

Abstract
The anaerobic metabolism of indoleacetate (indole-3-acetic acid [IAA]) in the denitrifying betaproteobacterium Azoarcus evansii was studied. The strain oxidized IAA completely and grew with a generation time of 10 h. Enzyme activities that transformed IAA were present in the soluble cell fraction of IAA-grown cells but were 10-fold downregulated in cells grown on 2-aminobenzoate or benzoate. The transformation of IAA did not require molecular oxygen but required electron acceptors like NAD+ or artificial dyes. The first products identified were the enol and keto forms of 2-oxo-IAA. Later, polar products were observed, which could not yet be identified. The first steps likely consist of the anaerobic hydroxylation of the N-heterocyclic pyrrole ring to the enol form of 2-oxo-IAA, which is catalyzed by a molybdenum cofactor-containing dehydrogenase. This step is probably followed by the hydrolytic ring opening of the keto form, which is catalyzed by a hydantoinase-like enzyme. A comparison of the proteome of IAA- and benzoate-grown cells identified IAA-induced proteins. Owing to the high similarity of A. evansii with strain EbN1, whose genome is known, we identified a cluster of 14 genes that code for IAA-induced proteins involved in the early steps of IAA metabolism. These genes include a molybdenum cofactor-dependent dehydrogenase of the xanthine oxidase/aldehyde dehydrogenase family, a hydantoinase, a coenzyme A (CoA) ligase, a CoA transferase, a coenzyme B12-dependent mutase, an acyl-CoA dehydrogenase, a fusion protein of an enoyl-CoA hydratase and a 3-hydroxyacyl-CoA dehydrogenase, a beta-ketothiolase, and a periplasmic substrate binding protein for ABC transport as well as a transcriptional regulator of the GntR family. Five predicted enzymes form or act on CoA thioesters, indicating that soon after the initial oxidation of IAA and possibly ring opening, CoA thioesters are formed, and the carbon skeleton is rearranged, followed by a CoA-dependent thiolytic release of another CoA thioester. We propose a scheme of an anaerobic IAA metabolic pathway that ultimately leads to 2-aminobenzoyl-CoA or benzoyl-CoA.
INTRODUCTION
Two-thirds of all known organic compounds contain heterocyclic structures. Tryptophan, indole, and derivatives containing a pyrrole ring are abundant N-heterocyclic aromatic compounds in nature. The indole ring of tryptophan can be released by beta-elimination; this reaction is catalyzed by tryptophan-indole lyase and is used in bacterial diagnostics. Tryptophan can also be oxidized via indolylpyruvate to indole-3-acetate (IAA) and CO2. IAA is known as the plant hormone auxin. The neurotransmitter serotonin, the blue dye indigo, and indole alkaloids are further natural products containing an indole ring. The aerobic metabolism of these and similar compounds has been fairly well studied (17). For instance, tryptophan 2,3-dioxygenase cleaves the N-heterocyclic ring with molecular oxygen and forms N-formylkynurenine. This discovery was the starting point of oxygenase research (21). Further metabolism proceeds via the chinoline pathway or via 2-aminobenzoate. All of these pathways involve oxygenases and require molecular oxygen (17, 29, 31).
In contrast, very little is known about the anaerobic metabolism of N-heterocyclic aromatic compounds like tryptophan or IAA. The incomplete metabolism of tryptophan can lead to indolepropionate, IAA, and indole, and the decarboxylation of IAA yields skatole (3-methylindole) (5, 27). Tryptophan or IAA can be oxidized completely by various anaerobic bacteria, which can use such compounds as the sole carbon and electron sources for growth. Early studies with sewage sludge indicated a methanogenic degradation of tryptophan via IAA, indole, 2-aminobenzoate, and benzoate (4). The anaerobic transformation of indole to oxindole was demonstrated previously by others (6, 7, 20, 36, 37, 47). Indole was transformed most likely via 3-oxindole, isatin/dioxindole, and 2-aminobenzoate. To our knowledge, no studies with pure cultures exist, although various bacteria are known to metabolize tryptophan, indole, or IAA under anaerobic conditions (1, 3, 32).
In this work, we investigated the anaerobic metabolism of IAA by the denitrifying betaproteobacterium Azoarcus evansii (formerly Pseudomonas sp. strain KB740) (1, 11). According to the results obtained with sewage sludge or enrichment cultures (4, 6, 7, 20, 36, 37, 47), indole and IAA are probably first oxidized at the N-heterocyclic pyrrole ring, implying water as the oxygen donor and probably a molybdenum cofactor-containing dehydrogenase (anaerobic hydroxylase). These enzymes are very common in the metabolism of heterocyclic compounds, both in aerobic and in anaerobic metabolisms (17); one example is xanthine dehydrogenase. Fortunately, the genome of a related species, strain EbN1 (now referred to as “Aromatoleum aromaticum”), is known (40; for taxonomic relationships, see reference 42). This enabled us to identify a gene cluster for anaerobic IAA metabolism that codes for a set of seven enzymes, one periplasmic binding protein of an ABC transporter, and one transcription regulator.
We propose a metabolic pathway of IAA that ultimately leads to 2-aminobenzoyl-coenzyme A (CoA) or benzoyl-CoA plus products derived from three of the four carbons of the pyrrole ring and the side chain. Benzoyl-CoA and 2-aminobenzoyl-CoA are both substrates of ring-reducing benzoyl-CoA reductase (8), and their further metabolism leads to three molecules of acetyl-CoA and one molecule of CO2 (24). After having elucidated the anaerobic metabolism of phenylalanine and tyrosine (see reference 24 and literature cited therein), we finally provide a work plan for studying the individual enzymes/steps of anaerobic tryptophan metabolism.
MATERIALS AND METHODS
Materials.
Chemicals were obtained from Fluka (Neu-Ulm, Germany), Sigma-Aldrich (Deisenhofen, Germany), Merck (Darmstadt, Germany), Serva (Heidelberg, Germany), or Roth (Karlsruhe, Germany). Biochemicals were obtained from Roche Diagnostics (Mannheim, Germany), Applichem (Darmstadt, Germany), or Gerbu (Craiberg, Germany). [Phenyl-14C(U)]tryptophan was obtained from Hartmann Analytic (Braunschweig, Germany), and indole-[1-14C]acetic acid was obtained from Biotrend (Köln, Germany) and Hartmann Analytic. Gases and gas mixtures (Formiergas [5% H2, 95% N2]) were obtained from Sauerstoffwerke Friedrichshafen (Friedrichshafen, Germany).
Syntheses.
[Phenyl-14C(U)]IAA was synthesized enzymatically from [phenyl-14C(U)]tryptophan. Reaction mixtures containing 0.1 M potassium phosphate (pH 6.5), 0.4 mg [phenyl-14C(U)]tryptophan (183 kBq), and 0.1 mg (0.03 U) l-amino acid oxidase (Sigma-Aldrich) in a total volume of 0.6 ml were incubated at 37°C for 3 h. Afterwards, a solution (0.12 ml) containing 1% H2O2 in 6 M NaOH was added. After 5 min at room temperature, the pH was adjusted to 3.0 by the addition of HCl, and the mixture was extracted three times with equal volumes of ethyl acetate. The solvent was evaporated under reduced pressure. The yield was 50%. For purification, samples were separated by use of thin-layer chromatography (TLC) plates (see below). We also used corresponding 100-fold-larger assay volumes (60 ml). For the purification of labeled indoleacetate from large-scale assays, we used a 120 ODS HE Grom-Sil column (11 μm, 250 mm by 2 mm; Grom, Herrenberg-Kayh, Germany) at a flow rate of 8 ml min−1. The column was equilibrated at room temperature with 4% (vol/vol) acetonitrile in 0.01% (vol/vol) acetic acid. After the application of 2 ml of sample, the column was washed with 80 ml of this solvent. IAA was eluted with a 20-min linear gradient of 4 to 20% acetonitrile in 0.01% acetic acid, followed by a 5-min isocratic 20% acetonitrile elution. IAA eluted after 160 ml of the linear gradient. The fraction was freeze-dried, and the dry residue was dissolved in 0.5 ml ethanol.
