

Abstract
Tsetse flies (Diptera: Glossinidae) are vectors for African trypanosomes (Euglenozoa: kinetoplastida), protozoan parasites that cause African trypanosomiasis in humans (HAT) and nagana in livestock. In addition to trypanosomes, two symbiotic bacteria (Wigglesworthia glossinidia and Sodalis glossinidius) and two parasitic microbes, Wolbachia and a salivary gland hypertrophy virus (SGHV), have been described in tsetse. Here we determined the prevalence of and coinfection dynamics between Wolbachia, trypanosomes, and SGHV in Glossina fuscipes fuscipes in Uganda over a large geographical scale spanning the range of host genetic and spatial diversity. Using a multivariate analysis approach, we uncovered complex coinfection dynamics between the pathogens and statistically significant associations between host genetic groups and pathogen prevalence. It is important to note that these coinfection dynamics and associations with the host were not apparent by univariate analysis. These associations between host genotype and pathogen are particularly evident for Wolbachia and SGHV where host groups are inversely correlated for Wolbachia and SGHV prevalence. On the other hand, trypanosome infection prevalence is more complex and covaries with the presence of the other two pathogens, highlighting the importance of examining multiple pathogens simultaneously before making generalizations about infection and spatial patterns. It is imperative to note that these novel findings would have been missed if we had employed the standard univariate analysis used in previous studies. Our results are discussed in the context of disease epidemiology and vector control.
INTRODUCTION
Mixed or coinfections, the coexistence of two or more pathogens in a single host, are common (14). However, most of our knowledge on host-pathogen interactions is based on the paradigm of one host-one pathogen, with less known about interactions among parasites, symbionts, and their host (1, 14). Since coinfections in a single host are likely to impact one another both directly (e.g., see reference 18) and indirectly (e.g., see reference 46), multiple infections and their host are likely to be coevolving. Thus, understanding the dynamics and interactions between infections and their hosts has implications in community assembly, patterns of genetic variation and biodiversity, and, when the host is a disease vector, epidemiology.
Tsetse (Glossina), the sole vector for human African trypanosomiasis (HAT), harbor a variety of microorganisms, including at least two microbial symbionts (6). The obligate mutualist found in all tsetse species, Wigglesworthia glossinidia, is essential for adult host fecundity and for proper immune maturation (51, 59). Some natural populations of tsetse also harbor the commensal symbiont Sodalis glossinidius, which has been suggested to promote the establishment of trypanosome infections (20). Additionally, some tsetse species are infected by the intracellular parasitic bacterium Wolbachia (13). Wolbachia-associated reproductive manipulation in the form of cytoplasmic incompatibility (CI) can alter host patterns of genetic diversity (17). CI can be unidirectional or bidirectional. Mating between infected males and uninfected females can result in unidirectional CI, while bidirectional CI is expressed when males and females carry two different Wolbachia strains. Expression of unidirectional CI has been shown in Glossina morsitans morsitans in the laboratory (7). Tsetse can also be infected by two other pathogens: the HAT-causing trypanosomes (Trypanosoma) that compromise tsetse physiology, in particular fecundity (56), and a salivary gland hypertrophy virus (SGHV; first described from Glossina pallidipes as GpSGHV) that causes salivary gland hypertrophy (SGH) and abnormalities in reproductive organs that reduce fecundity and fertility (4, 31) and therefore may also alter patterns of genetic diversity. While Wolbachia is transovarially transmitted, Wigglesworthia and Sodalis are maternally transmitted to the intrauterine larva in milk secretions. SGHV is also vertically transmitted either transovum or through milk gland secretions (33, 52). In contrast, tsetse adults acquire trypanosomes through feeding on trypanosome-infected vertebrates.
Each of these agents (Wolbachia, Sodalis, SGHV, and trypanosomes) has various impacts on their host's biology, but little is known about their within-community relationships. Both the single and collective impacts of these agents on tsetse may influence patterns of host genetic diversity and vectorial capacity. Hence understanding the dynamics of host, parasite, and symbiont coinfections in natural populations may have considerable impact on disease epidemiology and the development of vector control strategies. Indeed, in the absence of the obligate symbiont, Wigglesworthia, colony tsetse flies are rendered highly susceptible to trypanosome infections (51). Tsetse morphological studies suggest that the presence of Wolbachia, when coupled with SGHV infection, may have a synergistic effect causing degradation of the salivary glands (36, 41). In Drosophila, infections with some Wolbachia strains confer a general antiviral effect (25, 49). Similarly, transinfection of Wolbachia in mosquitoes prevents infection by dengue virus as well as by eukaryotic pathogens, including Plasmodium (45), and filarial nematodes (34). Although the presence of Sodalis, Wolbachia, and SGHV virus infections has been reported in natural tsetse populations, information on coinfections and their effect on parasite transmission is lacking.
Glossina fuscipes fuscipes is an ideal candidate to investigate the potential interactions between host genotypes and pathogens because of the unexpected partitioning of genetic diversity uncovered in Uganda (5, 9). Maternally inherited mitochondrial DNA (mtDNA) markers identified two geographically distinct haplogroups (sets of genetically related haplotypes), one to the north and a second to the south of Lake Kyoga (5). The results of a nuclear DNA survey (microsatellite loci) are only partly congruent with the mtDNA findings, as it also identified a third, western group (Fig. 1) (9). Around Lake Kyoga, mixed populations, with individuals belonging to either mtDNA haplogroup, can be found. This suggests that channels of dispersal through a suitable habitat are likely to exist or have existed in this area, although it is unclear how distinct genetic boundaries persist. A possible explanation is the occurrence of Wolbachia-induced CI, which drives patterns of mtDNA variation regardless of the nuclear DNA background. Furthermore, sleeping sickness epidemics have been historically associated with limited foci throughout sub-Saharan Africa, including regions of Uganda. The nature of the ecological and genetic determinants that shape such foci has been of interest yet remains largely unknown. It is possible that the foci may be shaped by environmental factors, such as the availability of hosts and reservoirs, or various tsetse host genetic factors, such as antiparasite immune responses. It is also possible, however, that the diverse resident microbial fauna associated with the different tsetse host populations may be influencing trypanosome transmission dynamics.
Fig 1.

Geographic distribution, sampling localities, and assignment to genetic groups based on tsetse mitochondrial DNA (mtDNA) and microsatellites. Light shading on the map represents the geographic distribution of tsetse in Uganda. Sampling localities are illustrated as site abbreviations. Letters in the large circles next to the sites show assignments to genetic groups. Sites with northern mtDNA (N), southern mtDNA (S), or both are distinguished by hatching within the large circles. Microsatellite genetic groups are abbreviated (n, north; s, south; and w, west) and positioned as shown in the legend. The inset map shows the location of Uganda within continental Africa.
In this paper, we investigated multiple tsetse populations that span the range of host genetic and spatial diversity in Uganda to describe the prevalence of tsetse pathogens (Wolbachia, SGHV, and trypanosomes) and symbionts (Sodalis) and to understand the coinfection dynamics operating on G. fuscipes fuscipes populations. We then exploited different G. fuscipes fuscipes genetic backgrounds to understand if host genetic makeup impacted interactions among the pathogens. Specifically, we looked at mitochondrial DNA to examine vertically transmitted infections and nuclear DNA markers (microsatellites) to assess whether neutral processes shape host genetic structure and impact parasite prevalence. Finally, we focused on coinfection dynamics using a multivariate statistical approach to determine if interactions between pathogens enhance or diminish the capacity of infection when other pathogens are present. We discuss our results relative to other single-pathogen studies and in the context of Wolbachia-based vector control.
