

Abstract
The purpose of this study was to test the hypothesis that diabetes aggravates periodontal destruction induced by Aggregatibacter actinomycetemcomitans infection. Thirty-eight diabetic and 33 normal rats were inoculated with A. actinomycetemcomitans and euthanized at baseline and at 4, 5, and 6 weeks after inoculation. Bone loss and the infiltration of polymorphonuclear leukocytes (PMNs) in gingival epithelium were measured in hematoxylin-eosin-stained sections. The induction of tumor necrosis factor alpha (TNF-α) was evaluated by immunohistochemistry and of apoptotic cells by a TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay. After A. actinomycetemcomitans infection, the bone loss in diabetic rats was 1.7-fold and the PMN infiltration 1.6-fold higher than in normoglycemic rats (P < 0.05). The induction of TNF-α was 1.5-fold higher and of apoptotic cells was up to 3-fold higher in diabetic versus normoglycemic rats (P < 0.05). Treatment with a caspase-3 inhibitor significantly blocked noninflammatory cell apoptosis induced by A. actinomycetemcomitans infection in gingival epithelium and connective tissue (P < 0.05). These results provide new insight into how diabetes aggravates A. actinomycetemcomitans-induced periodontal destruction in rats by significantly increasing the inflammatory response, leading to increased bone loss and enhancing apoptosis of gingival epithelial and connective tissue cells through a caspase-3-dependent mechanism. Antibiotics had a more pronounced effect on many of these parameters in diabetic than in normoglycemic rats, suggesting a deficiency in the capacity of diabetic animals to resist infection.
INTRODUCTION
Periodontitis is one of the most prevalent infectious diseases worldwide. It is characterized by loss of supporting connective tissue and alveolar bone around the teeth (36). Although triggered by a bacterial infection, the destruction of periodontal tissue is caused by the inflammatory response to pathogenic bacteria. Immune mediators such as interleukin-1 (IL-1), tumor necrosis factor alpha (TNF-α), IL-6, and RANKL have been found to be abundantly expressed in humans with periodontal disease, and increased levels have been shown in the crevicular fluid from patients with periodontitis (4, 13, 32). Animal studies have established cause-and-effect relationships between these cytokines and periodontal breakdown (13, 24).
There are several types of periodontal diseases ranging from chronic periodontitis that affects adults to a form of aggressive periodontitis that primarily affects adolescents, localized aggressive periodontitis (LAgP). LAgP is characterized by severe and rapid destruction of the supporting apparatus of the teeth, which may lead to tooth loss early in life (3). Aggregatibacter actinomycetemcomitans is commonly linked to LAgP (3, 16), and studies have shown that periodontal treatment leads to a reduction in its levels (5, 31). The presence of A. actinomycetemcomitans in periodontal pockets has also been considered indicative of future disease progression (9, 16). A. actinomycetemcomitans has virulence factors, such as leukotoxin and cytolethal distending toxin (CDT), that may contribute to its capacity to induce rapid tissue destruction by promoting apoptosis of a number of host cell types (20).
A rat model has been developed to study the pathogenic mechanisms of A. actinomycetemcomitans-induced periodontal tissue destruction (8, 28, 38, 39). This model is characterized by infecting the animals with a rough strain of A. actinomycetemcomitans that adheres to the oral epithelium and teeth (7). Although it may not mimic the specific form of periodontal disease found in localized aggressive periodontitis in humans, this model has provided insight into the colonization of the oral cavity by this bacterium and inflammation-induced periodontitis (28). However, relatively little is known about the local changes that are induced by this bacterium in vivo.
Periodontal disease is triggered by bacterial infection, but the local inflammatory response has been shown to mediate the actual destruction of periodontal tissue. This response can be modulated by systemic conditions such as diabetes. Diabetes has been identified as one of the important risk factors for periodontitis, increasing both its prevalence and severity (26, 27, 40). One mechanism through which diabetes increases periodontal tissue loss and other diabetic complications is by exacerbating the inflammatory response to periodontal pathogens through increased oxidative stress, advanced glycation end products, and expression of cytokines such as TNF-α (12, 25, 33, 37).
Apoptosis is thought to contribute to periodontal disease progression. It has been suggested that apoptosis of epithelial cells may contribute to the loss of epithelial barrier function (6). Moreover, loss of gingival fibroblasts has been shown to be one of the largest cellular changes that occurs with periodontal disease progression and may be associated with a loss of connective tissue attachment (29, 43). Infection by A. actinomycetemcomitans has been shown to induce apoptosis in vitro (21–23). However, relatively little is known about how it induces apoptosis in vivo and how a systemic condition such as diabetes affects A. actinomycetemcomitans-induced apoptosis and periodontal tissue destruction. The experiments described here address these issues using diabetic and matched normoglycemic rats, which are natural hosts of A. actinomycetemcomitans. The results indicate that the effect of A. actinomycetemcomitans infection on bone loss, TNF-α expression, and apoptosis of epithelial cells and nonleukocytic gingival connective tissue cells is aggravated by diabetes. Moreover, we demonstrate that apoptosis is induced by a caspase-3-dependent mechanism.
MATERIALS AND METHODS
Animals.
Goto-Kakizaki (GK) and normoglycemic control matched Wistar rats (5 to 10 weeks old) weighing 150 to 250 g were purchased from Charles River Laboratories (Wilmington, MA). The GK rat is a nonobese Wistar substrain that develops type 2 diabetes mellitus at age approximately 8 weeks. Rats were considered to be diabetic when glycated hemoglobin (HbAlc) levels exceeded 7.0%. During the experiments, the HbAlc level in GK rats was typically 7.0 to 10.5%. All normoglycemic rats had HbAlc levels that ranged from 4.3 to 4.8%. All animal procedures were approved by the Institutional Animal Care and Use Committee.
