

Abstract
The virulence of many Gram-negative pathogens is associated with type III secretion systems (T3SSs), which deliver virulence effector proteins into the cytoplasm of host cells. Components of enteropathogenic Escherichia coli (EPEC) T3SS are encoded within the locus of enterocyte effacement (LEE). While most LEE-encoded T3SS proteins in EPEC have assigned names and functions, a few of them remain poorly characterized. Here, we studied a small LEE-encoded protein, Orf15, that shows no homology to other T3SS/flagellar proteins and is only present in attaching and effacing pathogens, including enterohemorrhagic E. coli and Citrobacter rodentium. Our findings demonstrated that it is essential for type III secretion (T3S) and that it is localized to the periplasm and associated with the inner membrane. Membrane association was driven by the N-terminal 19 amino acid residues, which were also shown to be essential for T3S. Consistent with its localization, Orf15 was found to interact with the EPEC T3SS outer membrane ring component, EscC, which was previously shown to be embedded within the outer membrane and protruding into the periplasmic space. Interestingly, we found that the predicted coiled-coil structure of Orf15 is critical for the protein's function. Overall, our findings suggest that Orf15 is a structural protein that contributes to the structural integrity of the T3S complex, and therefore we propose to rename it EscA.
INTRODUCTION
Diarrhea is the second leading cause of death of children younger than 5 years globally, accounting for 1.3 million deaths annually (4). One of the most prevalent causes of infantile diarrhea in developing countries is enteropathogenic Escherichia coli (EPEC). EPEC belongs to a family of pathogens called attaching and effacing (A/E) pathogens, which colonize the gut epithelium and induce characteristic histopathological lesions by intimate adherence to the epithelial surface and effacement of host enterocyte brush border microvilli (24). Many important EPEC virulence factors are encoded within a 35-kb chromosomal pathogenicity island termed the locus of enterocyte effacement (LEE) (22). The LEE contains 41 genes, most of which are organized in five operons that encode components of (i) the type III secretion (T3S) apparatus, (ii) E. coli-secreted proteins and translocators, (iii) type III-specific chaperones, and (iv) transcriptional regulators.
The type III secretion system (T3SS) is a protein complex dedicated to the injection of effector proteins from the cytosol of Gram-negative pathogens into their target cell. These effectors modify signaling pathways within the host cell to benefit the pathogen. The main proteins that form the T3S export apparatus (also called nano-syringe or injectisome) are conserved among several pathogens and share significant similarity with components of the flagellar system (6, 10, 36, 38). The T3S apparatus in EPEC is comprised of about 20 different proteins that assemble into a multiring base structure, which transverses both inner and outer membranes as well as the periplasmic space, and a protruding needle (35, 37).
The structural core of the injectisome assembles through the formation of inner and outer membrane (IM and OM, respectively) rings. The OM ring in EPEC is composed mainly of a homomultimeric protein, EscC, which belongs to the secretin family (30, 34). This family of proteins represents a distinct class of integral OM proteins that participate in various transport machineries, such as the type II secretion system (T2SS), T3SS, and the type IV pilus biogenesis pathway. Electron microscopy studies have shown that secretins from unrelated transport pathways form similar ring structures composed of 12 to 14 subunits that enable the passage of unfolded or partially unfolded proteins (5, 18, 20, 26). The secretin family contains a variable N-terminal periplasmic region and a highly conserved protease-resistant C-terminal region embedded in the OM. The inner membrane ring of the T3S apparatus is composed of two proteins from the PrgK/MxiJ/EscJ and the PrgH/MxiG/EscD families. The exact mechanism of how these ring structures that are formed in different membranes find each other is still not clear.
While many of the T3SS components in EPEC are known, there are still a few poorly characterized LEE-encoded proteins. One example is the protein encoded by the orf15 gene. A large screening study aimed to identify critical components of the T3SS in the related murine pathogen Citrobacter rodentium found orf15 to be critical for virulence (8). Interestingly, Orf15 has no homologs in other secretion system/flagellar proteins outside the A/E pathogen group and has no homologs among general bacterial proteins. These observations suggest that Orf15 performs a critical function for the T3SS of A/E pathogens.
Based on our experimental results, we conclude that Orf15 functions as a critical structural protein within the T3SS, and we therefore suggest renaming it EscA, according to the standard type III nomenclature, where Esc stands for E. coli secretion apparatus component. We use this terminology throughout the manuscript.
MATERIALS AND METHODS
Bacterial strains.
Wild-type (WT) EPEC O127:H6 strain E2348/69 (streptomycin resistant) was used in this study. Strains were grown in Luria-Bertani (LB) broth supplemented with the appropriate antibiotics at 37°C. Antibiotics were used at the following concentrations: streptomycin, 50 μg/ml; ampicillin, 100 μg/ml; kanamycin, 50 μg/ml, and chloramphenicol, 30 μg/ml.
Construction of nonpolar mutants.
A nonpolar deletion mutant of the escA gene in the streptomycin-resistant (Smr) EPEC E2348/69 strain was generated using the sacB-based allelic exchange method (16). Briefly, two PCR fragments were generated using the primer pairs escA-01F/escA-01R and escA-02F/escA-02R (Table 1); the fragments were cloned into pCR2.1-TOPO (Invitrogen), verified by DNA sequencing, and then subcloned as a KpnI/NheI fragment and an NheI/SacI fragment, respectively, into the suicide vector pRE112 (9) digested by KpnI/SacI. The resulting plasmid, containing the flanking regions of escA with 73% of escA deleted, was transformed into E. coli SM10λpir by electroporation and introduced into the EPEC strain by conjugation. After sucrose selection, EPEC colonies that were resistant to sucrose and susceptible to chloramphenicol were screened for the deletion of escA by PCR. The ΔescC ΔescA double deletion mutant was generated using a similar method as described above by introducing the suicide plasmid carrying the ΔescC mutation (described in reference 13) into the ΔescA EPEC strain. The strains used in this study are listed in Table 2.
Table 1.