MATERIALS AND METHODS
Sampling and DNA extractions.
Glossina fuscipes fuscipes flies were trapped in Uganda over a 5-year period (2005 to 2010). Biconical tsetse traps were placed in a 1- to 5-km radius around the riverine and lacustrine habitat favored by G. fuscipes fuscipes (12). Tsetse were collected at 18 sites that comprised the genetic variation recovered from both nuclear and mitochondrial DNA (5, 9, 16). Table 1 lists collection sites, their abbreviations, and their geographic coordinates. Individual flies were preserved in 100% ethanol. For each individual, total genomic DNA (gDNA) was prepared from whole flies (legs, wings, and heads removed) using the DNeasy blood and tissue kit (Qiagen). Data for symbionts and pathogens were collected from 510 individuals. As a positive DNA control, we amplified a 365-bp fragment of the Wigglesworthia thiC gene (thiamine biosynthesis factor) (51) since all natural populations of G. fuscipes fuscipes carry the obligate mutualist. All samples screened in this study were Wigglesworthia positive.
Table 1.
List of collection sites, abbreviations for each site, and geographic coordinatesa
| Site code | Collection site | Longitude | Latitude |
|---|---|---|---|
| AR | Arua | 31.140 | 3.270 |
| BK | Pallisa | 33.875 | 1.020 |
| BN | Bunghazi | 33.977 | 0.934 |
| BU | Busime | 33.971 | 0.251 |
| BV | Buvuma | 33.300 | 0.310 |
| DK | Dokolo | 33.160 | 1.908 |
| JN | Junda | 32.737 | 1.331 |
| KB | Kabunkanga | 30.550 | 0.980 |
| KF | Kafu bridge | 32.042 | 1.542 |
| KK | Kakoga | 30.280 | 0.370 |
| KL | Kalengera | 32.767 | 0.167 |
| KR | Karuma bridge | 32.239 | 2.243 |
| MS | Masindi | 31.685 | 1.625 |
| MY | Moyo | 31.650 | 3.600 |
| NA | Nkumba | 32.530 | 0.060 |
| OS | Osuguro | 33.497 | 1.525 |
| PT | Puti-Puti | 33.797 | 1.151 |
| SS | Ssese | 32.188 | -0.317 |
Latitude and longitude are given in decimal degrees.
Wolbachia detection.
Initially, the heat shock protein 60 (Hsp60) gene (groEL) was PCR amplified using the published primers (WgroFor1 and WgroRev1) and conditions (11). The amplification intensity was found to be very low. To obtain sufficient PCR amplification, gel slices in the expected 795-bp region were excised and DNA was purified using the Qiagen gel purification kit and eluted in 30 μl of the template. We carried out a second sequential PCR with 1 μl of undiluted and 1 or 2 μl of 1:10 diluted template DNA to find the optimal amplification conditions for the different samples. For 20 individuals, the eventual product was gel purified and cloned into the pGEM-T vector plasmid (Promega Corporation) and a minimum of 3 clones was sequenced for each PCR product (Yale DNA Analysis Facility on Science Hill [DAFSH]). We employed BLAST using the NCBI GenBank database to verify PCR product identity. To improve our diagnostics, a new Wolbachia-specific forward primer [GffWgro(F), 5′-TTTGATCGC GGTTATC-3′] was designed from the conserved region based on the verified sequence using PrimerSelect (version 1.8.4) (7). The new forward primer was used with WgroRev1 under the following conditions: 40 cycles of 94°C for 1 min, 47°C for 1 min, and 72°C for 1 min, with a final extension at 72°C for 10 min. This generated a shorter 410-bp fragment. The shorter fragment was validated as Wolbachia groEL by cloning and sequencing analysis. Using the newly developed primer pair, all DNA samples were screened by two independent PCRs using the sequential PCR procedure described. A sample was scored positive when both independent PCRs generated the expected 410-bp fragment.
To verify the low Wolbachia density, a subset of the G. fuscipes fuscipes samples that were identified as positive (n = 12) and negative (n = 4) was screened by quantitative PCR (qPCR). Wolbachia density was quantified by qPCR using the groEL-specific primers F (5′CAGAGGATATCGAAGGTGAA 3′) and R (5′ CCTGGAGCTTTTACTGCGG 3′) for 40 cycles at 60°C and normalized to the host β-tubulin gene (F, 5′ CCATTCCCACGTCTTCACTT 3′, and R, 5′ GACCATGACGTGGATCACAG 3′) for 40 cycles at 60°C on the iCycler iQ real-time PCR detection system (Bio-Rad, Hercules, CA). Negative controls were included in all amplification reaction mixtures, and Wolbachia-infected G. morsitans morsitans gDNA served as the positive control. Relative densities are presented as means. Statistical significance was determined using a one-way analysis of variance (ANOVA).
Sodalis infection prevalence.
We amplified a 650-bp fragment of the Sodalis glossinidius hemolysin gene using a published protocol (51). Like the Wolbachia screens described above, sequential PCRs with products from the first round were performed to rule out possible low-density infections.
SGHV and trypanosome detection.
The presence of SGHV and Trypanosoma infections was detected using PCR amplification assays. For SGHV, we used the published primers and conditions of Abd-Alla et al. (2) to amplify a 401-bp fragment of the SGHV-2 gene. For trypanosome detection, we amplified a 621-bp fragment of the α-tubulin gene using the general primers Trypalphatub(F) (5′-CTC GAC ACA CTC ACT TCT GGA G-3′) and Trypalphatub(R) (5′-CGA ATT TGT GGT CAA TAC GAG-3′) and the following cycling conditions: 40 cycles of 94°C for 1 min, 55°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. To serve as a control for trypanosome infection prevalence data obtained from whole-fly DNA, we provided 12 newly emerged G. morsitans morsitans flies a blood meal supplemented with 105 trypanosomes/ml. We extracted gDNA from flies that had taken a blood meal 48 h postinfection by using the same DNA extraction methodology applied to field G. fuscipes fuscipes flies. We then performed PCR amplification using the same conditions as described above for the presence of trypanosomes. We could detect parasite DNA in only about 50% of the flies, which is also the infection prevalence we observed with this colony of flies. Thus, we deduce that the trypanosome-positive samples we noted from field flies reflect true infections rather than residual parasite DNA or infections that will be subsequently cleared.
Statistical analyses.
In order to investigate spatial pathogens and symbiont prevalence patterns, we used host groups genetically defined at different spatial scales. We then used simple logistic regression to look at differences in prevalence across groups. To evaluate the interactions among pathogens, we used multiple logistic regressions and multiple correspondence analysis (MCA). To explore the effect of geographic distances on pathogen prevalence differences between populations, we used the Mantel test (40).
In order to examine spatial patterns, groups were defined at different scales. At the broad scale, tsetse flies were grouped based on previous work (5, 9). Nuclear markers assigned G. fuscipes fuscipes populations to three geographically separate groups (north, south, and west), while mtDNA data defined two main groups (north and south) (Fig. 1). Most populations have individuals that are assigned to just one mtDNA haplogroup (a group of related haplotypes), but in five populations (BN, JN, KF, KR, and MS), individuals from the northern and southern haplogroups cooccur in the same population. At the fine scale, populations were compared based on sampling sites (Fig. 1). We also compared sexes across all samples (within genetic groups and within populations). For these analyses, we used a reduced data set, as only 477 individuals could be sexed (230 female and 247 males). The MY population was excluded from this analysis, as many individuals were not sexed.
