

Abstract
Staphylococcus aureus is a major human pathogen that is capable of producing an expansive repertoire of cell surface-associated and extracellular virulence factors. Herein we describe an S. aureus regulatory RNA, SSR42, which modulates the expression of approximately 80 mRNA species, including several virulence factors, in S. aureus strains UAMS-1 and USA300 (LAC) during stationary-phase growth. Mutagenesis studies revealed that SSR42 codes for an 891-nucleotide RNA molecule and that the molecule's regulatory effects are mediated by the full-length transcript. Western blotting and functional assays indicated that the regulatory effects of SSR42 correlate with biologically significant changes in corresponding protein abundances. Further, in S. aureus strain LAC, SSR42 is required for wild-type levels of erythrocyte lysis, resistance to human polymorphonuclear leukocyte killing, and pathogenesis in a murine model of skin and soft tissue infection. Taken together, our results indicate that SSR42 is a novel S. aureus regulatory RNA molecule that contributes to the organism's ability to cause disease.
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) is a leading cause of nosocomial and community-acquired infections, both of which range in severity from superficial skin infections to conditions with high morbidity such as endocarditis (19). Most United States hospital-acquired MRSA infections are caused by the pulsed-field type (PFT) USA100 and USA200 lineages, whereas community-acquired MRSA (CA-MRSA) infections can be attributed predominantly to strains belonging to the USA300 PFT. Strains of the USA300 PFT are generally regarded as hypervirulent and are a leading cause of illness in otherwise healthy individuals (14, 15, 61).
The ability of S. aureus to cause infection is due, in large part, to its ability to adapt to host and environmental stresses and to the coordinated expression of a vast repertoire of virulence factors. Most S. aureus virulence factors can be broadly divided into cell surface-associated and extracellular factors and are generally regulated in a growth phase-dependent manner under laboratory culture conditions (11, 47). Cell surface virulence factors, including adhesion and immune avoidance molecules, are expressed predominantly during exponential-phase growth, whereas their expression decreases as cells transition to stationary-phase growth (23). Conversely, extracellular virulence factors, such as tissue-degrading and immunomodulatory proteins, are generally expressed at low levels during exponential-phase growth and are subsequently induced as populations reach late-exponential/early-stationary-phase growth (23). Ostensibly, this growth phase-dependent transition in the expression of S. aureus cell surface and extracellular virulence factors is thought to recapitulate what occurs upon the infection of a human host, allowing an increased opportunity for cell surface factor-mediated attachment and subsequent colonization of host tissue(s), followed by the expression of extracellular virulence factors that limit host defenses and allow the organism to disseminate to secondary sites of infection.
Coordinated expression of virulence factors has been historically attributed to transcriptional regulation and is modulated by at least 17 two-component regulatory systems (TCRS), the DNA-binding protein SarA, and the SarA family of homologs (11, 33, 47). Of these, the one best characterized to date is the accessory gene regulator (Agr) TCRS. The agr locus produces two divergent transcripts, RNAII and RNAIII, during late-exponential-phase growth. RNAII encodes four proteins, AgrB, AgrD, AgrC, and AgrA (48). Of these, AgrD is presumably processed to the mature/functional form, known as the autoinducing peptide (AIP), and shuttled to the extracellular environment via AgrB. Once AIP reaches an extracellular threshold, it activates the AgrC signal receptor which, in turn, activates the AgrA response regulator, which consequently induces RNAII and RNAIII transcription (30). RNAIII contains the open reading frame (ORF) for δ-hemolysin (hld; 26 amino acids), yet the RNA molecule, as opposed to the Hld protein, represses cell surface virulence factor expression and stimulates the expression of many of the organism's extracellular virulence determinants (49).
Until recently, the mechanism(s) by which RNAIII governs S. aureus virulence factor expression has been poorly understood. In a series of studies, it was found that RNAIII can base pair with the mRNA species that it regulates, consequently affecting the stability and translation properties of the target transcripts (10, 17, 26, 29, 45). For instance, RNAIII binding to protein A mRNA (spa; cell surface virulence factor) creates a substrate for RNase III-mediated spa mRNA degradation and decreased protein A production (29). Conversely, RNAIII binding to the α-hemolysin transcript (hla; extracellular virulence factor) liberates the transcript's Shine-Dalgarno box, allowing increased Hla production (45). Thus, S. aureus RNAIII is a regulatory RNA molecule that binds and affects the stability and consequently the translation of target mRNA species.
Several subsequent studies have revealed that additional regulatory RNA molecules do, or are likely to, exist within S. aureus (reviewed in reference 24). Chabelskaya and colleagues recently identified a small pathogenicity island RNA, SprD, which, like RNAIII, base pairs and subsequently affects the expression properties of IgG-binding protein (Sbi; virulence factor) transcripts (13). Further, RNA sequencing and bioinformatic approaches have suggested that S. aureus produces an array of noncoding RNA molecules, any one of which may play important regulatory roles (27, 37). Likewise, our laboratory has identified a subset of RNA molecules with no discernible ORF that are specifically produced and/or stabilized in response to growth phase, SOS, stringent, heat shock, alkaline, acidic, or cold shock inducing conditions (3, 4, 50, 52). These molecules have collectively been termed small stable RNAs (SSRs) to distinguish them from the plethora of nonstable putative noncoding RNAs that have been identified in the organism. It has been hypothesized that SSRs represent regulatory molecules that participate in the organism's ability to adapt to otherwise deleterious conditions (3, 4, 50). Herein we describe the regulatory effects of one of these molecules, SSR42, which is produced and stabilized predominantly during stationary-phase growth. We show that SSR42 is indeed a regulatory RNA molecule that regulates the expression of approximately 80 mRNA species in two genetically divergent S. aureus strains, UAMS-1 (methicillin-susceptible S. aureus; USA200) and LAC (CA-MRSA; USA300). We also show that SSR42 regulates the expression of several virulence factors, including protein A, capsule, α-hemolysin, and Panton-Valentine leukocidin (PVL), but that the specific genes that it affects are strain dependent. Further, consistent with the molecule's predicted regulatory effects, we demonstrate that SSR42 contributes to LAC's abilities to lyse erythrocytes and survive the antimicrobial effects of primary human polymorphonuclear cells and the bacterial burden in a murine model of skin and soft tissue infection.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. All strains were grown in Luria-Bertani broth (BD, Sparks, MD) or tryptic soy broth (TSB; BD). Where indicated, the medium was supplemented with erythromycin (10 μg ml−1; Fisher Scientific, Fair Lawn, NJ), chloramphenicol (10 μg ml−1; Sigma-Aldrich, St. Louis, MO), or ampicillin (50 μg ml−1; Fisher Scientific). For S. aureus growth phase experiments, overnight cultures of cells were diluted 1:100 in 25 ml of fresh TSB medium with a flask volume ratio of 5:1 and grown to an optical density at 600 nm (OD600) of 0.250 (exponential phase) or incubated for 16 h (stationary phase) at 37°C at 225 rpm. For RNA studies, 5-ml aliquots of cells were removed, added to an equal volume of ice-cold 1:1 acetone-ethanol, and stored at −80°C. RNA turnover studies were performed as previously described (3, 52). Rifampin (200 μg ml−1; Sigma-Aldrich) was added to exponential- or stationary-phase cultures, and aliquots of cells were removed and added to an equal volume of ice-cold 1:1 acetone-ethanol at various time points after transcriptional arrest.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype and/or characteristic(s) | Source or reference(s) |
|---|---|---|
| S. aureus strains | ||
| UAMS-1 | Methicillin-susceptible osteomyelitis isolate (USA200) | 12, 28 |
| LAC | Community-associated methicillin-resistant isolate (USA300) | 43 |
| RN4220 | Restriction-deficient derivative of RN450 | 18 |
| U151 | UAMS-1 containing pCN51 | This study |
| EWM14 | UAMS-1 ΔSSR42 | This study |
| EWM15 | UAMS-1 ΔSSR42 containing pCN51 | This study |
| EWM16 | UAMS-1 ΔSSR42 containing pCN51::SSR42 | This study |
| EWM22 | UAMS-1 containing pEWM21 | This study |
| EWM24 | EWM14 containing pEWM21 | This study |
| EWM25 | UAMS-1 containing pEWM22 | This study |
| EWM28 | EWM14 containing pEWM22 | This study |
| JMM1 | LAC ΔSSR42 | This study |
| JMM2 | LAC ΔSSR42 containing pJMM1 (SSR42+) | This study |
| JMM3 | LAC ΔSSR42 containing pJMM2 (SSR42.mut+) | This study |
| JMM4 | LAC ΔSSR42 containing pJMM4 (SSR42.1+) | This study |
| JMM5 | LAC ΔSSR42 containing pJMM5 (SSR42.2+) | This study |
| KLA16 | UAMS-1 Δspa | 34 |
| VJT26.90 | LAC containing pXen-Phla-lux | This study |
| VJT26.91 | LAC containing pXen-Ppvl-lux | This study |
| VJT26.92 | LAC containing pXen-PlogCB-lux | This study |
| E. coli strains | ||
| DH5α | Invitrogen | |
| INVα | Invitrogen | |
| Plasmids | ||
| pCN51 | Shuttle vector | 16 |
| pCN38 | Shuttle vector | 16 |
| pEWM20 | pCN51 containing SSR42 | This study |
| pCRII | Shuttle vector | Invitrogen |
| pJMM1 | pCN38 containing SSR42 | This study |
| pJMM2 | pCN38 containing SSR42 with T-to-A transversion | This study |
| pJMM3 | pCN51 containing SSR42 with T-to-A transversion | This study |
| pJMM4 | pCN38 containing nt 1–445 of SSR42 | This study |
| pJMM5 | pCN38 containing nt 446–891 of SSR42 | This study |
| pKOR1 | Allelic replacement vector | 5 |
| pXen-1 | Luciferase reporter plasmid | Xenogen |
| pXen-Phla-lux | hla promoter fused to lux operon | This study |
| pXen-Ppvl-lux | pvl promoter fused to lux operon | This study |
| pXen-PhlgCB-lux | hlgCB promoter fused to lux operon | This study |
RNA isolation.