Sequences of primers used in this study
| Target and primer name | Sequencea |
|---|---|
| escA deletion mutant | |
| ESCA-01F | CGGGTACCAAAAGCGCGTATTGGTGC |
| ESCA-01R | CGGCTAGCTAAAATTCTGTCCAACATATTCA |
| ESCA-02F | TTTGCTAGCAAGGAACATTACTTTGACTAGCA |
| ESCA-02R | CAGAGCTCTACGGTTGCTAAAGGGTTATTAA |
| pEscA (pCR2.1-TOPO) | |
| EscA-F1 | AGAAGCTTCGAAAAAACGATTGAAAGCC |
| EscA-2HA-R | AACTCGAGGTCAAAGTAATGTTCCTTTATGG |
| Δ19EscA (pCR2.1-TOPO) | |
| Δ19EscA-F | AAAGCTTAGCGAACCGATTGAGAG |
| pEscA (pACYC184) | |
| EscA-F2 | CTGGATCCAACAAAGCACCAAAGATATC |
| EscA-HSV-R1 | AGAAGCTAGCGTCAAAGTAATGTTCCTTTATGG |
| EscA-HSV-R2 | GGTCGACTCAATCCTCGGG |
| pEscC (pCR2.1-TOPO) | |
| pEscC-F | CCAAGCTTGTTTTCTGATATAGGACG |
| pEscC-2HA-R | GGCTCGAGTTCGCTAGATGCAG |
| Δ21PhoA-2HA (pMAC2) | |
| Δ21PhoA-F | GTACGGTACCATTAAAGAGGAGAAATTAACTGTGCGGACACCAGAAATGCC |
| PhoA-R | TTAAGCGTAATCTGGAACATCGTATGGGTATTTCAGCCCCAGAGCGGC |
| HA-R | TAGAGCTCTTAAGCGTAATCTGGAACATCGTA |
| EscA19PhoA-2HA (pMAC2) | |
| EscA19PhoA-F1 | GTTGGACAGAATTTTATCTATTCGTAAAAGCAGAGCGAACCGATTGAGAGAATCACGGACACCAGAAATGCCTGT |
| EscA19PhoA-F2 | GTACGGTACCATTAAAGAGGAGAAATTAACTATGTTGGACAGAATTTTATCTATTCG |
Restriction sites are underlined.
Table 2.
Strains and plasmids used in this study
| Strain or plasmid | Reference or source |
|---|---|
| Strains | |
| Wild-type EPEC | 19 |
| ΔescA strain | This study |
| ΔescA ΔescC strain | This study |
| Plasmids | |
| pEscA-2HA | This study |
| pEscA-HSV | This study |
| pEscC-2HA | This study |
| pEmpty-2HA | 3 |
| Δ19EscA-2HA | This study |
| Δ21PhoA-2HA | This study |
| EscA19PhoA-2HA | This study |
| pCR2.1-TOPO | Invitrogen |
| pET27b(+) | Novagen |
| pTOPO-2HA | 8 |
| pACYC184 | 32 |
Construction of plasmids expressing escA and escC.
The escA gene was amplified using the primer pair EscA-F1/EscA-2HA-R (where 2HA indicates two copies of a hemagglutinin tag) (Table 1) or Δ19EscA-F/EscA-2HA-R (where Δ19EscA indicates a 19-amino-acid [aa] N-terminal deletion in EscA), cloned into pCR2.1-TOPO (Invitrogen), verified by DNA sequencing, and then subcloned as a HindIII/XhoI fragment into HindIII/XhoI-digested pTOPO-2HA (8). The resulting constructs, pEscA-2HA and the truncated version of EscApΔ19EscA-2HA, express a fusion protein of EscA with a double HA tag at its C terminus. Similarly, escA was amplified from the EPEC strain by PCR using the primer pair EscA-F2/EscA-HSV-R1 (Table 1) and cloned as a BamHI/NheI fragment into BamHI/NheI-digested pET27b(+) (Novagen) that contains a herpes simplex virus (HSV) tag. Then, a second PCR using the primer pair EscA-F2/EscA-HSV-R2 (Table 1) was performed, and the product was cloned as a BamHI/SalI fragment into BamHI/SalI-digested pACYC184 (32). The resulting construct, pEscA-HSV, expressed a fusion protein of EscA and an HSV tag at its C terminus. Mutations in the escA gene were introduced using site-directed mutagenesis (Stratagene).
To generate an expression vector of EscC fused to the 2HA tag, the entire coding region of escC from EPEC E2348/69 was amplified using the primer pair EscC-F/EscC-R (Table 1) and cloned it into HindIII/XhoI-digested pTOPO-2HA.
An HA-tagged phoA gene lacking the first 21 amino acids was PCR amplified from genomic DNA of E. coli MG1655 using the primer pair Δ21PhoA-F and PhoA-R (Table 1). The HA tag was added by using the above PCR product as the template with Δ21PhoA-F and HA-R primers (Table 1). The resulting product was digested with KpnI/SacI and cloned into a KpnI/SacI-digested pMAC2 vector (described below), yielding Δ21PhoA-2HA (pMAC2). An HA-tagged phoA construct fused with the first 19 amino acids of EscA (EscA19) was PCR amplified from MG1655 with EscA19PhoA-F1 and PhoA-R. This product served as the template in a second PCR with EscA19PhoA-F2 and HA-R (Table 1). The resulting product was cloned into the KpnI/SacI sites of pMAC2, yielding EscA19PhoA-2HA (pMAC2). The pMAC2 vector was designed based on the pACYC origin of replication (p15a) for compatibility with ColE1 replicons and contained a chloramphenicol resistance gene, a lac promoter, and universal sequencing sites flanking the multiple cloning sites. Briefly, an ∼572-bp fragment encompassing p15a was digested from pACYC184 (32) with AseI and SacII, followed by T4 polymerase treatment to blunt the ends. The ColE1 origin of replication was removed from pBC (Stratagene) by digestion with AgeI and AflIII, followed by T4 polymerase treatment to fill in the ends. The p15a origin of replication was blunt-end cloned into the remaining pBC backbone, yielding pMAC2. The plasmids used in this study are listed in Table 2.
