

Abstract
Bordetella pertussis and Bordetella bronchiseptica establish respiratory infections with notorious efficiency. Our previous studies showed that the fhaB genes of B. pertussis and B. bronchiseptica, which encode filamentous hemagglutinin (FHA), are functionally interchangeable and provided evidence that FHA-deficient B. bronchiseptica induces more inflammation in the lungs of mice than wild-type B. bronchiseptica. We show here that the robust inflammatory response to FHA-deficient B. bronchiseptica is characterized by the early and sustained influx of interleukin-17 (IL-17)-positive neutrophils and macrophages and, at 72 h postinoculation, IL-17-positive CD4+ T cells, suggesting that FHA allows the bacteria to suppress the development of an IL-17-mediated inflammatory response. We also show that the cyaA genes of B. pertussis and B. bronchiseptica, which encode adenylate cyclase toxin (ACT), are functionally interchangeable and that ACT, specifically its catalytic activity, is required for B. bronchiseptica to resist phagocytic clearance but is neither required for nor inhibitory of the induction of inflammation if bacteria are present in numbers sufficient to persist during the first 3 days postinoculation. Incubation of bone marrow-derived macrophages with a ΔcyaA strain caused decreased production of IL-1β and increased production of tumor necrosis factor alpha (TNF-α) and IL-12, while incubation with a ΔcyaA ΔfhaB strain caused increased production of IL-23. These data suggest that FHA and ACT both contribute to suppress the recruitment of neutrophils and the development of an IL-17-mediated immune response. To our knowledge, this is the first demonstration of a microbial pathogen suppressing IL-17-mediated inflammation in vivo as a strategy to evade innate immunity.
INTRODUCTION
Bordetella bronchiseptica is a Gram-negative bacterium that infects a broad range of mammalian species, typically colonizing the nasopharynx and trachea of its hosts chronically and asymptomatically (9, 10, 19, 26). Bordetella pertussis, which diverged from a B. bronchiseptica-like ancestor approximately 3 million years ago, infects only humans and causes whooping cough (or pertussis), an acute respiratory disease that is especially severe in young children and infants (72). Both of these organisms are capable of establishing respiratory infections in a highly efficient manner (48), suggesting that they possess specific strategies for resisting or overcoming innate immune clearance mechanisms operative in the respiratory tract. Molecular mechanisms underlying these strategies are not well understood.
Despite differences in host range and disease-causing propensity, B. pertussis and B. bronchiseptica produce a similar set of virulence factors that include filamentous hemagglutinin (FHABp and FHABb, respectively) and adenylate cyclase toxin (ACT) (55). ACT is a member of the RTX (repeat in toxin) family of bacterial pore-forming toxins. It is a unique fusion of a cytolysin with an N-terminal adenylate cyclase (AC) enzyme (67). ACT is first synthesized as a protein called CyaA (encoded by the cyaA gene) and then acylated by the product of the cyaC gene to become the active toxin. ACT can bind to the β2 integrin of Mac-1 (CD11b/CD18) expressed on myeloid cells, such as macrophages, neutrophils, dendritic cells, and natural killer cells (7, 21, 28, 29). Upon entry into the cell, ACT is activated by binding to eukaryotic calmodulin (71) and catalyzes unregulated conversion of ATP to cyclic AMP (cAMP). By elevating cAMP levels, ACT dysregulates cellular signaling pathways and suppresses superoxide production (oxidative burst), chemotaxis, and phagocytosis in macrophages and neutrophils (8, 15). ACT-deficient B. pertussis and B. bronchiseptica are cleared from the lungs of mice faster than wild-type bacteria (11, 32), presumably because they are unable to inhibit the bactericidal activities of phagocytic cells. Recent studies, however, suggest that ACT may also contribute to infection by affecting secretion of cytokines and chemokines in epithelial cells and macrophages. For example, purified ACT can inhibit production of interleukin-12 (IL-12) and tumor necrosis factor alpha (TNF-α) and augment production of IL-6 and IL-10 in lipopolysaccharide (LPS)-activated human monocyte-derived dendritic cells (MDDC) and macrophages (5, 13, 33, 60, 67), suggesting that ACT functions as a down-regulator of inflammation. Perkins et al., however, showed that purified ACT synergizes with LPS to induce cyclooxygenase 2 (COX-2) in epithelial cells and murine macrophages, suggesting that ACT may potentiate the inflammatory response to B. pertussis infection (57). Consistent with a proinflammatory role, Dunne et al. recently reported that ACT promotes robust IL-1β production by dendritic cells via activation of the NALP3 inflammasome complex and consequently polarizes the T cell response toward the Th17 subtype (20), which would cause enhanced neutrophil infiltration and activity (41).
FHA, encoded by the fhaB gene, is a large, rod-shaped, immunogenic protein that is both surface associated and secreted into the extracellular milieu (39, 51). FHA is a prototypic member of the two partner secretion (TPS) pathway. It is synthesized as an ∼370-kDa preproprotein (FhaB) with a 71-amino-acid (aa) signal sequence and a C-terminal ∼130-kDa prodomain that is removed at some point during translocation across the outer membrane, resulting in the mature ∼240-kDa FHA protein (50). FHA is required for colonization of the lower respiratory tract by B. bronchiseptica and mediates bacterial attachment to a variety of cell lines in vitro (17, 38, 43, 64, 66, 69). Recent studies using a mouse model suggest that FHA plays an important role in modulating the host innate immune response early during infection (36, 40). In vitro studies with B. pertussis or FHA purified from B. pertussis have also suggested an immunomodulatory role for FHA. For example, prolonged exposure of U-937 macrophages and human monocytes to FHA resulted in the cytosolic accumulation of IκBα and the inability of TNF-α to activate NF-κB (1), and Mills and colleagues showed that purified FHA could mitigate IL-12 secretion by a macrophage cell line, ex vivo alveolar macrophages, and bone marrow-derived dendritic cells and could induce a subset of IL-10-secreting regulatory T cells in vitro (52, 53).
Several studies have suggested that ACT function may depend, at least in part, on FHA. Weiss and Falkow demonstrated that more ACT is released from FHA-deficient B. pertussis strains than from wild-type B. pertussis (68). Zaretzky et al. showed that ACT can interact with FHA and that the presence of ACT on the bacterial surface correlates with the presence of FHA (74). It was also demonstrated that the bacteria-associated form of ACT generated greater levels of cAMP than toxin isolated from the medium of FHA mutant strains (74), and the presence of ACT in the outer membrane has been proposed to play a role in the adherence function of FHA (56). These data have led to the hypothesis that FHA acts as a delivery mechanism for ACT, increasing local concentrations for more robust intoxication of target cells (74). Subsequent work revealed that although a direct physical interaction between FHA and ACT was not required, toxin delivery was enhanced by close association of the bacteria with target cells (27).
The goal of this study was to investigate the contribution of FHA and ACT to the early inflammatory response to Bordetella infection. A majority of the studies investigating the involvement of these virulence factors in influencing the host response have been conducted using B. pertussis and/or purified B. pertussis proteins in in vitro assays. B. bronchiseptica's broad host range, however, allows the roles of various Bordetella virulence factors to be studied in vivo in the context of a natural bacteria-host interaction using laboratory animals such as rats and mice. We demonstrated previously that FHA from B. pertussis (FHABp) can substitute for FHA from B. bronchiseptica (FHABb) in vitro and in vivo (40). In this study, we set out to determine if the ACT proteins of B. pertussis and B. bronchiseptica are also functionally interchangeable and if ACT and FHA cooperate to control chemokine and cytokine production in the lower respiratory tract as a strategy to facilitate persistence.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Our protocol was approved by the University of North Carolina (UNC) IACUC (09-326 and 10-134) and the University of California, Santa Barbara (UCSB), IACUC (6-04-601). All animals were properly anesthetized for inoculations, monitored regularly, and euthanized when moribund, and efforts were made to minimize suffering.
Growth media and bacterial strains.
Wild-type B. bronchiseptica RB50 and mutant derivatives were grown at 37°C on Bordet-Gengou (BG) agar (Becton, Dickinson Microbiology Systems) supplemented with 7.5% defibrinated sheep blood (Colorado Serum Co., Denver, CO) or in Stainer-Scholte (SS) broth supplemented with 100 mg/ml (2,6-O-dimethyl)-β-cyclodextrin (63). Escherichia coli strains were cultured in LB agar or broth. Where appropriate, medium was supplemented with gentamicin (Gm; 30 μg/ml), streptomycin (Sm; 20 μg/ml), diaminopimelic acid (DAP; 20 μg/ml), or sucrose (8.3%). For E. coli, 100 μg/ml ampicillin (Ap) was used for plasmid selection.
All strains and relevant plasmids used in this study are listed in Table S1 in the supplemental material. Strains containing unmarked, in-frame chromosomal deletions or mutations in the cyaA and fhaB genes were constructed using allelic exchange methods described previously (2, 37). All strains are derivatives of B. bronchiseptica strain RB50, a wild-type isolate recovered from the nares of a naturally infected 3-month-old New Zealand White rabbit (16). The ΔcyaA strain, RB515, was generated using allelic exchange plasmid pCI56, which contains an in-frame deletion of codons 5 to 1701 of B. bronchiseptica cyaA (cyaABb). The ΔfhaB strain, RBX9, was described previously (17). Strain RB509 produces a catalytically inactive form of CyaA and was generated using plasmid pCI54, which contains two mutations (encoding H63A and K65A substitutions) in the AC domain-encoding region of cyaABb. To create a B. bronchiseptica strain expressing cyaA from B. pertussis, the entire cyaA gene and promoter from B. pertussis Tohama I was cloned into a pBR322 derivative (pEG7) that can be used as a suicide vector in Bordetella. The resulting plasmid, designated pDB05, was introduced into RB515, the B. bronchiseptica strain containing a deletion of the cyaA gene. Strain RBX11Δ28 produces an FhaB protein containing which contains a 28-aa deletion near the C terminus of mature FHA as described previously (40). All strains were confirmed to be constructed as intended by PCR and DNA sequence analysis.
Immunoblotting.