RNA isolation from S. aureus cells was performed as previously described (3, 52). Briefly, 5-ml cell suspension samples containing 1 × 109 CFU ml−1 in 1:1 acetone-ethanol were pelleted by centrifugation at 3,000 rpm at 4°C. Cell pellets were washed twice in TE buffer (10 mM Tris, 1 mM EDTA, pH 7.6) and resuspended in 500 μl of TE buffer (exponential-phase cells) or RLT buffer (Qiagen) plus 1% (vol/vol) 2-mercaptoethanol (stationary-phase cells). Cell suspensions were then transferred to BIO 101 Lysing Matrix B tubes (MP Biomedical, Pasadena, CA) and processed for 20 s each at 5.0 m s−1 and 4.5 m s−1 in a FastPrep24 machine (MP Biomedical). The cell debris was collected by centrifugation at 13,000 rpm for 15 min at 4°C, and the supernatants were used for RNA purification with a Qiagen RNeasy Mini kit in accordance with the manufacturer's recommendations (Qiagen, Valencia, CA). RNA concentrations were determined spectrophotometrically (an OD260 of 1.0 equals 40 μg ml−1). RNA used for real-time PCR (RT-PCR) or quantitative RT-PCR (qRT-PCR) was then treated with 10 U of RNase-free DNase I (Ambion, Austin, TX) at 37°C for 1 h and repurified using a Qiagen RNeasy Mini kit in accordance with the manufacturer's recommendation for RNA cleanup.
Generation of SSR42 deletion-containing strain.
SSR42 was deleted from the S. aureus UAMS-1 and LAC chromosomes by using the pKOR1 allelic replacement vector as previously described (5). Briefly, 500-bp regions upstream and downstream of the SSR42 transcriptional unit were amplified with primers EWM110/EWM111 and EWM112/EWM113 (Table 1) harboring either 3′ SacII restriction sites or 5′ att phage lambda attachment sites. Following PCR amplification, the products were gel purified and digested with the SacII restriction enzyme (New England BioLabs, Ipswich, MA) and then ligated using T4 DNA ligase (Invitrogen). The ligated products containing a deletion of SSR42 were then inserted into the pKOR1 vector using in vitro recombination via BP Clonase (Invitrogen), propagated in Escherichia coli strain DH5α, and purified using Plasmid Miniprep kits (Qiagen, Valencia, CA). The resultant pKOR1::SSR42 knockout vector was then electroporated into competent, restriction-deficient S. aureus RN4220 (1.8 kV, 500 Ω, 10 μF), and transformants were selected for on TSB agar plates in the presence of chloramphenicol (10 μg ml−1) at 30°C. The pKOR1::SSR42 vector was subsequently transferred to S. aureus strain UAMS-1 or LAC via φ11-mediated transduction. To promote recombination-mediated integration of the plasmid into the bacterial chromosome, transductants were propagated at 43°C (nonpermissive temperature for the pKOR1 vector) under the selection of chloramphenicol to select for homologous single recombination events. Individual colonies were then inoculated into fresh TSB supplemented with anhydrotetracycline to counterselect for double-recombination events. Transformants were screened for successful deletion of SSR42 via PCR amplification of the chromosomal regions upstream and downstream of SSR42 using primers EWM110 and EWM113.
Complementation of SSR42.
Complementation was performed using the pCN51 and pCN38 E. coli-S. aureus shuttle vectors for S. aureus strains UAMS-1 and LAC, respectively (16). Briefly, SSR42 and 500 bp upstream and downstream of the transcriptional unit were amplified with primers EWM147/EWM148 and gel purified as described above. Next, the purified insert and pCN51 or pCN38 plasmids were each digested with the KpnI and EcoRI enzymes and gel purified. Ligated plasmid pCN51::SSR42 (pEWM20) or pCN38::SSR42 (pJMM1) was electroporated into S. aureus RN4220, which was grown on TSB agar under selection with chloramphenicol (10 μg ml−1). The plasmids were then electroporated into S. aureus RN4220 and then transferred to S. aureus strain EWM14 (UAMS-1:ΔSSR42) via Φ11 phage-mediated transduction (pEWM20) or to strain LAC:ΔSSR42 via electroporation (pJMM1).
Construction of SSR42.mut plasmid.
Plasmids pJMM2 and pJMM3 are pCN38 and pCN51 derivatives, respectively, containing a mutated SSR42 locus; the transcript's putative ORF has been eliminated by site-specific mutagenesis. To create pJMM2 and pJMM3, the SSR42 locus including 500 bp upstream of the transcriptional unit (corresponding to nucleotides [nt]2540179 to 2538789; S. aureus MRSA252, GenBank) was commercially synthesized with a T-to-A transversion at nt 2539166 and placed into the multicloning site of pUC57 (GenScript USA Inc., Piscataway, NJ). The T → A change created a stop codon at the first amino acid of the putative SSR42 ORF (SSR42.mut). pUC57::SSR42.mut was transformed into E. coli DH5α for propagation. Following plasmid purification, the SSR42.mut insert was liberated via restriction digestion with KpnI and EcoRI. The 1,391-bp insert was gel purified and ligated into KpnI/EcoRI-digested pCN38 or pCN51 (16) to create pJMM2 and pJMM3, respectively. The plasmids were then electroporated into S. aureus RN4220 and then transferred to S. aureus strain EWM14 (UAMS-1:ΔSSR42) via Φ11 phage-mediated transduction (pJMM2) or to strain JMM1 (LAC:ΔSSR42) via electroporation (pJMM3).
Construction of plasmids containing mutagenized SSR42 transcriptional units.
Plasmids pJMM4 and pJMM5 are pCN38 derivatives containing the first (SSR42.1) and second (SSR42.2) halves of the SSR42 transcriptional unit, respectively. To synthesize pJMM4, nt 1 to 445 of SSR42 and the 500 bp upstream of the transcriptional unit were amplified using primers JMM4/JMM5, and the 500 bp downstream of SSR42 were amplified with primers JMM6/JMM7 containing 5′ and 3′ SacII restriction sites, respectively. The resulting fragments were purified and digested with SacII, and the ligated products were subsequently digested with KpnI/EcoRI and inserted into digested plasmid pCN38. Ligated plasmid pCN38::SSR42.1 (pJMM4) was propagated in E. coli DH5α cells, electroporated into S. aureus RN4220, and then transferred to S. aureus LAC:ΔSSR42 cells as described above. Plasmid pJMM5 was constructed using an identical approach utilizing primers JMM4/JMM8 and JMM7/JMM9 to link the 500-bp region upstream to the SSR42 transcriptional unit with nt 446 to 891 of SSR42 and 500 bp downstream of the unit.
Microarray studies.
Ten micrograms of total bacterial RNA from each sample was labeled and hybridized to S. aureus GeneChip arrays by following the manufacturer's recommendations for antisense prokaryotic arrays (Affymetrix, Santa Clara, CA). Average GeneChip signal intensities for biological replicates (two or more) for each condition were obtained from values normalized to the total GeneChip values. Differentially expressed genes exhibiting a 2-fold difference in expression (Student's t test; P < 0.05) in LAC wild-type and pSSR42+ cells in comparison with that in ΔSSR42 cells were determined using GeneSpring 7.2 software (Agilent Technologies, Redwood City, CA) as previously described (3, 4, 6, 52).
qRT-PCR.
qRT-PCR was performed with a MyiQ RT-PCR detection system (Bio-Rad, Hercules, CA). Briefly, 250 ng of total bacterial RNA was reverse transcribed using iScript reverse transcriptase and iScript reaction mix in accordance with the manufacturer's protocol. qRT-PCR amplification of the cDNA was then performed with iQ SYBR green Supermix and 1 pmol of the primers listed in Table 2 in accordance with the manufacturer's recommendations. Amplification was performed with an initial denaturation of 95°C for 10 min and 40 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s. Threshold cycles were calculated using the iQ5 Optical System Software 2.1 (Bio-Rad). Experimental cycle threshold values were normalized to those of 16S rRNA and are presented as n-fold changes in the expression of the gene of interest.
Table 2.
Primers used in this study
| Primer | Nucleotide sequence (5′ → 3′)a | Targetb |
|---|---|---|
| EWM-01 | GGATCCCTATTTTTTAATAAAACCTCAGCACATTATG | spa promoter (F) |
| EWM-05 | GCTAGCCATATGACCTAGTTTACGAATTGAATAAATGTTTTTCTTTTTC | spa promoter (R) |
| EWM-03 | CCGCGGTTGCAACACTCTATTATCATTTTAT | SSR42 complement (F) |
| EWM-04 | CCGCGGGGTAATAGTGAAAAATAAAAGAAAT | SSR42 complement (R) |
| EWM-52 | GCTTTATAAGCAAGGGGTGGTCAAC | pCN51 (F) |
| EWM-53 | GAAAATAAATCTCGAAAATAATAGAGGG | pCN51 (R) |
| EWM-11 | GGGGACCACTTTGTACAAGAAAGCTGGGTTTGCAACACTCTATTATCATTTTAT | SSR42 upstream (F) |
| EWM-111 | GTTTCACCGCGGATCTATCTCTTTCTTTTTGTGTTTA | SSR42 upstream (R) |
| EWM-112 | TAAACGCCGCGGGCTTTATTTTTTAGTAGATTGTTTA | SSR42 downstream (F) |
| EWM-113 | GGGGACAAGTTTGTACAAAAAAGCAGGCTGGTAATAGTGAAAAATAAAAGAAAT | SSR42 downstream (R) |
| EWM-147 | GGTACCTTGCAACACTCTATTATCATTTTAT | SSR42 (F) |
| EWM-148 | GAATTCGGTAATAGTGAAAAATAAAAGAAAT | SSR42 (R) |
| EWM-179 | CCTCAGTTTATGGCGCAAGAGGTTC | cap5A (F) |
| EWM-180 | GTGCGACTTTAACTGCTGTACCGTC | cap5A (R) |
| EWM-221 | GCAGAAGCTAAAAAGCTAAATGATG | spa (F) |
| EWM-222 | GCTCACTGAAGGATCGTCTTTAAGG | spa (R) |
| 16S-L | TAACCTACCTATAAGACTGGGATAA | 16S (F) |
| 16S-R | GCTTTCACATCAGACTTAAAA | 16S (R) |
| JMM-1 | GTGCATTGGACAACGAAATTCACCATCAA | SSR42 (F) |
| JMM-2 | CGCATCTCTTGATGTAAAATCTAAGATGTTTATG | SSR42 (R) |
| JMM-3 | GGGGAGTAATTTTAAGTAATATCTTGTTGCTGCT | SSR42 (R) |
| JMM-4 | GCTAACACCTAACATCAAAGAATATTCATCAA | SSR42 (F) |
| JMM-5 | GGGAGTAATTTTAAGTAATATCTTGTTGCT | SSR42 (R) |
| JMM-6 | GAATTCGGTAATAGTGAAAAATAAAAGAAAT | SSR42 (F) |
| JMM-7 | GTTTCACCGCGGATGCCTGTCTTGATGAAGTTATCTATGAT | SSR42 (R) |
| JMM-8 | TAAACGCCGCGGTTTGTGTTTAAAACACGACTTTGTAGT | SSR42 (F) |
| JMM-9 | GGTACCATTAACATTAATTATCGGAGGTAATTTTATG | SSR42 (R) |
| JMM-10 | GTTTCACCGCGGCTTTATTTTTTAGTAGATTGTTTAAAATTTCTTAGT | SSR42 (R) |
| JMM-11 | TAAACGCCGCGGCCACATCTAAGAAACTCATCTAGAA | SSR42 (F) |
| JMM-12 | GAGCACTTTATCACCAGCAGCATTAG | aur (F) |
| JMM-13 | CGGATTTAGGCAATTCTTTTAATGCTTTGA | aur (R) |
| JMM-14 | GGGCACACTGATTGGAGTAACAGTT | isaB (F) |
| JMM-15 | CCAGAATAAAATTAGCATTATTACCAATATAACCATT | isaB (R) |
| JMM-16 | GGTTATTGGTGGTGCATTCACGGTA | sraP (F) |
| JMM-17 | CCGAACTACTATTTTTCGTTACACTTGTGGAAT | sraP (R) |
| JMM-18 | CACCTGTAAGTGAGAAAAAGGTTGATGAT | lukF-PV (F) |
| JMM-19 | GCCAGAATAAATGTTTCCAGCAGCTTT | lukF-PV (R) |
| JMM-22 | CCGGTACTACAGATATTGGAAGCAATA | hla (F) |
| JMM-23 | CCAGCAATGGTACCTTTCGTTCTAATAA | hla (R) |
| JMM-24 | GGGGCGTGACTCAAAATATTCAATTTGA | hlgC (F) |
| JMM-25 | GGCCATCGCATAGCTTTAACATGATTA | hlgC (R) |
Nucleotides in italics indicate attB sites, and underlined nucleotides represent indicated restriction sites.