Secretion assay.
EPEC strains were grown overnight in LB broth in a shaker at 37°C. The cultures were diluted 1:50 into Dulbecco's modified Eagle's medium (DMEM) that was prewarmed in a CO2 tissue culture incubator overnight and grown for 6 h to an optical density at 600 nm (OD600) of 0.7 in a tissue culture incubator (with 5% CO2) with the tubes standing. The cultures were centrifuged for 5 min at 16,000 relative centrifugal force (RCF) to remove the bacteria; the supernatant was collected and then filtered through a 0.22-μm-pore-size filter (Millipore). The supernatant was then precipitated with 10% trichloroacetic acid ([TCA] vol/vol) to concentrate proteins secreted into the culture medium. The secreted proteins precipitated were dissolved in sodium dodecyl sulfate (SDS)-PAGE sample buffer, and TCA was neutralized with saturated Tris. The volumes of buffer used to resuspend the secreted proteins were normalized relative to the OD600 of the cultures to ensure equal loading of the samples. The proteins were analyzed by SDS–12% PAGE and stained with Coomassie blue.
Bacterial fractionation.
Bacterial cell fractionation was based on previously described procedures (11, 15, 25). Briefly, EPEC from an overnight culture was subcultured at 1:50 in 50 ml of DMEM for 6 h at 37°C under 5% CO2. The culture was harvested, washed in phosphate-buffered saline (PBS), and resuspended in 1 ml of buffer A (50 mM Tris [pH 7.5], 20% [wt/vol] sucrose, protease inhibitor cocktail [Roche Applied Science], and 100 μg/ml lysozyme) and incubated for 30 min at room temperature (RT) to generate spheroplasts. MgCl2 was then added to final a concentration of 20 mM and spun for 10 min at 5,000 RCF. The supernatants containing the periplasmic fractions were collected. The pellet containing the cytoplasm and the membrane fractions was resuspended in 1 ml of lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, and 2 mM β-mercaptoethanol with protease inhibitors). All subsequent steps were carried out at 4°C. RNase A at 10 μg/ml and 10 μg/ml DNase I were added, and the samples were sonicated three times for 15 s each time (Fisher Sonicator). Intact bacteria were removed by centrifugation (at 2,300 RCF for 15 min), and the cleared supernatant containing cytoplasmic and inner and outer membrane proteins was transferred to new tubes. To obtain the cytoplasmic fraction, supernatants were centrifuged (in a Beckman TLA 100 Ultracentrifuge with a TLA100.3 rotor) for 30 min at 100,000 RCF to pellet the membranes. The supernatant containing the cytoplasmic fraction was collected; the membrane pellet was washed once with 2 ml of lysis buffer, resuspended in 0.1 ml of lysis buffer with 0.5% N-lauroylsarcosine, which selectively solubilizes the IM, and centrifuged at 100,000 RCF for 1 h. The supernatant containing the IM fraction was removed, and the OM pellet was washed in lysis buffer with 0.5% N-lauroylsarcosine. The final pellet was resuspended in 0.1 ml of lysis buffer with 0.5% N-lauroylsarcosine and 0.1% sodium dodecyl sulfate (SDS). The protein content of all samples was determined using a Coomassie Plus protein assay (Thermo Scientific) before addition of SDS sample buffer with β-mercaptoethanol.
Protease accessibility assay.
The protease accessibility assay was based on a previously described procedure (23). Briefly, EPEC strains expressing EscA-2HA were grown under T3SS-inducing conditions, and two identical samples of cells were pelleted. One sample was used to prepare spheroplasts, while the second was used as a whole-cell control. For spheroplast preparation, cells were first resuspended in 90 μl of resuspension buffer (100 mM Tris, pH 8.2, 500 mM sucrose, 0.5 mM EDTA, 2.5 mg/ml lysozyme) and incubated at room temperature for 5 min. One volume of ice-cold water was then added, and the sample was mixed and placed on ice. MgCl2 was next added to a final concentration of 10 mM. After incubation on ice for 5 min, spheroplasts were pelleted at 500 RCF for 5 min, and the supernatant (corresponding to the periplasmic fraction) was removed. The pellet (containing spheroplasts) was resuspended in 90 μl of resuspension buffer and divided into three 30-μl aliquots. Three microliters of 10% Nonidet P-40 and 50 μg/ml proteinase K was added to one aliquot. Three microliters of resuspension buffer plus proteinase K was added to the second aliquot, while the third aliquot was left untreated (resuspended in buffer to maintain volume). Proteinase K digestion was allowed to proceed for 30 min on ice. Protein was then precipitated from each sample by addition of 3.5 μl of 100% TCA, and pellets were resuspended in SDS-PAGE loading buffer for electrophoresis and Western blotting with anti-HA, anti-maltose binding protein (MBP), and anti-DnaK antibodies. Whole-cell samples were processed alongside spheroplast samples (with PBS replacing resuspension buffer).
Stable isotope labeling of amino acids in cell culture (SILAC) and mass spectrometry.