Growth conditions.
Azoarcus evansii strain KB740 (DSMZ6898) (1, 11) was grown anaerobically at 30°C on a small scale and up to a 200-liter-fermentor scale on mineral medium (pH 7.6 to 7.8) (14). The growth medium was thoroughly gassed with nitrogen gas before inoculation, and no redox indicator and no reducing agent were added. The medium contained IAA or other organic compounds (routinely 4 mM) as the sole carbon source and nitrate (16 mM) as the electron acceptor. After growth in the 200-liter fermentor had started, the fermentor was operated in a continuous fed-batch mode (14), using a stock solution of 0.4 M indoleacetate and 2.6 M KNO3. A. evansii produces nitrite first and consumes this intermediate only if the nitrate is completely used up. Care was taken to have a slight excess of IAA to avoid incomplete denitrification and accumulation of toxic nitrite. Growth was monitored by measuring the optical density (OD) at 578 nm (OD578) (light path, 1 cm). Cells grew initially with a generation time of 11 h, and from an OD of 0.35 on, they grew with a generation time of 21 h, owing to a growth-limiting feeding rate (average generation time of 15.5 h). Cells were harvested while they were still in the exponential growth phase at an OD578 of 1.3. The culture was cooled down to 16°C before centrifugation, and the cell paste was kept frozen in liquid nitrogen until use. A. aromaticum strain EbN1 (DSMZ9506) was grown at 30°C under denitrifying conditions (10 mM nitrate) with 2 mM IAA as the sole substrate in anoxic 2-liter bottles as described previously (41). Nitrate and IAA were refed several times when the previous portions were exhausted.
Preparation of cell extracts.
Frozen cells of A. evansii were suspended in the same volume of 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS)–K+ buffer (pH 7.8) with 0.1 mg DNase I ml−1. The cell suspensions were passed through a chilled French pressure cell at 137 MPa. The cell lysate was ultracentrifuged (1 h at 100,000 × g at 4°C), and the supernatant was used immediately or stored frozen at −20°C. Some extracts were prepared under anaerobic conditions under an N2 headspace and using an anaerobic glove box under 95% N2–5% H2. The protein concentration was determined by use of the Bradford method (10).
Experiments with cell extracts.
The transformation of IAA was tested at 30°C routinely by monitoring the formation of radiolabeled products from indole-[1-14C]acetate- or [phenyl-14C(U)]-labeled IAA, either by TLC or by high-performance liquid chromatography (HPLC). The standard assay mixture (total volume, 0.2 ml) contained approximately 7 mg protein, 60 mM MOPS-K+ (pH 7.8), and 0.2 or 4 mM 14C-labeled IAA (2.7 kBq). In addition, 10 mM pyruvate, 2 mM NAD+, and 50 units of lactate dehydrogenase were added to immediately regenerate NAD+ from NADH. When a series of artificial electron acceptors was tested, the NAD+-regenerating system was omitted. Instead, the artificial electron acceptor (5 mM) was added. The reaction was started by the addition of the cell extract to the mixture. Samples (24 μl) were taken at intervals, added to 30 μl cold methanol, and kept frozen. After a centrifugation of the thawed sample, it was applied onto TLC plates. For HPLC analysis, a larger assay mixture volume was used, and 200-μl samples were added to 12 μl of 20% (vol/vol) formic acid. An aliquot was analyzed by HPLC. If products were to be analyzed by mass spectrometry (MS), an extract was prepared by using 10 mM ammonium bicarbonate buffer (pH 7.8), and the MOPS in the assay mixture was replaced with 60 mM ammonium bicarbonate (pH 7.8). Benzoyl-CoA reductase activity was tested as described previously, using [ring-14C]benzoate as the substrate (30). The assay mixture included 7 mM MgATP, 1 mM CoA, and 1 mM sodium dithionite, and endogenous benzoate-CoA ligase produced [ring-14C]benzoyl-CoA. CoA ligase activities were tested by a coupled spectrophotometric assay (35, 49).
Gel electrophoresis.
Standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (8 to 12.5%) were used (33). Protein was stained with Coomassie R 250. Two-dimensional gels were run according to methods described previously (18), using isoelectric focusing at pH 3 to 10 in the first dimension and SDS-PAGE (11%) in the second.
Spectroscopic methods.
UV-visible spectra were recorded by using a Lambda 2S UV/VIS spectrophotometer (Perkin-Elmer, Norwalk, CT). Samples for mass spectrometry were prepared by extracting the acidified samples for 5 h with diethyl ether by using a Kutscher-Steudel apparatus. For analysis by mass spectrometry (MS), fractions of product peaks from reverse-phase HPLC (RP-HPLC) runs were transferred by a syringe pump into the electrospray ionization (ESI) source of a Finnigan LTQ-FT mass spectrometer (Thermo Electron Corporation, Waltham, MA) for online mass detection assembled from a linear ion trap and an ion cyclotron (7-T magnet) with Fourier transform (FT) analysis. MS spectra (from m/z 250 to 1,000) were acquired in the FT-ion cyclotron resonance (ICR) with high resolution in the full-scan and secondary ion mass (SIM) scan modus with a mass accuracy of <2 ppm. The detected masses were then fragmented in the linear ion trap with the tandem MS (MS/MS) scan modus manually. The IAA-induced protein spots were excised from Coomassie brilliant blue R-stained gels, digested with trypsin, and identified by peptide mass fingerprinting using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) MS.
Chromatographic methods.
For TLC, samples were applied onto Silica Gel 60 F254-aluminum plates (20 by 20 cm, 0.2 mm; Merck) at room temperature, using 80% isobutanol–5% methanol–15% water (vol/vol/vol) as the solvent (46). The staining of indole derivatives and of organic acids was done according to methods described in a Merck brochure (38). For preparative purposes, we used 1-mm plates, 2-ml samples, and a 5-h developing time. The detection of the substrate and products was performed by staining or UV detection at 254 nm, and radiodetection was done by phosphorimaging. For isolation, the product bands were scraped off, and the material was eluted three times each with a double volume of ethanol and with centrifugation after each elution step. The sample volume was reduced to 0.1 ml by rotavaporation, and the sample was freeze-dried. For HPLC, different systems were used. For system 1 (product analysis), an analytical C18 column 5 μm, (125 mm by 4 mm) (LiChrospher 100; Merck) was used. The pumping rate was 1 ml min−1. Solvent A contained 100% acetonitrile, and solvent B contained 0.01% (vol/vol) aqueous acetic acid. Equilibration was done with 4% solvent A and 96% solvent B, followed by 10 min of 4% solvent A and rest with solvent B. A linear gradient was run for 20 min from 4% to 20% solvent A with rest with solvent B and for 5 min with 20% solvent A and rest with solvent B. Washing was done with 40% solvent A and rest with solvent B. The sample size was 50 to 100 μl. For system 2 (product analysis), an analytical C18 column (EC 125/4 Nucleosil 120-5; Macherey-Nagel, Düren, Germany) was used. Solvent A consisted of 40 mM formic acid, and solvent B consisted of 90% methanol and 40 mM formic acid. Equilibration was done with 95% solvent A and 5% solvent B, followed by 10 min with 95% solvent A and 5% solvent B, with a 25-min linear gradient to 60% solvent A and 40% solvent B. Washing was done with 100% solvent B. For system 3 (analysis of CoA thioesters), the column was the same as that used for system 2. Solvent A contained 100% acetonitrile, and solvent B contained 40 mM ammonium acetate (pH 4.5). Equilibration was done with 0.5% solvent A and 99.5% solvent B, with a 0- to 30-min linear gradient from 0.5% to 10% solvent A and rest with solvent B, followed by a 10-min linear gradient from 10% solvent A to 45% solvent A and rest with solvent B. Washing was done with 60% solvent A and rest with solvent B. Detection was performed by use of a photodiode array detector and/or by solid scintillation detection of radioactivity.