A. actinomycetemcomitans inoculation.
Both diabetic (GK) and normal (Wistar) rats were inoculated with A. actinomycetemcomitans as previously described (39). To depress the “natural” resident flora, rats received in their water a daily dose of kanamycin (20 mg) and ampicillin (20 mg) for 4 days. During the last 2 days of antibiotic treatment, the oral cavities of the rats were swabbed with a 0.12% chlorhexidine gluconate rinse (Peridex; Procter and Gamble, Cincinnati, OH). After a subsequent period of 3 days without antibiotic treatment, the rats were divided into six groups of approximately seven rats each. The adherent A. actinomycetemcomitans strain, Columbia University A. actinomycetemcomitans clinical isolate 1000 (CU1000NRif), was incubated in A. actinomycetemcomitans growth medium with 35 mg of rifampin (Sigma-Aldrich, St. Louis, MO)/ml for 2 days. Adherent cells in culture dishes were scraped into a solution of phosphate-buffered saline (PBS) plus 3% sucrose, and minor adjustment was made by the addition of buffer to obtain 108 cells/ml (optical density at 560 nm = 0.80). After fasting for 3 h, rats received 108 A. actinomycetemcomitans cells in 1 g of powdered food supplemented with 3% sucrose. This protocol was followed for 4 days and was repeated the following week for a total of eight A. actinomycetemcomitans inoculations in food (39). During the first 4 days of the feeding, the rats also received 108 A. actinomycetemcomitans in PBS by oral gavage. After 1 h, the inoculated food was removed and replaced with regular powdered food. Rats were euthanized 4, 5, and 6 weeks after the inoculation period was completed. Baseline animals did not receive A. actinomycetemcomitans in their food and were not inoculated with A. actinomycetemcomitans but did receive powered food supplemented with 3% sucrose under the same conditions as the experimental rats.
Treatment with antibiotics and caspase-3 inhibitor.
At 4 weeks after A. actinomycetemcomitans inoculation, two groups of rats received in their water a daily dose of kanamycin (20 mg) and ampicillin (20 mg) for 4 days with the intention to reduce the infection. Concomitantly, the oral cavities of the rats were swabbed with a 0.12% chlorhexidine gluconate rinse (Peridex).
Caspase-3 inhibitor (Z-DEVD-FMK; SM Biochemicals, Anaheim, CA) was administered by intraperitoneal injection (1.5 mg/kg). Control animals were injected with the same volume of vehicle (2% dimethyl sulfoxide; MP Biomedicals, Solon, OH). The caspase-3 inhibitor was administrated 1 week before euthanasia, and it was injected daily until the animals were euthanized.
Sampling of total anaerobic bacteria (CFU) and detection of A. actinomycetemcomitans by PCR.
Two microbial samples were collected: one after the inoculation of A. actinomycetemcomitans and the other at the time of euthanasia. The rats were anesthetized, and their oral microflora was sampled using a cotton tip swab for soft tissue sampling and a toothpick (Johnson & Johnson, Piscataway, NJ) for hard tissue sampling. Both samples were combined in tubes containing 1 ml of PBS. Serial 10-fold dilutions were made and plated on Trypticase soy agar with 5% sheep blood (BD Biosciences, San Jose, CA) for total anaerobe counts. Trypticase soy agar plates were incubated in an anaerobic atmosphere at 37°C for 7 days to obtain total bacterial counts. To detect whether A. actinomycetemcomitans was present in the samples, DNA was prepared directly from the collected oral samples with a DNA extraction kit (Qiagen, Valencia, CA) and subjected to PCR analysis using forward and reverse primers (5′-GGAATTCCTAGGTATTGCGAAACAATTTGATC-3′ and 5′-GGAATTCCTGAAATTAAGCTGGTAATC-3′, respectively), which amplified a 262-bp PCR product from the A. actinomycetemcomitans leukotoxin gene as previously described (10).
Level of antibody to A. actinomycetemcomitans.
IgG antibody reactive with A. actinomycetemcomitans was assessed by enzyme-linked immunosorbent assay (ELISA). Blood was collected by cardiac puncture and serum was obtained and stored at −20°C. An A. actinomycetemcomitans lysate was prepared and used to coat the wells of microtiter dishes (Nunc-ImmunoPlate with a Maxi Sorp surface; Thermo Fisher Scientific, Rochester, NY). A standard curve was generated using purified rat IgG (Sigma-Aldrich) in carbonate-bicarbonate buffer (pH 9.6; Sigma-Aldrich). Rat serum diluted 1/5 and 1/10 in blocking buffer was added to the wells coated with A. actinomycetemcomitans pellet lysate. The serum dilutions were added in duplicate wells, washed, incubated with rabbit anti-rat IgG-Fc conjugated to alkaline phosphatase (Bethyl Laboratories, Montgomery, TX), and quantified with p-nitrophenyl phosphate substrate (Sigma-Aldrich). Absorbance was read on a microplate reader at 405 nm.
Histomorphometric analysis of hematoxylin-eosin-stained sections.
Right maxillas were fixed in 4% paraformaldehyde at 4°C for 48 h and decalcified in 10% EDTA (pH 7.0) for 12 weeks. Paraffin-embedded sagittal sections were prepared at a thickness of 5 μm. The mid-interproximal region between first and second molars was examined in each specimen and was established by being sectioned to a level where the root canal systems in adjacent teeth were visible. Two randomly chosen sections of each interproximal area were examined at ×200 magnification. All data were analyzed by a blinded examiner who did not know the group to which an animal belonged. Bone loss was measured as the distance between the cemento-enamel junction (CEJ) and the highest peak of the interproximal bone. The number of polymorphonuclear (PMNs) leukocytes was counted in the gingival epithelium at ×600 magnification. The identification of these cells was confirmed by an experienced examiner.
Histomorphometric analysis of TNF-α immunohistochemistry-stained sections.
To evaluate the number of cells expressing TNF-α, sections were stained by immunohistochemistry with an antibody against TNF-α (IHC World, Woodstock, MD). The number of positive cells was evaluated 1 mm down from the level of bone crest apically in an area of periodontal ligament between the first and second molars. Cell counts were obtained by one examiner and confirmed by a second independent examiner with similar results. The numbers of positive cells in epithelium and gingival connective tissue were scored based on the following scale: 0, no positive cells; 1, three to four positive cells per field with weak immunostaining; 2, four to ten positive cells per field with strong immunostaining; and 3, more than ten positive cells per field with strong immunostaining. Sections were examined at ×600 magnification.
Detection of apoptotic cells.
Apoptotic cells were detected by an in situ TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay (DeadEnd fluorometric TUNEL system kit; Promega, Madison, WI) according to the manufacturer's instructions. This kit detects double-strand breaks in genomic DNA and identifies most stages of apoptosis. The fluorescein-12-dUTP-labeled DNA was then visualized directly by fluorescence microscopy. Additional counts were made to specifically avoid counting apoptotic leukocytes. This was accomplished by using the TUNEL assay, followed by immunofluorescence with an anti-CD18 antibody (Novus Biological, Littleton, CO). The number of nonleukocytic apoptotic cells (TUNEL+ CD18−) was counted at ×200 magnification with an immunofluorescence microscope using NIS Elements software (Nikon, Melville, NY) in epithelium or connective tissue above the alveolar bone crest. Cells counts were obtained by one examiner and confirmed by a second independent examiner with similar results.