Simple binomial regressions were used to test whether predefined host groups predict pathogen prevalence. In these analyses, each pathogen is used as the dependent variable and spatial categories are the independent variables. Significance was determined via z-scores but was not computed for comparisons involving populations with a prevalence of either 0% or 100%, as logistic regression resulted in meaningless confidence intervals, thus affecting z-scores and P values.
To test whether there are interactions among pathogens, we used multiple logistic regression for two- and three-pathogen comparisons. Like described above, we tested for differences between host groups for a given pathogen, but in multiple logistic regression analyses, we considered one or two other pathogens as independent variables. We explored all different possible combinations (see Table S3 in the supplemental material).
Differences in infection and coinfection patterns among groups were evaluated via the contrast procedure implemented in the “Design” package in the R environment (24). Significance was determined based on P values obtained from the z distribution. Additionally, to correct for multiple comparisons, we implemented a logically constrained multiplicity adjustment of P values as described in reference 60. This multiplicity adjustment incorporates dependence structures and logical constraints, thereby achieving greater power than simple step-down methods, such as that in reference 26. When comparisons involved populations, the “free-combination” (i.e., unconstrained) procedure (61) was used. These correction methods were implemented in the “multcomp” package (27) in R. Due to colinearity between infections (i.e., perfect or near-perfect correlation) within several populations, we used multiple correspondence analysis (MCA), as implemented in the R package “ca” (22), to determine whether Wolbachia, SGHV, and trypanosome infections cooccurred in any possible combination within any population. MCA is a generalization of principal components analysis (PCA) for categorical instead of quantitative data. We performed MCA using the R package ca (22). Presence/absence infection data and population membership of individual flies were used in the analysis and plotted onto the first two MCA axes that explain most of the variation. Microsatellites, mtDNA, and sex were used as supplementary variables, meaning that they were not included in the analysis but were plotted postanalysis onto the MCA space defined by populations and infections in order to aid in data interpretation. To account for overrepresentation of variables, the Greenacre method (21) was used to calculate corrected eigenvalues and percentages of explained inertia (variance).
We tested the effect of geographic distance on Wolbachia in the context of the other two pathogens by using Mantel tests (40), as implemented in the “adegenet” package (32) in R. Coinfection distance values between populations were obtained from the MCA scores. Microsatellite data (9) were used to calculate genetic distances between populations (47). Geographic distances were calculated from the site coordinates listed in Table 1. Since Wolbachia-induced CI is expected to result in a negative correlation with geographic distance, we tested for a correlation between geographic distance and the pathogens, including and excluding Wolbachia.
RESULTS
Infection prevalence and coinfection dynamics.
Wigglesworthia-specific DNA fragment amplification was used to rule out potential DNA extraction problems since all natural populations of tsetse carry Wigglesworthia (data not shown). While all flies had Wigglesworthia infections as expected, none of the 222 flies screened in our study carried Sodalis despite using sequential PCRs to detect low-density infections (data not shown). This was similar to another study performed on G. fuscipes fuscipes populations in Kenya where no Sodalis infections were detected (39).
The Wolbachia density in natural G. fuscipes fuscipes populations was unusually low and required an assay in which two sequential amplifications were performed for detection. Based on qPCR, the Wolbachia density in G. fuscipes fuscipes-infected individuals was found to be at least 20-fold lower than that in females from a laboratory line of G. morsitans morsitans (Fig. 2). Furthermore, flies shown to be negative by PCR amplification had no detectable levels of Wolbachia by a qPCR assay.
Fig 2.

Comparison of Wolbachia density in G. fuscipes fuscipes (Gff) with that in a laboratory strain of G. morsitans morsitans (Gmm). Wolbachia densities from female Wolbachia-positive G. fuscipes fuscipes flies (GffWol+) (n = 12), Wolbachia-negative G. fuscipes fuscipes flies (GffWol−) (n = 4), and Wolbachia-positive G. morsitans morsitans flies (GmmWol+) (n = 4) were measured by qPCR and normalized by tsetse β-tubulin (n ≥ 4). *, P < 0.05; ***, P < 0.0001.
To confirm that the parasite infection assay we performed on DNA extracted from whole flies reflects true trypanosome infections as opposed to those newly acquired in the last blood meal, we performed an experiment where we provided an infectious blood meal to laboratory flies, extracted the DNA from whole bodies, and amplified the DNA for parasites. We could detect trypanosomes in only 50% of the flies, which is similar to the level of mature parasite infections we typically obtain when we provide a trypanosome blood meal to newly emerged laboratory flies (see Fig. S1 in the supplemental material) (23). Hence, the PCR-positive flies likely represent those that would have given rise to mature midgut infections. In fact, the levels of parasitemia in the infected mammalian hosts that flies typically feed on are likely to be significantly less than the level of 105 parasites/ml that we provided in the laboratory. Furthermore, the majority of flies that are trapped in the field have little residual blood meal in their midgut, unlike those we analyzed in the laboratory 48 h after the infected blood meal. Hence, although we did not perform midgut infections in the field, our prevalence data based on a whole-fly DNA amplification assay likely reflect true trypanosome infections in the field.
Prevalence of Wolbachia, SGHV, and trypanosomes was heterogeneous throughout the landscape (see Table S1a, b, and c in the supplemental material). Across Uganda, Wolbachia prevalence was 44.3%, while SGHV and trypanosome prevalences were 12% and 18%, respectively. Wolbachia prevalence in different populations ranged from uninfected to near fixation, whereas the prevalences for SGHV and trypanosomes ranged from 0% to 47% and from 0% to 40%, respectively.
In general, Wolbachia prevalence in different populations was higher than the corresponding prevalence of either SGHV or trypanosomes. However, across the 18 populations in our study, when Wolbachia infection prevalence was below 30%, SGHV and/or trypanosome infections tended to be at the same level or higher than that noted for Wolbachia (Fig. 3). Prevalences of Wolbachia and SGHV were more negatively correlated (r = −0.408, P = 0.046) than the prevalences of Wolbachia and trypanosomes (r = −0.176, P = 0.242), while prevalences of SGHV and trypanosomes were positively correlated (r = 0.257, P = 0.152). These results suggest that Wolbachia, on one hand, and SGHV and trypanosomes, on the other hand, have a negative impact on each other.
Fig 3.
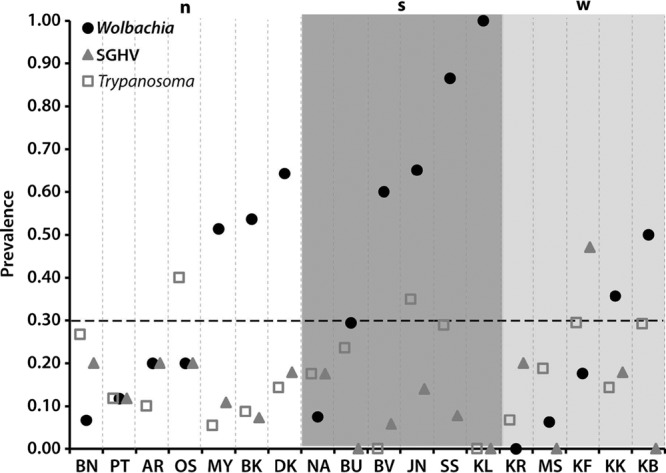
Prevalence of Wolbachia, SGHV, and Trypanosoma within G. fuscipes fuscipes populations. Populations, on the x axis, are arranged in ascending order of Wolbachia prevalence within the groups defined by microsatellites (n, north; s, south; w, west) (9). Above 30% prevalence (dotted line), Wolbachia prevalence is higher than that of both SGHV and Trypanosoma.