Radioisotope methods.
Liquid scintillation counting of 0.1-ml samples was performed by use of 3 ml of the scintillation cocktail Rotiscint 2200 (Roth, Karlsruhe, Germany) and scintillation counting with the channel ratio method to determine the counting efficiency. Phosphorimaging was done by use of Fuji BAS/MS 20-by-40 plates (Fuji, Osaka, Japan). The 14C in the HPLC runs was detected by using a Ramona 2000 yttrium-silicate solid scintillation system (Raytest, Straubenhardt, Germany).
Further methods.
The concentration of IAA in the culture medium was routinely determined by spectrophotometry at 280 nm (ε280 = 5,460 M−1 cm−1). Concentrations of nitrate and nitrite were semiquantitatively determined by using Quantofix tests (Macherey-Nagel, Düren, Germany).
Database analyses.
DNA and amino acid sequences were analyzed with the BLAST network service at the National Center for Biotechnology Information (Bethesda, MD). BLAST searches were performed by using the genome sequence of Aromatoleum aromaticum, which was formerly called Azoarcus sp. strain EbN1 and is closely related to A. evansii (GenBank accession number NC_006513) (40, 42). BLAST searches were performed with maximum target settings of up to 1,000 and at an expect threshold of 10 and a word size of 3. The tree of large subunits of B12-dependent mutases was generated by the distance tree program provided in BLAST from 1,000 sequences, which all showed query coverages of >90% and E values lower than e−110.
RESULTS AND DISCUSSION
Anaerobic growth on indoleacetate, 2-aminobenzoate, and benzoate.
A. evansii was grown under anaerobic conditions with nitrate as the electron acceptor and indoleacetate (IAA) as the carbon source. For comparison, cells were also grown with 2-aminobenzoate and benzoate, because we expected 2-aminobenzoate (or 2-aminobenzoyl-CoA) and benzoyl-CoA to be possible intermediates in the metabolic pathway. Normally, cells were grown with 4 mM organic substrate and 15 mM nitrate. In a large-scale 200-liter fermentor, we started with lower substrate concentrations and continuously fed the culture with the two substrates. The culture produced 234 g cell mass (wet mass) while consuming 1.52 mol IAA and 9.93 mol nitrate. The yield of 234 g fresh cells corresponds to approximately 70 g cell dry mass, assuming a 30% dry mass content of fresh cell paste. Seventy grams cell dry mass corresponds to 35 g cell carbon (50% of cell dry mass is carbon), which is equivalent to 2.9 mol carbon or 0.29 mol IAA assimilated as the sole carbon source (because IAA contains 10 carbon atoms, and no other carbon source was added). The molar ratio of IAA to nitrate consumed was 1:6.5. A molar growth yield of 46 g cell dry mass formed per mol IAA consumed was calculated.
The closely related bacterium A. aromaticum strain EbN1 was also tested for growth on IAA as the sole substrate under denitrifying conditions. It reached an OD600 of 1.2 while metabolizing 10 mM IAA and 50 mM nitrate, indicating highly similar IAA metabolic capacities for the two bacteria.
Growth stoichiometry and indoleacetate transformation rate.
The complete oxidation of 1 mol IAA follows the equation C10H9NO2 + 18 H2O→10 CO2 + NH3 + 42 [H]. Denitrification follows the equation 42 [H] + 8.4 HNO3→4.2 N2 + 25.2 H2O. The cell growth of A. evansii (formerly Pseudomonas strain KB740) under anaerobic denitrifying conditions consumed some 28% of the organic substrate for biosynthesis (49), corresponding to 0.42 mol IAA out of a total 1.52 mol IAA consumed. The observed stoichiometry (9.93 mol nitrate consumed per 1 mol IAA consumed) corresponds well with the expectation of 9.2 mol nitrate consumed, when 1.52 minus 0.42 mol IAA is completely oxidized. The specific substrate consumption rate, dS/dt, can be calculated as 32 nmol IAA consumed min−1 mg protein−1. This calculation is based on the equation dS/dt = (μ/Y) X, with a specific growth rate, μ, of 0.00075 min−1 (corresponding to an average generation time of 15.5 h); a dry mass cell yield, Y, of 46 g cell dry mass formed per mol IAA consumed; and a cell mass, X, of 1 g cell dry mass (corresponding to 500 mg protein). Under optimal growth conditions (30°C at pH 7.7), the doubling times were 10 h with IAA, 9 h with 2-aminobenzoate, and 4 to 6 h with benzoate.
Transformation of indoleacetate by cell extracts using different electron acceptors.
It is likely that initial reactions in the anaerobic metabolism of IAA are either the anaerobic oxidation of IAA to 2-oxoindoleacetate (2-oxo-IAA) or, alternatively, the activation of the carboxy group of the side chain to the CoA thioester. We therefore studied the transformation of IAA by extracts of IAA-grown cells using different electron acceptors, both in the presence and in the absence of MgATP and coenzyme A. Under strict anaerobic conditions, NAD+ (2 mM) worked best but could be replaced by methylene blue or cytochrome c from beef heart (each 5 mM), yielding nearly the same activity and the same product. The reaction also proceeded in the absence of such electron acceptors when oxygen was present.
To identify products, we used either commercially available IAA 14C labeled at C1 of the acetate side chain or synthesized phenyl-14C(U)-labeled IAA starting from [phenyl-14C(U)]tryptophan. In either case, labeled IAA had to be further purified before use, since both sources contained labeled contaminants. The consumption of IAA and the formation of products were monitored either by thin-layer chromatography and autoradiography (Fig. 1A), by HPLC coupled to UV detection (Fig. 1B), or by different HPLC systems coupled to radiodetection using a flowthrough solid scintillation detector (Fig. 1C to F). The initial IAA consumption rate was 6 to 7 nmol min−1 mg−1 (Fig. 1B), and several relatively apolar early intermediates were detected (Fig. 1C to F). Polar products that may be CoA thioesters were observed in significantly increased amounts when MgATP and CoA were added (Fig. 1F). The addition of MgATP and CoA hardly stimulated the initial IAA transformation process, but it must be taken into account that these cofactors were possibly supplied by the large amounts of cell extract used. However, CoA thioester formation is clearly not involved in the first few steps of anaerobic IAA metabolism.
Fig 1.