F, forward; R, reverse.
Northern blotting.
Northern blotting was performed using a Roche digoxigenin (DIG)-labeled cDNA probe complementary to a 543-bp region of SSR42. To synthesize the probe, UAMS-1 chromosomal template DNA was PCR amplified using primers JMM1 and JMM3, reamplified, and labeled with DIG using the Roche DIG DNA Labeling kit in accordance with the manufacturer's recommendations (Roche Diagnostics, Indianapolis, IN). The probe was boiled for 5 min, placed immediately on ice, and resuspended in 10 ml of DIG Easy Hyb buffer (Roche). Northern blotting was performed by separating 5 μg of total bacterial RNA by gel electrophoresis in a 1.2% agarose gel in 1× FA buffer (20 mM morpholinepropanesulfonic acid [MOPS], 5 mM sodium acetate, 1 mM EDTA, 2% [vol/vol] 37% formaldehyde). Next, the RNA gel was washed twice for 15 min in 20× SSC buffer (3 M NaCl, 300 mM trisodium citrate dehydrate, pH 7.0) and then transferred to a positively charged nylon membrane (Roche) overnight. The RNA was then UV cross-linked to the membrane in the presence of 2× SSC buffer and prehybridized in 10 ml of DIG Easy Hyb buffer at 50°C for 8 h. The expended buffer was then decanted, and 10 ml of the SSR42-DIG probe was added to the membrane and hybridized at 50°C overnight. The probe was again decanted, and the membrane was washed twice for 5 min in 2× SSC containing 0.1% SDS at room temperature and yet another two times for 15 min in 0.1× SSC buffer containing 0.1% SDS at 50°C. The membrane was then washed for 2 min in washing buffer (0.1 M maleic acid, 0.15 M NaCl, 0.3% [vol/vol] Tween 20, pH 7.5) and then blocked for 30 min in 1× Blocking Reagent (Roche). Next, the membrane was washed for 1 h with antibody solution (a 1:10,000 dilution of anti-DIG-alkaline phosphatase Fab fragments [Roche] in 1× Blocking Buffer). The membrane was then incubated with CSPD reagent (Roche), and SSR42 products were detected by autoradiography.
Protein A Western blotting.
Western blotting was used to measure the protein A (Spa) levels of S. aureus strains U151 (UAMS-1 pCN51), EWM15 (ΔSSR42 pCN51), and EWM16 (ΔSSR42 pEWM20) as previously described (42). Briefly, each strain was grown as described above, 5-ml samples of cell suspensions were normalized to equal cell numbers and pelleted by centrifugation, and cell pellets were washed twice in sterile phosphate-buffered saline (PBS) solution and resuspended in 100 μl sterile PBS containing 26% raffinose (Sigma-Aldrich). To liberate protein A from the cell surface, cell suspensions were incubated with 20 μM lysostaphin at 37°C for 2 h with shaking. Cellular debris was removed by centrifugation at 20,000 × g for 30 min, centrifugation was repeated, and the final protein concentration was determined using a Bradford assay. Next, 750 ng total protein was separated by 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane. The membrane was blocked using Tris-buffered saline with 1% Tween 20 (TBST) containing 5% milk–5% bovine serum albumin (Thermo Fisher) for 1 h at room temperature. Membranes were blotted with peroxidase-conjugated Affinity-Pure rabbit anti-sheep IgG (1:20,000 dilution; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and washed six times with TBST for 5 min each, and Spa protein levels were visualized using the enhanced-chemiluminescence Western blotting system (GE Healthcare).
Exoprotein profiles.
Exoproteins were produced and processed as described before (8). Briefly, S. aureus strains were grown in TSB overnight at 37°C with shaking at 225 rpm. The overnight cultures were then diluted 1:100 into a new 10-ml culture in a 50-ml flask. Cultures were grown to stationary phase and then normalized to the same OD600. Bacterial cells were collected by centrifugation at 400 × g for 15 min, and the proteins in the supernatant were precipitated using 10% (vol/vol) trichloroacetic acid at 4°C overnight. The precipitated proteins were sedimented by centrifugation, washed, dried, resuspended in 1× SDS loading buffer, and boiled for 10 min. Proteins were separated by 15% (wt/vol) SDS-PAGE and stained with Coomassie blue.
Immunoblotting.
Immunoblot assays were performed as described previously (8, 22). Briefly, S. aureus cultures were grown and supernatants were prepared as described for the exoprotein profile protocol. For cytoplasmic extracts, cell pellets were treated with lysostaphin and the resulting protoplasts were disrupted using a FastPrep Tissue and Cell Homogenizer (MP Biomedicals, Solon, OH). Proteins were resolved by 10 or 15% SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated primary antibody. An Alexa Fluor 680-conjugated anti-rabbit antibody was used as a secondary antibody. Membranes were then scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Generation of luciferase reporter plasmids and reporter assay.
The promoterless vector pXen-1, which contains the Photorhabdus luminescens luxABCDE operon, was obtained from Xenogen (25). The promoter regions of hla, lukSF-PV, and hlgCB were cloned into pXen-1 to make the transcriptional reporter plasmids. Plasmids were subsequently transformed into strain USA300-LAC (LAC) and grown as indicated above, and luminescence was recorded with a Perkin-Elmer Envision 2103 Multilabel Reader.
Quantification of hemolysis activity.
Quantification of hemolysis was performed as previously described but with slight modifications (9). Briefly, supernatants from overnight cultures were collected, standardized according to culture densities, and filter sterilized with a 0.45-μm-pore-size filter. Filtered supernatants were then combined with 2% rabbit blood (BD Diagnostic Systems, Hunt Valley, MD) in 0.9% NaCl supplemented with 10 mM Tris-HCl (pH 7.5). The supernatant-rabbit blood suspension was then incubated for 15 min at 37°C, and unlysed cells were removed via centrifugation. The amount of hemolysis activity was determined by spectrophotometrically measuring the OD (A405) of the supernatant fluid, and all samples were normalized to wild-type densities.
Ex vivo infection assays.
Blood samples were obtained from anonymous healthy donors as buffy coats (New York Blood Center). The New York Blood Center obtained written informed consent from all of the participants involved in this study. Because all of the samples were sent as anonymous, the Institutional Review Board at the New York University School of Medicine determined that our study was exempt from further ethics approval requirement. Primary human polymorphonuclear leukocytes (PMNs) were purified as described previously (22, 57). PMN-dependent killing of S. aureus was examined as described previously (22, 57, 58). Briefly, S. aureus was cultured to stationary-phase growth as described above. The bacteria were then washed with 1× PBS and added to approximately 1 × 105 pooled primary PMNs from three healthy donors at a multiplicity of infection (MOI) of 10 and seeded into individual wells of a 96-well flat-bottom tissue culture-treated plate. Cells were cultured in RPMI supplemented with 10% fetal bovine serum and incubated at 37°C with 5% CO2 for 3 h. Neutrophils were then lysed with 0.1% saponin, and the viability of S. aureus cells was determined by serial dilution onto fresh agar plates and counting of CFU.
Mouse model of skin and soft tissue infection.
Overnight cultures of S. aureus strains in TSB were diluted 1:100 and grown at 37°C with gyratory shaking until mid-exponential phase. Bacteria were washed 3× and adjusted to the desired inocula by OD in PBS. Murine skin infections were carried out as previously described (60). Eight-week-old female Swiss Webster mice were injected subcutaneously with 1 × 107 CFU of S. aureus LAC, ΔSSR42, or SSR42-complemented cells (n = 5 per strain). At 5 days postinfection, the mice were imaged for documentation of the pathology caused by the different strains. Following that, the mice were euthanized and abscesses were harvested for bacterial enumeration.
RESULTS
S. aureus SSRs.
As part of a comprehensive RNA profiling study, 142 SSRs were identified as a class of unique RNA molecules with relatively short sequences (average of 390 nt in length) in comparison with mRNAs. They are stabilized only under certain growth and/or environmental stress conditions and do not contain an obvious ORF (3, 4, 50, 52). Of these SSRs, six are expressed and stabilized primarily during stationary-phase growth, corresponding to a period during which the organism's virulence factors undergo dramatic changes in expression (50). We hypothesized that one or more of these six stationary-phase SSRs may play a role in modulating growth phase-dependent virulence factor expression.
To prioritize SSRs for further evaluation of the ability to regulate virulence factors, the sequence of each stationary-phase SSR transcriptional unit was determined by 5′ and 3′ rapid amplification of cDNA ends (RACE) (50). Each SSR transcriptional unit was subsequently compared with S. aureus strains in the National Center for Biotechnology Information database (NCBI; http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi) using the Basic Local Alignment Search Tool (BLAST) to determine their overall sequence conservation across S. aureus strains (2). SSR42 was found to be the most highly conserved (98% sequence identity; data not shown), suggesting that it might represent an important genetic locus.
SSR42 affects S. aureus virulence factor expression.