An EPEC ΔlysA ΔargH strain expressing the double HA tag (pEmpty-2HA) was labeled with the heavy amino acids l-[13C6]arginine (l-Arg6) and l-lysine-4,4,5,5-D4 (l-Lys4) by subculturing the bacteria grown in LB at 1:1,000,000 in DMEM without l-arginine and l-lysine supplemented with 87 μg/ml l-Arg6 and 150 μg/ml l-Lys4 for overnight growth. The culture was then diluted 1:5 in 50 ml of DMEM with l-Arg6 and l-Lys4 and grown for 3 h in a 5% CO2 incubator at 37°C. The same growth conditions were applied to an EPEC strain expressing EscA-2HA but using DMEM containing the normal (light) amino acids l-arginine and l-lysine. Cells were harvested by centrifugation at 5,000 RCF for 10 min, washed three times with PBS, and resuspended in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 1 mM Na3VO4, 10 mM NaF, 50 mM Na4P2O7, 2 mM β-mercaptoethanol, and protease inhibitor cocktail [Roche]). The cells were lysed using a probe sonicator, and then 0.1% NP-40 was added. Unlysed bacteria were removed by centrifugation for 10 min at 8,000 RCF. Protein concentrations of heavy and light lysates were measured using a Coomassie Plus protein assay (Pierce Scientific) according to the manufacturer's directions. Then, equal quantities of protein from clarified heavy and light cell lysates were mixed and precleared on N-(hydroxysuccinimide) NHS-activated Sepharose beads (GE Healthcare) charged with mouse IgG (Jackson ImmunoResearch Laboratories) at 4°C for 30 min with rocking. Precleared lysates were then spun, and the supernatant was added to Sepharose beads with mouse anti-HA antibody (Covance) and incubated at 4°C for 2 h with rocking. Beads were washed three times with 1 ml of cold lysis buffer, and all remaining buffer was drawn from the beads using GELoader Tips (Eppendorf, Westbury, NY). Next, the beads of the heavy and the light samples were combined and treated with elution buffer (6 M urea–2 M thiourea) for 30 min at room temperature to elute proteins from the beads. The eluted proteins were then precipitated using ethanol-acetate and digested in solution as previously described (12). Peptides were acidified and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on an LTQ-OrbitrapXL as previously described in (31).
Immunoprecipitation.
EPEC strains were subcultured at 1:40 in DMEM and grown for 3 h in a 5% CO2 incubator at 37°C, followed by centrifugation at 5,000 RCF for 10 min to harvest the cultures. The pellet was washed with PBS and resuspended in 1 ml of lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 2 mM β-mercaptoethanol, and protease inhibitor cocktail). The cells were lysed using a probe sonicator, and then 0.1% NP-40 was added. Unlysed bacteria were removed by centrifugation for 10 min at 8,000 RCF, and the supernatant representing the cleared lysate was collected. Cell lysates were precleared on protein A-Sepharose beads (GE Healthcare) by incubation on a rotary wheel for 30 min at 4°C. The beads were removed by centrifugation for 4 min at 1,000 RCF, and the precleared lysates were transferred to a clean tube. For the immunoprecipitation, 5 μg of anti-HSV antibody and 20 μl of washed protein G-Sepharose beads were added simultaneously to the lysates. Samples were incubated for 2 h at 4°C with rotation, and then the beads were pelleted by centrifugation for 4 min at 1,000 RCF, followed by three washes with 1 ml of ice-cold lysis buffer (plus 0.1% NP-40). The washed beads were resuspended in SDS-PAGE sample buffer, which resulted in elution of the precipitated protein complexes, boiled for 10 min, and subjected to Western blot analysis.
RESULTS AND DISCUSSION
EscA is essential for T3S.
The assigned function of many of the LEE genes was originally based on sequence homology as a prediction tool. A BLAST search revealed that EscA shares significant homology with components of an A/E pathogen secretion system but none with components of other secretion systems (Fig. 1A). Therefore, it was hard to predict its role within the T3SS. To examine if EscA is an essential component for T3S, we analyzed the secretion of the T3SS translocators EspA, EspB, and EspD in an EPEC escA null (ΔescA) strain by SDS-PAGE and Coomassie blue staining. The null strain did not secrete type III translocators, indicating that EscA is essential for T3S (Fig. 1). A similar result was found in the related murine A/E pathogen, C. rodentium (8). Complementation of the mutant strain with full-length escA in trans restored secretion of the translocators, thus confirming that the deletion of escA is nonpolar. Overall, this result demonstrates that EscA is an essential component for T3S in EPEC. To determine whether EscA is a secreted structural component, like the inner-rod or needle proteins, we assessed the ability of an EPEC ΔescA strain complemented with HA-tagged EscA to secrete the EscA protein when grown under T3S-inducing conditions. Whole-cell lysates and supernatants were analyzed using immunoblotting with a mouse anti-HA antibody. EscA was not detected in the secreted sample of the ΔescA strain complemented with HA-tagged EscA although it was highly expressed in the whole-cell lysate (Fig. 1C). These results demonstrated that EscA is a structural nonsecreted component of the T3SS.
Fig 1.

EscA is essential for T3S. (A) BLAST (http://blast.ncbi.nlm.nih.gov) and multiple alignment (using CLUSTAL W) of Orf15 from EPEC with T3S proteins of the related A/E pathogens, enterohemorrhagic (EHEC) E. coli (O157:H7) and C. rodentium (ICC1680). Accession numbers are as follows: EPEC Orf15, gb|AAC38384.1; EHEC (E. coli O157:H7) L0033, NP_290267.1; C. rodentium [CB] Orf15, gb|AF311901.1. An asterisk indicates positions which have a single, fully conserved residue; a colon indicates conservation between groups of strongly similar properties; and a period indicates conservation between groups of weakly similar properties. (B) Protein secretion profiles of WT EPEC, a ΔescA strain, and a ΔescA strain complemented with escA in trans. Secreted proteins were concentrated from supernatants of bacterial cultures grown in DMEM and analyzed by Coomassie staining of an SDS–12% PAGE gel. The locations of the translocators EspA, EspB, and EspD are indicated at the right of the gel. Also indicated is the location of EPEC EspC, which is not secreted via the LEE-encoded T3SS. (C) Whole-cell lysates (WCL) and secreted proteins (sup) of an EPEC WT and a ΔescA strain complemented with escA grown under T3S-inducing conditions were examined for EscA secretion. Samples were analyzed by SDS–16% PAGE and immunoblotting using an anti-HA antibody. Molecular size markers (kDa) are indicated at the left of the gel.
Subcellular localization of EscA.