Systemic leukocyte analysis.
Rat lymph leukocytes were isolated and analyzed as previously described (28). Single-cell suspensions were obtained from the submandibular and cervical lymph nodes. Lymphocyte populations were isolated by Ficoll-Hypaque density gradient centrifugation. Flow cytometry was conducted using anti-CD32 (clone D34-485) for blocking FcγII receptors, phycoerythrin (PE)-conjugated anti-CD4 (clone OX-38), fluorescein isothiocyanate (FITC)-conjugated anti-CD3 (clone G4.18; BD Biosciences); FITC-labeled anti-FoxP3 (clone FJK-16S), and PE-conjugated anti-CD25 (clone OX39) from eBioscience (San Diego, CA), and anti-IA (clone 14-4-4S) from the American Type Culture Collection (Manassas, VA). Blood was analyzed by HemaTrue hematology analyzer (Heska, Loveland, CO). The total numbers of white blood cells, the numbers of lymphocytes, monocytes, and granulocytes, and the percentages of lymphocytes, monocytes, and granulocytes were analyzed.
Statistical analysis.
Differences between two groups such as diabetic and normal rats were determined by using the Student t test and between time points within a group by one-way analysis of variance except for the evaluation of TNF-α. Differences in TNF-α values were determined by nonparametric analysis with Mann-Whitney U test. Significance levels were set at 5%.
RESULTS
Induction of periodontal disease.
At baseline, the antibody titer level in diabetic rats was low and increased after A. actinomycetemcomitans infection so that at 6 weeks it was 32-fold higher than at baseline (P < 0.01) (Fig. 1A). Moreover, for all A. actinomycetemcomitans infected diabetic rats, the antibody titers were 18-fold higher than for the noninfected animals (P < 0.01) (Fig. 1B). Also, after infection diabetic the animals had antibody titers level that were 2.3-fold higher than normoglycemic infected animals (P < 0.05) (Fig. 1B). Diabetic rats also showed a significant decrease in antibody titer level after antibiotic treatment (P < 0.05) (Fig. 1C).
Fig 1.
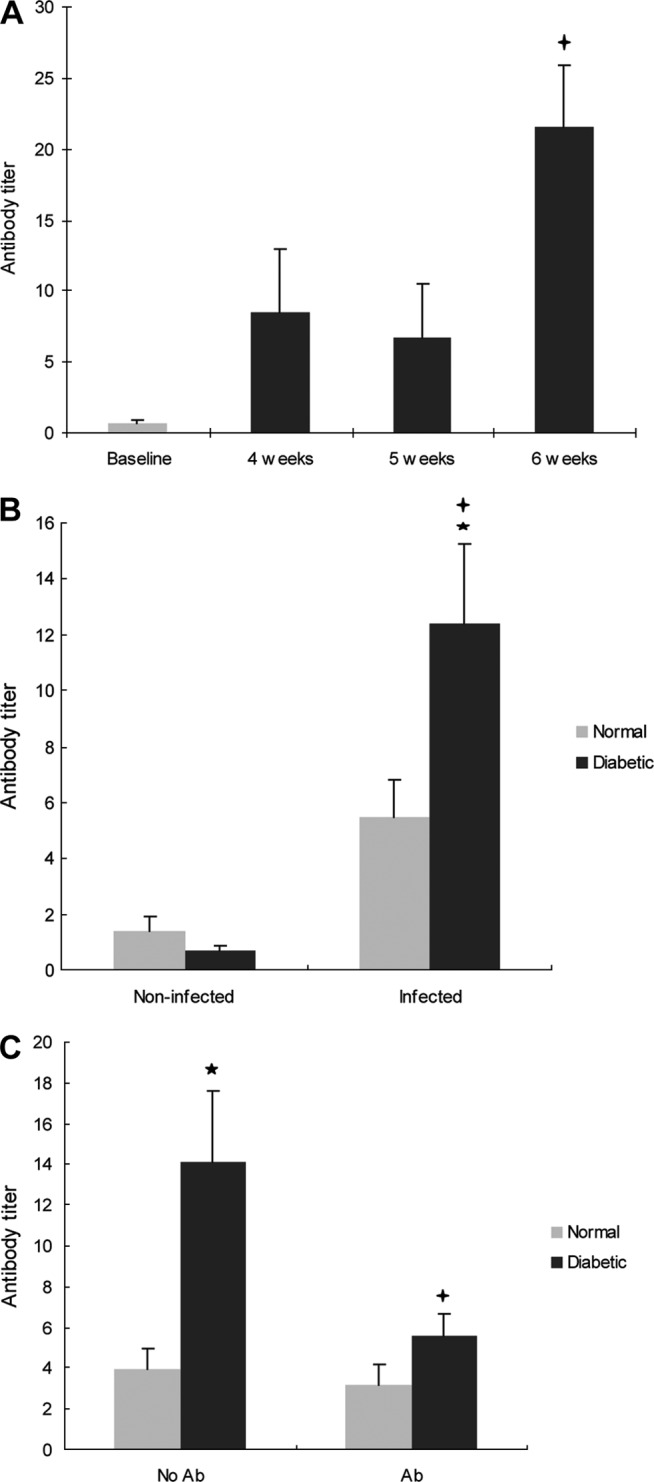
Diabetes increases the antibody titer to A. actinomycetemcomitans in infected rats. Diabetic and normal rats were infected orally with A. actinomycetemcomitans, and antibody (IgG) reactive with A. actinomycetemcomitans was assessed by ELISA. At 4 weeks postinfection, one group of rats was treated with antibiotics or equivalent vehicle alone. Rats were euthanized at baseline and at 4, 5, and 6 weeks after A. actinomycetemcomitans inoculation was completed. (A) Antibody titer levels in diabetic rats over time. (B) Antibody titer in noninfected (baseline) and infected (4 to 6 weeks) normoglycemic and diabetic rats. (C) Effect of antibiotic treatment on antibody titer in normoglycemic and diabetic rats. Each value in panels A, B, and C is the mean of five to seven rats ± the standard error of the mean (SEM). *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic rats in different groups (P < 0.05).
The impact of A. actinomycetemcomitans infection on total anaerobic bacteria levels in noninfected normoglycemic and diabetic rats, as well as infected diabetic rats, was also examined. Despite a trend toward increased levels of anaerobic bacteria in infected diabetic rats compared to normoglycemic rats, the differences were not significant (see Table S1 in the supplemental material). Similarly the percentage of rats exposed to A. actinomycetemcomitans that had detectable infection was not higher between the normoglycemic and diabetic groups (see Table S2 in the supplemental material).