Single infections were more frequent than coinfections, with only 1% of flies harboring all three pathogens and 40% lacking all three pathogens. In contrast, 33%, 6%, and 8% of flies were singly infected with Wolbachia, SGHV, and trypanosomes, respectively. In terms of coinfections, 8% of flies were infected with Wolbachia and trypanosomes, 3% with Wolbachia and SGHV, and 2% with SGHV and trypanosomes. Among the pathogens, Wolbachia is the most common of the three, and its presence correlates most often with the presence of trypanosomes.
Prevalence patterns of individual pathogens in relation to host groups.
Simple binomial logistic regression independently tested the effects of genetic groups, populations, and sex on the occurrence of one of the three infections. The contrast technique determined the statistical significance of individual infection differences between the 18 host populations, the three microsatellite groups, the two mtDNA groups, and the sexes (see Table S2a in the supplemental material). The following populations (white circles in Fig. 4) were excluded from comparisons due to uninformative confidence intervals: KR and KL (Wolbachia), BU, KL, KB, and MS (SGHV), and BV and KL (trypanosomes).
Fig 4.

Mean infection probabilities (circles) within G. fuscipes fuscipes populations and groups for Wolbachia (1), SGHV (2), and Trypanosoma (3), as determined by simple binomial logistic regression. Each panel shows infection probabilities within each population (abbreviations are as in Table 1) (A) as well as within groups defined by microsatellites (n, north; s, south; w, west) (9) (B) and mtDNA (N, north; S, south) (5) (C). Black circles are shown with 95% confidence intervals, while white circles represent populations that have only infected or uninfected individuals and thus do not have associated CIs.
Wolbachia prevalence did not differ significantly between the mtDNA haplogroups. However, the host microsatellite groups showed large differences in prevalence of Wolbachia (south, 55%; north, 42%; west, 26%) (Table 2). The infection probabilities in these groups were significantly different (north-south, z = −2.49, P = 0.013; north-west, z = 2.73, P = 0.006; south-west, z = 4.63, P = 0.000) (see Table S2a in the supplemental material). At the population level, SS (87% prevalence) had a significantly higher infection probability than any other population (see Table S2a in the supplemental material). DK (64% prevalence) and KB (50% prevalence), despite being in lower-prevalence regions (north and west, respectively), had a significantly higher infection probability than some southern populations, namely, BU (29%) and NA (8%) (DK-BU, z = 2.68, P = 0.007; DK-NA, z = 4.32, P = 0.000; KB-NA, z = 3.46, P = 0.001). Males and females did not significantly differ for Wolbachia infections, whether compared across Uganda or within microsatellite and mtDNA groups.
Table 2.
Female and male infection prevalence by geographic region
| Population groupa and sex | Sample size (n) | % infected withb: |
||
|---|---|---|---|---|
| Wolbachia | SGHV | Trypanosoma | ||
| n | ||||
| Female | 86 | 38.4 | 11.6 | 16.3 |
| Male | 89 | 40.4 | 16.9 | 14.6 |
| Total | 196 | 42.3 | 12.8 | 13.8 |
| s | ||||
| Female | 95 | 51.6 | 4.2 | 14.7 |
| Male | 107 | 55.1 | 14 | 29 |
| Total | 214 | 54.7 | 8.9 | 21 |
| w | ||||
| Female | 49 | 28.6 | 20.4 | 16.3 |
| Male | 51 | 23.5 | 11.8 | 23.5 |
| Total | 100 | 26 | 16 | 20 |
n, northern population group; s, southern population group; w, western population group. As determined by microsatellite data. Seven populations constitute the north, while six and five populations make up the south and west, respectively.
Infection status was determined by PCR of Wolbachia groEL, salivary gland hypertrophic virus SGHV-2, and trypanosome α-tubulin genes.
The lack of a difference in Wolbachia prevalence between the mtDNA haplogroups suggests that unidirectional CI is not responsible for maintaining the genetic division identified between the mtDNA haplogroups (Fig. 1) because Wolbachia is not absent from one of the two haplogroups. The significant differences in Wolbachia prevalence between the microsatellite groups and the high variation among populations within these groups suggest that the spread of the pathogen is dependent on the dispersal ability of the vector.
SGHV prevalence differed significantly (z = 2.28, P = 0.022) (see Table S2a in the supplemental material) between the north (15%) and south (8%) mtDNA haplogroups, but it did not differ based on microsatellite groups. At the population level, KF was the only population with a significantly higher infection probability (higher than eight other populations) (see Table S2a in the supplemental material). While in the population with the highest SGHV infection rate (KF [west microsatellite group]), most of the infected individuals were females, in the south, SGHV prevalence was highest in males (z = 2.25, P = 0.024) (see Table S2a in the supplemental material).
The significant difference we observed in SGHV prevalence between the mtDNA haplogroups and the lack of an association with microsatellite groups is not surprising, as it reflects vertical transmission coupled with reduced female dispersal (9). Given that SGHV can affect tsetse reproduction, the biological significance of these associations between virus prevalence and host genetic makeup deserves further investigation. Furthermore, given that viral density can influence both the pathogenesis and fecundity outcomes (3), future studies measuring viral densities from different populations are important.
Trypanosome prevalences were not significantly different between either mtDNA or microsatellite groups. At the population level, all significant differences in infection probability were found in pairwise comparisons of populations with higher prevalence than BK (9%) or lower prevalence than OS (40%) (see Table S2a in the supplemental material). The only other significant difference in infection probability was observed between males and females in the southern microsatellite group (higher in males; z = 2.39, P = 0.017).
Pathogen coinfection patterns in relation to host groups.
Multiple binomial logistic regression analyses independently tested the effect of populations and genetic groups (nuclear and mitochondrial) on an infection in the context of a second infection in two-pathogen comparisons. Multiple regressions also tested, independently, the effect of nuclear and mitochondrial groups on an infection in the context of the other two infections in three-pathogen comparisons.
In two-pathogen comparisons, the influence of Wolbachia was such that differences in prevalence between host groups of either SGHV or trypanosomes were significant only when uninfected with Wolbachia. SGHV prevalence was higher in the north than in the south mtDNA group (P = 0.019), and trypanosome prevalence was higher in three out of five mixed populations in the region around Lake Kyoga (BN, JN, and KF) (see Table S2b in the supplemental material). When uninfected with SGHV, we found that the prevalence differences in Wolbachia, identified by simple logistic regressions (see Table S2a in the supplemental material), persisted between the microsatellite groups and populations. When infected with SGHV, however, the only significant difference was found in trypanosome prevalence between the south and north microsatellite groups (z = 2.17, P = 0,030) (see Table S2b in the supplemental material). When uninfected with trypanosomes, Wolbachia prevalence differences found using simple logistic regression (see Table S2a in the supplemental material) persisted. For SGHV, significant differences were found between microsatellite groups (west-south, z = 2.1, P = 0.035; north-south, z = 1.98, P = 0.048) (see Table S2b in the supplemental material). At the population level, the significantly higher SGHV prevalence in KF persisted. When infected with trypanosomes, however, the only significant differences were found with respect to Wolbachia prevalence (BK-OS, z = 1.98, P = 0.048; SS-JN, z = 2.17, P = 0.030; SS-OS, z = 2.84, P = 0.005) (see Table S2b in the supplemental material).