The subcellular localization of HA-tagged EscA expressed in the ΔescA strain grown under T3SS-inducing conditions was examined in whole-cell extract that was fractionated into cytoplasm, periplasm, and membrane fractions. EscA was detected using an HA antibody. We found that EscA was enriched in the membrane and periplasmic fractions (Fig. 2A, left panel). Western blots were probed with cytoplasmic, periplasmic, and membrane markers to confirm proper fractionation (Fig. 2A, right panels). This result suggested that although EscA is predicted to be cytosolic according to PLSpred (2), it is present predominantly in the membrane and the periplasmic fractions. A detergent-selective solubilization assay was used to further fractionate the membranes and to determine whether EscA is enriched in a specific membrane. EscA was shown to be mainly localized to the IM fraction (Fig. 2B). Western blots were probed with IM and OM markers, and this confirmed proper fractionation (Fig. 2B, right panels). To examine whether the localization of EscA was dependent on the T3SS, we expressed EscA-2HA in E. coli DH10B, which does not carry the T3SS, and fractionated it into cytoplasm, periplasm, and membrane fractions. We found that the localization of EscA in DH10B was similar to what we found for the EscA protein expressed in EPEC grown under T3S-inducing conditions (Fig. 3C). These results suggested that the localization of EscA was independent of the T3SS.
Fig 2.

Subcellular localization of EscA. (A) An EPEC ΔescA strain expressing EscA-2HA was grown under T3S-inducing conditions and was fractionated into periplasmic (P), cytoplasmic (C), and membrane (M) fractions. Two micrograms of protein from each fraction was loaded on SDS–15% PAGE gels, transferred to nitrocellulose membrane, and blotted with an anti-HA antibody to detect EscA-2HA. To confirm correct fractionation, the Western blots were also probed with anti-MBP (periplasmic marker), anti-DnaK (cytoplasmic marker), and anti-βATPase (membrane marker) antibodies. (B) The membrane fraction was further separated into IM and OM using selective membrane solubilization detergents to examine the exact location of EscA. (C) Fractionation of EscA-2HA expressed in DH10B under conditions similar to those described in panel A.
Fig 3.
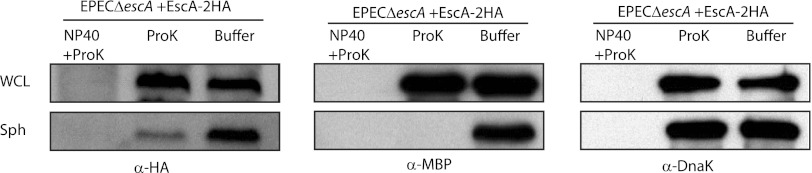
EscA is susceptible to protease digestion in spheroplasts but not in whole cells. The accessibility of EscA to proteinase K digestion in spheroplasts (Sph) and whole-cell lysates (WCL) was compared by Western blotting using antibody against HA. Western blots were also probed with anti-MBP and anti-DnaK antibodies.
To determine whether EscA associates with the cytoplasmic leaflet of the IM or with its periplasmic side, we performed a protease accessibility assay (23) that compares the digestion of a tagged protein between whole-cell lysates and spheroplasts. In this assay, cytoplasmic proteins are protected from protease digestion in both whole-cell extracts and spheroplasts in the absence of detergent. However, soluble or membrane-associated proteins that are localized to the periplasm are susceptible to digestion in spheroplasts but not in the whole-cell fraction in detergent-free solution. We found that EscA was susceptible to protease digestion in spheroplasts but not in whole-cell extracts in the absence of detergent (Fig. 3). This digestion pattern of EscA was similar to that of the periplasmic MBP protein that was susceptible to proteinase degradation in spheroplasts, unlike the cytoplasmic DnaK protein (Fig. 3). These results suggested that most EscA is localized to the periplasmic space, either as a soluble protein or in association with the IM.
Examination of the EscA sequence did not identify a classical N-terminal signal peptide sequence that directs secretion of the protein into the periplasm. To determine whether EscA contains a nonclassical N-terminal signal peptide that facilitates periplasmic localization, we examined the subcellular localization of HA-tagged EscA with a truncation of the N-terminal 19 amino acids (Δ19EscA-2HA). This truncated protein was found mostly in the periplasm and cytoplasm fractions but not in the membrane fraction (Fig. 4A). This result suggested that while the N-terminal 19-amino-acid sequence of EscA was critical for protein association with the membrane, is was not essential for periplasmic localization. To examine whether this short sequence is sufficient for membrane association, we fused it to alkaline phosphatase protein (PhoA) lacking its own export signal (Δ21PhoA) and examined the subcellular localization of the chimeric protein. The truncated version of PhoA (Δ21PhoA) localized largely to the cytoplasm fraction, as expected (Fig. 4B). However, when the PhoA truncated protein was fused to the N-terminal 19 amino acids of EscA (EscA19Δ21PhoA-2HA), the localization of the chimeric protein altered drastically, and much of the protein was found in the membrane fraction (Fig. 4B). Fractionation of the inner and outer membranes indicated that the chimeric protein localized mainly to the IM, similarly to EscA-2HA. Markers of specific cell fractions indicated correct fractionation (Fig. 4C). This result suggested that the N-terminal sequence of EscA (19 aa) is involved in membrane association. Examination of the protease accessibility profile of HA-tagged Δ21PhoA demonstrated that this truncated protein is protected from digestion, confirming that it is mostly cytoplasmic (Fig. 4D). However, EscA19Δ21PhoA-2HA was susceptible to proteinase degradation in spheroplasts but not in whole-cell extracts (Fig. 4D). This digestion profile was similar to the one observed for EscA-2HA, suggesting that the N-terminal 19 amino acid residues of EscA are critical and sufficient for IM association. In contrast to the localization results of the EscA protein (Fig. 4A) that suggested that the N terminus of EscA is not necessary for periplasmic localization, we observed that fusing the N-terminal 19-amino-acid sequence of EscA to the PhoA protein contributes to its periplasmic localization. Thus, we can postulate that EscA has redundant periplasm export signals: the N terminus and one other part of EscA. To clarify if membrane association is essential for EscA function, we examined the ability of the Δ19EscA-2HA construct to complement T3S of the ΔescA strain. The truncated protein could not complement T3S of the mutant strain, thus implying that the first 19 amino acids of EscA are crucial for the protein's function (Fig. 4E). The effect of these 19 amino acids on protein localization and membrane association suggests that EscA must associate, directly or through additional components, with the IM for its function.
Fig 4.