A number of parameters was evaluated to examine the impact of A. actinomycetemcomitans infection on the systemic leukocyte populations in diabetic animals. After A. actinomycetemcomitans infection there was no change in A. actinomycetemcomitans-infected rats compared to uninfected rats of major histocompatibility complex (MHC) class II-positive cells, T cells, B cells, or Treg cells for either normoglycemic or diabetic rats (Tables 1 and 2). However, there was a slight reduction in the percentage of lymphocytes in the peripheral circulation of infected diabetic rats compared to infected normoglycemic rats and a 1.5-fold increase in the percentage of granulocytes in infected diabetic rats compared to infected normoglycemic rats (P < 0.05) (Table 2).
Table 1.
Lymphocyte populations of noninfected and A. actinomycetemcomitans-infected ratsa
| Type | Mean ± SEM |
|||
|---|---|---|---|---|
| Normal rats |
Diabetic rats |
|||
| Noninfected | Infected | Noninfected | Infected | |
| MHC II | 55.9 ± 11.7 | 44.1 ± 14.8 | 38.3 ± 6.8* | 37.0 ± 12.0 |
| CD4+ | 31.5.1 ± 11.4 | 32.4 ± 13.0 | 42.1 ± 9.3 | 42.7 ± 13.9 |
| CD8+ | 24.6 ± 6.9 | 35.2 ± 10.7 | 29.6 ± 9.4 | 31.4 ± 8.9 |
| CD25+ | 5.2 ± 0.9 | 6.7 ± 2.8 | 5.4 ± 0.8 | 7.9 ± 2.4 |
| FoxP3+ | 4.3 ± 0.8 | 5.3 ± 3.1 | 7.0 ± 3.4 | 7.2 ± 2.4 |
Lymphocyte populations from draining cervical and submandibular lymph nodes were analyzed as described in Materials and Methods. The rats described in Fig. 1 were examined for lymphocyte populations according to infection status. Each value is the mean of five to seven rats.
, P < 0.05, compared to normal rats.
Table 2.
Leukocytic cells of noninfected and A. actinomycetemcomitans-infected ratsa
| Parameter | Mean ± SEM |
|||
|---|---|---|---|---|
| Normal |
Diabetic |
|||
| Noninfected | Infected | Noninfected | Infected | |
| No. (103 UI/ml) | ||||
| WBC | 7.9 ± 2.9 | 7.8 ± 4.7 | 6.9 ± 3.2 | 8.0 ± 4.0 |
| Lymphocytes | 6.2 ± 2.5 | 5.8 ± 3.3 | 4.4 ± 1.0 | 5.2 ± 2.8 |
| Monocytes | 0.4 ± 0.1 | 0.3 ± 0.2 | 0.4 ± 0.3 | 0.4 ± 0.2 |
| Granulocytes | 1.3 ± 0.5 | 1.7 ± 1.5 | 2.2 ± 2.0 | 2.3 ± 1.4 |
| Amt (%) | ||||
| Lymphocytes | 77.0 ± 5.5 | 76.0 ± 10.5 | 69.0 ± 13.8 | 65.6 ± 13.5* |
| Monocytes | 4.8 ± 1.0 | 3.5 ± 1.2 | 4.8 ± 1.4 | 4.5 ± 1.8 |
| Granulocytes | 18.1 ± 4.6 | 20.6 ± 9.9 | 26.2 ± 12.5 | 30.0 ± 13.4* |
Leukocytic cells from peripheral blood were analyzed as described in Materials and Methods. The rats described in Fig. 1 were examined for leukocytic cells according to infection status. Each value is the mean of five to seven rats.
, P <0.05, compared to normal rats. WBC, white blood cells.
Bone loss was induced in the diabetic rats, as evidenced by an increase in the distance from the CEJ to the alveolar bone crest after 5 weeks (P < 0.05) (Fig. 2A and see Fig. S1 in the supplemental material). In A. actinomycetemcomitans-infected diabetic animals there was a 1.8-fold increase in bone loss compared to noninfected diabetic rats at baseline. The bone loss was 1.7-fold higher in the infected diabetic rats compared to infected normoglycemic rats (P < 0.05) (Fig. 2B). Antibiotic treatment significantly decreased the bone loss in the diabetic rats (P < 0.05) (Fig. 2C).
Fig 2.
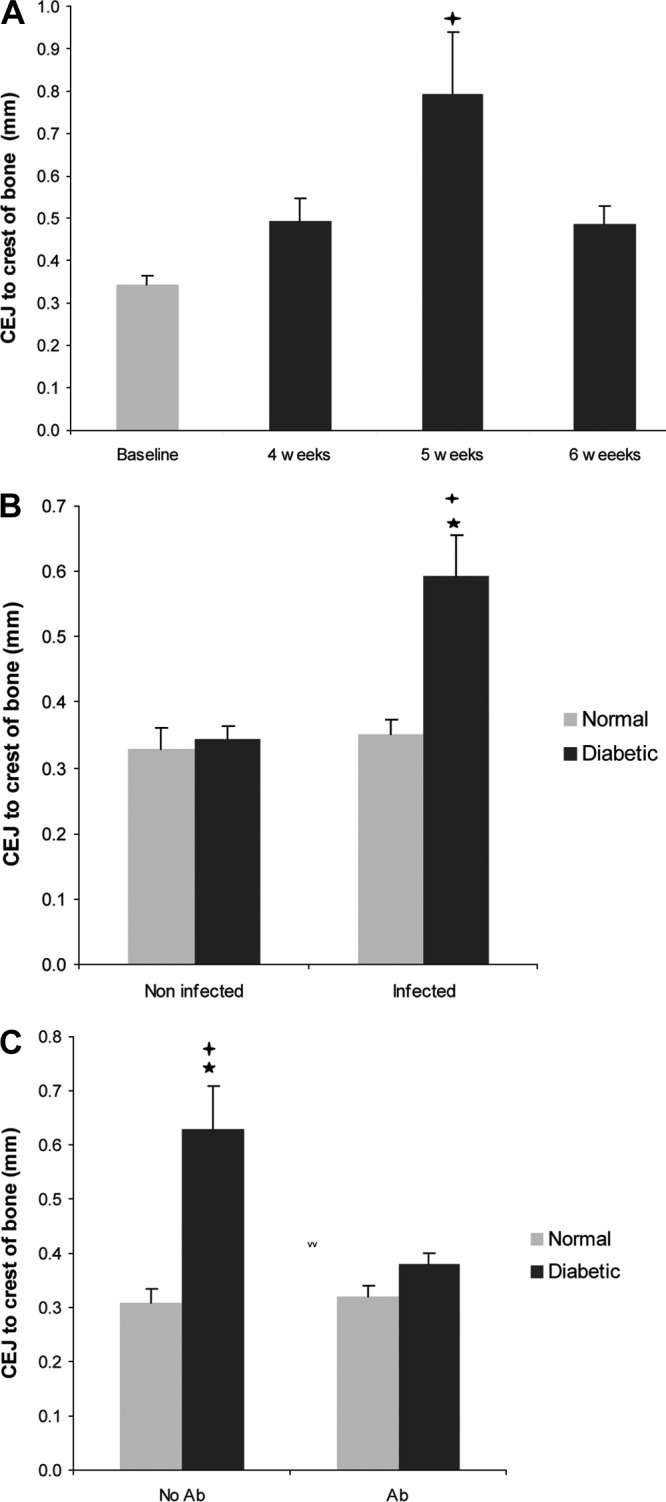
Diabetes increases bone loss in A. actinomycetemcomitans-infected rats. The distance between the CEJ and the alveolar bone crest was measured. (A) CEJ-to-bone distance in diabetic rats. (B) CEJ-to-bone distance in noninfected (baseline) and infected (4 to 6 weeks) normoglycemic and diabetic rats. (C) Effect of antibiotic treatment on CEJ-to-bone distance in normoglycemic and diabetic rats. Each value is the mean of five to seven rats ± the SEM. *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic rats in different groups (P < 0.05).