To evaluate production of ACT and FHA, proteins were prepared from B. bronchiseptica cultures grown overnight in SS broth to stationary phase. For whole-cell lysates, protein was extracted from a 1-ml culture by boiling in phosphate-buffered saline (PBS)–2× sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer with dithiothreitol (DTT). Proteins present in filtered culture supernatants were precipitated with trichloroacetic acid and resuspended in 10 mM Tris (pH 8.0)–2× SDS-PAGE sample buffer with DTT. Proteins were separated using SDS-PAGE (5% polyacrylamide), transferred to nitrocellulose, and probed with a mouse monoclonal antibody that was generated against CyaA (3D1; Santa Cruz Biotechnology) and a rabbit polyclonal antibody that was generated against a polypeptide corresponding to the mature C-terminal domain (MCD) of B. bronchiseptica FHA (40). Goat anti-mouse secondary antibody conjugated to IRdye 680 and goat anti-rabbit secondary antibody conjugated to IRdye 800 (Molecular Probes) were used to detect antigen-antibody complexes. The anti-CyaA antibody was used at a dilution of 1:5,000, the anti-FHA antibody was used at a dilution of 1:1,000, the anti-mouse secondary antibody was used at a dilution of 1:15,000, and the anti-rabbit secondary antibody was diluted 1:25,000. Antigen-antibody complexes were visualized using an Odyssey infrared imaging system (LiCor Biosciences).
Culture of J774A.1 cells.
J774A.1 cells (murine macrophage cell line) were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose (Gibco) plus 10% heat-inactivated fetal bovine serum (Gibco) in 5% CO2 at 37°C.
Intoxication of J774 cells.
J774 cells were grown overnight in 96-well tissue culture plates at 40,000 cells/well. Bacterial strains were grown overnight in liquid broth (SS), diluted to an optical density at 650 nm (OD650) of 0.1, and grown to logarithmic phase. Cultures were centrifuged and separated into supernatant and pellet fractions, and the bacterial pellets were resuspended to their original volume in medium. An equal volume (equivalent to ∼107 to 108 bacteria) of each fraction was added to the J774 cells and incubated for 30 min at 37°C. Cells were washed three times with Hank's balanced salt solution (HBSS) and lysed for 30 min at room temperature, and intracellular cAMP was measured with a Tropix chemiluminescent enzyme-linked immunosorbent assay (ELISA) system (Applied Biosystems). Intoxication is reported as a function of total J774 cell protein.
Intranasal inoculation and cytokine and histological analysis. (i) Mouse experiments performed at UCSB.
Three- to four-week-old BALB/c mice (Charles River Laboratories, Wilmington, MA) were inoculated intranasally with 5 × 105 CFU of B. bronchiseptica in 50 μl of PBS. The lungs were harvested at 3 h, 3 days, and 11 days postinoculation (p.i.) (see Fig. 4B), and the numbers of CFU from right lungs were determined by plating lung tissue homogenates on BG agar as described previously. These experiments were performed as described in the animal use protocols that have been approved by the UCSB IACUC (6-04-601).
Fig 4.
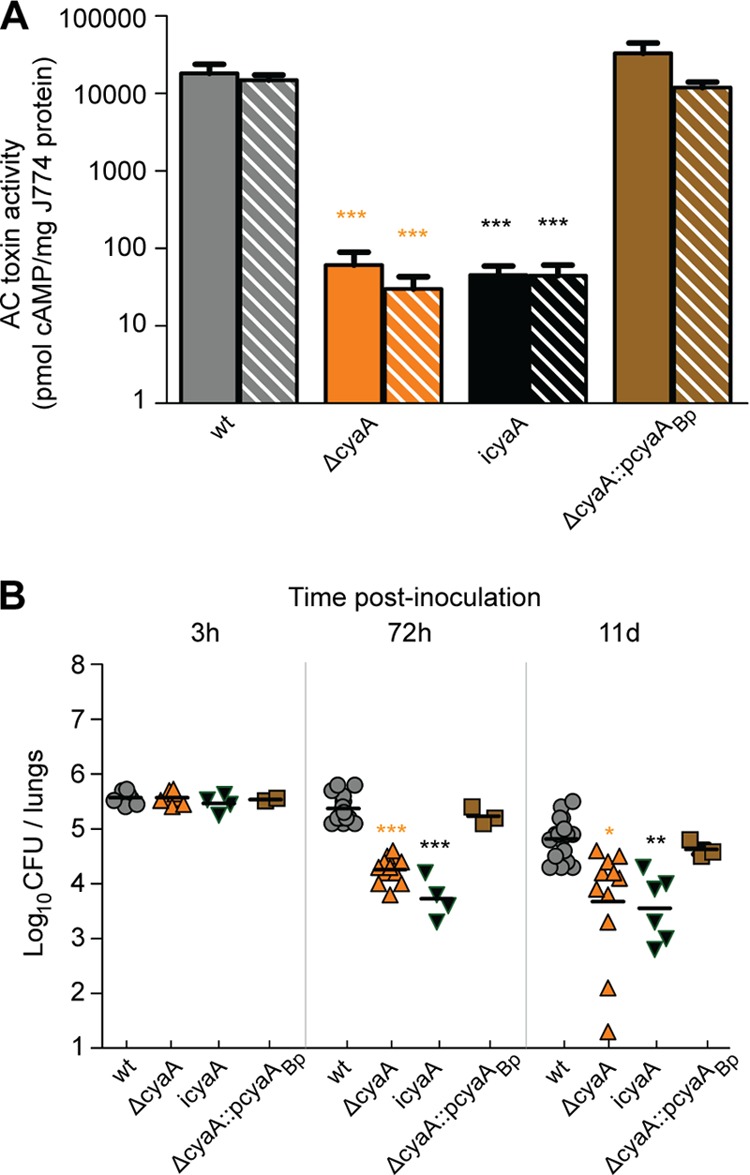
Functional interchangeability of ACT. (A) Toxin activity of ACT was measured by the production of cAMP. Bars represent pmol of cAMP per mg of protein of J774.1 macrophages, with solid bars representing supernatant-incubated cells and striped bars representing cells incubated with resuspended bacterial pellets. Asterisks indicate a statistically significant difference from wild-type bacteria and the complemented strain RB515::pDB505 (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). icyaA, catalytically inactive cyaA gene. (B) BALB/c mice were inoculated intranasally with 50 μl of PBS containing 5 × 105 CFU of B. bronchiseptica strains as indicated, and the numbers of CFU in the right lungs were determined at 3 h, 72 h, and 11 days p.i. Each point represents the log CFU recovered from a single animal. Horizontal black lines show the mean for each group. Asterisks indicate a statistically significant difference from wild-type bacteria (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
(ii) Mouse experiments performed at UNC.
Four-week-old BALB/c mice from Jackson Laboratories (Bar Harbor, ME) were inoculated intranasally with 1 × 105 or 5.5 × 105 CFU of B. bronchiseptica in 50 μl of PBS (see Fig. 1, 2, 3, and 5). Mice were infected with strains RB50, RBX9, RB515, RB516, and RBX11Δ28 (or PBS as a negative control). Where indicated in the figure legends, mice were inoculated with high doses (106 or 107 CFU) of strain RB516 (see Fig. 6). Lungs were harvested from infected mice at 3 h, 12 h, 24 h, 48 h, 72 h, or 11 days p.i. Right lungs were homogenized in 1 ml of PBS containing a protease inhibitor cocktail (Roche). Serial dilutions of homogenized lung tissue were plated on BG agar to assess bacterial burden. At the times indicated on the figures, the cytokine response to infection was evaluated using a Milliplex bead-based immunoassay (Millipore) run on a Luminex detector. Undiluted right lung homogenates were centrifuged at 3,800 × g for 10 min, and the supernatants were passed through a 0.22-μm-pore-size filter. Filtrates were analyzed as per the manufacturer's instructions for using serum/plasma. Cytokines/chemokines of interest were selected from mouse panel I (Millipore) and included the following: TNF-α, IL-1β, IL-6, IL-17, keratinocyte-derived chemokine (KC), macrophage inflammatory protein 1α (MIP-1α), MIP-2, IL-12p70, and monocyte chemotactic protein 1 (MCP-1). Cytokine concentrations were calculated using standard curve data for each analyte (range of detection was 3.2 to 25,000 pg/ml). To evaluate gross pulmonary inflammation, left lung lobes harvested from mice at 72 h p.i. were inflated with 10% formalin, embedded in paraffin, sectioned at 5 μm, stained with hematoxylin and eosin (H&E), and examined by microscopy. Immunohistochemistry was performed by following instructions for deparaffinization and epitope recovery according to Abcam specifications. Antibody for primary binding to Ly6G was purchased from Abcam, and Alexa Fluor 568-conjugated secondary antibody (Invitrogen) was used for detection on an Olympus FV1000. Animals that became moribund were immediately euthanized. The data shown in Fig. 2A and 5A are from two independent experiments, with all strains being used in both experiments. The data are shown in separate figures for clarification of the response to mutant strains. The results of the wild-type strain, however, are shown in both figures for reference. These experiments were performed as described in the animal use protocols that have been approved by the UNC IACUC (protocols. 09-326 and 10-134).
Fig 1.