In three-pathogen comparisons, when uninfected with both SGHV and trypanosomes (see Table S2b in the supplemental material, SGHV− and Tryp−), Wolbachia prevalence was significantly different between microsatellite host groups (north-south, z = −2.01, P = 0.045; north-west, z = 2.57, P = 0.010; south-west, z = 4.04, P = 0.000). When infected with Wolbachia but uninfected with trypanosomes (see Table S2a in the supplemental material, Wol+ and Tryp−), SGHV infection prevalence was higher in the northern than the southern mtDNA haplogroup (z = 2.16, P = 0.031).
We used MCA to simultaneously investigate cooccurrence patterns of the three pathogens at the population level, with microsatellite and mtDNA host groups and host sex plotted post hoc to facilitate the interpretation of similarities or dissimilarities between populations (Fig. 5). The two axes of MCA represent 63% of the total variation (proportions of explained inertia, τ1 = 46.9% and τ2 = 16.1%). Infection status (presence or absence) (Fig. 5) of Wolbachia varied along the first axis, trypanosomes varied along the second, and SGHV differed along both axes. Given the variance explained by each axis (46.9% versus 16.1%), we can infer that a majority of the differences between populations can be attributed to differences in Wolbachia prevalence rather than differences in SGHV or trypanosomes. MCA showed a strong association between the absence of SGHV and the absence of trypanosome infections across populations. Although there is an association between SGHV and trypanosome infections, this is mostly driven by two populations (KF and OS) (Fig. 5). MCA revealed exceptions to coinfection patterns found in microsatellite- and mtDNA-defined regions. For instance, BK and DK (north) and KB (west) were more associated with the presence of Wolbachia infection than with its absence, a pattern which was not generally true of the northern or the western microsatellite population groups. The opposite was true for BU (south), which was an exception to the rest of the southern microsatellite group, inasmuch as it was associated with the absence rather than the presence of Wolbachia infection. In concordance with the general pattern of the southern microsatellite group, the two southern populations JN and SS were associated with the presence of Wolbachia infection, but they also had a higher probability of SGHV and trypanosome infection than did the other populations in the southern microsatellite group.
Fig 5.

Multiple correspondence analysis plot. The first two axes, representing 63% of the total variation, are plotted. Eigenvalues (λ1 = 0.038 and λ2 = 0.013) and proportions of explained inertia (τ1 = 46.9% and τ2 = 16.1%) have been corrected using the Greenacre (21) method. Populations (abbreviations from Table 1), microsatellite genotype clusters (n, north; s, south; w, west), mtDNA haplogroups (N, north; S, south), and sex (F, female; M, male) are depicted to show their association with presence (+) or absence (−) of Wolbachia (Wgro), Glossina pallidipes salivary gland hypertrophy virus (SGHV), and Trypanosoma (Ttub). Triangles represent supplementary elements (microsatellite, mtDNA, and sex, plotted post hoc), while infections and populations are denoted with squares and circles, respectively.
The Mantel test between coinfection dissimilarity and genetic distance was not significant (r = −0.111). However, the Mantel test between coinfection and geographic distance revealed a significant negative correlation (Pearson's correlation coefficient; r = −0.261; α = 0.01). When Wolbachia data were excluded, geography and coinfection were not significantly negatively correlated (r = −0.125). Thus, geographically proximate populations are more dissimilar in coinfections than are distant populations. The fact that the exclusion of Wolbachia influences the negative correlation between coinfection dissimilarity and geographic distance indicates that no spatial heterogeneity between close populations exists without Wolbachia.
DISCUSSION
Here we examined interactions between three distinct microbes (a DNA virus, a parasitic bacterium, and a protozoan parasite) in the context of the tsetse host mitochondrial and nuclear genetic background in natural populations in Uganda. Although earlier studies had reported heterogeneity in the prevalence of individual microbes in tsetse populations, our study demonstrates that coinfections play a synergistic role in generating microbial community heterogeneity. Our results of multivariate comparisons reveal patterns of association between either hosts or pathogens that were not obvious in univariate comparisons, as only a few of the patterns observed in simple logistic regression hold in multipathogen comparisons. This suggests that there are complex interactions among the pathogens, highlighting the importance of using a multivariate approach. These interactions are particularly evident for Wolbachia and SGHV, both vertically transmitted to tsetse progeny. For instance, host groups are inversely correlated for Wolbachia and SGHV prevalence. On the other hand, trypanosome infection prevalence is more complex and covaries with the presence of the other two pathogens, highlighting the importance of examining multiple pathogens simultaneously before making generalizations about infection and spatial patterns.
This is particularly relevant when looking at pathogens that may be used for vector control. For instance, the negative correlation between Wolbachia and SGHV implies that if Wolbachia is used for vector control, SGHV infection prevalence must be considered in regions targeted for control. Additionally, when multiple pathogens are considered, the differences between host (vector) genetic groups enable us to identify regions where control efforts would be more successful. For example, regions that have low SGHV infection prevalence, such as the southern microsatellite group in Uganda, are better suited for Wolbachia-based control methods.
Patterns of variation in individual microfauna infections.
Our results show high levels of spatial heterogeneity in Wolbachia and SGHV prevalence among populations. Similar geographic influences on Wolbachia prevalence have been reported for other Glossina species, although over a much broader spatial scale. In Glossina austeni populations collected in the same year from Kenya and South Africa, Wolbachia prevalence ranged from 48% to 98%, respectively (13). Likewise, variation in the number of symptomatic flies with SGHV has been observed by location and even trap site in other Glossina species, such as G. morsitans morsitans and G. pallidipes from Kenya (48). Given that both Wolbachia and SGHV are vertically transmitted, it is possible that imperfect maternal transmission from mother to progeny may result in the uninfected populations observed.
Trypanosome infection prevalence is also not uniform among Ugandan G. fuscipes fuscipes populations and varied from 0% to 40% (see Table S1a, b, and c in the supplemental material). Given that tsetse acquire trypanosome infections as adults through feeding on infected vertebrate hosts, the observed heterogeneity in trypanosome prevalence may arise from spatial differences in the availability of trypanosome-infected vertebrate hosts and/or reservoirs and seasonal migrations (43) or livestock movements across Africa (55). The higher trypanosome prevalence in southern Uganda may be due to the greater availability of domestic animals in this region that has many cattle ranches (15). However, in addition to cattle, many other vertebrate hosts, including pigs and various wild game, can serve as reservoirs for trypanosomes. Alternatively, the higher trypanosome prevalence in the south may be due to host genetic differences, microfauna influences on host vector competence, or coinfection outcomes. In this study, we have not analyzed the status of trypanosome infections (midgut versus salivary glands) or the species of parasites with which flies are infected, but these factors are being investigated in our ongoing work. We chose not to investigate the trypanosome species, as they are unlikely to be statistically significant in our analysis given the relatively low overall parasite infection prevalence we observed in the field.
Apart from heterogeneity in infection, we also report high infection rates for SGHV and trypanosomes. The prevalence of SGHV in G. fuscipes fuscipes (12%) is much higher than the 0.1 to 7% reported in the natural populations of other Glossina species in which this virus has been detected (48). This difference may be due to the reliance on visual detection methods for the salivary gland hypertrophy (SGH) trait in dissected flies rather than the PCR assay we used in this study. The 18% overall trypanosome prevalence we report here is similar to that detected in G. pallidipes from Zambia (11%) (43), for which a DNA amplification-based assay was also used.
