However, it is conceivable that the enrichment of Hgl and Lgl in raft regions, which already contain Igl, facilitates the assembly of the lectin into a functional trimer. This, in turn, may serve to activate subsequent raft-based signaling pathways related to virulence.
Exposure to hRBCs or collagen was correlated with the enrichment of Hgl and Lgl subunits in lipid rafts. Interestingly, these raft populations differed slightly in their buoyant densities. For example, after binding to hRBCs, Hgl and Lgl associated with rafts that were more buoyant than the rafts harboring these same subunits after collagen binding. It is possible that there are multiple types of lipid rafts within the parasite membrane, and binding to collagen or hRBCs causes the lectin to localize to distinct and separate lipid raft domains. In other systems, there is evidence for distinct raft populations. For example, purification of rafts from Madin-Darby canine kidney cells, using a variety of detergents, resulted in the isolation of distinct lipid raft domains with different protein residents (41). Immunogold labeling and electron microscopy have shown that all lipid raft markers do not colocalize. These data from other systems support the notion that multiple lipid raft domains exist within the plasma membrane (55). Our data suggest that the same is true in E. histolytica.
Differences in the buoyant density of rafts containing the lectin may be due to the association of the lectin with a different set of signaling proteins or cytoskeletal proteins in a ligand-specific manner. In neutrophils, heavier detergent-resistant membranes were found to contain more cytoskeletal proteins (34). Adhesion plaques, which contain actin, myosin I and II, α-actinin, vinculin, and tropomyosin (49), have been observed in E. histolytica upon attachment to ECM components but have not been observed upon attachment to hRBCs. Thus, the formation of a Gal/GalNAc lectin-containing adhesion plaque after exposure to collagen may explain why the lipid rafts harboring the lectin after collagen exposure are less buoyant than those harboring the lectin after hRBC exposure.
We showed that ligand binding was not correlated with the enrichment of Hgl and Lgl in rafts in a transgenic cell line with reduced levels of PIP2. We also showed that addition of exogenous PIP2 to this cell line partially rescued the phenotype. Together, these data provide strong genetic evidence for a role for PIP2 in regulating the submembrane distribution of the lectin subunits in E. histolytica. To our knowledge, this is the first study, in any system, to use a PIP2-deficient mutant to illustrate the role of PIP2 in protein-lipid raft interactions.
In the current study, intracellular calcium levels were increased upon exposure to collagen but not fibronectin. Others have shown that calcium levels increase when trophozoites are exposed to fibronectin (5). One explanation for this difference is that we exposed cells to fibronectin-coated coverslips instead of fibronectin in solution (5); adhesion to the solid ECM surface may initiate different signaling pathways. It is currently unknown whether the increased intracellular calcium levels are directly related to PIP2 hydrolysis in the cell or are attributable to other mechanisms related to calcium influx. In mammalian cells, the physical interaction between αIIβ3 integrin, sodium-proton exchangers, and sodium-calcium exchangers occurs simultaneously with integrin binding to ligand and results in increased intracellular calcium levels (56). Additionally, in phagocytes, extracellular calcium influx was shown to be essential for movement of an integrin bound to adenylate cyclase toxin from Bordetella into lipid rafts (3). Similarly, in the current study, the increase in calcium levels was shown to be necessary for ligand-induced enrichment of Hgl and Lgl in lipid raft domains.
Other studies, with mammalian cells as well as with E. histolytica, have supported the connection between calcium, PIP2, regulation of actin cytoskeleton, regulation of transcription, and virulence. For example, in B cells, calcium signaling has been shown to activate transcriptional regulators, such as NF-κB and NFAT (13). Likewise, attachment to collagen by trophozoites induces an increase in the binding of transcriptional regulators AP-1, STAT1, and STAT3 to DNA (7, 36) and an increase in the expression of several important virulence factors, including amoebapore and cysteine proteases (10). In E. histolytica, actin remodeling occurs during attachment to collagen (32) and hRBCs (1), and calcium mobilization can affect actin organization (5). In mammalian cells, calpain, a calcium-dependent protease, has been shown to cleave the cytoskeletal elements talin, filamin, and α-actinin, thereby releasing integrins from the actin cytoskeleton (45). It has been proposed previously that this cleavage of talin may be responsible for freeing proteins to allow their recruitment to lipid raft domains (3). PIP2 also contributes to actin cytoskeletal reorganization by guiding and activating actin binding proteins (20, 31). PIP2 plays an important role in mammalian cells by binding to talin, thereby targeting it to focal adhesions, where it can interact with and activate integrins (25). Together with our data, these findings suggest an intriguing link between parasite-host interactions, raft association of the Gal/GalNAc lectin, calcium mobilization, the cytoskeleton, and changes in gene expression.