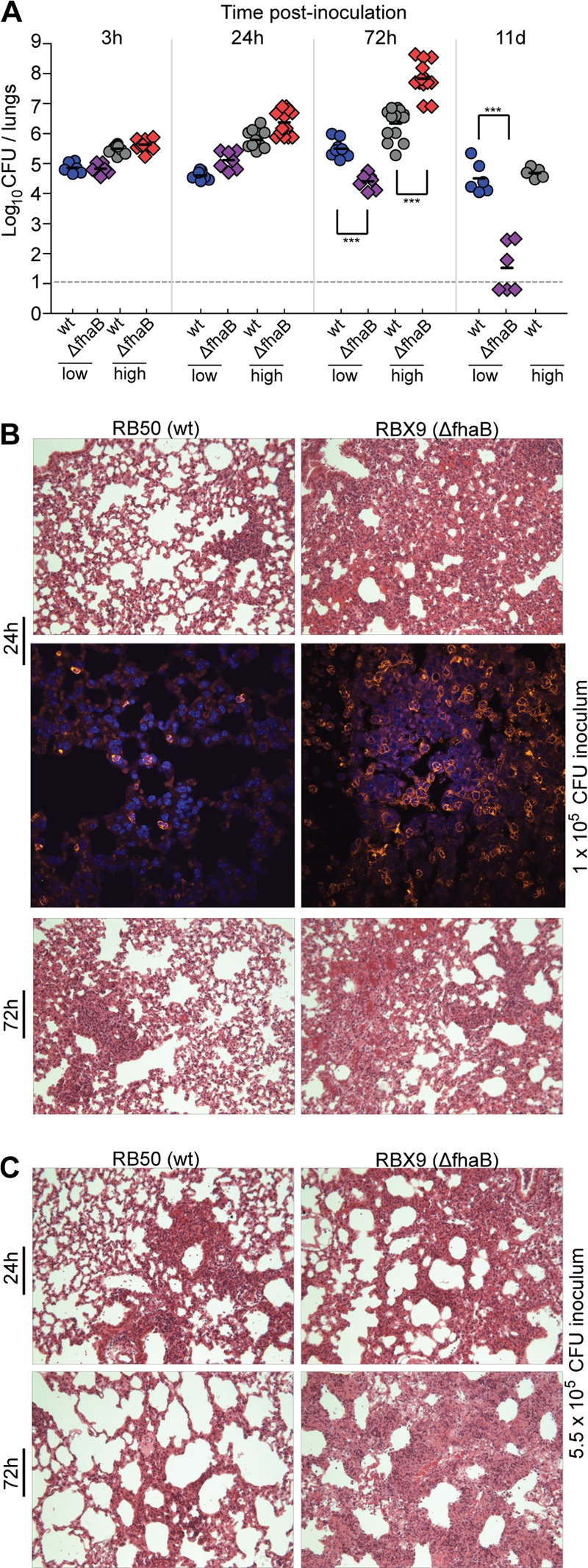
Bacterial burden and lung pathology in mice inoculated with low and high numbers of CFU of wild-type and ΔfhaB B. bronchiseptica bacteria. (A) Bacterial burden in mice at 3 h, 24 h, 72 h, and 11 days postinoculation following delivery of 50 μl of PBS containing doses of 1 × 105 (low) or 5.5 × 105 (high) CFU of B. bronchiseptica strains as indicated. Each symbol represents an individual animal. Horizontal black lines show the mean for each group. The horizontal dashed line represents the lower limit of detection. Asterisks indicate a statistically significant difference where compared (*, P < 0.05; **, P < 0.001; ***, P < 0.0001). The data are from two independent experiments. (B) Inflammation of mouse lung tissue at 24 and 72 h p.i.; animals were infected with 1 × 105 CFU. H&E-stained 5-μm lung sections were examined by light microscopy for peribronchiolar and perialveolar infiltrate and by assessing hypertrophy of bronchiolar epithelial cells. Representative sections were viewed at a magnification of ×40. Neutrophil infiltration at 24 h p.i. from animals infected with 1 × 105 CFU of wild-type or FHA-deficient bacteria at a magnification of ×60. Antibody against Ly6G and 4′,6′-diamidino-2-phenylindole nuclear stain were used to identify cells. (C) H&E-stained sections from animals infected with 5.5 × 105 CFU of wild-type (RB50) or ΔfhaB (RBX9) B. bronchiseptica bacteria at 24 and 72 h p.i. at a magnification of ×40. d, days; wt, wild type.
Fig 2.

Impact of FHA on the early chemokine/cytokine response to infection. (A) Bacterial burden in mouse lungs at indicated times p.i., in hours. Each point represents the number of CFU recovered from the right lung of a single animal. Horizontal black lines show the mean for each group. (B) Chemokine and cytokine production after infection with wild-type strain RB50 (gray circles) and the ΔfhaB strain RBX9 (red diamonds) or PBS (control; open diamonds with a center dot). Levels of TNF-α, IL-1β, IL-6, IL-17, KC, MIP-1α, MIP-2, IFN-γ, and MCP-1 (pg/ml) present in mouse lungs at 3, 12, 24, 48, and 72 h postinoculation of 5.5 × 105 CFU of B. bronchiseptica strains were measured with a Multiplex immunoassay. The lower limit of detection for all samples was 3.2 pg/ml. Significant differences between levels for animals infected with wild-type bacteria and PBS-inoculated controls are indicated with daggers (†, P < 0.05; ††, P < 0.001; †††, P < 0.0001), and significant differences between animals infected with the ΔfhaB strain and wild-type bacteria are indicated with asterisks (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
Fig 3.

Cellular environment and IL-17 production. (A) Bacterial burden in mouse lungs through 3 days of infection. Animals were infected with 1 × 105 CFU of the B. bronchiseptica strain as indicated, and burden was assessed via multiple dilutions and enumeration of bacteria. Δ28, strain RBX11Δ28. (B) Histological sections (H&E stained) taken from the left lungs at 72 h p.i. at a magnification of ×40. (C) Graphs represent percentages of identified cell types; inner, lighter-shaded bars represent the portions of those cells staining positive for IL-17. Leukocyte subsets were identified as follows, with indicated levels of expression: neutrophils, CD11b mid and Ly6G/C high; alveolar macrophages, CD11b mid-low, CD11c mid-low, and F4/80 low; monocytes/macrophages, CD11b high, F4/80 mid-high, and CD11c negative; gamma delta (γδ) T cells, γδ TCR positive; CD4 T cells, CD3 and CD4 positive; CD8 T cells, CD3 and CD8 positive; dendritic cells, CD11b low-negative, CD11c high, and CD 8 positive/negative. All cells were first gated as being of appropriate forward and side scatter dimensions, CD45 positive, and negative for the succinimidyl ester stain. Percentages are based on 500,000 events gated in the forward and side scatter gates. Unidentified cells are those that expressed any of the markers used for detection other than CD45 or CD45 marker alone. Statistics are calculated as significant variation from wild type (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
Fig 5.

Impact of ACT on the early chemokine/cytokine response to infection. (A) Bacterial burden in mouse lungs at labeled times p.i., in hours. Each point represents the number of CFU recovered from the right lung of a single animal. Horizontal black lines show the mean for each group. (B) Histological sections (H&E stained) taken from the left lungs at 72 h p.i. at magnifications of ×2 (inset) and ×40. Photographs are representative samples of each population. (C) Chemokine and cytokine production after infection with wild-type strain RB50 and the ΔcyaA strain RB515, ΔfhaB ΔcyaA strain RB516, or PBS (control). Levels of TNF-α, IL-1β, IL-6, IL-17, KC, MIP-1α, MIP-2, and MCP-1 (pg/ml) present in mouse lungs at 3, 12, 24, 48, and 72 h postinoculation of 5.5 × 105 CFU of B. bronchiseptica strains were measured with a Multiplex immunoassay. The lower limit of detection for all samples was 3.2 pg/ml. Statistics are calculated as significant variation from wild type and where black bars indicate (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
Fig 6.

Murine cytokine/chemokine production in response to infection with high-dose ACT- and FHA-deficient bacteria. BALB/c mice were intranasally inoculated with 50 μl of PBS containing 5.5 × 105, 1 × 106, or 1 × 107 CFU of the ΔcyaA ΔfhaB strain (RB516) and 5.5 × 105 CFU from the ΔfhaB strain (RBX9). (A) Bacterial burden. Each point represents the number (log10) of CFU recovered from the right lungs of a single animal, and the data are from two independent experiments. (B) Levels of TNF-α, IL-1β, IL-6, IL-17, KC, MIP-1α, MIP-2, and MCP-1 were measured at 3 h, 48 h, and 72 h p.i. in lung homogenates of mice inoculated with 5.5 × 105 CFU of the ΔfhaB strain or the ΔfhaB ΔcyaA strain or 1 × 107 of the ΔfhaB ΔcyaA strain. Cytokines were assayed using Luminex technology and standardized to a range of 3.2 to 22,000 pg/ml, depending on the analyte assayed. Dashed gray lines represent the upper limit of detection. The cytokine profile of animals infected with the intermediate dose of 1 × 106 is not shown. Statistics are calculated as significant variation from the ΔfhaB strain (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
Lung digestion and intracellular cytokine staining.
Animals were deeply (sublethal) anesthetized via intraperitoneal injection of 340 mg/kg Avertin and subsequently euthanized by severing the renal artery and performing a thoracotomy. Lungs were transcardially perfused with PBS. The trachea was ligated using silk suture, and lungs were infused with 1 ml of dispase II (Roche, Indianapolis, IN) via tracheal injection and allowed to incubate. Lungs were excised, and tissue was macerated in PBS-DNase (Sigma-Aldrich) and passed through a 70-μm-pore-size strainer. Cells were stimulated for 1 h with phorbol myristate acetate (PMA)-ionomycin (Sigma), then incubated with brefeldin A (Sigma) for 5 h in RPMI medium (Gibco). Hybridoma 2.4G2 supernatant was added to block Fc receptors for 1 h, and then Pacific orange succinimidyl ester (Invitrogen) was used at a concentration for discerning live and dead cells. Cells were next surface stained for CD11b, CD11c, F4/80, Ly6G/C, CD8, CD4, CD3, CD49b, CD45, and γδ T cell receptor (TCR) (BD Pharmingen, eBioscience). Cells were permeabilized, fixed, and then stained with anti-IL-17A, according to the manufacturer's instructions (BD Pharmingen). Analysis was performed on a Beckman Coulter CyAn Flow cytometer (Brea, CA) using Summit, version 4.3, software (Beckman Coulter).
Bone marrow-derived macrophages.
Bone marrow-derived macrophages were prepared by culturing cells harvested from the femur and tibia of C57BL/6 mice in DMEM (Sigma) supplemented with 10% fetal bovine serum (FBS; PAA). L929 cell supernatants (DMEM) were used as a source of growth factors and were added to the medium at 25% of the total volume. Macrophages were cultured for 7 days, with fresh medium being added every 3 days; adherent cells were infected, harvested, and analyzed by flow cytometry to verify purity prior to immunoassay.
In vitro cytokine immunoassay.
Bone marrow-derived macrophages were seeded at 1 × 106 cells/ml into a 24-well tissue culture treated dish (BD Biosciences) on day 6 of culture and allowed to adhere overnight. Prior to bacterial infection, medium was harvested, and 200 μl of fresh medium was added. B. bronchiseptica strains were added at a multiplicity of infection (MOI) of 10 in 100 μl of PBS and were cultured for 2 h. Bacterial supernatants were then aspirated, and cells were washed once with PBS, followed by the addition of 1 ml of fresh medium supplemented with 30 μg/ml gentamicin sulfate (Fisher Scientific). Supernatants were harvested and analyzed via ELISA for TNF-α, IL-1β, IL-12p70, IL-6, IL-10 (BD Biosciences), IL-23(p19/p40), and TGF-β1 (R&D Systems) according to the manufacturer's instructions. The lowest level of detection was calibrated to 15.1 pg/ml. Absorbance was determined using a Molecular Devices plate reader and analyzed by Softmax Pro software (Molecular Devices). After removal of supernatants for the ELISA, the macrophages were washed twice with PBS containing 2.5% FBS and then incubated with antibodies for CD11b, CD80, CD86, F4/80, CD11c (eBioscience), and major histocompatibility complex (MHC) class II I-Aβ (BD Pharmingen) for 1 h. Cells were washed twice and analyzed on a CyAN using Summit, version 4.3, software (Dako). Live cells were gated via forward and side scatter profiles, and single-cell events were determined via pulse width. Purity was determined to be ∼93 to 97%.