The expression and stability of SSR42 were verified by growing S. aureus strain UAMS-1 (USA200 PFT) to exponential or stationary phase and measuring the transcript's titers following transcriptional arrest. As expected, Northern blotting and qRT-PCR revealed that SSR42 was expressed and stabilized predominantly during stationary-phase growth (Fig. 1A and B). The transcript exhibited a half-life of <5 min during exponential phase but was stabilized during stationary-phase growth with a half-life of >30 min, which directly correlated with previous results. To empirically determine whether S. aureus SSR42 exhibits any regulatory effects, the SSR42 locus of S. aureus strain UAMS-1 was deleted. The resulting strain, EWM14 (ΔSSR42), was subsequently used to generate two additional strains, EWM15 (UAMS-1ΔSSR42 harboring the shuttle vector pCN51) and complementation strain EWM16 (UAMS-1ΔSSR42 harboring pEWM20 [pCN51 containing a wild-type copy of SSR42]). Using microarrays, the transcriptional profiles of these strains were compared to that of wild-type cells (harboring vector pCN51) during both exponential- and stationary-phase growth.
Fig 1.
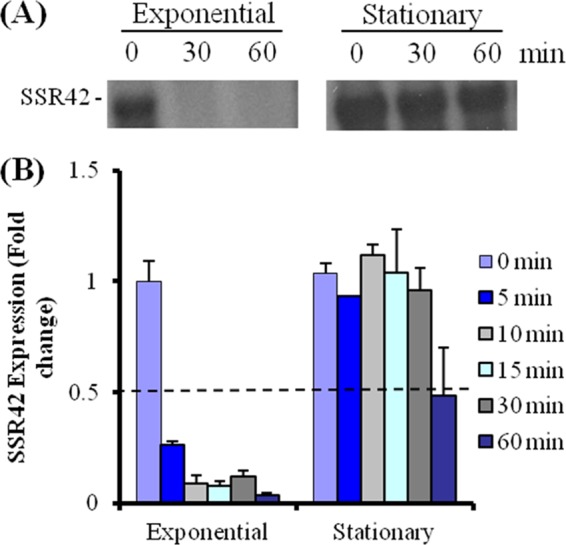
SSR42 expression and stability. (A) Northern blotting measurements of SSR42 transcript abundances of RNA extracted from exponential- and stationary-phase wild-type S. aureus at 0, 30, or 60 min after transcriptional arrest. (B) qRT-PCR-based measurements of SSR42 transcript titers at 0, 5, 10, 15, 30, and 60 min after transcriptional arrest during the indicated growth phase. Error bars represent 1 standard deviation from the mean.
Microarray analyses revealed that SSR42 did not exhibit any regulatory effects during exponential-phase growth (data not shown), but that the molecule affected the expression of 82 mRNA species during stationary-phase growth. More specifically, two RNA species were upregulated (≥2-fold; Student's t test; P ≤ 0.05) in an SSR42-dependent manner during stationary-phase growth. As expected, SSR42 was one of those two transcripts; the molecule was undetectable in ΔSSR42 cells but was detectable at nearly equal amounts in wild-type and SSR42-complemented cells, verifying that the three strains represent an appropriate strain set to evaluate what, if any, regulatory effects the SSR42 locus may elicit in S. aureus cells. The second transcript was cap5B, a member of the capsule biosynthesis operon (cap). Although other members of the cap operon were not significantly differentially expressed, reduction of the stringency of the test statistic to P ≤ 0.1 identified two additional SSR42-regulated genes, cap5D and cap5O, both of which are members of the capsule operon. As shown in Fig. 2, qRT-PCR and qualitative RT-PCR verified that the transcript titers of capsule cap5A correlated with the presence of SSR42, suggesting that the molecule may be required for wild-type levels of capsule expression.
Fig 2.
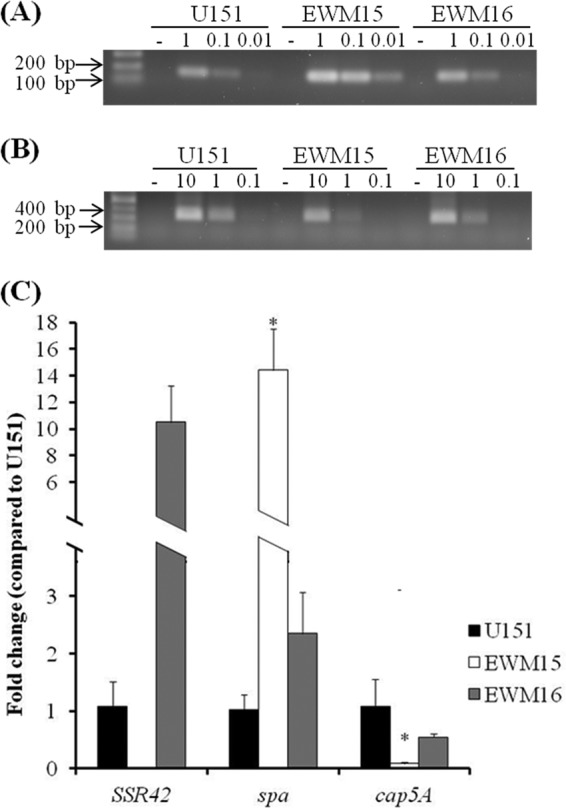
Effects of SSR42 on spa and cap5A transcript titers. Shown are qRT-PCR and qualitative RT-PCR results for wild-type (U151/pCN51), SSR42 deletion (EWM15; ΔSSR42/pCN51), and SSR42-complemented (EWM16; ΔSSR42/pEWM20) cells. Qualitative RT-PCR was used to measure spa (A) and cap5A (B) mRNA levels. Panel C shows qRT-PCR-based measurements of SSR42, spa, and cap5A; all values were normalized to 16S rRNA and are expressed as relative to wild-type (U151) levels, which were set to 1.0. Error bars represent standard deviations (n = 3). The asterisk indicates a statistically significant difference (Student's t test; P < 0.001).
While SSR42 induced the expression of only two stationary-phase mRNA species, the molecule repressed the expression of 80 stationary-phase transcripts; each exhibited higher titers (≥2-fold; Student's t test; P ≤ 0.05) in stationary-phase cells lacking SSR42 than in wild-type or SSR42-complemented cells (see Table S3 in the supplemental material). Among these were transcripts coding for several known or putative S. aureus cell surface-associated virulence factors, including protein A (spa; 8.5-fold repression), IgG-binding protein (sbi; 4-fold repression), and the cell wall-associated protein (SA0119; 7-fold repression). As shown in Fig. 2, the effects of SSR42 on spa transcript titers were experimentally verified by qRT-PCR and qualitative RT-PCR. Western blotting revealed that the SSR42 deletion-containing strain exhibited an approximately 10-fold increase in protein A production in comparison with both wild-type and SSR42-complemented cells (Fig. 3), indicating that SSR42's regulatory effects result in a biologically significant change in protein production. Taken together, these results suggest that SSR42 is an S. aureus strain UAMS-1 regulatory RNA that predominantly represses the expression of many genes, including several cell surface virulence factors, during stationary-phase growth.
Fig 3.

Effect of SSR42 on protein A production. (A) Liberated cell surface proteins from stationary-phase (UAMS-1 pCN51), EWM15 (ΔSSR42 pCN51), EWM16 (ΔSSR42 pEWM20), and KLA16 (UAMS-1 Δspa) cells were separated by SDS-PAGE and stained with Coomassie blue for assessment for equally loaded amounts of protein. (B) Western blotting-based detection of Spa levels of stationary-phase U151. Commercially available Spa protein was used as a positive control.
SSR42 is expressed and stabilized predominantly during stationary-phase growth in a CA-MRSA strain.
To determine whether SSR42's regulatory effects are conserved across S. aureus strains, we expanded our studies to strain LAC, a representative of the USA300 PFT that is the leading etiologic agent of CA-MRSA infections in the United States (32, 41, 43, 44). As a first step, we evaluated whether the molecule's expression and RNA turnover properties mimic those of S. aureus strain UAMS-1. Accordingly, S. aureus strain LAC was grown to early exponential or stationary phase and the transcript titers were measured following transcriptional arrest by qRT-PCR. As was observed for strain UAMS-1, SSR42 was found to be expressed and stabilized predominantly during stationary-phase growth within S. aureus strain LAC. SSR42's half-life was determined to be between 5 and 10 min during exponential-phase growth, whereas the molecule's half-life increased to ≥30 min during stationary-phase growth (Fig. 4). These results suggest that the expression and RNA turnover properties of SSR42 are conserved across two genetically distinct, clinically relevant S. aureus backgrounds.
Fig 4.
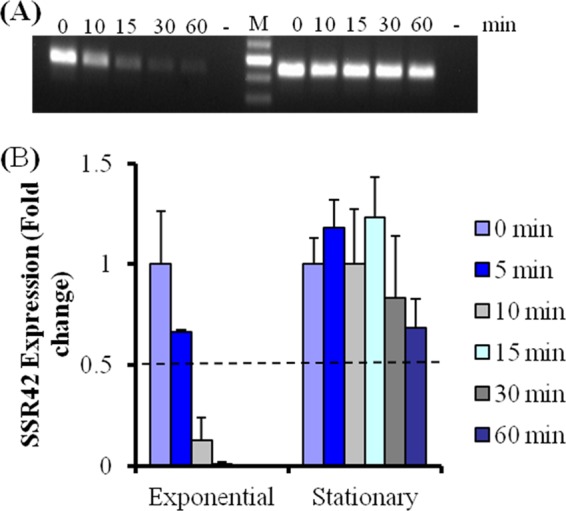
Expression profile of SSR42 in S. aureus strain LAC. Detection of SSR42 titers using qualitative RT-PCR (A) and qRT-PCR (B) at various time points after transcriptional arrest during the indicated growth phase. Error bars represent 1 standard deviation (n = 3).
SSR42 affects S. aureus virulence factor transcript titers in a strain of CA-MRSA.
Based on the conservation of SSR42 expression across both strains, we next evaluated whether SSR42's regulatory effects are also conserved. Accordingly, the SSR42 locus was deleted from S. aureus strain LAC and microarrays were used to compare the expression properties of wild-type, ΔSSR42, and SSR42-complemented cells during both the exponential and stationary phases of growth.
Consistent with what was observed with S. aureus strain UAMS-1, microarray results revealed that SSR42 acts predominantly as a repressor of gene expression within S. aureus strain LAC during stationary-phase growth. Indeed, only 12 exponential-phase transcripts were regulated in an SSR42-dependent manner, whereas 90 transcripts were regulated by SSR42 during stationary-phase growth. Among the stationary-phase SSR42-regulated transcripts, 79 (88%) were repressed by the molecule; their transcript titers decreased in wild-type and complemented cells (P < 0.05) in comparison with ΔSSR42 cells (see Table S4 in the supplemental material). Included among this list of transcripts were three known virulence factors, sraP (encodes a serine-rich cell surface protein), aur (encodes aureolysin), and isaB (encodes immunodominant surface antigen B), which were increased 6.1-, 6.8-, and 4.3-fold, respectively, in ΔSSR42 cells in comparison with wild-type cells and were restored to wild-type levels in complemented cells (53, 54). As shown in Fig. 5A, these results were confirmed by qRT-PCR.