Patterns of pathogen prevalence relative to host genetics. (i) SGHV.
Our data suggest that SGHV presence is significantly associated with the north mtDNA haplogroup (see Table S2a in the supplemental material). We also found a significant association between SGHV and fly sex, but only in the southern microsatellite group, in which SGHV presence is significantly higher in males than in females (Table 2). A male bias has been shown in several other insects, such as the house fly Musca domestica, in which infection is up to 2-fold higher in males than in females (19). The biological significance of these associations between virus prevalence and host genetic makeup is unclear and deserves further investigation, as it suggests that genetic variation in the hosts can modulate the prevalence rate of this viral infection, a hypothesis that can be experimentally tested.
(ii) Trypanosomes.
Similar to the SGHV virus, trypanosome prevalence is higher in males than in females and is also dependent on the host genetic background, but this pattern holds only in the southern host microsatellite group (see Table S2a in the supplemental material). The higher prevalence rates in males may be due to variations in life history traits between the sexes. Interestingly, a different result has been reported in G. pallidipes populations from Gabon, where prevalence rates were 2.6-fold higher in females than in males (35). However, a sample bias in the study (significantly more females than males were analyzed) may render a comparison with that study difficult. Although different life history traits between males and females may explain some of our results, it is hard to understand why the significant sex differences in trypanosome prevalence rates we report in this study are evident in only one of the host genetic groups. One explanation is that the two genetic host groups differ in traits that affect the capacity of the different sexes to respond to the parasite infection, a hypothesis that can be experimentally tested.
Coinfections and host background.
The multivariate analyses illustrate that prevalence differences between host groups are influenced by infection with either two or three pathogens. Significant differences between host genetic groups in Wolbachia prevalence persist only when the host is uninfected with one (two-pathogen comparisons) or both (three-pathogen comparisons) of the other pathogens. Significant differences between host genetic groups in SGHV persist only when uninfected with Wolbachia. While trypanosomes do not have significant differences between host genetic groups regardless of the presence or absence of infection by other pathogens, trypanosome prevalence is positively correlated with SGHV infection (r = 0.257) and negatively correlated with Wolbachia infection (r = −0.176). Wolbachia is significantly negatively correlated with SGHV (r = −0.408). This implies that infection by Wolbachia may prevent infection by the other two pathogens. Wolbachia infections introduced into novel vectors have been shown to induce host resistance to malaria parasites, dengue virus, Brugia filarial nematodes, and RNA viruses (reviewed in reference 59). Such host manipulations can be mutually beneficial, as they enhance Wolbachia presence by preventing the establishment of other competitors and, as a result, lead to increased host fitness. Wolbachia-mediated pathogen resistance in insects has been found to result from induction of a variety of host immune responses, including the major antimicrobial signaling pathways and the complement system (34). In addition to enhancing host immunity, Wolbachia presence has been shown to downregulate host metabolic and/or redox transcripts. One group of candidate molecules includes the heat shock proteins (HSP), which have been suggested to diminish vector competence in Anopheles gambiae (38). Downregulation of HSP transcripts by two Wolbachia strains, wRi (a Wolbachia pipientis strain) and wAlbB (an Aedes albopictus-infecting Wolbachia strain), has been demonstrated (29). In such a scenario, Wolbachia-induced downregulation of tsetse HSP may decrease trypanosome infection and act synergistically in preventing SGHV infections.
Some of these same mechanisms, including antimicrobial activities, have also been shown to reduce tsetse's trypanosome transmission ability (23, 57). However, we found Wolbachia and trypanosome coinfection rates (8%) to be higher than those for any other coinfection. But we found a nonsignificant negative correlation between trypanosome and Wolbachia infection prevalence which may indicate a complex interaction between the host, Wolbachia, and trypanosome transmission ability.
Wolbachia infection and CI in G. fuscipes fuscipes.
Our results show the presence of Wolbachia infections in G. fuscipes fuscipes (see Table S1a, b, and c in the supplemental material), albeit at an unusually low density (Fig. 2). This can explain why our earlier investigations that relied on single PCR amplification methods could not identify these infections from G. fuscipes fuscipes populations (54). This result echoes some recent studies reporting low-density Wolbachia infections in other insects (the European cherry fruit fly, Rhagoletis cerasi [8], and Drosophila paulistorum [44]). Higher densities of Wolbachia have been reported in several other Glossina species, including G. morsitans morsitans (13). However, such high densities and expanded tissue tropisms have recently been shown to be an artifact of chromosomal insertions of the wsp gene used in the screens (54).
Wolbachia density may be important for determining CI occurrence (30). CI expression has been documented in G. morsitans morsitans laboratory cultures, but in these cultures, Wolbachia infection densities are higher than they are in G. fuscipes fuscipes (Fig. 2). It remains to be seen if the low Wolbachia densities we observed in G. fuscipes fuscipes can induce CI expression. Wolbachia infections in G. fuscipes fuscipes, if they confer CI, can prevent mixing of mtDNA lineages, resulting in an unexpected genetic divide similar to what we observe in mtDNA haplotypes of G. fuscipes fuscipes populations in southern and northern Uganda (5, 9). We initially expected to detect Wolbachia infections limited to one or the other mtDNA haplogroup. However, our results indicate Wolbachia presence throughout the landscape and no significant differences between the two mtDNA haplogroups (Fig. 3). Bidirectional CI, however, may maintain genetic differences in G. fuscipes fuscipes mtDNA haplogroups if each haplogroup is infected with multiple and different strains of Wolbachia (superinfected). Such superinfections have been noted in other arthropods (28). Our preliminary studies using the groEL gene sequence suggest the presence of multiple genotypes, and thus, superinfections are possible and are being further examined in these populations (data not shown). The Wolbachia prevalence assay we used here, however, does not allow us to identify the presence of different strains in the different populations screened. Alternatively, Wolbachia infections in G. fuscipes fuscipes may be commensalistic or mutualistic in their association. Laboratory studies in Drosophila and under in vitro culture conditions have shown that Wolbachia strains tend to modify their relationship with the host from a pathogenic to a commensalistic and/or mutualistic one over 2 years (about 50 generations) (42, 58), suggesting that mutualism may arise relatively fast.
Although mtDNA data do not support the existence of CI in G. fuscipes fuscipes populations (Fig. 4), unidirectional CI may still be responsible for the observed pattern of spatial heterogeneity in prevalence, as suggested by the Mantel test results. This test identified a negative correlation between geographic distance and coinfection dissimilarities (r = −0.261) that disappeared when Wolbachia was excluded from the test. In our G. fuscipes fuscipes data, populations only 15 km apart differed significantly in Wolbachia prevalence (see Table S1a in the supplemental material) (BN, 7%; BK, 54%). In unidirectional CI, infected females are expected to have higher reproductive success than uninfected females because they can mate with both infected and uninfected males without a reduction in fertility (53). Wolbachia can then spread among populations through female dispersal. However, when dispersal is male biased, spatial heterogeneity in Wolbachia prevalence among populations may result. In infected populations, immigrating males do not negatively impact female reproductive success. Thus, Wolbachia prevalence will continue to increase. However, even if infected males disperse out of infected populations, Wolbachia does not spread among populations because Wolbachia is maternally transmitted. As a result, neighboring populations can vary in Wolbachia prevalence. Male-biased dispersal has been observed in G. pallidipes (50) and in G. fuscipes fuscipes (37). Furthermore, females are known to have multiple matings (10). Therefore, a large difference in Wolbachia prevalence is expected between neighboring G. fuscipes fuscipes populations.