Transformation of indoleacetate by cell extracts of Azoarcus evansii. IAA, indoleacetate; P1, enol form of 2-oxo-IAA; P2, keto form of 2-oxo-IAA; P3, unknown product. (A) Thin-layer chromatographic analysis. The assay was performed at 30°C with a total volume of 125 μl containing 0.1 M MOPS-K+ (pH 7.8), 0.2 mM [1-14C]IAA (2.75 kBq), 10 mM pyruvate, 2 mM NAD+, 6 U lactate dehydrogenase, and 75 μl cell extract (5.6 mg protein). Samples (24 μl) were taken at intervals, and the reaction was stopped by the addition of 30 μl of methanol to the mixture. (B) Time course of substrate consumption and formation of products. The assay was performed at 30°C with a total volume of 1.6 ml containing 0.1 M ammonium bicarbonate (pH 7.8), 4 mM IAA, 10 mM pyruvate, 2 mM NAD+, 60 U lactate dehydrogenase, and 0.96 ml cell extract (75 mg protein). Analysis was done by HPLC and UV detection at 260 nm. Note that the IAA concentration was 20-fold higher than that for panel A. IAA was quantified by UV detection at 260 nm using a standard. The amounts of the two main products that were combined in the graph were estimated by using the same absorption coefficient as that for IAA. This approximation is probably not exact and apparently causes a slightly larger amount of the main products formed than IAA consumed. (C) HPLC analysis and UV detection of the main products. Assay conditions were the same as those described above for panel B except that 146 kBq indole-[1-14C]acetate was added. Data for the 10-min sample (50 μl) are shown. HPLC system 2 was used. Note that in panels C and D, data for time zero to 22.5 min were omitted. (D) HPLC analysis and radiodetection of the main products. The sample, HPLC system, and assay conditions were the same as those described above for panel C. (E) HPLC analysis and radiodetection. Assay conditions were same as those described above for panel B except that 0.5 mM [phenyl-14C(U)]IAA (19 kBq) was added and the assay mixture volume was 0.5 ml. Data for the 30-min sample (70 μl) are shown. HPLC system 2 was used. (F) HPLC analysis and radiodetection. Assay conditions were same as those described above for panel B except that 0.5 mM [phenyl-14C(U)]IAA (20 kBq) was added; the assay mixture additionally contained 2 mM CoA, 2 mM ATP, and 5 mM MgCl2; and the assay mixture volume was 0.75 ml. Data for the 30-min sample (70 μl) are shown. HPLC system 3 was used for a better separation of CoA thioesters.
Effect of oxygen and inhibitors.
The transformation of IAA in the presence of methylene blue (5 mM) or NAD+ (2 mM) was equally effective when oxygen was present or excluded; at most, oxygen slightly stimulated the process. Also, no difference was observed between strictly anaerobic assay mixtures and the cell extract preparation and tests in the presence of oxygen. High concentrations of cyanide (5 mM) inhibited the reaction, whereas nitrite, azide, formate, thiocyanate, sulfite, sulfide, hydroxylamine, methanol, or NO (each 1 mM) had no significant effect. The IAA transformation activity of the cell extract was slowly lost during the storage of the extract. Storage with glycerol (10%) or under anaerobic conditions had no positive effect. Storage for 1 week at −20°C was appropriate and resulted in almost no loss of activity.
Characterization of the system.
The strictly anaerobic transformation of IAA required an electron acceptor. For the standard assay mixture, we used NAD+ (2 mM) in MOPS-K buffer (pH 7.8), and activity was tested with the membrane fraction (100,000 × g pellet) and the soluble fraction (supernatant). Two main products were formed by the soluble fraction, but none were formed with the membrane fraction, indicating that the involved initial enzymes were soluble. The same products were formed at a similar rate by the cell extract (40,000 × g supernatant) that contained both soluble and membrane-bound proteins. Optimal transformation was obtained at pH values of between 7.3 and 7.8. For the standard radioactive assay, we used indole-[1-14C]acetate, in which the carboxy group was labeled. To test whether volatile 14CO2 was formed by a decarboxylation event, we determined the total label in the substrate and products and found that the radioisotope balance was still practically at 100% when 80% of the [14C]IAA was transformed into products. Moreover, the same pattern of labeled products was seen when the alternatively labeled [phenyl-14C(U)]IAA was used. These findings exclude that the C2 side chain or the carboxyl group of IAA is lost early in the formed products.
Products formed.
Two labeled products, product 1 (P1) and P2 (Fig. 1A to D), were formed early from IAA either labeled at C1 of the side chain or uniformly labeled at the benzene ring. Product 1 was transiently formed and gave rise to product 2. The radioactive products were extracted with diethyl ether and concentrated. The recovery of 14C-labeled IAA extraction was 95%. HPLC-mass spectrometric analysis confirmed IAA as the substrate (mass, 176.07 Da) and 2-oxo-IAA (mass, 192.06 Da) as product 1. Product 2 had the same mass as product 1. MS/MS analysis indicated that product 1 was the enol form and that product 2 was the keto form of 2-oxo-IAA. These two tautomers characteristically differ in the relative amounts of the 174- and 146-kDa subfragments obtained in the second MS procedure. Apparently, the interconversion of the two tautomeric forms is rather slow. It remains to be shown whether this tautomerization occurs spontaneously or is catalyzed as a side reaction by the hydantoinase-like hydrolase IaaCE (see below). Product 3 had a mass of 191.9 Da as 2-oxo-IAA but has a different retention time by HPLC. The postulated product of the hydrolytic ring cleavage of 2-oxo-IAA, 2(2′-aminophenyl)succinate (mass, 210.07 Da), could not be detected. A minor product with a mass of 166.09 Da was observed in HPLC runs, which would correspond to the decarboxylation product of this ring cleavage product or of a later intermediate of the suggested pathway, 2-aminobenzylmalonyl-CoA (more chemically plausible after thioester hydrolysis). It is known that derivatives of oxindole are very reactive, giving rise to colored products (indigo and others). When the product mixture was acidified, the sample indeed reversibly turned red with an absorption maximum at 505 nm.
HPLC analysis by two different systems revealed that polar products were formed after prolonged incubation (Fig. 1E). When MgATP and CoA were added, the amount of polar products increased (Fig. 1F).
Differential regulation.
We tested whether extracts of cells grown on IAA, 2-aminobenzoate, and benzoate transformed IAA and found that only IAA-grown cells were active, indicating that the enzymes of the pathway were induced by IAA. The transformation rates of IAA in extracts of 2-aminobenzoate- and benzoate-grown cells were less than 10% of those obtained with IAA-grown cells. Extracts of IAA-grown cells catalyzed the ATP-dependent reduction of [ring-14C]benzoyl-CoA with sodium dithionite nearly as effectively as those of benzoate-grown cells. These findings indicate that the enzymes of the central anaerobic benzoyl-CoA degradation pathway are coinduced in IAA-grown cells. However, this coincidence does not necessarily imply that 2-aminobenzoate and/or benzoate (or the respective CoA thioesters) is an intermediate of the pathway. Extracts of IAA-grown cells also contained benzoate-CoA ligase activity (16 nmol min−1 mg−1 at 30°C) and 2-aminobenzoate-CoA ligase activity (24 nmol min−1 mg−1 at 30°C), but no IAA-CoA ligase activity was detected.
The differential induction of pathway-specific enzymes was also seen when SDS-PAGE gels of extracts of cells (100,000 × g supernatant) grown on these three substrates were compared (Fig. 2). Several IAA-induced bands were visible for IAA-grown cells that were missing in benzoate- or 2-aminobenzoate-grown cells, whereas all cells showed a common basic protein pattern. The only exceptions were a few bands that were present in both IAA- and 2-aminobenzoate-grown cells but that were lacking in benzoate-grown cells. This difference may be taken as a hint that 2-aminobenzoyl-CoA may be an intermediate in IAA metabolism. The clear differences in the protein patterns prompted us to use a proteomic approach to identify specific enzymes and genes involved in the IAA pathway.
Fig 2.