Fig 5.
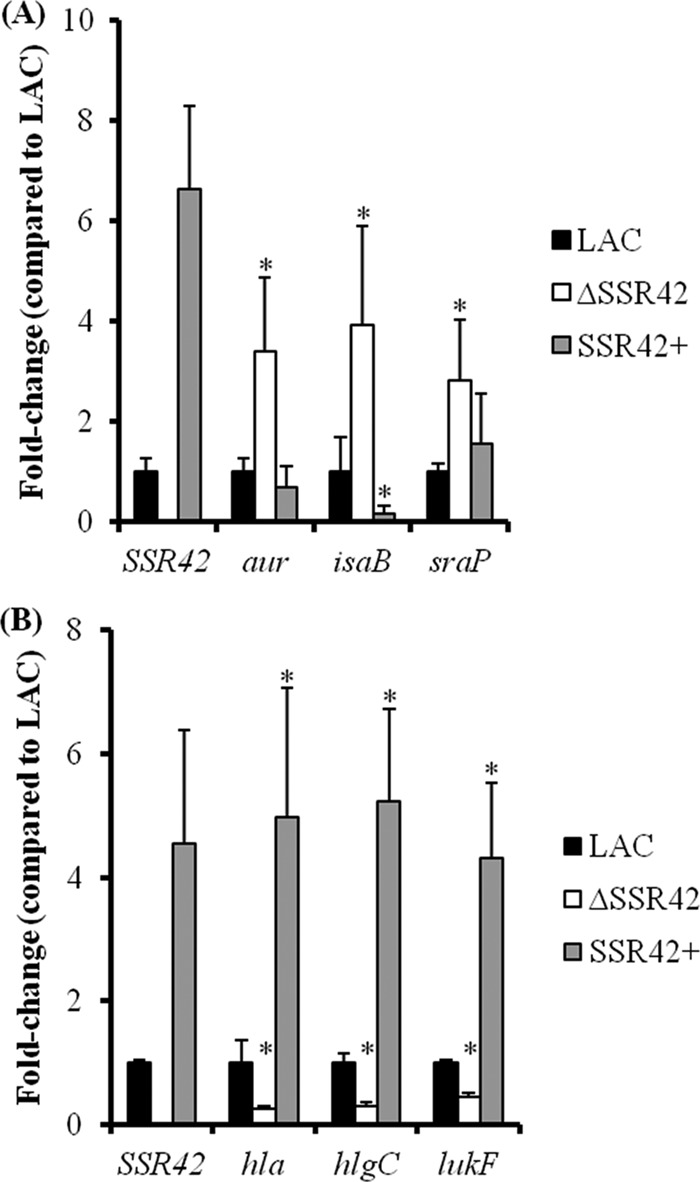
SSR42 affects the expression of several virulence factors in S. aureus strain LAC during stationary-phase growth. Virulence factor mRNA titers were determined by qRT-PCR for genes found to exhibit increased (A) or decreased (B) expression levels in ΔSSR42 cells using GeneChips. Results are presented as a n-fold changes in ΔSSR42 (white) and SSR42+ (gray) mRNA titers with respect to LAC (black) levels, which were set to 1.0. An asterisk represents statistical significance (P < 0.05) determined by multiple comparisons of means using the Bonferroni-Holm correction. Error bars represent 1 standard deviation from the mean of each sample.
Although the molecule functioned predominantly as a repressor of stationary-phase transcripts in both strain backgrounds, SSR42 also modestly induced the expression of 11 genes (≥1.5-fold; Student's t test; P ≤ 0.05) during the stationary-phase growth of strain LAC (see Table S5 in the supplemental material). Of interest, many of these genes have been associated with the organism's ability to cause disease and have been hypothesized to contribute to USA300 hypervirulence, suggesting that SSR42 may contribute to the lineage's increased ability to cause disease. More specifically, the mRNA titers for two extracellular virulence factors were upregulated in the presence of SSR42; hla (α-hemolysin) and hlgA (γ-hemolysin) titers increased 1.6- and 1.8-fold, respectively, in wild-type and complemented cells compared to those in ΔSSR42 cells. As shown in Fig. 5B, qRT-PCR confirmed that SSR42 induces the expression of α-hemolysin and γ-hemolysin approximately 4.1-fold. Because SSR42 appeared to affect the expression of extracellular factors and two known USA300 extracellular virulence factors are not represented on the commercially available microarray used in this study (lukA and PVL), qRT-PCR studies were expanded to evaluate whether these genes were also regulated in an SSR42-dependent manner. Results revealed that SSR42 did not affect lukA expression (data not shown) but that leukotoxin lukF-PV expression was regulated in an SSR42-dependent manner (Fig. 5B). Western blotting confirmed that SSR42's regulatory effects correlated with changes in α-hemolysin, γ-hemolysin, and LukFS/PVL protein production (Fig. 6A).
Fig 6.
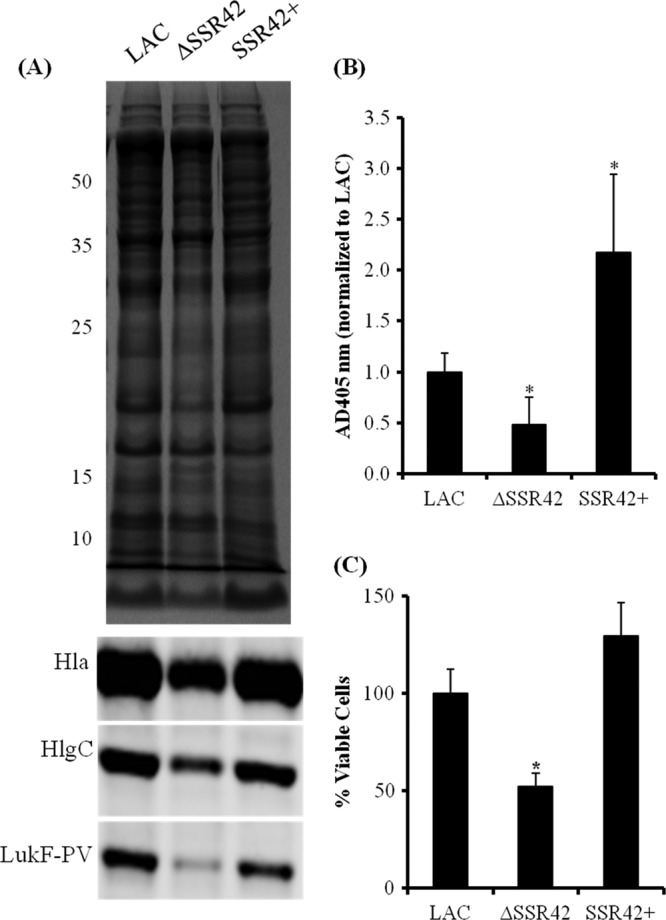
SSR42 affects extracellular virulence factor expression and pathogenesis phenotypes. (A) Stationary-phase culture supernatants from LAC, ΔSSR42, and SSR42+ cells were separated using SDS-PAGE and stained with Coomassie blue for assessment for equally loaded amounts of protein. The supernatants were subsequently probed for LukF-PV, Hla, and HlgC. (B) Hemolytic capabilities of stationary-phase culture supernatants from LAC, ΔSSR42, and SSR42+ were quantified as measurements of A405 due to heme liberated from lysed rabbit erythrocytes. Error bars represent 1 standard deviation from the mean (n = 5). (C) S. aureus neutrophil survival assay assessed the abilities of LAC, ΔSSR42, and SSR42+ cells to survive in the presence of primary human PMNs. Bacterial cells were cultured in the presence of PMNs (MOI = 10) for 3 h, and viable S. aureus cells were determined by serial plating and CFU counting. Error bars represent the standard error of the mean (n = 3). In panels B and C, asterisks represent statistical significance (P < 0.05) determined by multiple comparisons of means with a Bonferroni-Holm correction. Error bars represent 1 standard deviation from the mean for each sample. The values to the left of panel A are molecular sizes in kilodaltons.
Despite the similarities in SSR42 sequence (100% identity), expression/stability characteristics, and regulatory phenotypes, SSR42 affected the expression of substantially different sets of genes within S. aureus strains UAMS-1 and LAC. Indeed, only three genes exhibited conserved SSR42 regulatory effects in both backgrounds (all were repressed by SSR42 during stationary phase): SAR2290/SAUSA300_2159 (aldo-ketose reductase family), SAR2468/SAUSA300_2326 (hypothetical transcriptional regulator belonging to the AraC family), and SAR2535/SAUSA300_2390 (glycine betaine/carinitine/choline transporter). Growth curve analyses did not reveal any overt differences between the growth characteristics of the two strains or their derivatives (see Fig. S1 in the supplemental material). Thus, the limited overlap between sets of SSR42-regulated genes of the two strains suggests that the molecule's regulatory capacity is dependent on the genetic background in which it resides, as has been observed for numerous other S. aureus regulatory loci, such as RNAIII. Nonetheless, taken together, these results indicate that SSR42 is a regulatory RNA that pleiotropically affects the expression of many S. aureus genes, including several well-established virulence factors.
SSR42 affects S. aureus hemolysis.
Given the importance of the S. aureus USA300 lineage from a health care perspective and that the molecular mechanisms by which this background elicits a hypervirulent phenotype are poorly understood, we set out to further characterize the effects of SSR42 on LAC's pathogenic potential. Accordingly, the observation that SSR42 induces α- and γ-hemolysin production in strain LAC (Fig. 6A) prompted us to investigate whether the molecule affects the organism's ability to lyse erythrocytes, a function thought to contribute to the invasiveness of USA300 isolates (40). In comparison with wild-type and SSR42-complemented cells, the LACΔSSR42 strain exhibited diminished zones of clearance when grown on rabbit blood agar, indicating that the molecule is required for wild-type levels of hemolysin production (data not shown). To quantify the levels of hemolysis of each strain, filtered supernatants from stationary-phase cultures of wild-type, ΔSSR42, and SSR42-complemented cells were incubated in the presence of 2% rabbit blood and then erythrocyte lysis was measured spectrophotometrically as a function of heme liberation (A405) as previously described (9). As shown in Fig. 6B, ΔSSR42 supernatants exhibited a significant decrease (P < 0.05) in hemolysis in comparison with the supernatants of wild-type cells. Moreover, supernatants from SSR42-complemented cells, which slightly overexpress SSR42 (Fig. 5B), exhibited an approximately 2.5-fold increase in hemolysis (P < 0.05) compared to that of wild-type cells. Taken together, these data suggest that SSR42 affects S. aureus strain LAC's potential to lyse erythrocytes.