PMN infiltration.
The formation of a PMN infiltrate in gingival epithelium was assessed (see Fig. S2 in the supplemental material). PMNs increased 7-fold (P < 0.05) 4 weeks after A. actinomycetemcomitans inoculation in diabetic rats (Fig. 3A). Both normal and diabetic noninfected rats had similar levels of PMNs. After A. actinomycetemcomitans infection PMN numbers increased 3.4-fold in normal rats, while it increased 5.3-fold in diabetic rats (P < 0.05) (Fig. 3B). The greater increase in PMNs in infected diabetic rats, compared to infected normal rats, is consistent with the significant increase (P < 0.05) in blood granulocytes in infected diabetic rats. Antibiotic treatment significantly reduced the PMN infiltration in the diabetic rats (P < 0.05) (Fig. 3C).
Fig 3.

Diabetes increases the number of PMNs of A. actinomycetemcomitans-infected rats. The number of PMNs infiltrating the gingival epithelium was measured. (A) PMN infiltration in diabetic rats over time. (B) PMNs in noninfected (baseline) and infected (4 to 6 weeks) normoglycemic and diabetic rats. (C) Effect of antibiotic treatment on PMN infiltration in normoglycemic and diabetic rats. Each value in panels A, B, and C is the mean of five to seven rats ± the SEM. *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic rats in different groups (P < 0.05).
TNF-α.
TNF-α was measured in the gingival epithelium and connective tissue. In the epithelium, the TNF-α values of diabetic rats were significantly higher in animals exposed to A. actinomycetemcomitans inoculation compared to noninfected diabetic rats (P < 0.05) and significantly higher than in infected normoglycemic rats (P < 0.05) (Fig. 4B and see Fig. S3 in the supplemental material). Antibiotic treatment resulted in a significant decrease in TNF-α expression in the epithelium of diabetic rats (P < 0.05) but had no effect in the normoglycemic group (Fig. 4C).
Fig 4.
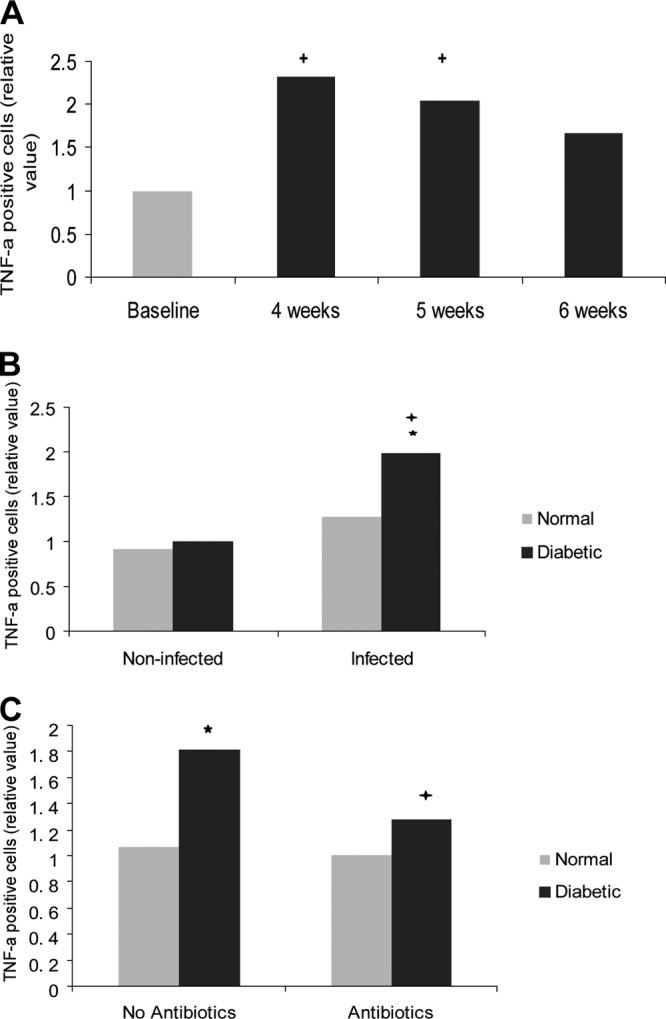
Diabetes increases the TNF-α expression in the gingival epithelia of A. actinomycetemcomitans-infected rats. TNF-α-positive cells were detected by immunohistochemistry in histologic specimens using a specific antibody. The rats described in Fig. 1 were examined for TNF-α expression using the following scale that took both the number of immunopositive cells and the intensity of the immunostaining into account: 0, no positive cells; 1, three to four positive cells per field with weak immunostaining; 2, four to ten positive cells per field with strong immunostaining; and 3, more than ten positive cells per field with strong immunostaining. (A) TNF-α expression in the gingival epithelia of diabetic rats. (B) TNF-α expression in noninfected (baseline) and infected (4 to 6 weeks) gingival epithelia in normoglycemic and diabetic rats. (C) Effect of antibiotic treatment in normoglycemic and diabetic rats. Each value represents the mean of 5 to 7 rats ± the SEM. *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic or normal rats in different groups (P < 0.05).