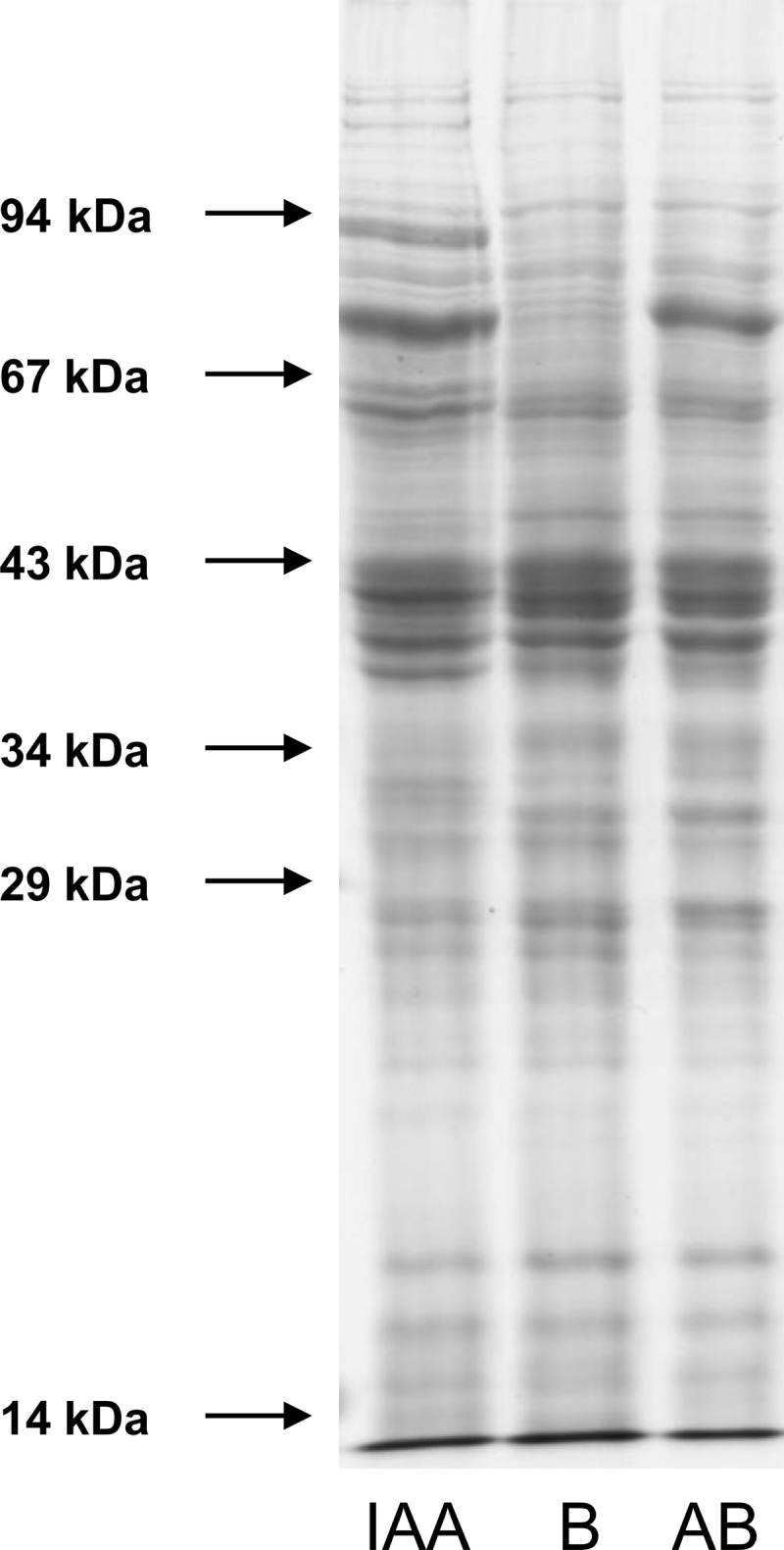
SDS-PAGE of extracts (100,000 × g supernatant) of A. evansii cells grown anaerobically with nitrate and benzoate (B), indoleacetate (IAA), and 2-aminobenzoate (AB). Thirty micrograms of protein each was applied per lane (11-cm gel separation length).
Indoleacetate-induced proteins and identification of a corresponding gene cluster.
Two-dimensional gel electrophoresis was used to identify IAA-induced proteins. Extracts of cells grown on IAA were compared to extracts of cells grown on benzoate (see Fig. S1 in the supplemental material) and 2-aminobenzoate (not shown). A set of more than a dozen proteins was clearly present in IAA-grown cells which were lacking in benzoate-grown cells. A few of these spots were also present in 2-aminobenzoate-grown cells but were lacking in benzoate-grown cells; these spots were not considered IAA induced but may be specific for 2-aminobenzoate metabolism. Again, the presence of these proteins in IAA-grown cells may indicate that 2-aminobenzoate or its CoA thioester might be an intermediate in IAA degradation.
Mass spectrometric analysis of IAA-induced spots led to the identification of a gene cluster in the genome of the closely related strain EbN1 of A. aromaticum (40) that had not been correlated with a function previously (Table 1). The sequence coverage for the proteins analyzed from A. evansii and compared to the predicted gene products from A. aromaticum strain EbN1 was between 10 and 23%. Because of the extraordinarily well-fitting masses of the tryptic fragments of the proteins from A. evansii with the predicted gene products from A. aromaticum, we assume that both bacteria contain essentially identical gene clusters to provide both of them with the IAA catabolic module. We indeed verified the prediction that A. aromaticum grows anaerobically on IAA as well as A. evansii.
Table 1.
Gene cluster of Aromatoleum aromaticum strain EbN1 (GenBank accession number NC_006513.1) correlated with identified IAA-induced protein spots in Azoarcus evansiia
| Gene | Aromatoleum aromaticum EbN1 gene | Corresponding protein spot no.b | Putative annotation | Calculated size (amino acids) | Intergenic distance to next gene (bp) |
|---|---|---|---|---|---|
| iaaP | ebA2038 | Fusion of enoyl-CoA-hydratase and 3-hydroxyacyl-CoA dehydrogenase | 671 | 200 | |
| iaaQ | ebA2046 | TetR family transcriptional regulator | 302 | 247 | |
| iaaR | ebA2047 | GntR family transcriptional regulator | 229 | −1 | |
| iaaA | ebA2049 | 20, 22 | Beta-oxoacyl-CoA thiolase | 401 | 151 |
| iaaB | ebA2050 | 5, 6 | CoA ligase similar to o-benzoylsuccinyl-CoA synthase | 510 | 2 |
| iaaC | ebA2051 | 3, 4 | Hydantoin utilization protein A (HyaA), similar to 5-oxoprolinase | 707 | −1 |
| iaaD | ebD51 | C terminus identical to that of IaaC, possibly from partial gene duplication | 68 | −1 | |
| iaaE | ebA2053 | 15, 16 | Hydantoin utilization protein B (hyaB) | 564 | 2 |
| iaaF | ebA2055 | Acyl-CoA dehydrogenase similar to isovaleryl-CoA dehydrogenase | 377 | −1 | |
| iaaG | ebD52 | Methylmalonyl-CoA mutase C-terminal-domain-containing protein | 139 | −1 | |
| iaaH | ebA2060 | Methylmalonyl-CoA mutase subunit A | 537 | 13 | |
| iaaI | ebA2062 | 11, 14 | Medium FAD-binding subunit of molybdenum enzyme (xanthine dehydrogenase family) | 288 | −14 |
| iaaJ | ebA2063 | Small Fe/S-containing subunit of molybdenum enzyme | 157 | 14 | |
| iaaK | ebA2064 | 1,2 | Large molybdenum-containing subunit of molybdenum enzyme | 803 | 148 |
| iaaL | ebA2066 | 7 | CoA transferase family III | 397 | 220 |
| iaaM | ebA2070 | 12 | Branched-chain-amino-acid ABC transporter, periplasmic component, with N-terminal signal peptide sequence | 382 | 326 |
| ebA2073 | l-Sorbosone dehydrogenase | 752 |
The predicted functions of the proteins are indicated, along with their sizes and the distances of the respective gene to the following gene of the cluster. Negative numbers indicate overlapping genes (e.g., −1 for ATGA start/stop codon overlaps). Note that iaaQ and iaaR are divergent and that the 247-bp intergenic sequence between them should contain two promoters. FAD, flavin adenine dinucleotide.