Statistical analyses.
Statistical analyses were performed using Prism, version 5.0, software from GraphPad Software, Inc. Statistical significance was determined using an unpaired student's t test or analysis of variance (ANOVA).
RESULTS
FHA-deficient B. bronchiseptica induces a more robust inflammatory response than wild-type B. bronchiseptica, independent of dose and bacterial burden.
We showed previously that inoculation of BALB/c mice with 5 × 105 CFU of FHA-deficient B. bronchiseptica cells results in a “bimodal” response: half of the animals appear moribund by day 3 p.i. and have very high numbers of bacteria in their lungs (∼108 CFU) and massive influx of inflammatory cells in their lung tissues, and half of the animals appear healthy, contain ∼3 × 105 CFU in their lungs on day 3 p.i., and have only low levels of inflammatory cells in their lung tissues (36, 40). Animals that are healthy at day 3 p.i. clear the FHA-deficient bacteria from their lungs by day 11 p.i. We hypothesized that our standard dose, 5 × 105 CFU, is the 50% lethal dose (LD50) for FHA-deficient B. bronchiseptica and that a robust inflammatory response is generated in all animals inoculated with this strain. We reasoned that this robust inflammatory response either clears the infection quickly (animals that remain healthy) or causes tissue damage that allows for increased bacterial growth, more inflammation, and more tissue damage, ultimately resulting in the death of the mouse. To test this hypothesis, we inoculated mice with a slightly lower dose (1 × 105 CFU) or a slightly higher dose (5.5 × 105 CFU) and determined bacterial burden in the lungs and lung pathology at days 0, 1, 3, and 11 p.i. The slightly higher dose was determined empirically. We discovered that increasing the dose from 5.0 × 105 CFU to 5.5 × 105 CFU was sufficient to eliminate the bimodal response. Repeating the experiment twice more with more animals produced the same results, as shown here. All mice inoculated with the higher dose of the ΔfhaB strain (RBX9) had nearly 2 logs more CFU in their lungs than mice inoculated with wild-type B. bronchiseptica (RB50) at 72 h p.i. and were moribund (hence, all of these animals were euthanized, and so there are no data for this group at day 11 p.i.); in contrast, all mice inoculated with the lower dose of RBX9 remained overtly healthy and had substantially fewer CFU in their lungs at 72 h p.i., and many cleared the bacteria from their lungs by day 11 p.i. (Fig. 1A). None of the mice inoculated with wild-type bacteria showed signs of illness or respiratory distress at any time p.i. For these animals, bacterial numbers in the lungs increased during the first 72 h p.i. and then decreased to about 104 to 105 CFU by day 11 p.i. (Fig. 1A). The left lungs of each animal were fixed, sectioned, stained with H&E, and examined microscopically (Fig. 1B and C). Sections from animals inoculated with 1 × 105 CFU (Fig. 1B) were also stained with anti-Ly6G antibody followed by Alexa Fluor 568-conjugated secondary antibody and examined by fluorescence microscopy to identify neutrophils. Sections from the lungs of animals inoculated with the lower dose of wild-type bacteria showed only very mild inflammation. At 72 h p.i., the majority of cellular infiltrate was localized in the alveolar interstitium, causing mild atelectasis. Few fusions of alveolar septae were observed, and perivascular inflammation was moderate. Small accumulations of mononuclear leukocytes (most likely lymphocytes) were present but no significant microscopic lesions. The proportion of cells that stained positively with the anti-Ly6G antibody was small. Regardless of dose, the lungs of mice inoculated with the ΔfhaB strain showed substantially more inflammation, characterized by bronchial cuffing and increased infiltration of neutrophils (as shown by staining with anti-Ly6G antibody), macrophages, and mononuclear leukocytes, than the lungs of mice inoculated with wild-type bacteria (Fig. 1B and C). FHA-deficient B. bronchiseptica, therefore, induced a more robust inflammatory response than wild-type bacteria, independent of bacterial burden, supporting the hypothesis that FHA allows B. bronchiseptica to suppress the initial inflammatory response to infection.
Increased production of proinflammatory cytokines, especially IL-17, in the lungs of animals infected with FHA-deficient B. bronchiseptica.
To characterize the initial inflammatory response to wild-type and FHA-deficient B. bronchiseptica more thoroughly, we inoculated BALB/c mice with 5.5 × 105 CFU and evaluated bacterial burden and chemokine and cytokine levels in lung homogenates at 3, 12, 24, 48, and 72 h p.i. As expected, wild-type B. bronchiseptica increased in number by approximately 1 log during the first 72 h p.i. (Fig. 2A), and animals showed no signs of respiratory distress throughout the course of the experiment. We measured cytokine and chemokine levels in the lungs using multiplex technology (Fig. 2B; see also Fig. S2 in the supplemental material) Figure 2B shows cytokine levels per ml of lung homogenate while Fig. S2 shows cytokine levels per ml of homogenate per log10 CFU. Neither wild-type nor mutant B. bronchiseptica induced significant IL-10, gamma interferon (IFN-γ), or IL-12p70 production in these mice (data not shown). Lung homogenates from mice inoculated with wild-type bacteria contained appreciable levels of TNF-α at 3 h p.i. and lower levels at later time points. KC, MIP-2, and MIP-1α levels were increased at 72 h compared with levels in homogenates from PBS-inoculated mice. Lung homogenates from mice inoculated with wild-type bacteria also contained significantly increased levels of IL-1β and IL-6.
As expected, mice inoculated with 5.5 × 105 CFU of the ΔfhaB strain exhibited signs of respiratory distress and mild weight loss at 72 h p.i. and had significantly greater numbers of CFU in their lungs at 48 and 72 h p.i. than mice inoculated with wild-type bacteria (Fig. 2A). Lung homogenates from these mice had greater amounts of TNF-α at 3 h p.i. than lung homogenates from mice inoculated with wild-type bacteria. The chemokines MCP-1, KC, and, notably, IL-17 were significantly higher in lung homogenates from mice infected with the ΔfhaB strain than in homogenates from mice infected with wild-type bacteria at 72 h p.i. (Fig. 2B), suggesting a greater IL-17-mediated response in mice infected with FHA-deficient B. bronchiseptica.
FHA-deficient B. bronchiseptica induces a sustained inflammatory response in the lungs, characterized by increased proportions of IL-17-producing neutrophils, macrophages, and CD4+ T cells.
To characterize the inflammatory response to wild-type and ΔfhaB bacteria at the cellular level, we inoculated additional groups of mice with 1 × 105 CFU. Two animals per group were sacrificed at 3 h p.i. to determine the number of CFU delivered to the lungs (Fig. 3A). Of the animals sacrificed at 24, 48, and 72 h p.i., four per group were used to determine bacterial burden and histopathology, and the results were consistent with previous experiments (Fig. 3A and B). Lungs from the remaining three animals per group were harvested and digested with dispase to create a single suspension, and intracellular staining was performed to identify cell types and the presence of IL-17. At 24 h p.i. with wild-type B. bronchiseptica, the lungs contained a large proportion of alveolar macrophages (∼20%), monocyte-derived macrophages (∼30%), and neutrophils (∼20%) (Fig. 3C). Approximately 25% of the neutrophils and 4% of the monocyte-derived macrophages stained positively for IL-17. Approximately 12% of the cells recovered at 24 h p.i. were CD4+ T cells. Other cells identified included dendritic cells, γδ T cells, NK cells, and CD8+ T cells. Although γδ T cells composed only ∼1% of the cells recovered, 50% of them stained positively for IL-17. The cellular profile at 48 h p.i. was similar to that at 24 h p.i., except that the proportion of alveolar macrophages was greater and the proportion of monocyte-derived macrophages was decreased. By 72 h p.i., the cell types present in the lungs were consistent with recovery from mild inflammation; the proportion of neutrophils was less than 5%, the proportion of alveolar macrophages was about 20%, and the proportion of monocyte-derived macrophages was about 40%, with approximately 15% of those cells staining positive for IL-17. A majority of the IL-17 detected in lung homogenates at 72 h p.i. (Fig. 2B), therefore, appears to have been produced by activated macrophages.
Consistent with histological analyses (Fig. 3B), the lungs of mice inoculated with the ΔfhaB strain contained a significantly greater proportion of neutrophils at 24 h p.i. than lungs of mice inoculated with wild-type bacteria (Fig. 3C). Although the proportion of monocyte-derived macrophages was less in mice inoculated with the ΔfhaB strain than in mice inoculated with wild-type bacteria, the proportion of those staining positive for IL-17 was much greater. At 48 h p.i., the proportion of neutrophils in the lungs of mice infected with the ΔfhaB strain dropped to about 14%, but a majority of those cells produced IL-17. At 72 h p.i., the cellular profile in the lungs of mice inoculated with the ΔfhaB strain differed significantly from that of mice inoculated with wild-type B. bronchiseptica; neutrophils composed a substantial portion of the population with approximately 30% of those staining positively for IL-17, and the proportions of alveolar and monocyte-derived macrophages were reversed. In addition, approximately 10% of the cells recovered were CD4+ T cells, and nearly all of those cells stained positively for IL-17. This cellular profile indicates an ongoing neutrophil-mediated inflammatory response with IL-17 apparently being produced by neutrophils, activated macrophages, and CD4+ T cells. These data indicate that without FHA, B. bronchiseptica induces a robust and sustained IL-17- and neutrophil-mediated inflammatory response that leads to faster clearance of the bacteria from the lower respiratory tract than infection by wild-type bacteria. FHA, therefore, allows B. bronchiseptica to control the inflammatory response in a way that allows the bacteria to persist in the lower respiratory tract for extended periods of time.
FHA-dependent adherence in vitro does not correlate with FHA-dependent function in vivo.