TNF-α was also measured in the gingival connective tissue. It significantly increased in diabetic rats 5 weeks after A. actinomycetemcomitans infection (P < 0.05) (Fig. 5A) and was substantially higher than values found in infected normoglycemic rats (P < 0.05) (Fig. 5B). When rats were treated with antibiotic, there were no differences in TNF-α levels in the connective tissues of diabetic and normal rats (P > 0.05) (Fig. 5C).
Fig 5.

TNF-α expression is increased in gingival connective tissues of diabetic rats following A. actinomycetemcomitans infection. (A) TNF-α expression in gingival connective tissues of diabetic rats. (B) TNF-α expression in noninfected (baseline) and infected (4–6 weeks) gingival connective tissues in normoglycemic and diabetic rats. (C) Effect of antibiotic treatment in gingival connective tissues of normoglycemic and diabetic rats. Each value represents the mean of five to seven rats ± the SEM. *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic or normal rats in different groups (P < 0.05).
Induction of apoptosis.
Because apoptosis is thought to play an important role in periodontal disease progression, we sought to determine whether diabetic animals had significantly higher levels of apoptosis in the gingival epithelium (see Fig. S4 in the supplemental material) and whether the increase was mediated by caspase-3 in both. Prior to A. actinomycetemcomitans infection, the level of apoptosis was low in both diabetic and normoglycemic groups. The onset of A. actinomycetemcomitans infection significantly increased the level of apoptosis 2- to 3-fold in the normoglycemic rats and 12-fold in the diabetic rats, with the difference between the two groups being significant (P < 0.05) (Fig. 6A). The results were similar when presented as the percentage of gingival epithelial cells that were apoptotic or the number of apoptotic epithelial cells/μm2 (Fig. 6B). The principal leukocytic cell type infiltrating A. actinomycetemcomitans-infected gingiva was the granulocyte. Apoptosis was evaluated as the percentage of nonleukocytic TUNEL+ CD18− cells in the gingival epithelium. The number of TUNEL+ CD18− epithelial cells was significantly increased in both the normal and diabetic groups after A. actinomycetemcomitans infection (P < 0.05) (Fig. 6C). The percentage of apoptotic cells in diabetic animals was 2-fold greater than in normoglycemic animals (P < 0.05) (Fig. 6C). To assess the impact of inhibiting caspase-3/7, the specific caspase inhibitor DEVD was administered daily, starting at week 4, and the number of TUNEL+ CD18− cells was counted 1 week later. A. actinomycetemcomitans infection at this time point increased the apoptosis of epithelial cells by 2.6-fold compared to the baseline, but antibiotic treatment had no significant effect in reducing these levels (P > 0.05). Treatment with caspase inhibitor plus antibiotics reduced the number of apoptotic epithelial cells by reversing the impact of A. actinomycetemcomitans infection to baseline levels (P < 0.05) (Fig. 6D).
Fig 6.

Diabetes increases the apoptosis of epithelial cells of A. actinomycetemcomitans-infected rats in a caspase-3-dependent manner. Apoptotic cells were detected in gingival epithelium by TUNEL staining in the epithelia in the rats described in Fig. 1. In some groups, rats were treated with antibiotic or antibiotic plus capase-3 inhibitor starting at week 4. (A) Percentage of apoptotic gingival epithelial cells per total number of gingival epithelial cells; (B) total number of apoptotic gingival epithelial cells per area. (C and D) Nonleukocytic cells were identified as CD18 negative, and apoptotic cells were identified as TUNEL positive. (C) TUNEL+ CD18− cells per total number of CD18− cells. (D) Rats at week 4 were treated with antibiotic or antibiotic plus caspase-3 inhibitor and euthanized a week later. The TUNEL+ CD18− cells per total number of CD18− cells were counted. Each value is the mean of five to seven rats ± the SEM. *, significant difference between diabetic and normal rats (P < 0.05); +, significant difference between diabetic or normal rats in different groups (P < 0.05); ∧, significant difference between antibiotic and antibiotic plus caspase-3 inhibitor.
Apoptosis was also examined in gingival connective tissue. Both normal and diabetic rats showed an almost 3-fold increase in apoptotic cells after infection when examined as the percentage of positive cells or as the number of apoptotic cells per area (P < 0.05) (Fig. 7A and B). The total numbers of apoptotic cells in the gingival connective tissue of the diabetic group were >2-fold higher than for the normoglycemic rats (P < 0.05). The percentage of TUNEL+ CD18− cells in the gingival connective tissue was measured. After A. actinomycetemcomitans infection, the values increased 3.9-fold (P < 0.05) in diabetic animals but not in normal animals (Fig. 7C). At 5 weeks after A. actinomycetemcomitans infection, the percentage of TUNEL+ CD18− cells significantly increased in the diabetic group (P < 0.05) (Fig. 7D). Antibiotic treatment alone had no effect, but antibiotic treatment combined with caspase-3/7 inhibitor significantly blocked the increase in nongranulocytic cell apoptosis in the connective tissue (P < 0.05) (Fig. 7D).
Fig 7.

Diabetes increases apoptosis of cells in gingival connective tissue of A. actinomycetemcomitans-infected rats in a caspase-3-dependent manner. (A) Total apoptotic gingival connective tissue cells per total number of gingival connective tissue cells. (B) Total apoptotic gingival connective tissue cells per epithelial area. (C and D) Nonleukocytic cells were identified as CD18 negative, and apoptotic cells were identified as TUNEL positive. (C) TUNEL+ CD18− cells per total number of CD18− cells in connective tissue. (D) Rats at week 4 were treated with antibiotic or antibiotic and caspase-3 inhibitor and euthanized a week later. The TUNEL+ CD18− cells per total number of CD18− cells were counted. Each value is the mean of five to seven rats ± the SEM. *, significant difference between diabetics and normal rats (P < 0.05); +, significant difference between diabetics or normal rats in different groups (P < 0.05); ∧, significantly different between antibiotic and antibiotic plus caspase-3 inhibitor.
DISCUSSION
The results presented here demonstrate that A. actinomycetemcomitans infection significantly enhances PMN infiltration and TNF-α expression in both normal and diabetic rats. Moreover, each of these parameters was significantly greater in the diabetic animals, a finding which agrees with the increased bone loss observed in the diabetic group here as well as in other studies (17, 29, 30). Thus, diabetic rats exhibited greater inflammatory responses compared to the normoglycemic group in response to similar A. actinomycetemcomitans inocula.