The data presented here provide insights into signaling pathways in E. histolytica and, importantly, add to a developing model of the regulation of Gal/GalNAc lectin function. In the absence of ligand, GPI-anchored Igl subunits reside predominantly in raft-like domains, whereas Hgl-Lgl dimers are localized primarily to a different submembrane compartment. Binding to at least two biologically relevant ligands, hRBCs and collagen, brings all three subunits to the same raft fractions. Interestingly, our data are the first to show a correlation between the submembrane position of the lectin subunits and phosphoinositide-based signaling in this pathogen. In the future, it will be important to identify effectors that act downstream and in parallel with the Gal/GalNAc lectin after ligand binding and enrichment in lipid rafts. Fully understanding the behavior of this receptor after contact with extracellular ligands during invasion is necessary to fully appreciate virulence functions in E. histolytica.
ACKNOWLEDGMENTS
We thank William A. Petri, Jr. (University of Virginia School of Medicine, Charlottesville, VA), for antibodies specific to the Gal/GalNAc lectin subunits. We thank Brenda Welter for critical reading and editing of the manuscript and Terri Bruce and the Jordan Hall Imaging Facility (JHIF) for microscopy support.
The project described was supported by grant R01AI046414 from the National Institute of Allergy and Infectious Diseases to L.A.T. Additionally, this work was supported by a Grant-In-Aid of Research from Sigma Xi to A.B.K. This material is based upon work supported by NIFA/USDA under project number SC-1700312.
The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or the USDA.
Footnotes
Published ahead of print 13 April 2012
This article is Technical Contribution 5989 of the Clemson University Experiment Station.
REFERENCES
- 1. Bailey GB, Day DB, Gasque JW. 1985. Rapid polymerization of Entamoeba histolytica actin induced by interaction with target cells. J. Exp. Med. 162:546–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boettner DR, Huston CD, Sullivan JA, Petri WA., Jr 2005. Entamoeba histolytica and Entamoeba dispar utilize externalized phosphatidylserine for recognition and phagocytosis of erythrocytes. Infect. Immun. 73:3422–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bumba L, Masin J, Fiser R, Sebo P. 2010. Bordetella adenylate cyclase toxin mobilizes its β2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 6:e1000901 doi:10.1371/journal.ppat.1000901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byekova YA, Powell RR, Welter BH, Temesvari LA. 2010. Localization of phosphatidylinositol(3,4,5)-trisphosphate to phagosomes in Entamoeba histolytica achieved using glutathione S-transferase-and green fluorescent protein-tagged lipid biosensors. Infect. Immun. 78:125–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carbajal M, Manning-Cela R, Pina A, Franco E, Meza I. 1996. Fibronectin-induced intracellular calcium rise in Entamoeba histolytica trophozoites: effect on adhesion and the actin cytoskeleton. Exp. Parasitol. 82:11–20 [DOI] [PubMed] [Google Scholar]
- 6. Chadee K, Petri WA, Jr, Innes D, Ravdin J. 1987. Rat and human colonic mucins bind to and inhibit adherence lectin of Entamoeba histolytica. J. Clin. Invest. 80:1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cruz-Vera J, et al. 2003. Collagen-induced STAT family members activation in Entamoeba histolytica trophozoites. FEMS Microbiol. Lett. 229:203–209 [DOI] [PubMed] [Google Scholar]
- 8. Cybulsky AV, McTavish AJ, Papillon J. 1996. Extracellular matrix stimulates production and breakdown of inositol phospholipids. Am. J. Physiol.-Renal. 271:F579–F587 [DOI] [PubMed] [Google Scholar]
- 9. Czech MP. 2000. PIP2 and PIP3: complex roles at the cell surface. Cell 100:603–606 [DOI] [PubMed] [Google Scholar]
- 10. Debnath A, Das P, Sajid M, McKerrow JH. 2004. Identification of genomic responses to collagen binding by trophozoites of Entamoeba histolytica. J. Infect. Dis. 190:448–457 [DOI] [PubMed] [Google Scholar]
- 11. Diamond LS, Harlow DR, Cunnick CC. 1978. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans. R. Soc. Trop. Med. Hyg. 72:431–432 [DOI] [PubMed] [Google Scholar]