For spots identified by gel electrophoresis analysis of the A. evansii proteome, see Fig. S1 in the supplemental material.
The gene cluster comprises 14 genes that are oriented in the same direction on the chromosome with only minimal intergenic regions, indicating that these genes might form an operon. Two genes of the cluster have opposite orientations on the left flank, which may also be involved in the IAA metabolic pathway (see below). A gene coding for a predicted pyrrolo-quinoline quinone (PQQ)-dependent sorbosone dehydrogenase is located on the right flank on the same strand but is separated from the genes of the apparent operon by a larger intergenic distance (Fig. 3). We therefore assigned the products of the gene cluster to the IAA metabolic pathway and propose to name the genes accordingly (iaa, for indoleacetic acid) (Table 1 and Fig. 3).
Fig 3.

Gene cluster involved in anaerobic indoleacetate metabolism in Aromatoleum aromaticum strain EbN1. Annotated functions of the genes are indicated. Ech_adh, enoyl-CoA hydratase-alcohol dehydrogenase fusion; TetR and GntR, regulators of the respective protein families; Xdh, xanthine-dehydrogenase-like molybdoenzyme; DH, dehydrogenase.
Eight of the predicted gene products encoded by the gene cluster of A. aromaticum were unambiguously identified by the tryptic fragment masses of the IAA-induced proteins of A. evansii. The genes of the apparent operon without observed matching protein masses include those coding for the regulators (iaaR), a small hypothetical protein (iaaD), and the small subunits of the molybdenum protein (iaaI) and of the mutase (iaaG), which may be expected to be missed on two-dimensional gels because of a low abundance, deviating charges, or small sizes. Thus, only two gene products of the operon have not been identified as induced proteins without a plausible explanation (acyl-CoA dehydrogenase and the large mutase subunit, encoded by iaaF and iaaH, respectively).
Possible functions of the genes.
The first gene of the apparent operon, iaaR, codes for a member of the GntR family of transcriptional regulators, which may be responsible for the IAA-induced expression of the genes. The following genes code for at least seven enzymes potentially involved in IAA metabolism. They comprise (in the order of the putative operon) a beta-oxoacyl-CoA thiolase (iaaA), a CoA ligase acting on aromatic acids (iaaB), a hydantoin- or 5-oxoproline hydrolase (iaaCE), an acyl-CoA dehydrogenase (iaaF), a coenzyme B12-dependent mutase (iaaGH), a molybdenum enzyme of the xanthine dehydrogenase family (iaaIJK), and a CoA transferase of family III (iaaL). All these putative enzymes appear to be soluble. Finally, a periplasmic binding protein for an ABC transporter system is encoded by the last gene of the cluster (iaaM). The iaaD gene spans the gap between iaaC and iaaE, coding for the two subunits of the hydantoinase-like enzyme with overlapping start and stop codons, and has a good Shine-Dalgarno sequence. A closer inspection revealed that its 3′ end is almost identical to that of iaaC at both the DNA and protein levels, suggesting its origin from a recent partial gene duplication event. Therefore, iaaD is likely to be expressed, but it needs to be shown whether its gene product serves any function, e.g., as an additional subunit or regulatory factor for the hydantoinase-like enzyme. In addition to the apparent iaaRABCDEFGHIJKLM operon, the two genes located at the left flank of the apparent operon are also likely to be involved in the pathway. They code for a transcriptional regulator of the TetR family (iaaQ) and a fusion protein of an L-specific enoyl-CoA hydratase and a 3-hydroxyacyl-CoA dehydrogenase (iaaP). Both enzymatic functions are required in the proposed pathway but are not accounted for by genes in the apparent operon. Therefore, we speculate that IaaP may take over this role, although it was not identified as an induced protein. The regulator IaaQ may be involved in the induction of the iaaQP operon.
Proposal of a metabolic scheme.
The presence of a gene coding for a periplasmic substrate binding protein in the apparent IAA catabolic operon suggests that IAA is actively transported into the cell by an ABC-type transporter. The binding protein probably takes advantage of the membrane components of another transporter, for example, predicted ABC-type transporters encoded adjacent to the anaerobic benzoate or aerobic phenylacetate catabolic genes in the A. aromaticum genome (40). As indicated by the identified transformation products of IAA, the initial two reactions of IAA metabolism appear to be the hydroxylation of IAA to 2-oxo-IAA followed by the hydrolytic ring cleavage of the oxindole heterocycle. The enzymes found to be induced proteins in IAA-metabolizing cells and those encoded by the common iaa gene cluster can easily be included in a metabolic route starting with these reactions. We therefore propose the following scheme for an IAA metabolic pathway as a working hypothesis for future studies. The proposed IAA pathway ultimately leads to 2-aminobenzoyl-CoA.
Because initial IAA oxidation occurs both aerobically and anaerobically, it is very plausibly catalyzed by a molybdoenzyme of the xanthine dehydrogenase family, which is encoded by the gene cluster (iaaIJK). These enzymes typically catalyze oxygen-independent hydroxylation reactions at N-heterocyclic rings (25), and even the position of the hydroxylated atom in the indole ring is analogous to that in xanthine hydroxylation. The enzyme probably produces initially the enol form of the oxindole ring, which tautomerizes to the keto (lactam) form. The subsequent hydrolytic opening of the heterocycle at the lactam bond to produce (2-aminophenyl)succinate is analogous to the hydrolytic cleavage of other cyclic lactams, like oxoproline (pyroglutamate) or hydantoins. Therefore, it is likely catalyzed by an enzyme annotated as ATP-dependent hydantoinase (39), whose genes are also part of the gene cluster (iaaCE). Hydantoinases of this family are similar to ATP-dependent acetone or acetophenone carboxylases (28, 44, 45) and are thought to activate the lactam group by ATP-dependent phosphorylation to the corresponding tautomeric phospho-lactim, which is easier to hydrolyze (Fig. 4). It may be speculated that the enzyme additionally facilitates the preceding tautomerization between the enol and keto forms of the oxindole intermediates (Fig. 5).
Fig 4.

Comparison of the postulated hydrolytic ring cleavage reactions of ATP-dependent hydantoinases. Shown are reactions catalyzed by 5-oxoprolinases (A), hydantoinases (B), and the proposed 2-oxoindoleacetate hydrolase IaaCE (C).
Fig 5.

Proposed scheme of anaerobic metabolism of indoleacetate. Gene products of the A. aromaticum iaa gene cluster are indicated as proposed to catalyze the respective steps, as follows: step I, IAA; step II, 2-oxoindoleacetate, enol form; step III, 2-oxoindoleacetate, keto form; step IV, (2-aminophenyl)succinate; step V, 2(2′-aminophenyl)succinyl-CoA; step VI, 2-aminobenzylmalonyl-CoA.