Diabetes generally enhances inflammation by altering myeloid and lymphoid functions (12, 34). We found here that the local periodontal inflammatory response in diabetic animals was greater, as evidenced by an enhanced expression of TNF-α and a larger PMN infiltrate, and this finding is consistent with findings in other models (15, 33). These local findings were in agreement with the significant increase in the percentage of whole-blood granulocytes in diabetic rats postinfection. Elevated levels of antibody against A. actinomycetemcomitans were also found in diabetic rats postinfection compared to normal rats. The number of lymphocytes collected from whole blood, however, did not exhibit the same trend, showing a significant decrease compared to normoglycemic rats after A. actinomycetemcomitans infection. It is conceivable that this decrease in lymphocyte population could be a result of CDT-induced apoptosis. Alternatively, the decrease in lymphocyte population after A. actinomycetemcomitans infection may be due to a proportional increase in granulocytes.
A. actinomycetemcomitans infection has been shown to increase apoptosis in vitro but has not yet been tested in vivo in a periodontal model (20). We demonstrate here that inoculating animals with A. actinomycetemcomitans significantly stimulated apoptosis in both the gingival epithelium and connective tissue of rats, especially in diabetic animals. Other studies have also shown that apoptosis is significantly increased in diabetes when periodontal disease is induced in an animal model (14, 29). There are several mechanisms through which A. actinomycetemcomitans infection could enhance apoptosis in the rat. Our study indicates that the high rate of apoptosis in diabetic rats due to A. actinomycetemcomitans infection is largely blocked by a caspase-3/7 inhibitor. It is possible that A. actinomycetemcomitans through its cytolethal distending toxin (CDT) could stimulate apoptosis. CDT has been shown to induce apoptosis in epithelial cells, fibroblasts, and endothelial cells (19, 35). It has recently been shown that CDT induces apoptosis through a caspase-3-dependent pathway in immortalized gingival epithelial cells (1). However, the other apoptosis-inducing factor produced by A. actinomycetemcomitans, leukotoxin A, appears not to stimulate apoptosis in rat cells (20). Alternatively, A. actinomycetemcomitans could induce apoptosis through indirect mechanisms. Interestingly, diabetic rats had significantly higher TNF-α levels and more apoptotic cells compared to normal rats after A. actinomycetemcomitans infection. TNF-α has been shown to mediate both P. gingivalis- and lipopolysaccharide-induced apoptosis in vivo (2, 11). Thus, excessive production of TNF-α is another potential pathway through which diabetes could enhance the apoptosis of epithelial and connective tissue cells, thereby affecting the response to bacterial infection, and may occur simultaneously with CDT-induced apoptosis.
The impact of antibiotic treatment postinfection was also evaluated. Antibiotics have long been used as an adjunct therapy in the treatment of localized aggressive periodontitis (18, 41, 42). We also examined the impact of antibiotic treatment on A. actinomycetemcomitans antibody titer, alveolar bone resorption, PMN infiltration, TNF-α levels, and apoptosis in A. actinomycetemcomitans-infected periodontium. For A. actinomycetemcomitans antibody titer, PMN infiltration, TNF-α levels, and apoptosis, the diabetic rats showed a significant reduction with antibiotic treatment, whereas these parameters were not reduced by antibiotic treatment in normoglycemic rats. These results suggest that there are antibacterial deficits in diabetic animals that contribute to greater induction of proinflammatory events stimulated by periodontal pathogens at the local level that can be reversed by antibiotic treatment.
In summary, the impact of diabetes on the periodontium was investigated in a relatively new model of periodontitis, oral inoculation of A. actinomycetemcomitans in the rat, which has the advantage that the rat is a natural host of A. actinomycetemcomitans. In this model, diabetes affected A. actinomycetemcomitans-induced periodontal destruction by significantly increasing the inflammatory response, leading to increased bone loss and apoptosis of gingival epithelial and connective tissue cells. The excessive production of TNF-α and the impact of CDT could be potential mechanisms through which apoptosis was induced at higher levels in diabetic animals. Antibiotics were able to reverse many parameters of the local host response in diabetic animals compared to normoglycemic animals, suggesting that a component to the enhanced inflammatory response is due to a deficit in the capacity of diabetic animals to resist infection. This information provides valuable insight as to how diabetes may alter host-bacterium interactions in a way that promotes periodontal breakdown.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants DE018307 and DE017732 from the NIDCR. Beatriz de Brito Bezerra was the recipient of a fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brazil).
We thank Sunitha Batchu for helping with the preparation of the manuscript, the staff of the Comparative Medicine Facility of the New Jersey Medical School for their help with procedures involving the animal, and Zoltan Spolarics and Stephanie Federici of Department Surgery at New Jersey Medical School (UMDNJ) for their assistance with the hematology assays.