SSR42 affects S. aureus PMN survival.
Leukotoxins have been shown to target and kill PMNs and are thought to mediate S. aureus survival in the presence of these cells (1, 22, 38, 39, 56). Accordingly, we hypothesized that because SSR42 affects γ-hemolysin and PVL expression, it may also affect S. aureus strain LAC's ability to survive in the presence of primary human PMNs. To test this possibility, human primary PMNs were infected with wild-type, ΔSSR42, or SSR42-complemented cells at an MOI of 10:1 and bacterial survival was measured after 3 h by plating for CFU counts. As shown in Fig. 6C, the results revealed that the ΔSSR42 strain exhibited an approximately 50% reduction (P < 0.05) in comparison with both wild-type and SSR42-complemented cells, suggesting that the molecule is required for wild-type levels of resistance to primary neutrophils.
SSR42 effects S. aureus pathogenesis in a murine model of skin and soft tissue infection.
The ability of S. aureus USA300 to initiate invasive infection via the formation of skin and soft tissue infections is well recognized and is hypothesized to be mediated by the lineage's expression of extracellular virulence factors (31, 35, 57). Thus, we investigated whether SSR42 affects strain LAC's ability to cause skin abscesses. Mice were examined 5 days after intradermal infection with 107 CFU of each strain tested. Upon gross examination of the caudal region of the mice, animals infected with the SSR42-complemented strain exhibited increased signs of pathology in comparison with both the LAC (wild-type) and ΔSSR42 strains (Fig. 7A). This observation was not surprising, given that SSR42 was complemented on a high-copy-number plasmid, which resulted in increased expression of SSR42 by the complement strain compared to that by the wild type. These observations are consistent with the increased production of virulence factors by this strain. While the pathology due to the wild-type strain was not as striking, quantification of the bacterial burden in each abscess revealed that the strain with SSR42 deleted was attenuated in comparison with the wild-type and SSR42-complemented strains (∼5-fold lower bacterial burden; P < 0.05; Fig. 7B). Taken together, these data imply a pathogenic role for SSR42 in an infection setting.
Fig 7.

SSR42 is required for full virulence in a murine skin infection model. The effect of SSR42 in a murine skin infection model was examined by subcutaneous injection of female Swiss Webster mice (n = 5 for each strain) with 1 × 107 CFU of S. aureus LAC, ΔSSR42, or SSR42-complemented (SSR42+) cells. (A) At 5 days postinfection, photographs of the caudal regions of the mice were used to examine the gross pathology of mice infected with each strain. (B) Abscesses were homogenized, and bacterial burdens per abscess were determined by serial plating. Error bars represent the standard error of the mean for each strain.
Characterization of SSR42's mechanism of action.
Because SSR42 appeared to be an important regulator within strain LAC, we initiated a preliminary assessment of the mechanism(s) by which it regulates the strain's virulence factor expression. As shown in Fig. 8A, SSR42 is an 891-nt RNA molecule that maps to an S. aureus intergenic region. Although the SSR42 transcriptional unit is annotated as an intergenic region, detailed analysis indicated that it does contain a small (29-amino-acid) putative ORF, beginning with the alternative start codon TTG at position 524 and ending with a stop codon at position 614 (Fig. 8B). BLAST analysis of the putative amino acid product indicated that it did not code for a protein with any discernible function or significant homology to a known protein. To distinguish whether the SSR42 transcript and/or putative protein product accounted for the aforementioned regulatory effects, we created plasmid pJMM3, which harbors the SSR42 promoter region and the SSR42 transcriptional unit containing a T → A transversion in the second position of the start codon, thereby resulting in a stop codon and eliminating the putative ORF. pJMM3 was transferred to LACΔSSR42, and the regulatory effect of the mutated SSR42 transcript (SSR42.mut) was measured using microarrays. As shown in Fig. 8C, the transcriptional profile of cells expressing SSR42.mut was virtually identical to that of cells expressing wild-type copies of the transcript. Further, as elaborated below, ΔSSR42 cells expressing the second half of SSR42, which includes the putative 29-amino-acid sequence, did not exhibit regulatory effects. Taken together, these results established that the RNA molecule, as opposed to a putative SSR42-encoded protein, was responsible for the transcript's regulatory effects.
Fig 8.

Locus profile of SSR42. (A) Chromosomal architecture of the SSR42 locus of S. aureus strain USA300_FPR3757 (contains only 11 single nucleotide polymorphisms with respect to strain LAC [32]). (B) SSR42 transcript sequence as determined by 5′ and 3′ RACE. Underlined is the putative 29-amino-acid ORF beginning with the alternative start codon TTG (at position 527). (C) SSR42 deletion-containing LAC cells were transformed with a plasmid-borne copy of SSR42 containing a T-to-A transversion at the first codon of the transcript's putative 29-amino-acid ORF to create a stop codon at this site. Consequently, the production of any putative amino acid product was eliminated. Transcriptional analysis revealed that SSR42 deletion-containing cells complemented with either a wild-type copy (SSR42+) or the mutated copy (SSR42.mut+) were capable of complementing the regulatory effects of the RNA molecule.
Many regulatory RNAs, including RNAIII, modulate gene expression by directly binding to the transcripts that they regulate and affecting the target molecule's stability and/or translation potential. However, gel mobility shift assays and RNA turnover studies indicated that SSR42 does not directly bind to the virulence factor transcripts that it regulates nor does the molecule affect their RNA degradation properties (data not shown). Thus, it is likely that the molecule affects the expression and/or activities of an upstream regulatory molecule which, in turn, regulates virulence factor gene expression. As a first test of that hypothesis, transcriptional reporter assays were performed to establish whether SSR42 affects the transcript synthesis of S. aureus virulence factors. The endogenous promoter regions of hla, hlg, and lukSF were fused to a promoterless luciferase operon of P. luminescens and subsequently transformed into wild-type LAC and LACΔSSR42 cells; the promoter activity of each gene was then measured as a function of bioluminescence. In comparison with the promoter activities of hla, hlgCB, and lukSF in wild-type cells, those in ΔSSR42 cells were significantly reduced during stationary-phase growth, supporting the prediction that SSR42 indirectly affects the expression of these toxins (Fig. 9).
Fig 9.
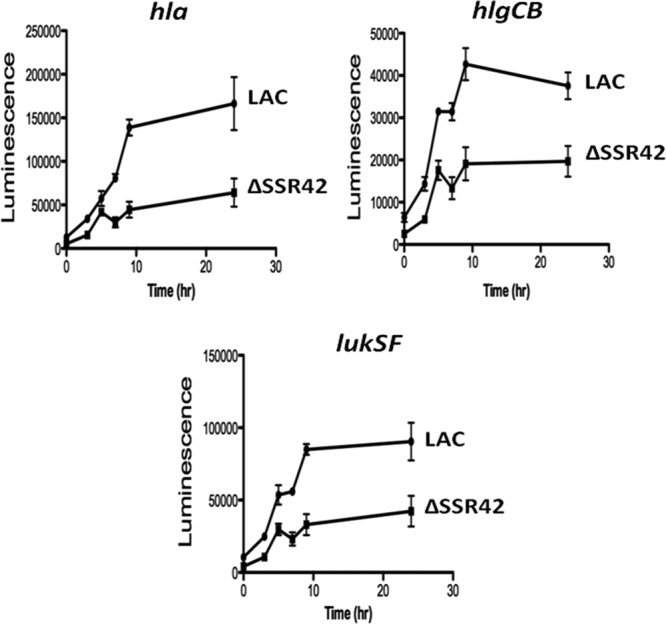
SSR42 is required for full activity of extracellular virulence factor promoters. The indirect effects of SSR42 on virulence factor expression were examined by using transcriptional fusions of virulence factor promoters to a luciferase bioluminescence reporter as described in Materials and Methods. Luminescence resulting from each fusion construct was measured in LAC (circle) and ΔSSR42 (square) cells over a period of 24 h. Data points represent the average of three samples, and error bars represent 1 standard deviation from the mean.
Full-length SSR42 is required for regulatory activity.
In preliminary efforts to identify the SSR42 sequences and/or structural elements that modulate the molecule's stability and regulatory functions, we measured the RNA turnover properties and regulatory capacity of fragments of SSR42. We individually linked plasmids containing bases 1 to 445 (SSR42.1) or 446 to 891 (SSR42.2), corresponding to the first and second halves of SSR42, respectively, to the predicted SSR42 endogenous promoter (500 bp upstream of the transcriptional unit) and transformed them into LACΔSSR42 cells, creating strains JMM4 (ΔSSR42 SSR42.1+) and JMM5 (ΔSSR42 SSR42.2+).
To determine the expression characteristics of each fragment, strains JMM4 and JMM5 were grown to exponential or stationary phase and qRT-PCR was used to measure the transcript levels and RNA turnover properties of each SSR42 fragment as described above. Results revealed that during exponential-phase growth, both halves of the transcript mimicked full-length SSR42 and were rapidly degraded (half-life, ≤5 min; data not shown). Conversely, while the first half of the transcript was rapidly degraded (half-life, ≤5 min) during stationary-phase growth, the second half of the transcript (SSR42.2) was stabilized (half-life, ≥30 min), suggesting that nt 446 to 891 are required for the stationary-phase-specific stabilization of full-length SSR42 (Fig. 10A). It should also be noted that SSR42.1 exhibited significantly (approximately 100-fold) decreased titers in comparison with those of ΔSSR42 cells complemented with full-length SSR42 or SSR42.2 but was still above the detection limit of the system and present at 1,000-fold greater levels than the measured background signal of the SSR42 deletion-containing strain (Fig. 10B). Sequencing of the promoter region within the complementation vector pJMM4 did not identify mutations that may have contributed to the difference in expression between SSR42.1 and SSR42.2. Thus, it is likely that the decreased stability of SSR42.1 accounts for the significantly lower titers of the RNA molecule.
Fig 10.