- 12. Dodson JM, Jr,et al. 1999. Infection and immunity mediated by the carbohydrate recognition domain of the Entamoeba histolytica Gal/GalNAc lectin. J. Infect. Dis. 179:460–466 [DOI] [PubMed] [Google Scholar]
- 13. Dolmetsch R, Lewis R, Goodnow C, Healy J. 1997. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386:855–858 [DOI] [PubMed] [Google Scholar]
- 14. Gray A, Olsson H, Batty IH, Priganica L, Peter Downes C. 2003. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 313:234–245 [DOI] [PubMed] [Google Scholar]
- 15. Guerrant RL, Brush J, Ravdin JI, Sullivan JA, Mandell GL. 1981. Interaction between Entamoeba histolytica and human polymorphonuclear neutrophils. J. Infect. Dis. 143:491–499 [DOI] [PubMed] [Google Scholar]
- 16. Hilgemann DW, Feng S, Nasuhoglu C. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001:1–8 [DOI] [PubMed] [Google Scholar]
- 17. Holleran BJ, Barbar É, Payet MD, Dupuis G. 2003. Differential recruitment of α2ß1 and α4ß1 integrins to lipid rafts in Jurkat T lymphocytes exposed to collagen type IV and fibronectin. J. Leukoc. Biol. 73:243–252 [DOI] [PubMed] [Google Scholar]
- 18. Insall RH, Weiner OD. 2001. PIP3, PIP2, and cell movement—similar messages, different meanings? Dev. Cell 1:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janmey PA. 1994. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu. Rev. Physiol. 56:169–191 [DOI] [PubMed] [Google Scholar]
- 20. Johnson CM, Chichili GR, Rodgers W. 2008. Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J. Biol. Chem. 283:29920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kolanus W, Seed B. 1997. Integrins and inside-out signal transduction: converging signals from PKC and PIP3. Curr. Opin. Cell Biol. 9:725–731 [DOI] [PubMed] [Google Scholar]
- 22. Kosek M, Bern C, Guerrant RL. 2003. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull. World Health Organ. 81:197–204 [PMC free article] [PubMed] [Google Scholar]
- 23. Laughlin RC, McGugan GC, Powell RR, Welter BH, Temesvari LA. 2004. Involvement of raft-like plasma membrane domains of Entamoeba histolytica in pinocytosis and adhesion. Infect. Immun. 72:5349–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leitinger B, Hogg N. 2002. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 115:963–972 [DOI] [PubMed] [Google Scholar]
- 25. Ling K, Schill NJ, Wagoner MP, Sun Y, Anderson RA. 2006. Movin' on up: the role of PtdIns(4,5)P2 in cell migration. Trends Cell Biol. 16:276–284 [DOI] [PubMed] [Google Scholar]
- 26. Liu J, et al. 2011. Structural basis of phosphoinositide binding to kindlin-2 pleckstrin homology domain in regulating integrin activation. J. Biol. Chem. 286:43334–43342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loftus B, et al. 2005. The genome of the protist parasite Entamoeba histolytica. Nature 433:865–868 [DOI] [PubMed] [Google Scholar]
- 28. Logan M, Mandato C. 2006. Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol. Cell 98:377–388 [DOI] [PubMed] [Google Scholar]
- 29. Martin TFJ. 2001. PI(4,5)P2 regulation of surface membrane traffic. Curr. Opin. Cell Biol. 13:493–499 [DOI] [PubMed] [Google Scholar]
- 30. McCoy JJ, Mann BJ. 2005. Proteomic analysis of Gal/GalNAc lectin-associated proteins in Entamoeba histolytica. Exp. Parasitol. 110:220–225 [DOI] [PubMed] [Google Scholar]
- 31. McLaughlin S, Wang J, Gambhir A, Murray D. 2002. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31:151–175 [DOI] [PubMed] [Google Scholar]
- 32. Meza I. 2000. Extracellular matrix-induced signaling in Entamoeba histolytica: its role in invasiveness. Parasitol. Today 16:23. [DOI] [PubMed] [Google Scholar]
- 33. Mittal K, Welter BH, Temesvari LA. 2008. Entamoeba histolytica: lipid rafts are involved in adhesion of trophozoites to host extracellular matrix components. Exp. Parasitol. 120:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nebl T, et al. 2002. Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. J. Biol. Chem. 277:43399. [DOI] [PubMed] [Google Scholar]
- 35. Pérez E, de Lourdes Muñoz M, Ortega A. 1996. Entamoeba histolytica: involvement of pp125FAK in collagen-induced signal transduction. Exp. Parasitol. 82:164–170 [DOI] [PubMed] [Google Scholar]