All the other five predicted enzymes encoded by genes of the IAA-induced gene cluster form or act on CoA thioesters. This indicates that the rest of the pathway uses CoA thioesters. It starts with 2(2′-aminophenyl)succinyl-CoA formation, followed by carbon skeleton rearrangement, beta-oxidation, and a CoA-dependent thiolytic cleavage reaction. The proposed reaction sequence is reminiscent of the proven or proposed pathways of anaerobic toluene and n-alkane oxidation (12, 19, 34, 48). The formation of 2(2′-aminophenyl)succinyl-CoA may be catalyzed by the CoA ligase (IaaB) or the CoA transferase (IaaL) of the CoA transferase III family (22), encoded by the apparent operon, or by both of them (Fig. 5). Similar cases of parallel CoA-thioester formation by both types of enzymes in the biotransformation pathways of carnitine to butyrobetaine or of cholate to deoxycholate are known (22, 43). Potential acyl-CoA-donor substrates for the CoA transferase would be succinyl-CoA, acetyl-CoA, or 2-aminobenzoyl-CoA, which occur as intermediates in the following pathway. These donors will be readily reactivated to CoA thioesters by the respective CoA ligases, which are expected to be induced under these conditions (see reference 23).
The pathway may continue by a reversible rearrangement of 2(2′-aminophenyl)succinyl-CoA to 2-aminobenzylmalonyl-CoA, catalyzed by a coenzyme B12-dependent mutase encoded by the iaaGH genes. The intermediate 2-aminobenzylmalonyl-CoA is suitable for further degradation by beta-oxidation, which would be initiated by the acyl-CoA dehydrogenase encoded by iaaF. It is possible that this first step of beta-oxidation is coupled with a decarboxylation of the α-carboxy group of 2-aminobenzylmalonyl-CoA (Fig. 5). A similar but reverse reaction is catalyzed by crotonyl-CoA carboxylase/reductase, an acyl-CoA dehydrogenase of the medium-chain dehydrogenase/reductase superfamily that transforms crotonyl-CoA plus CO2 into ethylmalonyl-CoA (16). The following steps are the hydration of the double bond and the dehydrogenation of the 3-hydroxyacyl-CoA intermediate to a 3-oxoacyl-CoA compound. Although there are no genes in the apparent iaa operon to account for these functions, the immediately adjacent iaaP gene oriented in the other direction codes for a fusion protein comprising both required activities and therefore is possibly also involved in IAA degradation. Its expression may be regulated separately by the TetR-type regulator IaaQ, encoded by a common two-gene operon (Fig. 3). The last step of IAA degradation would be catalyzed by a thiolase encoded by iaaA. The final intermediate, 2-aminobenzoyl-CoA, will then be either reductively deaminated or directly reduced to nonaromatic intermediates by benzoyl-CoA reductase, as known from previous studies of A. evansii and Thauera aromatica (8, 9).
Distribution of the pathway and relation of the enzymes and regulator to known proteins.
By inspecting the database for proteins related to the iaa gene products of A. aromaticum, we detected gene clusters coding for the key enzymes of the proposed pathway in three other microbes. In addition to the betaproteobacterium A. aromaticum, these microbes including the alphaproteobacterium Rhodopseudomonas palustris (only strain BisA3), the deltaproteobacterial strain NaphS2 (13), and the euryarchaeum Ferroglobus placidus (2). Although separated by a wide phylogenetic distance, it is remarkable that all these microorganisms are capable of anaerobic benzoate degradation (2, 13, 15, 26), which is the prerequisite for further metabolizing the intermediate 2-aminobenzoyl-CoA generated from IAA. The fact that IAA is a common substrate derived from a canonical amino acid is in stark contrast to the apparently sparse occurrence of the detected pathway in very few organisms, suggesting that there may be other strategies of anaerobic tryptophan and IAA degradation. In the supplemental material, we discuss and document in detail the relationships of the enzymes and regulator to known proteins. Notably, we document the presence of orthologous genes in IAA-related gene clusters of various microorganisms (see Table S1 in the supplemental material). Furthermore, we compare the conserved clusters of iaa-related genes in four species, A. aromaticum strain EbN1, R. palustris strain BisA3, the deltaproteobacterium strain NaphS2, and F. placidus (see Fig. S2 in the supplemental material). An analysis of the phylogenetic tree of the large subunits of coenzyme B12-dependent mutases shows that the new mutases encoded by the putative iaa gene clusters from all strains reported here form a distinct subgroup (see Fig. S3 in the supplemental material).
Outlook.
Taken together, the genomic evidence suggests strongly that all four organisms should be capable of anaerobic IAA degradation, although only A. aromaticum and R. palustris harbor genes for all steps of the suggested degradation pathway in the respective gene clusters. However, all crucial enzymes catalyzing the unusual reactions are accounted for in all four strains, if one accepts the quite likely substitution of a molybdoenzyme of the bacterial strains with a tungstoenzyme in the archaeal species. It seems that a few key enzymes, such as the molybdoenzyme (or tungsten enzyme), the coenzyme B12-containing mutase, and the IaaF-type acyl-CoA dehydrogenase, constitute the core of the pathway, whereas the other functions may be taken over by proteins of different origins during evolution.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Deutsche Forschungsgemeinschaft to G.F. and J.H. and from the Synmikro Loewe Center Marburg to J.H.
We thank C. Warth, Freiburg, Germany, for mass spectrometry analysis of products during the initial work and A. Schlosser, Zentrum für Biosystemanalyse der Universität Freiburg, for MS analysis of IAA-induced proteins.