The role of the first 19 aa of EscA in its localization. (A) Subcellular localization of Δ19EscA-2HA expressed in an EPEC ΔescA strain and grown under T3S-inducing conditions. Whole-cell extract was fractionated into periplasmic (P), cytoplasmic (C), and membrane (M) fractions. Two micrograms of protein from each fraction was loaded onto SDS–15% PAGE gels, transferred to nitrocellulose membrane, and blotted with an anti-HA antibody to detect Δ19EscA-2HA. (B) The subcellular localization of Δ21PhoA-2HA and EscA19Δ21PhoA-2HA constructs was examined using a procedure similar to that described in panel A. The membrane fraction was further separated into IM and OM using selective membrane solubilization detergent to examine the exact location of the fused proteins. (C) To confirm correct fractionation, the Western blots were also probed with anti-MBP (periplasmic marker), anti-DnaK (cytoplasmic marker), anti-βATPase (IM marker), and OmpA (OM marker) antibodies. (D) The accessibility of Δ21PhoA-2HA and EscA19Δ21PhoA-2HA to proteinase K digestion in spheroplasts (Sph) and whole-cell lysates (WCL) was examined by Western blotting using antibody against HA. (E) Protein secretion profiles of WT EPEC, a ΔescA strain, and a ΔescA strain complemented with Δ19EscA-2HA. Secreted proteins were concentrated from supernatants of bacterial cultures grown in DMEM and analyzed by Coomassie staining of an SDS–12% PAGE gel. The locations of the translocators EspA, EspB, and EspD are indicated at the right of the gel. Also indicated is the location of EPEC EspC, which is not secreted via the LEE-encoded T3SS.
EscA interacts with EscC.
In order to identify the interaction partners of EscA within the T3SS of EPEC, we employed a mass spectrometry-based stable isotope labeling of amino acids in cell culture (SILAC) technique. SILAC is a quantitative proteomic method for identification of protein-protein interactions with high confidence and without a priori knowledge of the interaction partners (27). The background strain, an EPEC ΔlysA ΔargH strain expressing the double HA tag without a protein attached (pEmpty-2HA), was grown in medium supplemented with stable (heavy) isotopes l-[13C6]arginine (l-Arg6) and l-lysine-4,4,5,5-D4 (l-Lys4). The EPEC ΔlysA ΔargH strain was shown to be completely auxotrophic for arginine and lysine; therefore, the proteome of this strain would completely incorporate heavy lysine and arginine derivatives. The EPEC ΔescA null strain, carrying a double HA-tagged EscA, was supplemented with normal (light) l-arginine and l-lysine. Lysates of heavy and light bacterial populations were mixed at a 1:1 ratio and immunoprecipitated using anti-HA antibodies. Eluted proteins were digested into peptides, separated by high-performance liquid chromatography, and analyzed by tandem mass spectrometry (LC-MS/MS) (Fig. 5A). The relative abundance of proteins present in two different samples can thus be quantified based on the ratios of the peak intensities of arginine and lysine containing peptide pairs and results in a heavy/light (H/L) ratio. Peptides with H/L ratios below 0.5 are considered significant, and the corresponding protein is considered a candidate interaction partner of the tagged protein.
Fig 5.

EscA interacts with the secretin protein, EscC. (A) Representative mass spectra for peptides recovered from a SILAC experiment with EscA-2HA. In the right panel, open and filled triangles indicate the expected mass/charge ratio (m/z) of light and heavy forms, respectively, of a peptide (VASLESITSGTLLR) from the specifically bound EscC from pulldowns with EscA-2HA and 2HA, respectively. The light peptide of EscC is present at a ratio of >2.0 higher than the heavy peptide, indicating a specific interaction with EscA. In the left panel, open and filled triangles indicate the expected m/z of light and heavy forms, respectively, of a peptide (AGTYATTLPAGR) from the nonspecifically bound FepA protein from pulldowns with EscA-2HA and 2HA, respectively. (B) EPEC ΔescA ΔescC strain cultures expressing EscC-2HA, EscA-HSV, or both proteins were grown under T3S-inducing conditions. Whole-cell extracts were subjected to immunoprecipitation using an antibody directed against HSV. Whole-cell extracts were analyzed by Western blotting probing for EscA (using anti-HSV antibody) and EscC (using anti-HA antibody). (C) Complementation of the EPEC ΔescA ΔescC strain by the EscC-2HA and EscA-HSV constructs. Protein secretion profiles of WT EPEC, a ΔescA ΔescC strain, and a ΔescA ΔescC strain complemented with EscC-2HA and EscA-HSV in trans. Secreted proteins were concentrated from supernatants of bacterial cultures grown in DMEM and analyzed by Coomassie staining of an SDS–12% PAGE gel. The locations of the translocators EspA, EspB, and EspD are indicated at the right of the gel. Also indicated is the location of EPEC EspC, which is not secreted via the LEE-encoded T3SS.
Immunoprecipitation of EscA-2HA showed an interaction with the LEE-encoded protein, EscC. Two unique peptides from EscC were identified. Both peptides showed an H/L ratio of <0.5 (one is presented in Fig. 5A, right panel), indicating specific binding to EscA. Peptides from nonspecific binding proteins, such as the OM receptor for ferric enterobactin FepA, showed an H/L ratio near 1.0 (Fig. 5A, left panel). Verification of EscA-EscC interaction was obtained by coimmunoprecipitation. The EPEC ΔescA ΔescC strain was complemented with pEscA-HSV and pEscC-2HA and grown under T3S-inducing conditions. Immunoprecipitations were performed using an anti-HSV antibody, while the eluted fractions were immunoblotted against the HA epitope. EscC tagged with the HA epitope coeluted with EscA-HSV (Fig. 5B). These data are in line with the SILAC results and confirm that EscA and EscC interact, either directly or through additional component(s) of the T3SS.
EscC has been previously shown to form the OM ring of the T3SS apparatus but also constitutes a structural platform that is crucial for interaction with other components of the export apparatus at the periplasmic space (35). Therefore, the EscA-EscC interaction is compatible with EscA localization, as described above.