B. bronchiseptica adheres to a wide range of mammalian cell lines in vitro in an FHA-dependent manner (40, 49). To explore the possibility that the hyperinflammatory response to infection by the ΔfhaB strain is due to lack of adherence-dependent delivery of immunomodulatory molecules to host cells, we inoculated mice with a strain producing FHA with a 28-aa deletion near the C terminus of the mature FHA protein (called RBX11Δ28) and compared the inflammatory response to that induced by RB50 (wild type) and RBX9 (ΔfhaB). RBX11Δ28 adheres to epithelial cells and macrophage-like cell lines in vitro similarly to wild-type B. bronchiseptica (40). The number of CFU of RBX11Δ28 recovered after inoculation with 1 × 105 CFU was similar to that of wild-type B. bronchiseptica at 24 and 48 h p.i. and was similar to that of the ΔfhaB strain at 72 h p.i. (Fig. 3A). Examination of H&E-stained lung sections showed increased infiltration of inflammatory cells at 24 and 72 h p.i., similar to the lungs of mice infected with the ΔfhaB strain (Fig. 3B). Investigation of the cell types present in the lungs at 24, 48, and 72 h p.i. indicated a cellular infiltrate profile similar to that induced in response to infection by the ΔfhaB strain (Fig. 3C). In general, experiments shown here as well as those published previously (40) indicate that although RBX11Δ28 is able to adhere to various cell lines in vitro, similar to wild-type B. bronchiseptica, it is defective for lower respiratory tract colonization and for suppressing inflammation in the lungs, similar to ΔfhaB B. bronchiseptica. These data suggest that FHA itself may affect signaling in host cells to modulate the inflammatory response, rather than functioning solely as an adhesin, although we cannot rule out the possibility that RBX11Δ28 is as defective as RBX9 at adhering to cells in vivo.
The cyaA gene from B. pertussis can substitute for the cyaA gene of B. bronchiseptica.
Our data suggest that FHA may be playing a direct role in suppressing inflammation. If so, one or more bacterial factors must be responsible for causing the robust inflammation that occurs in response to infection by FHA-deficient bacteria. Because ACT of B. pertussis has been proposed to function in a proinflammatory manner based on in vitro studies (57), we set out to test the hypothesis that ACT is proinflammatory in vivo. We began by determining if the cyaA gene from B. pertussis could substitute for the cyaA gene of B. bronchiseptica. The predicted amino acid sequences of the ACT proteins produced by B. pertussis and B. bronchiseptica are 96% identical (55). We cloned the cyaA gene from B. pertussis Tohama I on a suicide plasmid and introduced this plasmid into a ΔcyaA derivative strain of B. bronchiseptica RB50 (see Materials and Methods and Fig. S1A in the supplemental material). The resulting ΔcyaA::pcyaABp strain, designated RB515::pDB05, produced and secreted ACT in a manner indistinguishable from that of wild-type RB50 (see Fig. S1B). The adenylate cyclase activity of cell-associated and secreted protein was assayed by measuring the amount of intracellular cAMP from infected macrophage-like cells, and the data are displayed on a log scale (Fig. 4A). Activity values for the ΔcyaA strain were three orders of magnitude lower than for the wild-type strain, essentially at the background level for the assay. The ΔcyaA::pcyaABp strain caused production of equivalent levels of cAMP compared to the wild-type strain. To determine if cyaABp can substitute for cyaABb during infection, we inoculated BALB/c mice using our standard protocol (i.e., intranasal inoculation with 5 × 105 CFU). Consistent with previous data (32), fewer CFU of the ΔcyaA strain were recovered from the lungs at all time points p.i. after 3 h (Fig. 4B). These results confirm a role for ACT in resisting innate immune clearance. The number of CFU of the ΔcyaA::pcyaABp strain was similar to that of wild-type bacteria at all time points, demonstrating that the B. pertussis cyaA gene can substitute for the B. bronchiseptica cyaA gene in this murine model. These data suggest that the ACT proteins of B. pertussis and B. bronchiseptica are functionally interchangeable and that information gleaned about the function of ACT using B. bronchiseptica and natural host animal models may apply to B. pertussis ACT as well. Because pcyaABp essentially complements the ΔcyaA mutation, these data also show that the phenotype displayed by the ΔcyaA strain is due only to the lack of cyaA and not polar effects or other unintended mutations.
The catalytic activity of ACT is required for bacterial growth in the lungs.
To determine if the adenylate cyclase catalytic activity of ACT is required for ACT function in vivo, we used site-directed mutagenesis and allelic exchange to construct a B. bronchiseptica strain producing ACT in which the histidine at position 63 and the lysine at position 65 were replaced with alanine residues (67). The resulting strain, called RB509, which harbors a catalytically inactive cyaA gene, produced ACT protein in a manner indistinguishable from wild-type bacteria (see Fig. S1A in the supplemental material) but lacked measurable adenylate cyclase activity (Fig. 4A). The strain harboring the catalytically inactive cyaA gene was recovered in numbers similar to the ΔcyaA strain at days 3 and 11 p.i. (Fig. 4B), demonstrating that the adenylate cyclase activity of ACT is required for bacterial persistence in the lungs.
ACT is required to induce a sustained inflammatory response, even under hyperinflammatory conditions induced by FHA-deficient bacteria.
We inoculated mice with 5.5 × 105 CFU of wild-type bacteria, the ΔcyaA strain, and a ΔfhaB ΔcyaA double mutant and evaluated bacterial burden, lung pathology, and chemokine and cytokine levels in lung homogenates at 3, 12, 24, 48, and 72 h p.i. As expected, the number of CFU of the ΔcyaA strain decreased over the course of the experiment (Fig. 5A). Mice inoculated with the ΔfhaB ΔcyaA strain (RB516) had greater numbers of CFU in their lungs at 12 and 24 h p.i. than mice inoculated with the ΔcyaA strain (Fig. 5), similar to the way that mice infected with the ΔfhaB strain at the low dose had greater numbers of CFU at these time points than mice infected with wild-type bacteria at the same dose (Fig. 1 and data not shown). Whatever mechanism allows the ΔfhaB mutants to grow to higher numbers in the lungs than wild-type bacteria during the first 24 h p.i., therefore, also appears to be operative for bacteria that do not produce ACT. From 24 to 72 h p.i., the animals infected with the ΔfhaB ΔcyaA strain showed a decrease of 2 log CFU compared to those infected with the wild-type strain, suggesting that even though the initial growth rate/survival capacity was increased similar to the ΔfhaB strain, the lack of ACT rendered the bacteria incapable of persisting. Examination of H&E-stained lung sections of mice infected with the ΔcyaA strain revealed very mild multifocal alveolar hemorrhages (Fig. 5B). At 72 h p.i., perivascular accumulations of mononuclear leukocytes, composed mostly of lymphocytes with some macrophages, and few neutrophils were present. Histological analysis, as well as significantly lower cytokine production during the course of infection (Fig. 5C), suggests that a robust inflammatory response was not engendered, and the lack of ACT shaped this response. Animals that had been inoculated with the ΔfhaB ΔcyaA strain exhibited moderate inflammation at 12 h, primarily consisting of a large neutrophil infiltrate (data not shown). At 24 and 48 h p.i., histological examination revealed fusions of alveolar septae, marked by fibrotic processes, but these lesions were scattered diffusely rather than encompassing the entire lung as in the ΔfhaB mutant (data not shown). However, at 72 h p.i., lungs from mice infected with the ΔfhaB ΔcyaA double mutant exhibited mild multifocal pneumonia (Fig. 5B), with most of the lesions resolving, and nonsuppurative perivasculitis predominating the tissue. Cytokine analysis of the lungs from mice inoculated with ΔfhaB ΔcyaA B. bronchiseptica showed that bacteria elicited a modest inflammatory response compared to animals inoculated with the wild-type strain (Fig. 5C; see also Fig. S2 in the supplemental material), yet TNF-α levels were increased at 12 h p.i., and MIP-1α levels were increased from 3 to 48 h p.i. These data suggest that a substantial inflammatory response was generated in animals inoculated with the ΔfhaB ΔcyaA strain, yet the response was limited due to the fact that the bacterial burden was decreasing over time.
ACT is not absolutely required for induction of robust inflammation in response to FHA-deficient B. bronchiseptica.
We hypothesized that lack of substantial induction of IL-6, KC, IL-17, MIP-2, and MCP-1 in response to inoculation with the ΔcyaA and ΔfhaB ΔcyaA strains (Fig. 5) might be due to the relatively rapid clearance of these strains rather than reflecting an activity of ACT. We therefore inoculated mice with 5.5 × 105, 1 × 106, or 1 × 107 CFU of the ΔcyaA ΔfhaB (RB516) strain or 5.5 × 105 CFU of the ΔfhaB (RBX9) strain and measured bacterial burden and cytokine and chemokine levels at various times p.i. As expected, animals inoculated with the ΔfhaB strain at this dose were ill at 48 h p.i. and moribund by 72 h p.i. Bacterial load at these times increased 2 and 3 logs, respectively, from the initial CFU numbers (Fig. 6A). When administered at the lowest dose, the number of CFU of the ΔfhaB ΔcyaA strain recovered was ∼1.5 logs less at 12 h and 72 h p.i. than the level at 3 h p.i. Inoculation with the intermediate dose, 1 × 106 CFU, resulted in a bimodal response: half of the animals appeared healthy, and the number of CFU recovered decreased at 48 and 72 h compared with the number recovered at 3 h p.i.; in contrast, the other half showed evidence of acute disease, and the number of CFU recovered increased at 48 and 72 h p.i. Inoculation of 107 CFU of the ΔfhaB ΔcyaA strain caused respiratory distress within 24 h and mild weight loss (4 to 7%) (data not shown) by 48 h, and animals were moribund by 72 h p.i. The number of CFU in the lungs increased to ∼8.8 × 108 by 72 h after infection (Fig. 6A). Cytokine and chemokine levels in the lungs of mice inoculated with 5.5 × 105 CFU of the ΔfhaB ΔcyaA strain were low or undetectable at all time points, presumably because the bacterial numbers were too low to induce an appreciable response (Fig. 6B). TNF-α, MIP-1α, and MIP-2 levels were statistically higher in mice inoculated with 1 × 107 of the ΔcyaA ΔfhaB strain than in the mice inoculated with the ΔfhaB mutant. Although bacterial loads were higher in the double mutant-inoculated mice than in RBX9-inoculated mice at 3 and 48 h p.i., they were similar at 72 h p.i., suggesting that, at least at the 72-h time point, the increased cytokine levels were not simply due to increased bacterial numbers. Levels of IL-1β, MCP-1, and IL-17 were not significantly different between mice inoculated with 1 × 107 CFU of RB516 and mice inoculated with RBX9 at any time point, suggesting that ACT does not play a role in controlling the production of these cytokines at this dose. IL-6 and KC levels were dramatically higher in mice inoculated with 1 × 107 CFU of the ΔcyaA ΔfhaB strain, beginning with the earliest time point, than those of the lowest dose. These responses may simply reflect the increased bacterial numbers in the initial inoculum. Cytokines were also measured at the 1 × 106 dose, and production was correlated with bacterial burden, supporting the hypothesis that bacterial load drives robust cytokine production. Overall, the results show that an inflammatory response can be generated in response to bacteria that lack ACT if enough bacteria are present, and therefore ACT is not absolutely required for the development of a proinflammatory response.