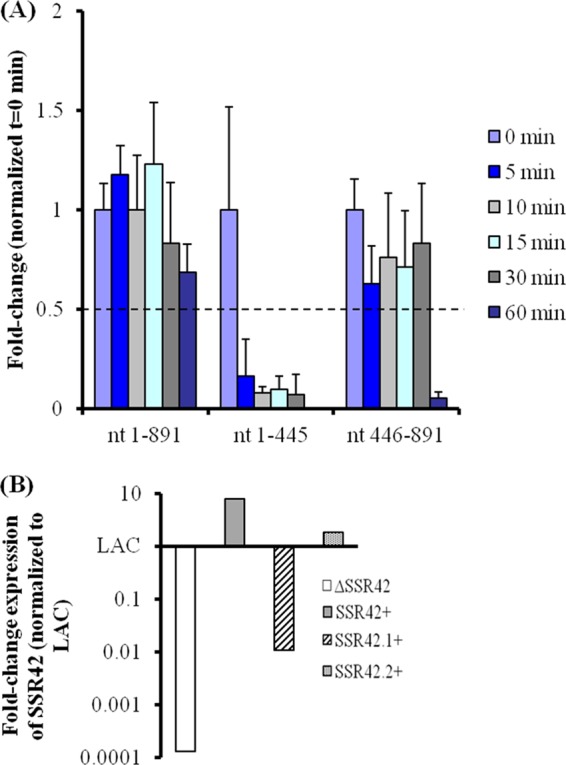
Stability of 5′ and 3′ fragments of SSR42. Bases 1 to 445 and 446 to 891 were individually linked to the SSR42 promoter region and expressed in LACΔSSR42 cells. (A) RNA titers of full-length SSR42 and the 5′ and 3′ fragments of SSR42 were measured by qRT-PCR at 0, 5, 10, 15, 30, and 60 min after transcriptional arrest. Values for each segment of SSR42 were normalized to their respective 0-min time points. Error bars represent 1 standard deviation from the mean (n = 3). (B) Relative expression levels in LACΔSSR42 cells complemented with full length SSR42 (gray) or bases 1 to 445 (diagonal) or 446 to 891 (punctate) of SSR42 in comparison with those in wild-type strain LAC cells.
We next evaluated the regulatory effects of SSR42.1 and SSR42.2 by using qRT-PCR to determine whether each half of SSR42 could complement the expression properties of JMM1 (LACΔSSR42) cells. As described earlier, SSR42 deletion results in increased expression of aur and sraP and decreased expression of hla, hlgC, and lukF within strain LAC during stationary-phase growth (Fig. 5). While the expression of each of these genes could be restored to wild-type levels via complementation with full-length SSR42, neither half of the molecule exhibited a similar phenotype, suggesting that the entire SSR42 transcript is required for its regulatory functions (Fig. 11). The only exception was isaB, which was not complemented by SSR42.1+-expressing cells but was complemented by SSR42.2+ expression (P < 0.05), indicating that the second half of SSR42 may harbor a direct or indirect regulatory capacity for controlling isaB gene expression. Taken together, although the second half of the transcript appears to mediate the regulatory RNA's stationary-phase stability, the full-length molecule is likely to be required for SSR42 regulatory effects.
Fig 11.
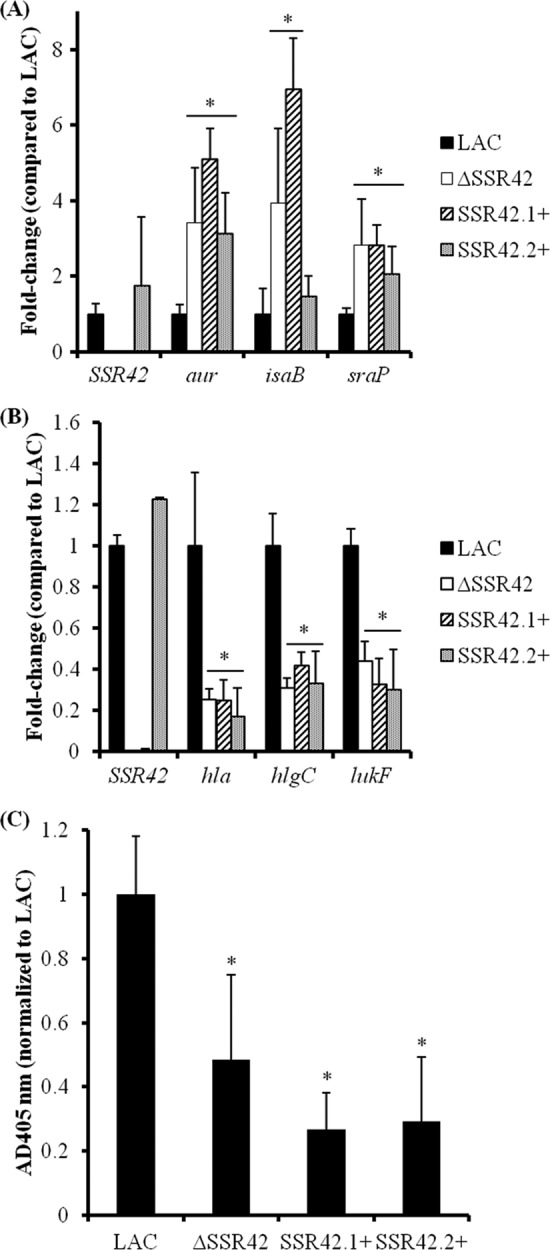
Full-length SSR42 is required for full regulatory capacity. LACΔSSR42 cells were complemented with full-length SSR42 or bases 1 to 445 or 446 to 891 of SSR42 and evaluated for the ability of each construct to complement virulence factor expression in USA300. Virulence factor mRNA titers were determined by qRT-PCR for genes found to exhibit increased (A) or decreased (B) expression levels in ΔSSR42 cells with GeneChips. Results are presented as n-fold changes in ΔSSR42 (white), SSR42.1+ (diagonal), or SSR42.2+ (punctate) mRNA titers over LAC (black) levels, which were set to 1.0. An asterisk represents a statistically significant difference between LAC and the complemented strains (P < 0.05) determined by multiple comparisons of means using the Bonferroni-Holm correction. Error bars represent 1 standard deviation from the mean of each sample (n = 5). (C) Hemolytic capabilities of supernatants from stationary-phase LAC, ΔSSR42, and SSR42+ cell cultures were quantified by the measurement of A405 due to heme liberated from lysed rabbit erythrocytes. Error bars represent 1 standard deviation from the mean of each sample (n = 5).
DISCUSSION
Regulatory RNAs are a newly described class of regulatory molecules that have been shown to have pleiotropic effects and have been associated with the modulation of bacterial adaptation to changing environmental conditions, plasmid maintenance, bacteriophage resistance, and pathogenesis (reviewed in references 55 and 59). Most bacterial regulatory RNAs can be classified on the basis of their molecular characteristics as cis-encoded (antisense), trans-encoded, or protein-modulatory RNAs.
cis-encoded antisense RNAs are produced adjacent to and in an antisense orientation with respect to the transcripts that they regulate, typically affecting the expression of one or, in polycistronic instances, a few genes. As the name implies, they exhibit considerable sequence complementarity (often 75 nt or more) with target transcripts; binding can promote degradation and/or repress translation. trans-encoded RNAs also act primarily by base pairing to target transcripts, but in comparison with cis-encoded RNAs, there is little correlation between the chromosomal locations of the regulatory RNA and its target mRNA. trans-encoded regulatory RNAs tend to have limited complementarity to their target transcripts and consequently bind to—and affect the stability and/or translation of—a broader number of mRNA molecules. Protein-modulatory RNAs have been shown to interact with—and affect the function of—bacterial transcription factors or the RNA synthesis machinery and consequently regulate gene expression.
A combination of in silico RNA sequencing and global DNA microarray studies has indicated that S. aureus is likely to produce an expansive repertoire of regulatory RNAs (3, 4, 24, 50, 52). The first of those studies was conducted by Pichon and Felden, who found, using a custom microarray to measure the expression properties of GC-rich intergenic regions, that the organism produces at least 12 putative regulatory RNAs, 5 of which were chromosomally encoded whereas 7 were localized to putative pathogenicity islands (51). The latter were termed small pathogenicity island RNAs (Spr), and subsequent work revealed that one of these, SprD, is a trans-encoded regulatory RNA that binds to the 5′ untranslated region of the Sbi immune evasion molecule transcripts. This, in turn, limits sbi mRNA translation and reduces S. aureus pathogenesis (13). A similar mechanism of regulation has been described for RNAIII, an RNA molecule that is well known to play a major role in S. aureus virulence but for which a regulatory mechanism was lacking until recently. Like SprD, RNAIII is a trans-encoded regulatory molecule that binds to and affects the stability and translation of target transcripts (7, 10, 17, 26, 29).
We previously used a microarray-based approach to demonstrate that S. aureus produces at least 142 extremely stable RNA species that are produced predominantly in a growth phase-dependent manner or in response to environmental stresses and map to the organism's intergenic regions (3, 4, 50, 52). While most S. aureus mRNA species exhibit a half-life of less than 2.5 min, these molecules are considerably stable, with measurable half-lives of ≥30 min, which is a hallmark of many regulatory RNAs. Accordingly, we have termed these 142 molecules SSRs to distinguish them from the plethora of the organism's nonstable RNAs that map to intergenic regions. Of direct relevance to this report, we hypothesized that SSRs are regulatory RNAs. As a preliminary test of that prediction, the regulatory effects of SSR42, which is detected and stabilized predominantly during stationary-phase growth, were examined. The reason we focused on this particular molecule is that its expression characteristics correspond to time points that correlate with massive changes in S. aureus physiology and, in particular, the expression properties of virulence factors. This means the expression and stabilization of SSR42 during stationary-phase growth correlates with the downregulation of many of the S. aureus cell surface virulence factors and upregulation of extracellular virulence determinants occurs (23). Thus, we considered that SSR42 may play a role in the regulation of S. aureus virulence factors.
To evaluate the potential of SSR42 as a novel regulatory molecule, we generated and characterized two genetically divergent deletion-containing strains of S. aureus lacking the SSR42 transcriptional unit. The first strain tested, UAMS-1, is a well-studied musculoskeletal isolate of the USA200 PFT (12, 28). The second strain, LAC, is a representative of the USA300 PFT as the leading etiologic agent responsible for CA-MRSA infections (31, 44). The expression properties of SSR42 were similar in UAMS-1 and LAC; during exponential phase, the half-life of the molecule is ∼5 min, whereas during stationary-phase growth, the half-life is increased to ≥30 min. The mechanism(s) responsible for the growth phase-dependent alterations in SSR42 stabilization are unknown.