The occurrence of CI in G. fuscipes fuscipes may be masked by numerous factors, such as low-density infections, sex-biased dispersal, the possibility of Wolbachia superinfections, and infection by multiple pathogens. Given the relevance of Wolbachia in the development of CI-based control methods and the role of G. fuscipes fuscipes in disease transmission in Uganda, it is imperative to experimentally tease apart these factors that may influence CI.
Summary and future directions.
We have shown that the prevalence rates for all three microbes are heterogeneous across the Ugandan landscape and that host genetic similarity and geographic proximity do not necessarily imply similarity of prevalence rates. These findings, together with the fact that heterogeneity in rates among populations occurs even over quite small geographic distances, underscore the need to avoid reporting prevalence without a clear reference to the geographic scale to which they apply.
The statistically significant associations we found between host genetic groups and pathogen prevalence are an unexpected novel result, given the few genetic markers at our disposal. To better explore the biological interpretation of these associations, we plan to develop a panel of single nucleotide polymorphisms (SNPs) that can provide more representative coverage of the G. fuscipes fuscipes genome than the few genetic loci at our disposal and thus allow for detailed association studies.
Additionally, although the biological significance of some of these microbial associations is currently unclear, this study shows the usefulness of analyzing pathogen coinfection dynamics together with the host genetic background. In doing so, we have generated a series of testable hypotheses not apparent from single-infection studies that may shed light on the pathogen and vector life history traits and, in turn, be exploited for disease control.
Supplementary Material
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Abbot P, Aviles AE, Eller L, Durden LA. 2007. Mixed infections, cryptic diversity, and vector-borne pathogens: evidence from Polygenis fleas and Bartonella species. Appl. Environ. Microbiol. 73:6045–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abd-Alla A, et al. 2007. Development of a non-destructive PCR method for detection of the salivary gland hypertrophy virus (SGHV) in tsetse flies. J. Virol. Methods 139:143–149 [DOI] [PubMed] [Google Scholar]
- 3. Abd-Alla AM, et al. 2009. Quantitative PCR analysis of the salivary gland hypertrophy virus (GpSGHV) in a laboratory colony of Glossina pallidipes. Virus Res. 139:48–53 [DOI] [PubMed] [Google Scholar]
- 4. Abd-Alla AM, et al. 2010. Dynamics of the salivary gland hypertrophy virus in laboratory colonies of Glossina pallidipes (Diptera: Glossinidae). Virus Res. 150:103–110 [DOI] [PubMed] [Google Scholar]
- 5. Abila PP, et al. 2008. High levels of genetic differentiation between Ugandan Glossina fuscipes fuscipes populations separated by Lake Kyoga. PLoS Negl. Trop. Dis. 2:e242 doi:10.1371/journal.pntd.0000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aksoy S. 2000. Tsetse—a haven for microorganisms. Parasitol. Today 16:114–118 [DOI] [PubMed] [Google Scholar]
- 7. Alam U, et al. 2011. Wolbachia symbiont infections induce strong cytoplasmic incompatibility in the tsetse fly Glossina morsitans. PLoS Pathog. 7:e1002415 doi:10.1371/journal.ppat.1002415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arthofer W, et al. 2009. Hidden Wolbachia diversity in field populations of the European cherry fruit fly, Rhagoletis cerasi (Diptera, Tephritidae). Mol. Ecol. 18:3816–3830 [DOI] [PubMed] [Google Scholar]
- 9. Beadell JS, et al. 2010. Phylogeography and population structure of Glossina fuscipes fuscipes in Uganda: implications for control of tsetse. PLoS Negl. Trop. Dis. 4:e636 doi:10.1371/journal.pntd.0000636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bonomi A, et al. 2011. Polyandry is a common event in wild populations of the Tsetse fly Glossina fuscipes fuscipes and may impact population reduction measures. PLoS Negl. Trop. Dis. 5:e1190 doi:10.1371/journal.pntd.0001190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casiraghi M, et al. 2005. Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology 151:4015–4022 [DOI] [PubMed] [Google Scholar]
- 12. Cecchi G, Mattioli RC, Slingenbergh J, de la Rocque S. 2008. Land cover and tsetse fly distributions in sub-Saharan Africa. Med. Vet. Entomol. 22:364–373 [DOI] [PubMed] [Google Scholar]
- 13. Cheng Q, et al. 2000. Tissue distribution and prevalence of Wolbachia infections in tsetse flies, Glossina spp. Med. Vet. Entomol 14:44–50 [DOI] [PubMed] [Google Scholar]
- 14. Cox FE. 2001. Concomitant infections, parasites and immune responses. Parasitology 122(Suppl):S23–S38 [DOI] [PubMed] [Google Scholar]
- 15. Davis S, Aksoy S, Galvani A. 2011. A global sensitivity analysis for African sleeping sickness. Parasitology 138:516–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Echodu R, et al. 2011. Temporal stability of Glossina fuscipes fuscipes populations in Uganda. Parasit. Vectors. 4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Engelstadter J, Telschow A. 2009. Cytoplasmic incompatibility and host population structure. Heredity 103:196–207 [DOI] [PubMed] [Google Scholar]
- 18. Garbutt J, Bonsall MB, Wright DJ, Raymond B. 2011. Antagonistic competition moderates virulence in Bacillus thuringiensis. Ecology Lett. 14:765–772 [DOI] [PubMed] [Google Scholar]
- 19. Geden CJ, Lietze VU, Boucias DG. 2008. Seasonal prevalence and transmission of salivary gland hypertrophy virus of house flies (Diptera: Muscidae). J. Med. Entomol. 45:42–51 [DOI] [PubMed] [Google Scholar]
- 20. Geiger A, Ravel S, Frutos R, Cuny G. 2005. Sodalis glossinidius (Enterobacteriaceae) and vectorial competence of Glossina palpalis gambiensis and Glossina morsitans morsitans for Trypanosoma congolense savannah type. Curr. Microbiol. 51:35–40 [DOI] [PubMed] [Google Scholar]
- 21. Greenacre M. 1993. Correspondence analysis in practice. Academic Press, London, United Kingdom [Google Scholar]
- 22. Greenacre M, Nenadic O. 2010. ca: simple, multiple and joint correspondence analysis. R package, version 0.33. [Google Scholar]
- 23. Hao Z, et al. 2001. Tsetse immune responses and trypanosome transmission: implications for the development of tsetse-based strategies to reduce trypanosomiasis. Proc. Natl. Acad. Sci. U. S. A. 98:12648–12653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrell F. 2009. Design: design package. R package, version 2.3–0. [Google Scholar]
- 25. Hedges LM, Brownlie JC, O'Neill SL, Johnson KN. 2008. Wolbachia and virus protection in insects. Science 322:702. [DOI] [PubMed] [Google Scholar]
- 26. Holm S. 1979. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6:65–70 [Google Scholar]
- 27. Hothorn T, Bretz F, Westfall P. 2008. Simultaneous inference in general parametric models. Biom. J. 50:346–363 [DOI] [PubMed] [Google Scholar]