Effects of ACT- and FHA-deficient B. bronchiseptica on cytokine production of unprimed bone marrow-derived macrophages.
Our data suggest that both FHA and ACT influence the inflammatory response to B. bronchiseptica infection, and we hypothesize that they do so, at least in part, by controlling cytokine and chemokine production in macrophages. To test this hypothesis, we cultured bone marrow-derived macrophages with live bacteria, added at an MOI of 10 into a 24-well plate containing 100 μl of medium per well (so that both FHA-producing and FHA-deficient bacteria would interact with the macrophages), for 2 h, washed samples to remove any unattached bacteria, incubated cultures for 24 h in medium containing gentamicin to kill remaining extracellular bacteria, and then measured various cytokines by ELISA. Enumeration of CFU at the end of the experiment indicated that in all cases only approximately 0.1% of the bacteria had been internalized and survived for 24 h (data not shown). Macrophages produced substantial amounts of IL-6 (8,919 ± 2,241 pg/ml), IL-10 (3,779 ± 890 pg/ml), and IL-1β (954 ± 285 pg/ml) after culture with wild-type bacteria, indicating that an effective proinflammatory response was generated. In contrast, wild-type bacteria did not induce levels of TNF-α, transforming growth factor β1 (TGF-β1), or IL-12 that were above the detection limit of the assay and induced only a modest amount of IL-23 (116 ± 16 pg/ml). Cytokines induced by the mutant strains were compared with those induced by wild-type B. bronchiseptica, levels of which were set to 1 (Fig. 7). We observed a substantial amount of macrophage cell death in response to infection by the ΔfhaB strain. Although we do not know the reason for the hypercytotoxicity of this strain compared with wild-type bacteria, the cell death is likely responsible for the large amount of TGF-β1 present in the supernatants of these cultures. No other cytokines were produced at significantly different levels in response to the ΔfhaB strain compared with the response to wild-type bacteria. RBX11Δ28 did not cause hypercytotoxicity, and cytokine levels produced in response to this strain were not statistically different from those produced in response to wild-type bacteria. These data indicate that if FHA causes changes in cytokine production in macrophages, those effects are not detectable in this assay. In contrast, significantly less IL-1β and more TNF-α and IL-12 were produced in response to infection by the ΔcyaA strain than in response to infection by wild-type bacteria. These data suggest that ACT may function in an anti-inflammatory capacity. Increased levels of IL-23 were produced in response to strains deficient in production of both ACT and FHA, suggesting that ACT and FHA may function together to suppress the production of this cytokine, which is involved in the development of Th17 cells.
Fig 7.

Cytokine secretion in bone marrow-derived macrophages following infection with B. bronchiseptica. Bone marrow-derived macrophages were harvested from 6-week-old C57BL/6J mice and allowed to culture for 7 days. Macrophages (106) were incubated for 2 h with 107 CFU of wild-type B. bronchiseptica or mutant bacteria (MOI of 10). After incubation, cells were washed once with PBS, and growth medium was then supplemented with 30 μg/ml gentamicin to kill any remaining bacteria. Medium was harvested 24 h later, and cytokine production was assayed via ELISA. The lowest limit of detection was 10 pg/ml. Average observed amounts of cytokines for RB50 (pg/ml) were as follows: IL-6, 8,919.56; IL-10, 3,779.35; TGF-β1, 15.97; IL-1β, 954.5; IL-23, 116.25; TNF-α, 35.79; IL-12, 15. Results are fold changes relative to wild-type RB50 and are the average of six independent experiments. Asterisks indicate significant variation from the wild type (*, P < 0.05; **, P < 0.001; ***, P < 0.0001).
DISCUSSION
Studies on immunity to Bordetella have traditionally focused on understanding the adaptive immune response to infection and vaccination, and many, based primarily on observing IFN-γ production by restimulated lymphocytes in vitro, led to the conclusion that B. pertussis induces a Th1-type immune response (4, 30, 47, 54). These results have been perplexing, however, as it is well established that Th1 responses control the development of cellular immunity and clearance of intracellular pathogens (58), and Bordetella remains predominantly extracellular during infection. The relatively recent discovery of Th17 cells and evidence that they function as important mediators of inflammation and the control of extracellular bacterial and fungal pathogens (18, 59, 73) have prompted studies to explore the role of this pathway in controlling Bordetella infection. Fedele et al. reported that human monocyte-derived dendritic cells (MDDC) incubated with B. pertussis or with lipooligosaccharide (LOS)/LPS from B. pertussis or human-restricted Bordetella parapertussis in vitro produce IL-1β, IL-6, and IL-23 (cytokines involved in maturation of Th17 cells) and that these MDDC could cause naïve lymphocytes to produce IL-17 (22, 23). Siciliano et al. similarly showed that macrophages pulsed with B. bronchiseptica could induce splenocytes to secrete IL-17, and they showed that lung tissue from mice inoculated with B. bronchiseptica could produce IL-17 when restimulated in vitro with B. bronchiseptica (61). Andreasen et al., using B. pertussis, also provided evidence of a Th17 response to Bordetella infection (3). Most recently, Dunne et al. showed that ACT could induce IL-1β production in LPS-activated macrophages, which could potentially polarize a Th cell response toward the Th17 lineage, and they also showed that IL-17−/− mice were less able to control B. pertussis infection than wild-type mice (20). Consistent with these studies, we showed in this study that wild-type B. bronchiseptica induces a mild inflammatory response in the lungs of mice that is characterized by the early induction of TNF-α and the recruitment of neutrophils and macrophages that stain positively for IL-17.
Our previous investigations into the role of FHA in vivo showed that intranasal inoculation of mice with our standard dose (5 × 105 CFU) of FHA-deficient B. bronchiseptica results in a bimodal response in which half of the animals become ill and are moribund by day 3 to 4 p.i. and half remain healthy throughout the course of the experiment (36, 40). We had hypothesized that this bimodal phenotype was due to the production of a more robust inflammatory response to infection by FHA-deficient bacteria that either clears the infection rapidly or causes tissue damage that facilitates bacterial growth and consequently more inflammation, more immunopathology, and ultimately death of the mouse. We reasoned that our standard dose was such that the inflammatory response was precisely at a balance that could be tipped, on a per animal basis, toward either bacterial clearance and recovery or immunopathology-mediated death. The results of inoculating mice with slightly higher and slightly lower doses support our hypothesis; the bimodal response is dose dependent, and, significantly, FHA-deficient bacteria induce a more robust inflammatory response than wild-type bacteria even when a lower dose is administered. These data support the conclusion that FHA functions, either directly or indirectly, in an immunomodulatory capacity in vivo.
Our characterization of inflammatory cells in the lungs in response to infection indicated that both wild-type and FHA-deficient B. bronchiseptica caused the recruitment of large numbers of neutrophils and monocyte-derived macrophages within 24 h postinoculation. By 72 h p.i., the response to wild-type bacteria shifted to one characterized by a large proportion of macrophages and very few neutrophils. In contrast, a predominant neutrophil response was sustained for at least 72 h in mice inoculated with FHA-deficient bacteria, and a substantial proportion of those neutrophils stained positively for IL-17. Production of IL-17 by neutrophils has been documented under certain conditions (35, 42), including upon interaction with Bordetella virulence factors (3). However, we cannot rule out the possibility that neutrophils that stained positively for IL-17 in our experiments did so because of IL-17 bound to surface receptors. Our data showed that mice inoculated with FHA-deficient bacteria contained a greater percentage of IL-17-positive γδ T cells and a significantly greater percentage of IL-17-positive CD4+ T cells in their lungs, suggesting the development of a Th17 response, which was not apparent in mice infected with wild-type bacteria. Consistent with the sustained neutrophil response, mice inoculated with FHA-deficient bacteria had fewer numbers of CFU in their lungs at 72 h p.i. than mice inoculated with wild-type bacteria, and we have now shown in several experiments that most mice inoculated with FHA-deficient B. bronchiseptica that remain healthy clear their infections completely (from the lungs) by day 11 p.i. (36, 40; also the present study). For mice inoculated with wild-type B. bronchiseptica, the low proportion of neutrophils in the lungs at 72 h p.i. correlated with higher numbers of bacteria at 72 h and a persistent lung infection that lasts for at least 30 days (31, 32). These data suggest that without FHA, B. bronchiseptica induces a Th17 response that leads to rapid bacterial clearance. With FHA, B. bronchiseptica causes a shift in the response to one that prevents the sustained recruitment and activation of neutrophils, resulting in a persistent infection. The fact that the host response and the outcome of infection were similar for animals infected with the ΔfhaB and RBX11Δ28 strains suggests that the role of FHA in vivo is not solely to mediate attachment of the bacteria to host cells—unless adherence to host cells in vivo does not correlate with adherence to host cells in vitro.
Our data indicate that FHA is involved in suppressing the development of an IL-17-mediated inflammatory response. We hypothesize that one way in which it may do so is by altering signaling pathways and, hence, cytokine and chemokine production in macrophages, key regulators of T cell development. Although our macrophage assay did not reveal a role for FHA in controlling production of most of the cytokines that we measured, it did indicate that both FHA and ACT are involved in suppressing production of IL-23, one of the key cytokines involved in the development of Th17 cells from naïve T cells (12, 34). Consistent with our data demonstrating FHA-dependent suppression of inflammation in vivo, McGuirk and Mills showed that purified FHA can suppress IL-12 production in macrophages that have been primed with E. coli LPS and/or IFN-γ in vitro (53), and Abramson et al. showed that prolonged exposure to purified FHA could block TNF-α-dependent NF-κB activation in U-937 macrophages (1). A major difference between our in vitro experiments and those of McGuirk and Mills and of Abramson et al. is that we investigated a role for FHA in the context of the whole bacterium while these other investigators used purified FHA. We were surprised to find that the ΔfhaB strain induced hypercytotoxicity in macrophages and are currently investigating the mechanism underlying this phenotype. If FHA-mediated adherence functions to facilitate the delivery of toxins to the host cell, we would expect the ΔfhaB strain to be hypocytotoxic, and therefore we hypothesize that the phenotype may reflect altered signaling in the macrophages due to direct binding by FHA.