To confirm that the tagged versions of EscC and EscA are functional, we expressed both proteins in the double-deletion background of an ΔescA ΔescC strain and checked for T3S. The ΔescA ΔescC mutant strain showed no T3S; however, expression of both tagged proteins rescued the T3S activity, as indicated by the levels of EspB, EspD, and EspA in the secreted fraction (Fig. 5C).
The predicted coiled-coiled structure of EscA is critical for its activity.
In an attempt to functionally characterize EscA, we searched for structural motifs within the EscA protein sequence. Using the Paircoil program, we observed that EscA is predicted to adopt a coiled-coil structure (Fig. 6A) (1). Coiled-coil structures contain two or more α-helices that wind around each other to form supercoils. These domains were shown to contribute to various aspects of structural and regulatory proteins mostly related to complex assembly and intermolecular interactions (7, 14, 21). Coiled-coil elements were previously shown to contribute to the structural flexibility of T3 secreted effectors (7, 29); however, their involvement in T3S apparatus assembly or in its activity was not shown.
Fig 6.

Multiple mutations in the coiled-coil region of EscA abolish T3S. (A) EscA sequence. Predicted helical regions are shown as gray rectangles. Positions of point mutations are indicated by arrows. (B) Helical wheel projection of residues 75 to 106 of the EscA protein. Hydrophilic residues are presented as circles, hydrophobic residues are represented by diamonds, potentially negatively charged amino acids are shown as triangles, and potentially positively charged amino acids are shown as pentagons. The residues that were mutated are shown in red. (C) Protein secretion profiles of an EPEC ΔescA strain complemented with WT EscA or EscA carrying triple lysine-to-glutamic acid mutations. Secreted proteins were concentrated from supernatants of bacterial cultures grown in DMEM and analyzed by Coomassie staining of an SDS–12% PAGE gel. The locations of the translocators EspA, EspB, and EspD are indicated at the right of the gel. Also indicated is the location of EPEC EspC, which is not secreted via the LEE-encoded T3SS. (D) Protein secretion profiles of an EPEC ΔescA strain complemented with WT EscA or single lysine-to-glutamic acid mutations. Secretion was examined using a procedure similar to that described for in panel C.
Visualization of the helical organization of the EscA protein (http://rzlab.ucr.edu/scripts/wheel) revealed a positively charged cluster (residues K77, K84, and K91) localized at a specific helical interface (Fig. 6B). A tertiary structure prediction model, based on the I-TASSER prediction software (33, 39), indicated that EscA folds into a coiled-coil structure with four helices (see Fig. S1A in the supplemental material), with the three lysines at positions 77, 84, and 91 pointing toward the protein's core and contributing to the overall stability of the coiled-coil structure. To assess the contribution of the coiled-coil structure on the function of EscA protein, we tried to force EscA to adopt an alternative conformation by replacing lysines with glutamic acid residues in these core amino acid positions (K77E, K84E, and K91E) and examined the ability of this triple EscA mutant to complement T3S of a ΔescA strain. We found that while the WT EscA protein was able to complement the T3S activity of a ΔescA strain, the triple EscA mutant could not (Fig. 6C). To confirm that the deficient activity of the triple mutant did not result from a single mutation of a functionally critical lysine, we introduced single lysine-to-glutamic acid mutations into EscA and examined the ability of the single EscA mutants to complement the T3S activity of the ΔescA strain. All three single mutants enabled T3S activity (Fig. 6D). Mutation of lysine at position 77 showed lower levels of the T3 secreted proteins than the WT; however, this mutant was still functional. These results show that none of the lysines at positions 77, 84, and 91 is critical for the function of the protein. However, the triple mutant resulted in a loss of function of the EscA protein, probably due to an extensive structural disruption of the proper coiled coil folding (see Fig. S1).
It was recently reported that coiled-coil structures enhance the membrane association of Salmonella T3S effectors (17). This raises the possibility that the disruption of the coiled-coil structures of EscA hinders its membrane association and therefore affects T3S. This possibility correlates well with the effect we observed for truncation of the first 19 amino acids of EscA that abolish membrane association and cause T3S malfunction (Fig. 4).
Conclusions.
The T3SS is a specialized transport machinery that has evolved to secrete virulence proteins into host cells. T3SSs have many common features, and they resemble the flagellar system. However, every T3SS has adapted to function best under specific environmental conditions and in a specific host. Although the main proteins that form the T3S export apparatus are highly conserved, there are a few proteins that do not have homologs in other systems. Here, we studied such a protein, EscA.
The EscA/Orf15 is a small protein (∼15 kDa) encoded by the LEE. Bioinformatics analysis of this protein, using standard BLAST search and even a more sophisticated PSI-BLAST search, found no homologs of this protein except in the related pathogens C. rodentium and enterohemorrhagic E. coli that belong to the same A/E pathogen family (28). The findings that this protein is crucial for T3S of EPEC and for the virulence of C. rodentium (8) suggested that this protein has a significant role in the secretion apparatus. Based on our results, we concluded that EPEC Orf15 is a structural protein involved in the assembly of the T3S, and we therefore suggested renaming it with the prefix Esc (for E. coli secretion apparatus component) and the suffix A. The biochemical characterization of EscA raises the possibility that this periplasmic protein serves as a bridge between the OM and IM rings. However, additional work is required to identify the exact protein within the IM ring with which EscA interacts and to determine the exact architecture of the connection between the OM and IM rings.
Supplementary Material
ACKNOWLEDGMENTS
We thank Wanyin Deng for critical reading of the manuscript.
Work in B.B.F.'s laboratory is funded by the Canadian Institutes of Health Research (CIHR). The work done in L.J.F.'s laboratory was funded by CIHR (MOP-77688). B.B.F. is the University of British Columbia Peter Wall Distinguished Professor. L.J.F. is the Canada Research Chair in Quantitative Proteomics. N.S.-M. and E.B.-O. are supported by postdoctoral fellowships from the Michael Smith Foundation for Health Research. N.S.-M. is also supported by the Natural Sciences and Engineering Research Council of Canada and the Rothschild Foundation. M.A.C. is supported by a Canadian Association of Gastroenterology/CIHR/Ferring Pharmaceuticals fellowship.