Microarray analyses of UAMS-1 and LAC and their corresponding isogenic ΔSSR42 and complementation derivatives revealed that SSR42 pleiotropically represses approximately 80 genes in either strain background. Further, the effects of SSR42 on gene expression were largely confined to stationary-phase growth. These data strongly suggest that SSR42 is a regulatory RNA that predominantly represses gene expression irrespective of the genetic background. However, the results indicated that the identities of the specific transcripts that it regulated were largely strain specific. The limited overlap between SSR42 regulons is presumably due to inherent differences in the genomic composition and corresponding gene expression properties of each strain. Indeed, it is recognized that significant differences in RNAIII production exist between the USA300 PFT and other S. aureus lineages (20, 21, 36). We also observed that SSR42 titers are elevated in the LAC strain in comparison with those in UAMS-1, which may partially account for the difference in SSR42 regulatory effects within these two strains (data not shown). In addition to the molecule's repressor activities, SSR42 also induced the expression of 11 transcripts within strain LAC during stationary-phase growth, and many of these mRNA species code for extracellular virulence factors, including α-hemolysin (hla), γ-hemolysin (hlgC), and PVL (lukF-PV). Although the transcript titers of these factors were only mildly affected by SSR42, Western blotting and functional assays confirmed that these changes were biologically significant and that the molecule contributes to LAC's ability to cause disease in a murine model of skin and soft tissue infection.
The data presented here also establish that SSR42's regulatory capacity is due to the RNA molecule itself, as opposed to putative peptides encoded within the transcriptional unit. Indeed, elimination of a small (29-amino-acid) putative ORF within the SSR42 transcript sequence did not alter the molecule's regulatory capacity. Likewise, overexpression of the second half of the molecule harboring a wild-type copy of this putative ORF, which would presumably allow the protein's expression, did not elicit a regulatory effect.
Because SSR42 appears to modulate the expression of an expansive repertoire of genes, we predict that it behaves as a trans-encoded—or protein-modulatory—regulator, as opposed to an antisense regulatory RNA. Consistent with that prediction, SSR42 does not exhibit significant complementarity to other S. aureus genomic sequences within the NCBI genomic database. Accordingly, we hypothesized that SSR42 functions by directly interacting with mRNA and/or protein targets, as has been described for other bacterial regulatory RNAs (46, 59, 62). Electrophoretic mobility shift assays, mRNA turnover studies, and promoter activity measurements indicated that SSR42 does not directly bind to or modulate the mRNA turnover properties of virulence factor transcripts that are under its control within strain LAC. Rather, the molecule appears to affect the promoter activities of hla, hlgCB, and lukSF. Thus, it is likely that SSR42 influences S. aureus toxin expression by affecting the expression and/or activity of a transcriptional regulator or RNA polymerase. Likewise, assays assessing the spa promoter activity in the S. aureus strain UAMS-1 background revealed that SSR42 activated transcription at the spa promoter (data not shown). Studies are under way to characterize the SSR42 regulatory network.
As a preliminary effort to characterize the molecular mechanism by which SSR42 modulates S. aureus gene expression, we began an assessment of regions of the molecule that mediate its stationary-phase-associated stability and regulatory features. The approach we took was based, in part, on the observation that the interactions between RNAIII and its target mRNAs are not necessarily dependent on the presence of the full-length RNA molecule; Benito and colleagues demonstrated that a 3′ fragment of RNAIII is sufficient for the molecule's regulation of spa translation (7). This, in turn, facilitated characterization of the mechanism by which RNAIII affects spa expression. Accordingly, fragments of SSR42 consisting of bases 1 to 445 and 446 to 891 were linked to the transcript's endogenous promoter and examined for the respective ability to complement the alterations of gene expression in SSR42 deletion-containing cells. The results revealed that while full-length SSR42 is an extremely stable molecule (half-life, ≥30 min) during stationary-phase growth, bases 1 to 445 are relatively unstable (half-life, <5 min). In comparison, bases 446 to 891 are stable in ΔSSR42 cells and have a half-life of ≥30 min. It is unknown whether this stability is due to a stable secondary structure that is formed or a trans-acting factor (such as an RNA-binding protein) that stabilizes the factor through direct interaction with the molecule. As the 5′ end of SSR42 was unstable and expressed at lower levels than the full-length molecule, we hypothesized that bases 1 to 445 would exhibit little or no ability to regulate gene expression. Indeed, the 5′ end was unable to complement the gene expression profile of ΔSSR42 cells, suggesting that either the 3′ end is required for regulation or an unstable molecule cannot sufficiently modulate gene expression. However, the inability of bases 446 to 891 to restore SSR42-dependent control over the virulence factors examined here (isaB is the only exception) is evidence against the former. One possibility is that the first half of SSR42 mediates the molecule's regulatory effects but does so only when present at a high concentration, which is achieved by stabilization involving the second half of the molecule. Regardless, the results of these studies indicate that full-length SSR42 is required for full regulatory functionality within S. aureus.
Taken together, the results presented here indicate that SSR42 is a novel regulatory RNA within S. aureus that modulates the expression of many genes, including several virulence factors, predominantly during stationary-phase growth. The molecule's effects on virulence factor expression correlate with biologically significant changes in the expression of those proteins and in the case of USA300 strain LAC contribute to the organism's ability to cause disease. This and future studies delineating the mechanisms by which SSR42 and other SSR regulatory molecules function in S. aureus will provide a better understanding of the complex regulatory network that appears vital for the success of CA-MRSA in establishing infection rates of epidemic proportions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI073780 (P.M.D.). J.M.M. was partially supported by American Heart Association predoctoral award 11PRE7420020 and a University of Nebraska Medical Center graduate assistantship.
Footnotes
Published ahead of print 6 April 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alonzo F, III, et al. 2012. Staphylococcus aureus leukocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 83:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 3.Anderson KL, et al. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 188:6739–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson KL, et al. 2010. Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol. Med. Microbiol. 60:208–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63 [DOI] [PubMed] [Google Scholar]
- 6.Beenken KE, et al. 2004. Global gene expression in Staphylococcus aureus biofilms. J. Bacteriol. 186:4665–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benito Y, et al. 2000. Probing the structure of RNAIII, the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in repression of protein A expression. RNA 6:668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benson MA, et al. 2011. Staphylococcus aureus regulates the expression and production of the staphylococcal superantigen-like secreted proteins in a Rot-dependent manner. Mol. Microbiol. 81:659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blevins JS, Beenken KE, Elasri MO, Hurlburt BK, Smeltzer MS. 2002. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect. Immun. 70:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boisset S, et al. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 21:1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronner S, Monteil H, Prevost G. 2004. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol. Rev. 28:183–200 [DOI] [PubMed] [Google Scholar]
- 12.Cassat JE, et al. 2005. Comparative genomics of Staphylococcus aureus musculoskeletal isolates. J. Bacteriol. 187:576–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chabelskaya S, Gaillot O, Felden B. 2010. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 6:e1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chambers HF. 2001. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis. 7:178–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambers HF. 2005. Community-associated MRSA—resistance and virulence converge. N. Engl. J. Med. 352:1485–1487 [DOI] [PubMed] [Google Scholar]
- 16.Charpentier E, et al. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 70:6076–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chevalier C, et al. 2010. Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog. 6:e1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Azavedo JC, et al. 1985. Expression of the cloned toxic shock syndrome toxin 1 gene (tst) in vivo with a rabbit uterine model. Infect. Immun. 50:304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deleo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diep BA, et al. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739 [DOI] [PubMed] [Google Scholar]
- 21.Diep BA, Otto M. 2008. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 16:361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumont AL, et al. 2011. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 79:814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunman PM, et al. 2001. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 183:7341–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felden B, Vandenesch F, Bouloc P, Romby P. 2011. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 7:e1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francis KP, et al. 2000. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect. Immun. 68:3594–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol. Microbiol. 61:1038–1048 [DOI] [PubMed] [Google Scholar]
- 27.Geissmann T, et al. 2009. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 37:7239–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gillaspy AF, et al. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 63:3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huntzinger E, et al. 2005. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 24:824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji G, Beavis RC, Novick RP. 1995. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc. Natl. Acad. Sci. U. S. A. 92:12055–12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy AD, et al. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202:1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy AD, et al. 2008. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc. Natl. Acad. Sci. U. S. A. 105:1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolar SL, et al. 2011. NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology 157:2206–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuechenmeister LJ, Anderson KL, Morrison JM, Dunman PM. 2009. The use of molecular beacons to directly measure bacterial mRNA abundances and transcript degradation. J. Microbiol. Methods 76:146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, et al. 2010. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J. Infect. Dis. 202:1866–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M, et al. 2009. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 106:5883–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livny J, Teonadi H, Livny M, Waldor MK. 2008. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS One 3:e3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Löffler B, et al. 2010. Staphylococcus aureus Panton-Valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 6:e1000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malachowa N, et al. 2011. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6:e18617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCaskill ML, et al. 2007. Increase of the USA300 clone among community-acquired methicillin-susceptible Staphylococcus aureus causing invasive infections. Pediatr. Infect. Dis. J. 26:1122–1127 [DOI] [PubMed] [Google Scholar]
- 41.McDougal LK, et al. 2003. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J. Clin. Microbiol. 41:5113–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merino N, et al. 2009. Protein A-mediated multicellular behavior in Staphylococcus aureus. J. Bacteriol. 191:832–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller LG, et al. 2005. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N. Engl. J. Med. 352:1445–1453 [DOI] [PubMed] [Google Scholar]
- 44.Moran GJ, et al. 2006. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 355:666–674 [DOI] [PubMed] [Google Scholar]
- 45.Morfeldt E, Taylor D, von Gabain A, Arvidson S. 1995. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 14:4569–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrison JM, Dunman PM. 2011. The modulation of Staphylococcus aureus mRNA turnover. Future Microbiol. 6:1141–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429–1449 [DOI] [PubMed] [Google Scholar]
- 48.Novick RP, et al. 1995. The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol. Gen. Genet. 248:446–458 [DOI] [PubMed] [Google Scholar]
- 49.Novick RP, et al. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 12:3967–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olson PD, et al. 2011. Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog. 7:e1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pichon C, Felden B. 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. U. S. A. 102:14249–14254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts C, et al. 2006. Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J. Bacteriol. 188:2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanchez CJ, et al. 2010. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6:e1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siboo IR, Chambers HF, Sullam PM. 2005. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 73:2273–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomason MK, Storz G. 2010. Bacterial antisense RNAs: how many are there, and what are they doing? Annu. Rev. Genet. 44:167–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ventura CL, et al. 2010. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5:e11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voyich JM, et al. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175:3907–3919 [DOI] [PubMed] [Google Scholar]
- 58.Voyich JM, et al. 2006. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J. Infect. Dis. 194:1761–1770 [DOI] [PubMed] [Google Scholar]
- 59.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoong P, Pier GB. 2010. Antibody-mediated enhancement of community-acquired methicillin-resistant Staphylococcus aureus infection. Proc. Natl. Acad. Sci. U. S. A. 107:2241–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zetola N, Francis JS, Nuermberger EL, Bishai WR. 2005. Community-acquired meticillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 5:275–286 [DOI] [PubMed] [Google Scholar]
- 62.Zhou Y, Xie J. 2011. The roles of pathogen small RNAs. J. Cell. Physiol. 226:968–973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.