The robust inflammatory response that occurs in the absence of FHA-mediated suppression indicates that one or more Bordetella factors function as potent activators of inflammation. Several studies suggest that ACT functions in a proinflammatory way. Mills and colleagues showed that purified ACT could induce IL-1β production in LPS-stimulated dendritic cells in vitro and that lymph node cells from immunized mice produce IL-17 in response to restimulation with heat-inactivated ACT (20). Perkins et al. showed that thioglycolate-elicited peritoneal macrophages had higher transcript levels of COX-2 and IL-6 following treatment with purified ACT (57), and Fedele et al. showed that MDDC incubated with ACT-deficient B. pertussis were less able to stimulate IL-17 production in cocultured T lymphocytes than MDDC that had been incubated with wild-type B. pertussis (23). Siciliano et al. similarly showed that less IL-17 was produced when splenocytes were incubated with macrophages that had been pulsed with ACT-deficient B. bronchiseptica than when incubated with macrophages pulsed with wild-type B. bronchiseptica (61). Together, these data suggest that ACT functions to induce inflammation, possibly by promoting the development of a Th17 response. Apparently consistent with a proinflammatory role for ACT, the lungs of mice inoculated with ACT-deficient B. bronchiseptica display less inflammation than those of mice infected with wild-type bacteria (32) (Fig. 3). However, lack of a detectable Th17 response in animals inoculated with ACT-deficient bacteria could be due simply to the fact that these bacteria are cleared rapidly, and therefore inflammation may already be resolving in these mice at the times p.i. when IL-17 is detectable in the lungs of mice infected with wild-type or FHA-deficient B. bronchiseptica. We addressed this possibility by comparing the inflammatory response to bacteria unable to produce FHA with that to bacteria unable to produce both FHA and ACT administered at a critical range of doses. When present in sufficiently high numbers, ACT-deficient bacteria are able to induce a robust inflammatory response, and both IL-1β and IL-17 are produced at increased levels. ACT is therefore not absolutely required for the production of IL-17 in animals infected with FHA-deficient bacteria. Furthermore, when bacterial numbers were similar, greater cytokine production was detected in mice infected with ΔfhaB ΔcyaA double mutant bacteria than those infected with the ΔfhaB single mutant. Consistent with this result, our in vitro experiments indicated that macrophages infected with the ΔfhaB ΔcyaA double mutant secreted many cytokines at greater levels than macrophages infected with the strain lacking only FHA. Although these data do not rule out a possible proinflammatory role for ACT, they implicate another factor as the driving force for the production of IL-17 in vivo.
There is evidence that Bordetella LOS/LPS may be involved in the induction of a Th17 response. Fedele et al. have shown that human dendritic cells secrete IL-1β, IL-6, and IL-23 following treatment with LOS/LPS from B. pertussis and human-restricted B. parapertussis and that when these cells were cocultured with purified T cells, IL-17 was detected in supernatants (22). Several other studies have also demonstrated proinflammatory activities of Bordetella LOS/LPS (24, 25, 44, 46, 62). The importance of Toll-like receptor 4 (TLR4) signaling, which occurs in response to LPS, in the host response to Bordetella has also been demonstrated (6, 45, 70). We hypothesize, therefore, that Bordetella LOS/LPS (and possibly other surface molecules) stimulates a proinflammatory response via TLR signaling, resulting in the early recruitment of neutrophils, macrophages, and dendritic cells to the site of infection. However, we along with others have shown that FHA-deficient B. pertussis strains are indistinguishable from wild-type B. pertussis in mice, and no increase in lung inflammation in response to the ΔfhaB strain was observed (40, 65). A possible explanation for the apparent discrepancy with results obtained from studies with B. bronchiseptica is that fact that murine macrophages are substantially less sensitive to B. pertussis LOS than human macrophages (46). Thus, the roles of FHA and ACT may not be apparent from studies using B. pertussis and mice because of inherent differences in the ability of mice and humans to respond to the LOS of B. pertussis.
Together with our previous work, the data presented in this study support roles for ACT both in surviving a neutrophil-mediated response and in inactivation of macrophages, which would lead to a decreased inflammatory response. While in vitro experimentation provides substantial information about functional capabilities of ACT and its potential effects on host responses, in vivo studies reveal that ACT is likely performing a myriad of functions during the course of infection and may in fact be proinflammatory under some circumstances and anti-inflammatory in others. Our data suggest that both ACT and FHA may alter signaling in the host such that cytokines that would lead to the development of a Th17 response are suppressed. Since FHA and ACT have been reported to bind CR3, an integrin involved in signal transduction, it is possible that this interaction is necessary for immune modulation. Studies are presently being conducted to determine if this receptor is involved in controlling IL-17 production in the lungs. ACT also suppresses cytokines that would push the T cell response toward a Th1 profile. The result is a muted influx of inflammatory cells that allows Bordetella to establish infection and persist in the respiratory tract during the first week or so postinoculation.
To our knowledge, our study is the first to demonstrate suppression of IL-17-mediated inflammation in vivo as a mechanism to promote infection by a microbial pathogen. Moreover, we have identified two bacterial factors involved, FHA and ACT. Although Cheng et al. have proposed that Candida albicans downregulates IL-17 production as a mechanism to promote commensal-like colonization, their conclusions were drawn from in vitro studies (14). While in vitro analyses using wild-type and mutant organisms as well as purified proteins are essential for determining how specific factors function mechanistically, they are necessarily reductionist, and the results obtained must be interpreted within the context of the analysis. Moreover, functions identified for specific factors using in vitro analyses are likely to represent only a subset of the functions actually performed by those factors in their native context during the course of a natural infection. Reciprocally, determining the roles of specific factors in pathogenesis from in vivo studies is limited by the sheer complexity of the systems under study, i.e., both the microbe and the host. While our current study may have served to highlight our limited understanding of the mechanisms underlying the Bordetella-host interaction, we believe that it represents a significant step toward closing the gap between our knowledge of what specific factors can do in vitro and what they actually do in vivo.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carlton Anderson at the UNC Immunotechnologies Core for performing multiplex cytokine analyses and Virginia Godfrey for her professional analysis of tissue sections. We thank members of the Tisch and Cotter labs for many helpful discussions.
This work was supported by funds from the NIH (AI43986 to P.A.C. and AI18000 to E.L.H.) and startup funds from the UNC Chapel Hill School of Medicine.
Footnotes
Published ahead of print 2 April 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abramson T, Kedem H, Relman DA. 2008. Modulation of the NF-κB pathway by Bordetella pertussis filamentous hemagglutinin. PLoS One 3:e3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akerley BJ, Cotter PA, Miller JF. 1995. Ectopic expression of the flagellar regulon alters development of the Bordetella-host interaction. Cell 80:611–620 [DOI] [PubMed] [Google Scholar]
- 3. Andreasen C, Powell DA, Carbonetti NH. 2009. Pertussis toxin stimulates IL-17 production in response to Bordetella pertussis infection in mice. PLoS One 4:e7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ausiello CM, et al. 2000. Cell-mediated immunity and antibody responses to Bordetella pertussis antigens in children with a history of pertussis infection and in recipients of an acellular pertussis vaccine. J. Infect. Dis. 181:1989–1995 [DOI] [PubMed] [Google Scholar]
- 5. Bagley KC, Abdelwahab SF, Tuskan RG, Fouts TR, Lewis GK. 2002. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J. Leukoc. Biol. 72:962–969 [PubMed] [Google Scholar]
- 6. Banus HA, et al. 2006. Host genetics of Bordetella pertussis infection in mice: significance of Toll-like receptor 4 in genetic susceptibility and pathobiology. Infect. Immun. 74:2596–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barry EM, et al. 1991. Bordetella pertussis adenylate cyclase toxin and hemolytic activities require a second gene, cyaC, for activation. J. Bacteriol. 173:720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bassinet L, et al. 2000. Role of adhesins and toxins in invasion of human tracheal epithelial cells by Bordetella pertussis. Infect. Immun. 68:1934–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bemis DA. 1992. Bordetella and Mycoplasma respiratory infections in dogs and cats. Vet. Clin. North Am. 22:1173–1186 [DOI] [PubMed] [Google Scholar]
- 10. Bemis DA, Shek WR, Clifford CB. 2003. Bordetella bronchiseptica infection of rats and mice. Comp. Med. 53:11–20 [PubMed] [Google Scholar]
- 11. Betsou F, Sebo P, Guiso N. 1995. The C-terminal domain is essential for protective activity of the Bordetella pertussis adenylate cyclase-hemolysin. Infect. Immun. 63:3309–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bettelli E, Korn T, Oukka M, Kuchroo VK. 2008. Induction and effector functions of T(H)17 cells. Nature 453:1051–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyd AP, et al. 2005. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptive immune responses: distinct roles for acylation and enzymatic activity in immunomodulation and cell death. J. Immunol. 175:730–738 [DOI] [PubMed] [Google Scholar]
- 14. Cheng SC, et al. Candida albicans dampens host defense by downregulating IL-17 production. J. Immunol. 185:2450–2457 [DOI] [PubMed] [Google Scholar]
- 15. Confer DL, Eaton JW. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948–950 [DOI] [PubMed] [Google Scholar]
- 16. Cotter PA, Miller JF. 1994. BvgAS-mediated signal transduction: analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect. Immun. 62:3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cotter PA, et al. 1998. The filamentous hemagglutinin (FHA) of Bordetella bronchiseptica is required for efficient establishment of tracheal colonization. Infect. Immun. 66:5921–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Curtis MM, Way SS. 2009. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126:177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diavatopoulos DA, et al. 2005. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 1:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dunne A, et al. 2010. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J. Immunol. 185:1711–1719 [DOI] [PubMed] [Google Scholar]