- 36. Pérez E, Munoz ML, Ortega A. 1998. Entamoeba histolytica: collagen-induced AP-1 DNA binding activity. FEMS Microbiol. Lett. 159:187–192 [DOI] [PubMed] [Google Scholar]
- 37. Pike LJ, Miller JM. 1998. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 273:22298–22304 [DOI] [PubMed] [Google Scholar]
- 38. Powell R, et al. 2006. Entamoeba histolytica: FYVE-finger domains, phosphatidylinositol 3-phosphate biosensors, associate with phagosomes but not fluid filled endosomes. Exp. Parasitol. 112:221–231 [DOI] [PubMed] [Google Scholar]
- 39. Ravdin JI, Guerrant RL. 1981. Role of adherence in cytopathogenic mechanisms of Entamoeba histolytica: study with mammalian tissue culture cells and human erythrocytes. J. Clin. Invest. 68:1305–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rohacs T. 2009. Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium. 45:554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roper K, Corbeil D, Huttner WB. 2000. Retention of prominin in microvilli reveals distinct cholesterol-based lipid microdomains in the apical plasma membrane. Nat. Cell Biol. 2:582–592 [DOI] [PubMed] [Google Scholar]
- 42. Salim K, et al. 1996. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton's tyrosine kinase. EMBO J. 15:6241. [PMC free article] [PubMed] [Google Scholar]
- 43. Savarese DMF, Russell JT, Fatatis A, Liotta LA. 1992. Type IV collagen stimulates an increase in intracellular calcium. Potential role in tumor cell motility. J. Biol. Chem. 267:21928–21935 [PubMed] [Google Scholar]
- 44. Stenmark H, Aasland R, Driscoll PC. 2002. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 513:77–84 [DOI] [PubMed] [Google Scholar]
- 45. Stewart MP, McDowall A, Hogg N. 1998. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140:699–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Talamás-Rohana P, Hernández VI, Rosales-Encina JL. 1994. A β1 integrin-like molecule in Entamoeba histolytica. Trans. R. Soc. Trop. Med. Hyg. 88:596–599 [DOI] [PubMed] [Google Scholar]
- 47. Talamás-Rohana P, Hernández-Ramirez VI, Perez-García JN, Ventura-Juárez J. 1998. Entamoeba histolytica contains a β1 integrin-like molecule similar to fibronectin receptors from eukaryotic cells. J. Eukaryot. Microbiol. 45:356–360 [DOI] [PubMed] [Google Scholar]
- 48. Talamás-Rohana P, Meza I. 1988. Interaction between pathogenic amebas and fibronectin: substrate degradation and changes in cytoskeleton organization. J. Cell Biol. 106:1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vázquez J, Franco E, Reyes G, Meza I. 1995. Characterization of adhesion plates induced by the interaction of Entamoeba histolytica trophozoites with fibronectin. Cell Motil. Cytoskeleton 32:37–45 [DOI] [PubMed] [Google Scholar]
- 50. Villalba JDA, et al. 2007. Programmed cell death in Entamoeba histolytica induced by the aminoglycoside G418. Microbiology 153:3852–3863 [DOI] [PubMed] [Google Scholar]
- 51. Vines RR, Jr,et al. 1998. Regulation of adherence and virulence by the Entamoeba histolytica lectin cytoplasmic domain, which contains a β2 integrin motif. Mol. Biol. Cell 9:2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Waugh MG, Lawson D, Tan SK, Hsuan JJ. 1998. Phosphatidylinositol 4-phosphate synthesis in immunoisolated caveolae-like vesicles and low buoyant density non-caveolar membranes. J. Biol. Chem. 273:17115–17121 [DOI] [PubMed] [Google Scholar]
- 53. Welter BH, Goldston AM, Temesvari LA. 2011. Localization to lipid rafts correlates with increased function of the Gal/GalNAc lectin in the human protozoan parasite, Entamoeba histolytica. Int. J. Parasitol. 41:1409–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. WHO 2010. Progress on sanitation and drinking water: 2010 update. WHO/UNICEF Joint Monitoring Programme for Water Supply and Sanitation, Geneva, Switzerland [Google Scholar]
- 55. Wilson BS, et al. 2004. Markers for detergent-resistant lipid rafts occupy distinct and dynamic domains in native membranes. Mol. Biol. Cell 15:2580–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yi YH, et al. 2009. Membrane targeting and coupling of NHE1-integrinαIIbβ3-NCX1 by lipid rafts following integrin-ligand interactions trigger Ca2+ oscillations. J. Biol. Chem. 284:3855–3864 [DOI] [PubMed] [Google Scholar]