Footnotes
Published ahead of print 23 March 2012
This work is dedicated to August Böck on the occasion of his 75th birthday.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Anders H, Kaetzke A, Kämpfer P, Ludwig W, Fuchs G. 1995. Taxonomic position of aromatic-degrading denitrifying pseudomonad strains K 172 and KB 740 and their description as new members of the genera Thauera, as Thauera aromatica sp. nov., and Azoarcus evansii sp. nov., respectively, members of the beta subclass of the Proteobacteria. Int. J. Syst. Bacteriol. 45:327–333 [DOI] [PubMed] [Google Scholar]
- 2. Anderson I, et al. 2011. Complete genome sequence of Ferroglobus placidus AEDII12DO. Stand. Genomic Sci. 5:50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bak F, Widdel F. 1986. Anaerobic degradation of indolic compounds by sulfate-reducing enrichment cultures, and description of Desulfobacterium indolicum gen. nov., sp. nov. Arch. Microbiol. 146:170–176 [Google Scholar]
- 4. Balba M, Evans W. 1980. Methanogenic fermentation of the naturally occurring aromatic amino acids by a microbial consortium. Biochem. Soc. Trans. 8:625–627 [DOI] [PubMed] [Google Scholar]
- 5. Barker H. 1981. Amino acid degradation by anaerobic bacteria. Annu. Rev. Biochem. 50:23–40 [DOI] [PubMed] [Google Scholar]
- 6. Berry D, Francis A, Bollag J. 1987. Microbial metabolism of homocyclic and heterocyclic aromatic compounds under anaerobic conditions. Microbiol. Rev. 51:43–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berry D, Madsen E, Bollag J. 1987. Conversion of indole to oxindole under methanogenic conditions. Appl. Environ. Microbiol. 53:180–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boll M, Fuchs G. 1995. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain. K172. Eur. J. Biochem. 234:921–933 [DOI] [PubMed] [Google Scholar]
- 9. Boll M, et al. 2000. Nonaromatic products from anoxic conversion of benzoyl-CoA with benzoyl-CoA reductase and cyclohexa-1,5-diene-1-carbonyl-CoA hydratase. J. Biol. Chem. 275:21889–21895 [DOI] [PubMed] [Google Scholar]
- 10. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 11. Braun K, Gibson D. 1984. Anaerobic degradation of 2-aminobenzoate (anthranilic acid) by denitrifying bacteria. Appl. Environ. Microbiol. 48:102–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Callaghan AV, et al. 2012. The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ. Microbiol. 14:101–113 [DOI] [PubMed] [Google Scholar]
- 13. DiDonato RJ, Jr, et al. 2010. Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5:e14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ebenau-Jehle C, Boll M, Fuchs G. 2003. 2-Oxoglutarate:NADP+ oxidoreductase in Azoarcus evansii: properties and function in electron transfer reactions in aromatic ring reduction. J. Bacteriol. 185:6119–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Egland PG, Pelletier DA, Dispensa M, Gibson J, Harwood CS. 1997. A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc. Natl. Acad. Sci. U. S. A. 94:6484–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erb TJ, Brecht V, Fuchs G, Müller M, Alber BE. 2009. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Proc. Natl. Acad. Sci. U. S. A. 106:8871–8876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fetzner S. 2000. Enzymes involved in the aerobic bacterial degradation of N-heteroaromatic compounds: molybdenum hydroxylases and ring-opening 2,4-dioxygenases. Naturwissenschaften 87:59–69 [DOI] [PubMed] [Google Scholar]
- 18. Görg A, Postel W, Domscheit A, Günther S. 1988. Two-dimensional electrophoresis with immobilized pH gradients of leaf proteins from barley (Hordeum vulgare): method, reproducibility and genetic aspects. Electrophoresis 9:681–692 [DOI] [PubMed] [Google Scholar]
- 19. Grundmann O, et al. 2008. Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbiol. 10:376–385 [DOI] [PubMed] [Google Scholar]
- 20. Gu J, Berry D. 1991. Degradation of substituted indoles by an indole-degrading methanogenic consortium. Appl. Environ. Microbiol. 57:2622–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hayaishi O. 1994. Tryptophan, oxygen, and sleep. Annu. Rev. Biochem. 63:1–24 [DOI] [PubMed] [Google Scholar]
- 22. Heider J. 2001. A new family of CoA-transferases. FEBS Lett. 509:345–349 [DOI] [PubMed] [Google Scholar]
- 23. Heider J, et al. 1998. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch. Microbiol. 170:120–131 [DOI] [PubMed] [Google Scholar]
- 24. Heider J, Fuchs G. 1997. Anaerobic metabolism of aromatic compounds. Eur. J. Biochem. 243:577–596 [DOI] [PubMed] [Google Scholar]
- 25. Hille R. 2005. Molybdenum-containing hydroxylases. Arch. Biochem. Biophys. 433:107–116 [DOI] [PubMed] [Google Scholar]
- 26. Holmes DE, Risso C, Smith JA, Lovley DR. 2012. Genome-scale analysis of anaerobic benzoate and phenol metabolism in the hyperthermophilic archaeon Ferroglobus placidus. ISME J. 6:146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Honeyfield DC, Carlson JR. 1990. Assay for the enzymatic conversion of indoleacetic acid to 3-methylindole in a ruminal Lactobacillus species. Appl. Environ. Microbiol. 56:724–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jobst B, Schühle K, Linne U, Heider J. 2010. ATP-dependent carboxylation of acetophenone by a novel type of carboxylase. J. Bacteriol. 192:1387–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaiser J, Feng Y, Bollag J. 1996. Microbial metabolism of pyridine, quinoline, acridine, and their derivatives under aerobic and anaerobic conditions. Microbiol. Rev. 60:483–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koch J, Fuchs G. 1992. Enzymatic reduction of benzoyl-CoA to alicyclic compounds, a key reaction in anaerobic aromatic metabolism. Eur. J. Biochem. 205:195–202 [DOI] [PubMed] [Google Scholar]
- 31. Koenig K, Andreesen J. 1992. Aerober und anaerober Abbau von heterozyklischen, aromatischen Verbindungen durch Bakterien. Bio Engineering 8:78–84 [Google Scholar]
- 32. Kohda C, Ando T, Nakai Y. 1997. Isolation and characterization of anaerobic indole- and skatole-degrading bacteria from composting animal wastes. J. Gen. Appl. Microbiol. 43:249–255 [DOI] [PubMed] [Google Scholar]
- 33. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 34. Leuthner B, Heider J. 2000. Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of beta oxidation of the intermediate benzylsuccinate. J. Bacteriol. 182:272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lochmeyer C, Koch J, Fuchs G. 1992. Anaerobic degradation of 2-aminobenzoic acid (anthranilic acid) via benzoyl-coenzyme A (CoA) and cyclohex-1-enecarboxyl-CoA in a denitrifying bacterium. J. Bacteriol. 174:3621–3628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Madsen E, Bollag J. 1989. Pathway of indole metabolism by a denitrifying microbial community. Arch. Microbiol. 151:71–76 [Google Scholar]
- 37. Madsen E, Francis A, Bollag J. 1988. Environmental factors affecting indole metabolism under anaerobic conditions. Appl. Environ. Microbiol. 54:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Merck 1980. Anfärbereagenzien für Dünnschicht und Papier-Chromatographie. Merck, Darmstadt, Germany. [Google Scholar]
- 39. Ogawa J, et al. 1995. Purification and characterization of an ATP-dependent amidohydrolase, N-methylhydantoin amidohydrolase, from Pseudomonas putida 77. Eur. J. Biochem. 229:284–290 [DOI] [PubMed] [Google Scholar]
- 40. Rabus R, et al. 2005. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch. Microbiol. 183:27–36 [DOI] [PubMed] [Google Scholar]
- 41. Rabus R, Widdel F. 1995. Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch. Microbiol. 163:96–103 [DOI] [PubMed] [Google Scholar]
- 42. Reinhold-Hurek B, Tan Z, Hurek T. 2005. Genus II. Azoarcus Reinhold-Hurek, Hurek, Gillis, Hoste, Vancanneyt, Kersters and DeLey 1993b, 582VP, p 890–901 In Brenner DJ, Krieg NR, Staley JT, Garrity GM. (ed), Bergey's manual of systematic bacteriology, 2nd ed, vol 2, part C Springer, New York, NY [Google Scholar]
- 43. Ridlon JM, Hylemon PB. 2012. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J. Lipid Res. 53:66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schühle K, Heider J. 2012. Acetone and butanone metabolism of the denitrifying bacterium “Aromatoleum aromaticum” demonstrates novel biochemical properties of an ATP-dependent aliphatic ketone carboxylase. J. Bacteriol. 194:131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sluis MK, Ensign SA. 1997. Purification and characterization of acetone carboxylase from Xanthobacter strain Py2. Proc. Natl. Acad. Sci. U. S. A. 94:8456–8461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stahl E. 1967. Dünnschicht-Chromatographie, Ein Laboratoriumshandbuch. Springer-Verlag, Berlin, Germany [Google Scholar]
- 47. Wang Y-T, Suidan MT, Pfeffer JT. 1984. Anaerobic biodegradation of indole to methane. Appl. Environ. Microbiol. 48:1058–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilkes H, et al. 2002. Anaerobic degradation of n-hexane in a denitrifying bacterium: further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch. Microbiol. 177:235–243 [DOI] [PubMed] [Google Scholar]
- 49. Ziegler K, Braun K, Böckler A, Fuchs G. 1987. Studies on the anaerobic degradation of benzoic acid and 2-aminobenzoic acid by a denitrifying Pseudomonas strain. Arch. Microbiol. 149:62–69 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.