- 21. El-Azami-El-Idrissi M, et al. 2003. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 278:38514–38521 [DOI] [PubMed] [Google Scholar]
- 22. Fedele G, et al. 2008. Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J. Immunol. 181:208–216 [DOI] [PubMed] [Google Scholar]
- 23. Fedele G, et al. 2010. Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS One 5:e8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geurtsen J, et al. 2009. Identification of a novel lipopolysaccharide core biosynthesis gene cluster in Bordetella pertussis, and influence of core structure and lipid A glucosamine substitution on endotoxic activity. Infect. Immun. 77:2602–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geurtsen J, et al. 2006. Expression of the lipopolysaccharide-modifying enzymes PagP and PagL modulates the endotoxic activity of Bordetella pertussis. Infect. Immun. 74:5574–5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodnow RA. 1980. Biology of Bordetella bronchiseptica. Microbiol. Rev. 44:722–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gray MC, Donato GM, Jones FR, Kim T, Hewlett EL. 2004. Newly secreted adenylate cyclase toxin is responsible for intoxication of target cells by Bordetella pertussis. Mol. Microbiol. 53:1709–1719 [DOI] [PubMed] [Google Scholar]
- 28. Guermonprez P, et al. 2001. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the αMβ2 integrin (CD11b/CD18). J. Exp. Med. 193:1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. 1994. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 266:433–435 [DOI] [PubMed] [Google Scholar]
- 30. Hafler JP, Pohl-Koppe A. 1998. The cellular immune response to Bordetella pertussis in two children with whooping cough. Eur. J. Med. Res. 3:523–526 [PubMed] [Google Scholar]
- 31. Harvill ET, Cotter PA, Miller JF. 1999. Pregenomic comparative analysis between Bordetella bronchiseptica RB50 and Bordetella pertussis Tohama I in murine models of respiratory tract infection. Infect. Immun. 67:6109–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harvill ET, Cotter PA, Miller JF. 1999. Probing the function of a bacterial virulence factor by manipulating host immunity. Infect. Immun. 67:1493–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hickey FB, Brereton CF, Mills KH. 2008. Adenylate cycalse toxin of Bordetella pertussis inhibits TLR-induced IRF-1 and IRF-8 activation and IL-12 production and enhances IL-10 through MAPK activation in dendritic cells. J. Leukoc. Biol. 84:234–243 [DOI] [PubMed] [Google Scholar]
- 34. Hirota K, Martin B, Veldhoen M. 2010. Development, regulation and functional capacities of Th17 cells. Semin. Immunopathol. 32:3–16 [DOI] [PubMed] [Google Scholar]
- 35. Hoshino A, et al. 2008. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J. Autoimmun. 31:79–89 [DOI] [PubMed] [Google Scholar]
- 36. Inatsuka CS, Julio SM, Cotter PA. 2005. Bordetella filamentous hemagglutinin plays a critical role in immunomodulation, suggesting a mechanism for host specificity. Proc. Natl. Acad. Sci. U. S. A. 102:18578–18583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inatsuka CS, et al. 2010. Pertactin is required for Bordetella species to resist neutrophil-mediated clearance. Infect. Immun. 78:2901–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ishibashi Y, Claus S, Relman DA. 1994. Bordetella pertussis filamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/CD18). J. Exp. Med. 180:1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jacob-Dubuisson F, et al. 1996. Amino-terminal maturation of the Bordetella pertussis filamentous haemagglutinin. Mol. Microbiol. 19:65–78 [DOI] [PubMed] [Google Scholar]
- 40. Julio SM, et al. 2009. Natural-host animal models indicate functional interchangeability between the filamentous haemagglutinins of Bordetella pertussis and Bordetella bronchiseptica and reveal a role for the mature C-terminal domain, but not the RGD motif, during infection. Mol. Microbiol. 71:1574–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laan M, et al. 1999. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 162:2347–2352 [PubMed] [Google Scholar]
- 42. Li L, et al. 2010. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest. 120:331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Locht C, Bertin P, Menozzi FD, Renauld G. 1993. The filamentous haemagglutinin, a multifaceted adhesion produced by virulent Bordetella spp. Mol. Microbiol. 9:653–660 [DOI] [PubMed] [Google Scholar]
- 44. Mann PB, Elder KD, Kennett MJ, Harvill ET. 2004. Toll-like receptor 4-dependent early elicited tumor necrosis factor alpha expression is critical for innate host defense against Bordetella bronchiseptica. Infect. Immun. 72:6650–6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mann PB, Kennett MJ, Harvill ET. 2004. Toll-like receptor 4 is critical to innate host defense in a murine model of bordetellosis. J. Infect. Dis. 189:833–836 [DOI] [PubMed] [Google Scholar]
- 46. Marr N, et al. 2010. Substitution of the Bordetella pertussis lipid A phosphate groups with glucosamine is required for robust NF-κB activation and release of proinflammatory cytokines in cells expressing human but not murine Toll-like receptor 4-MD-2-CD14. Infect. Immun. 78:2060–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mascart F, et al. 2003. Bordetella pertussis infection in 2-month-old infants promotes type 1 T cell responses. J. Immunol. 170:1504–1509 [DOI] [PubMed] [Google Scholar]
- 48. Mattoo S, Cherry JD. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 18:326–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mattoo S, Miller JF, Cotter PA. 2000. Role of Bordetella bronchiseptica fimbria in tracheal colonization and development of a humoral immune response. Infect. Immun. 68:2024–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mazar J, Cotter PA. 2007. New insight into the molecular mechanisms of two-partner secretion. Trends Microbiol. 15:508–515 [DOI] [PubMed] [Google Scholar]
- 51. Mazar J, Cotter PA. 2006. Topology and maturation of filamentous haemagglutinin suggest a new model for two-partner secretion. Mol. Microbiol. 62:641–654 [DOI] [PubMed] [Google Scholar]
- 52. McGuirk P, McCann C, Mills KH. 2002. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J. Exp. Med. 195:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McGuirk P, Mills KH. 2000. Direct anti-inflammatory effect of a bacterial virulence factor: IL-10-dependent suppression of IL-12 production by filamentous hemagglutinin from Bordetella pertussis. Eur. J. Immunol. 30:415–422 [DOI] [PubMed] [Google Scholar]
- 54. Mills KH. 2001. Immunity to Bordetella pertussis. Microbes Infect. 3:655–677 [DOI] [PubMed] [Google Scholar]
- 55. Parkhill J, et al. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35:32–40 [DOI] [PubMed] [Google Scholar]
- 56. Perez Vidakovics ML, Lamberti Y, van der Pol WL, Yantorno O, Rodriguez ME. 2006. Adenylate cyclase influences filamentous haemagglutinin-mediated attachment of Bordetella pertussis to epithelial alveolar cells. FEMS Immunol. Med. Microbiol. 48:140–147 [DOI] [PubMed] [Google Scholar]
- 57. Perkins DJ, Gray MC, Hewlett EL, Vogel SN. 2007. Bordetella pertussis adenylate cyclase toxin (ACT) induces cyclooxygenase-2 (COX-2) in murine macrophages and is facilitated by ACT interaction with CD11b/CD18 (Mac-1). Mol. Microbiol. 66:1003–1015 [DOI] [PubMed] [Google Scholar]
- 58. Pulendran B. 2004. Modulating TH1/TH2 responses with microbes, dendritic cells, and pathogen recognition receptors. Immunol. Res. 29:187–196 [DOI] [PubMed] [Google Scholar]
- 59. Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P. 2008. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J. Immunol. 180:5157–5162 [DOI] [PubMed] [Google Scholar]
- 60. Ross PJ, Lavelle EC, Mills KH, Boyd AP. 2004. Adenylate cyclase toxin from Bordetella pertussis synergizes with lipopolysaccharide to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infect. Immun. 72:1568–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Siciliano NA, Skinner JA, Yuk MH. 2006. Bordetella bronchiseptica modulates macrophage phenotype leading to the inhibition of CD4+ T cell proliferation and the initiation of a Th17 immune response. J. Immunol. 177:7131–7138 [DOI] [PubMed] [Google Scholar]
- 62. Sisti F, et al. 2011. A deep rough type structure in Bordetella bronchiseptica lipopolysaccharide modulates host immune responses. Microbiol. Immunol. 55:847–854 [DOI] [PubMed] [Google Scholar]
- 63. Stainer DW, Scholte MJ. 1970. A simple chemically defined medium for the production of phase I Bordetella pertussis. J. Gen. Microbiol. 63:211–220 [DOI] [PubMed] [Google Scholar]
- 64. Urisu A, Cowell JL, Manclark CR. 1986. Filamentous hemagglutinin has a major role in mediating adherence of Bordetella pertussis to human WiDr cells. Infect. Immun. 52:695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vandebriel RJ, et al. 2003. Association of Bordetella pertussis with host immune cells in the mouse lung. Microbial pathogenesis. 35:19–29 [DOI] [PubMed] [Google Scholar]
- 66. van den Berg BM, Beekhuizen H, Willems RJ, Mooi FR, van Furth R. 1999. Role of Bordetella pertussis virulence factors in adherence to epithelial cell lines derived from the human respiratory tract. Infect. Immun. 67:1056–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vojtova J, Kamanova J, Sebo P. 2006. Bordetella adenylate cyclase toxin: a swift saboteur of host defense. Curr. Opin. Microbiol. 9:69–75 [DOI] [PubMed] [Google Scholar]
- 68. Weiss AA, Falkow S. 1983. Transposon insertion and subsequent donor formation promoted by Tn501 in Bordetella pertussis. J. Bacteriol. 153:304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weiss AA, Hewlett EL, Myers GA, Falkow S. 1985. Genetic studies of the molecular basis of whooping cough. Dev. Biol. Stand. 61:11–19 [PubMed] [Google Scholar]
- 70. Wolfe DN, Buboltz AM, Harvill ET. 2009. Inefficient Toll-like receptor-4 stimulation enables Bordetella parapertussis to avoid host immunity. PLoS One 4:e4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wolff J, Cook GH, Goldhammer AR, Berkowitz SA. 1980. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. U. S. A. 77:3841–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wood N, Isaacs D. 2006. Monitoring vaccine reactions in Australia. Med. J. Aust. 184:150. [DOI] [PubMed] [Google Scholar]
- 73. Ye P, et al. 2001. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 25:335–340 [DOI] [PubMed] [Google Scholar]
- 74. Zaretzky FR, Gray MC, Hewlett EL. 2002. Mechanism of association of adenylate cyclase toxin with the surface of Bordetella pertussis: a role for toxin-filamentous haemagglutinin interaction. Mol. Microbiol. 45:1589–1598 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.