- 28. Hughes GL, et al. 2011. Variable infection frequency and high diversity of multiple strains of Wolbachia pipientis in Perkinsiella planthoppers. Appl. Environ. Microbiol. 77:2165–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hughes GL, et al. 2011. Wolbachia infections in Anopheles gambiae cells: transcriptomic characterization of a novel host-symbiont interaction. PLoS Pathog. 7:e1001296 doi:10.1371/journal.ppat.1001296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikeda T, Ishikawa H, Sasaki T. 2003. Infection density of Wolbachia and level of cytoplasmic incompatibility in the Mediterranean flour moth, Ephestia kuehniella. J. Invertebr. Pathol. 84:1–5 [DOI] [PubMed] [Google Scholar]
- 31. Jaenson TG. 1978. Virus-like rods associated with salivary gland hyperplasia in tsetse, Glossina pallidipes. Trans. R. Soc. Trop. Med. Hyg. 72:234–238 [DOI] [PubMed] [Google Scholar]
- 32. Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405 [DOI] [PubMed] [Google Scholar]
- 33. Jura WG, Odhiambo TR, Otieno LH, Tabu NO. 1988. Gonadal lesions in virus-infected male and female tsetse, Glossina pallidipes (Diptera: Glossinidae). J. Invertebr. Pathol. 52:1–8 [DOI] [PubMed] [Google Scholar]
- 34. Kambris Z, Cook PE, Phuc HK, Sinkins SP. 2009. Immune activation by life-shortening Wolbachia and reduced filarial competence in mosquitoes. Science 326:134–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kohagne TL, M'eyi MP, Mimpfoundi R, Louis JF. 2010. Entomological patterns in the human African trypanosomiasis focus of Komo Mondah, Gabon. Afr. Health Sci. 10:341–348 [PMC free article] [PubMed] [Google Scholar]
- 36. Kokwaro ED, Nyindo M, Chimtawi M. 1990. Ultrastructural changes in salivary glands of tsetse, Glossina morsitans morsitans, infected with virus and rickettsia-like organisms. J. Invertebr. Pathol. 56:337–346 [DOI] [PubMed] [Google Scholar]
- 37. Kone N, N′Goran KE, Sidibe I, Kombassere AW, Bouyer J. 2011. Spatio-temporal distribution of tsetse and other biting flies in the Mouhoun River basin, Burkina Faso. Med. Vet. Entomol. 25:156–168 [DOI] [PubMed] [Google Scholar]
- 38. Lefevre T, et al. 2007. Malaria Plasmodium agent induces alteration in the head proteome of their Anopheles mosquito host. Proteomics 7:1908–1915 [DOI] [PubMed] [Google Scholar]
- 39. Lindh JM, Lehane MJ. 2011. The tsetse fly Glossina fuscipes fuscipes (Diptera: Glossina) harbours a surprising diversity of bacteria other than symbionts. Antonie Van Leeuwenhoek 99:711–720 [DOI] [PubMed] [Google Scholar]
- 40. Mantel N. 1967. The detection of disease clustering and a generalized regression approach. Cancer Res. 27:209–220 [PubMed] [Google Scholar]
- 41. Maudlin I, Ellis DS. 1985. Association between intracellular rickettsial-like infections of midgut cells and susceptibility to trypanosome infection in Glossina spp. Z. Parasitenkd. 71:683–687 [DOI] [PubMed] [Google Scholar]
- 42. McGraw EA, Merritt DJ, Droller JN, O'Neill SL. 2002. Wolbachia density and virulence attenuation after transfer into a novel host. Proc. Natl. Acad. Sci. U. S. A. 99:2918–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mekata H, et al. 2008. Prevalence and source of trypanosome infections in field-captured vector flies (Glossina pallidipes) in southeastern Zambia. J. Vet. Med. Sci. 70:923–928 [DOI] [PubMed] [Google Scholar]
- 44. Miller WJ, Ehrman L, Schneider D. 2010. Infectious speciation revisited: impact of symbiont-depletion on female fitness and mating behavior of Drosophila paulistorum. PLoS Pathog. 6:e1001214 doi:10.1371/journal.ppat.1001214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moreira LA, et al. 2009. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 139:1268–1278 [DOI] [PubMed] [Google Scholar]
- 46. Mouton L, et al. 2004. Virulence multiple infections and regulation of symbiotic population in the Wolbachia-Asobara tabida symbiosis. Genetics 168:181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nei M. 1972. Genetic distances between populations. Am. Nat. 106:283–292 [Google Scholar]
- 48. Odindo MO, Payne CC, Crook NE, Jarrett P. 1986. Properties of a novel DNA virus from the tsetse fly, Glossina pallidipes. J. Gen. Virol. 67(Part 3):527–536 [DOI] [PubMed] [Google Scholar]
- 49. Osborne SE, Leong YS, O'Neill SL, Johnson KN. 2009. Variation in antiviral protection mediated by different Wolbachia strains in Drosophila simulans. PLoS Pathog. 5:e1000656 doi:10.1371/journal.ppat.1000656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ouma JO, et al. 2011. Genetic diversity and population structure of Glossina pallidipes in Uganda and western Kenya. Parasit. Vectors 4:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pais R, Lohs C, Wu Y, Wang J, Aksoy S. 2008. The obligate mutualist Wigglesworthia glossinidia influences reproduction, digestion, and immunity processes of its host, the tsetse fly. Appl. Environ. Microbiol. 74:5965–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sang RC, Jura W, Otieno LH, Ogaja P. 1996. Ultrastructural changes in the milk gland of tsetse Glossina morsitans centralis (Diptera; Glissinidae) female infected by a DNA virus. J. Invertebr. Pathol. 68:253–259 [DOI] [PubMed] [Google Scholar]
- 53. Telschow A, Hammerstein P, Werren JH. 2002. The effect of Wolbachia on genetic divergence between populations: models with two-way migration. Am. Nat. 160(Suppl 4):S54–S66 [DOI] [PubMed] [Google Scholar]
- 54. Doudoumis V, et al. 2012. Detection and characterization of Wolbachia infections in laboratory and natural populations of different species of tsetse flies (genus Glossina). BMC Microbiol. 12(Suppl 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wacher TJ, Rawlings P, Snow WF. 1993. Cattle migration and stocking densities in relation to tsetse-trypanosomiasis challenge in The Gambia. Ann. Trop. Med. Parasitol. 87:517–524 [DOI] [PubMed] [Google Scholar]
- 56. Walshe DP, et al. 2009. The enemy within: interactions between tsetse, trypanosomes and symbionts, p 119–175. In Simpson SJ, Casas J. (ed), Advances in insect physiology, vol 37 Academic Press, Liverpool, United Kingdom. [Google Scholar]
- 57. Wang J, Wu Y, Yang G, Aksoy S. 2009. Interactions between mutualist Wigglesworthia and tsetse peptidoglycan recognition protein (PGRP-LB) influence trypanosome transmission. Proc. Natl. Acad. Sci. U. S. A. 106:12133–12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weeks AR, Turelli M, Harcombe WR, Reynolds KT, Hoffmann AA. 2007. From parasite to mutualist: rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 5:e114 doi:10.1371/journal.pbio.0050114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weiss B, Aksoy S. 2011. Microbiome influences on insect host vector competence. Trends Parasitol. 11:514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Westfall PH. 1997. Multiple testing of general contrasts using logical constraints and correlations. J. Am. Stat. Assoc. 92:299–306 [Google Scholar]
- 61. Westfall PH, Tobias RD, Rom R, Wolfinger RD, Hochberg Y. 1999. Multiple comparisons and multiple tests using the SAS system. SAS Institute, Cary, NC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.