Footnotes
Published ahead of print 23 March 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Berger B, et al. 1995. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. U. S. A. 92:8259–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bhasin M, Garg A, Raghava GP. 2005. PSLpred: prediction of subcellular localization of bacterial proteins. Bioinformatics 21:2522–2524 [DOI] [PubMed] [Google Scholar]
- 3. Biemans-Oldehinkel E, Sal-Man N, Deng W, Foster LJ, Finlay BB. 2011. Quantitative proteomic analysis reveals formation of an EscL-EscQ-EscN type III complex in enteropathogenic Escherichia coli. J. Bacteriol. 193:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black RE, et al. 2010. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375:1969–1987 [DOI] [PubMed] [Google Scholar]
- 5. Burghout P, et al. 2004. Structure and electrophysiological properties of the YscC secretin from the type III secretion system of Yersinia enterocolitica. J. Bacteriol. 186:4645–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cornelis GR. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4:811–825 [DOI] [PubMed] [Google Scholar]
- 7. Delahay RM, Frankel G. 2002. Coiled-coil proteins associated with type III secretion systems: a versatile domain revisited. Mol. Microbiol. 45:905–916 [DOI] [PubMed] [Google Scholar]
- 8. Deng W, et al. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. U. S. A. 101:3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edwards RA, Keller LH, Schifferli DM. 1998. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207:149–157 [DOI] [PubMed] [Google Scholar]
- 10. Fields KA, Plano GV, Straley SC. 1994. A low-Ca2+ response (LCR) secretion (ysc) locus lies within the lcrB region of the LCR plasmid in Yersinia pestis. J. Bacteriol. 176:569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Filip C, Fletcher G, Wulff JL, Earhart CF. 1973. Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J. Bacteriol. 115:717–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foster LJ, De Hoog CL, Mann M. 2003. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. U. S. A. 100:5813–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gauthier A, Puente JL, Finlay BB. 2003. Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect. Immun. 71:3310–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gazi AD, Charova SN, Panopoulos NJ, Kokkinidis M. 2009. Coiled-coils in type III secretion systems: structural flexibility, disorder and biological implications. Cell. Microbiol. 11:719–729 [DOI] [PubMed] [Google Scholar]
- 15. Ilan O, et al. 1999. Protein tyrosine kinases in bacterial pathogens are associated with virulence and production of exopolysaccharide. EMBO J. 18:3241–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaniga K, Delor I, Cornelis GR. 1991. A wide-host-range suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene 109:137–141 [DOI] [PubMed] [Google Scholar]
- 17. Knodler LA, Ibarra JA, Perez-Rueda E, Yip CK, Steele-Mortimer O. 2011. Coiled-coil domains enhance the membrane association of Salmonella type III effectors. Cell. Microbiol. 13:1497–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koster M, et al. 1997. The outer membrane component, YscC, of the Yop secretion machinery of Yersinia enterocolitica forms a ring-shaped multimeric complex. Mol. Microbiol. 26:789–797 [DOI] [PubMed] [Google Scholar]
- 19. Levine MM, et al. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 1:1119–1122 [DOI] [PubMed] [Google Scholar]
- 20. Linderoth NA, Simon MN, Russel M. 1997. The filamentous phage pIV multimer visualized by scanning transmission electron microscopy. Science 278:1635–1638 [DOI] [PubMed] [Google Scholar]
- 21. Lupas A. 1996. Coiled coils: new structures and new functions. Trends Biochem. Sci. 21:375–382 [PubMed] [Google Scholar]
- 22. McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mendrola JM, Berger MB, King MC, Lemmon MA. 2002. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J. Biol. Chem. 277:4704–4712 [DOI] [PubMed] [Google Scholar]
- 24. Moon HW, Whipp SC, Argenzio RA, Levine MM, Giannella RA. 1983. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41:1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nikaido H. 1994. Isolation of outer membranes. Methods Enzymol. 235:225–234 [DOI] [PubMed] [Google Scholar]
- 26. Nouwen N, et al. 1999. Secretin PulD: association with pilot PulS, structure, and ion-conducting channel formation. Proc. Natl. Acad. Sci. U. S. A. 96:8173–8177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ong SE, et al. 2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1:376–386 [DOI] [PubMed] [Google Scholar]
- 28. Pallen MJ, Beatson SA, Bailey CM. 2005. Bioinformatics analysis of the locus for enterocyte effacement provides novel insights into type-III secretion. BMC Microbiol. 5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pallen MJ, Dougan G, Frankel G. 1997. Coiled-coil domains in proteins secreted by type III secretion systems. Mol. Microbiol. 25:423–425 [DOI] [PubMed] [Google Scholar]
- 30. Pugsley AP. 1993. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57:50–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogers LD, et al. 2008. Identification of cognate host targets and specific ubiquitylation sites on the Salmonella SPI-1 effector SopB/SigD. J. Proteomics 71:97–108 [DOI] [PubMed] [Google Scholar]
- 32. Rose RE. 1988. The nucleotide sequence of pACYC184. Nucleic. Acids Res. 16:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Russel M. 1994. Phage assembly: a paradigm for bacterial virulence factor export? Science 265:612–614 [DOI] [PubMed] [Google Scholar]
- 35. Spreter T, et al. 2009. A conserved structural motif mediates formation of the periplasmic rings in the type III secretion system. Nat. Struct. Mol. Biol. 16:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van Gijsegem F, et al. 1995. The hrp gene locus of Pseudomonas solanacearum, which controls the production of a type III secretion system, encodes eight proteins related to components of the bacterial flagellar biogenesis complex. Mol. Microbiol. 15:1095–1114 [DOI] [PubMed] [Google Scholar]
- 37. Waksman G. 2009. Going round in circles: the structural biology of type III secretion systems. Nat. Struct. Mol. Biol. 16:459–460 [DOI] [PubMed] [Google Scholar]
- 38. Woestyn S, Allaoui A, Wattiau P, Cornelis GR. 1994. YscN, the putative energizer of the Yersinia Yop secretion machinery. J. Bacteriol. 176:1561–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.