Footnotes
Published ahead of print 26 March 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Alaoui-El-Azher M, et al. 2010. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 5:e11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alikhani M, et al. 2003. Lipopolysaccharides indirectly stimulate apoptosis and global induction of apoptotic genes in fibroblasts. J. Biol. Chem. 278:52901–52908 [DOI] [PubMed] [Google Scholar]
- 3. Armitage GC, Cullinan MP. 2010. Comparison of the clinical features of chronic and aggressive periodontitis. Periodontol. 2000 53:12–27 [DOI] [PubMed] [Google Scholar]
- 4. Boch JA, Wara-aswapati N, Auron PE. 2001. Interleukin 1 signal transduction: current concepts and relevance to periodontitis. J. Dent. Res. 80:400–407 [DOI] [PubMed] [Google Scholar]
- 5. Christersson LA. 1993. Actinobacillus actinomycetemcomitans and localized juvenile periodontitis: clinical, microbiologic, and histologic studies. Swed. Dent. J. Suppl. 90:1–46 [PubMed] [Google Scholar]
- 6. Dickinson BC, et al. 2011. Interaction of oral bacteria with gingival epithelial cell multilayers. Mol. Oral Microbiol. 26:210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fine DH, Furgang D. 2002. Lactoferrin iron levels affect attachment of Actinobacillus actinomycetemcomitans to buccal epithelial cells. J. Periodontol. 73:616–623 [DOI] [PubMed] [Google Scholar]
- 8. Fine DH, et al. 2001. Colonization and persistence of rough and smooth colony variants of Actinobacillus actinomycetemcomitans in the mouths of rats. Arch. Oral Biol. 46:1065–1078 [DOI] [PubMed] [Google Scholar]
- 9. Fine DH, et al. 2007. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J. Clin. Microbiol. 45:3859–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goncharoff P, Figurski DH, Stevens RH, Fine DH. 1993. Identification of Actinobacillus actinomycetemcomitans: polymerase chain reaction amplification of lktA-specific sequences. Oral Microbiol. Immunol. 8:105–110 [DOI] [PubMed] [Google Scholar]
- 11. Graves D, et al. 2001. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J. Dent. Res. 80:1875–1879 [DOI] [PubMed] [Google Scholar]
- 12. Graves DT, Kayal RA. 2008. Diabetic complications and dysregulated innate immunity. Front. Biosci. 13:1227–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Graves DT, Li J, Cochran DL. 2011. Inflammation and uncoupling as mechanisms of periodontal bone loss. J. Dent. Res. 90:143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graves DT, Liu R, Oates TW. 2007. Diabetes-enhanced inflammation and apoptosis: impact on periodontal pathosis. Periodontol. 2000 45:128–137 [DOI] [PubMed] [Google Scholar]
- 15. Graves DT, et al. 2005. Inflammation is more persistent in type 1 diabetic mice. J. Dent. Res. 84:324–328 [DOI] [PubMed] [Google Scholar]
- 16. Haubek D, et al. 2008. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet 371:237–242 [DOI] [PubMed] [Google Scholar]
- 17. He H, et al. 2004. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology 145:447–452 [DOI] [PubMed] [Google Scholar]
- 18. Heller D, et al. 2011. Impact of systemic antimicrobials combined with anti-infective mechanical debridement on the microbiota of generalized aggressive periodontitis: a 6-month RCT. J. Clin. Periodontol. 38:355–364 [DOI] [PubMed] [Google Scholar]
- 19. Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. 2011. Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157:1851–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kachlany SC. 2010. Aggregatibacter actinomycetemcomitans leukotoxin: from threat to therapy. J. Dent. Res. 89:561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kasai H, Yamamoto K, Koseki T, Yokota M, Nishihara T. 2004. Involvement of caspase activation through release of cytochrome c from mitochondria in apoptotic cell death of macrophages infected with Actinobacillus actinomycetemcomitans. FEMS Microbiol. Lett. 233:29–35 [DOI] [PubMed] [Google Scholar]
- 22. Kato S, et al. 1995. Evidence for apoptosis of murine macrophages by Actinobacillus actinomycetemcomitans infection. Infect. Immun. 63:3914–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kato S, Sugimura N, Nakashima K, Nishihara T, Kowashi Y. 2005. Actinobacillus actinomycetemcomitans induces apoptosis in human monocytic THP-1 cells. J. Med. Microbiol. 54:293–298 [DOI] [PubMed] [Google Scholar]
- 24. Kirkwood KL, Cirelli JA, Rogers JE, Giannobile WV. 2007. Novel host response therapeutic approaches to treat periodontal diseases. Periodontol. 2000 43:294–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lalla E, et al. 2000. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Invest. 105:1117–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lalla E, Papapanou PN. 2011. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat. Rev. Endocrinol. 7:738–748 [DOI] [PubMed] [Google Scholar]
- 27. Lamster IB, Lalla E, Borgnakke WS, Taylor GW. 2008. The relationship between oral health and diabetes mellitus. J. Am. Dent. Assoc. 139(Suppl):19S–24S [DOI] [PubMed] [Google Scholar]
- 28. Li Y, et al. 2010. Adaptive immune response in osteoclastic bone resorption induced by orally administered Aggregatibacter actinomycetemcomitans in a rat model of periodontal disease. Mol. Oral Microbiol. 25:275–292 [DOI] [PubMed] [Google Scholar]
- 29. Liu R, et al. 2006. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J. Dent. Res. 85:510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mahamed DA, et al. 2005. G(−) anaerobes-reactive CD4+ T cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes 54:1477–1486 [DOI] [PubMed] [Google Scholar]
- 31. Mandell RL, Socransky SS. 1988. Microbiological and clinical effects of surgery plus doxycycline on juvenile periodontitis. J. Periodontol. 59:373–379 [DOI] [PubMed] [Google Scholar]
- 32. Mogi M, et al. 1999. Interleukin 1 beta, interleukin 6, beta 2-microglobulin, and transforming growth factor-alpha in gingival crevicular fluid from human periodontal disease. Arch. Oral Biol. 44:535–539 [DOI] [PubMed] [Google Scholar]
- 33. Naguib G, Al-Mashat H, Desta T, Graves D. 2004. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J. Invest. Dermatol. 123:87–92 [DOI] [PubMed] [Google Scholar]
- 34. Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R. 2011. State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun. 12:239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ohara M, Miyauchi M, Tsuruda K, Takata T, Sugai M. 2011. Topical application of Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces cell cycle arrest in the rat gingival epithelium in vivo. J. Periodontal Res. 46:389–395 [DOI] [PubMed] [Google Scholar]
- 36. Pihlstrom BL, Michalowicz BS, Johnson NW. 2005. Periodontal diseases. Lancet 366:1809–1820 [DOI] [PubMed] [Google Scholar]
- 37. Salvi G, et al. 1997. Monocytic TNFalpha secretion patterns in IDDM patients with periodontal diseases. J. Clin. Periodontol. 24:8–16 [DOI] [PubMed] [Google Scholar]
- 38. Schreiner H, et al. 2011. Aggregatibacter actinomycetemcomitans-induced bone loss and antibody response in three rat strains. J. Periodontol. 82:142–150 [DOI] [PubMed] [Google Scholar]
- 39. Schreiner HC, et al. 2003. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc. Natl. Acad. Sci. U. S. A. 100:7295–7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taylor G, et al. 1998. Non-insulin-dependent diabetes mellitus and alveolar bone loss progression over 2 years. J. Periodontol. 69:76–83 [DOI] [PubMed] [Google Scholar]
- 41. Xajigeorgiou C, Sakellari D, Slini T, Baka A, Konstantinidis A. 2006. Clinical and microbiological effects of different antimicrobials on generalized aggressive periodontitis. J. Clin. Periodontol. 33:254–264 [DOI] [PubMed] [Google Scholar]
- 42. Yek EC, et al. 2010. Efficacy of amoxicillin and metronidazole combination for the management of generalized aggressive periodontitis. J. Periodontol. 81:964–974 [DOI] [PubMed] [Google Scholar]
- 43. Zappa U, Reinking-Zappa M, Graf H, Chase D. 1992. Cell populations associated with active probing attachment loss. J. Periodontol. 63:748–752 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.