

Abstract
Antimicrobial peptides (AMPs) have therapeutic potential, particularly for localized infections such as those of the lung. Here we show that airway administration of a pegylated AMP minimizes lung tissue toxicity while nevertheless maintaining antimicrobial activity. CaLL, a potent synthetic AMP (KWKLFKKIFKRIVQRIKDFLR) comprising fragments of LL-37 and cecropin A peptides, was N-terminally pegylated (PEG-CaLL). PEG-CaLL derivatives retained significant antimicrobial activity (50% inhibitory concentrations [IC50s] 2- to 3-fold higher than those of CaLL) against bacterial lung pathogens even in the presence of lung lining fluid. Circular dichroism and fluorescence spectroscopy confirmed that conformational changes associated with the binding of CaLL to model microbial membranes were not disrupted by pegylation. Pegylation of CaLL reduced AMP-elicited cell toxicity as measured using in vitro lung epithelial primary cell cultures. Further, in a fully intact ex vivo isolated perfused rat lung (IPRL) model, airway-administered PEG-CaLL did not result in disruption of the pulmonary epithelial barrier, whereas CaLL caused an immediate loss of membrane integrity leading to pulmonary edema. All AMPs (CaLL, PEG-CaLL, LL-37, cecropin A) delivered to the lung by airway administration showed limited (<3%) pulmonary absorption in the IPRL with extensive AMP accumulation in lung tissue itself, a characteristic anticipated to be beneficial for the treatment of pulmonary infections. We conclude that pegylation may present a means of improving the lung biocompatibility of AMPs designed for the treatment of pulmonary infections.
INTRODUCTION
Antimicrobial peptides (AMPs) are short ∼15- to ∼40-mer cationic peptides which are produced naturally by both eukaryotes and prokaryotes and which possess microbicidal activity against a range of bacterial and virus species. AMPs have attracted considerable interest as synthetic, molecularly tunable agents that may potentially subvert a number of microbial-resistance mechanisms (21). However, their physical properties (i.e., cationic charge, peptidic composition) present challenges in the creation of biologically stable agents and in balancing antimicrobial efficacy against host cell membrane toxicity.
In the treatment of respiratory tract infections, local inhaled delivery of antimicrobial agents may be particularly appropriate, and especially so for AMPs, whose stability may be poor when administered by other mucosal routes or indeed following intravenous administration. Exogenous delivery of natural AMPs into the lung has clinical precedence; e.g., colistin, a mixture of several natural peptides (polymyxins), is nebulized for the management of pulmonary infections in cystic fibrosis patients (31). Local lung delivery may also offer potential for greater antimicrobial concentrations in the lung epithelial lining fluid (ELF) to combat infection while concurrently minimizing systemic exposure. Nevertheless, concern over local AMP-induced pulmonary toxicities will be heightened.
In the few in vivo experimental studies that have examined lung antimicrobial efficacy of airway-administered AMPs (3, 6, 11, 23, 47), the effectiveness in clearing lung infection has been variable. While such mixed success likely reflects multifactorial differences in the modes of action of the various AMPs, the microbial burden, and the character of the lung infection model itself, another critical determinant about which little is known is the pulmonary disposition of AMPs following local delivery.
We have previously reported (15) on the design of a novel 21-mer peptide (CaLL) composed of fragments from the human cathelicidin peptide LL-37 and the silk moth peptide cecropin A. The synthetic hybrid CaLL displayed potent antimicrobial activity in vitro against the pulmonary pathogens Bacillus anthracis and Burkholderia cepacia; however, this was accompanied by high hemolytic activity. It is well documented that pegylation of proteins and peptides can elicit steric hindrance to reduce interactions with cells and enzymes (43). When CaLL was conjugated with polyethylene glycol (PEG), a modest but nevertheless significant decrease in hemolytic activity of CaLL was observed (15).
In this study, we tested the hypothesis that pegylation of an inhaled AMP would minimize pulmonary cell and lung tissue toxicity while nevertheless still affording maintenance of effective antimicrobial activity. Using both primary cultures of lung epithelial cells and an intact lung model, i.e., the isolated perfused rat lung (IPRL), we assessed the lung toxicity associated with LL-37 and cecropin A and that associated with the synthetic AMPs CaLL and CaLL N-terminally conjugated to low-molecular-weight PEG chains (PEG-CaLL). Antimicrobial activity was assessed against both Gram-negative and Gram-positive bacteria. The airway administration of the AMPs into the IPRL also provided unique information on the pulmonary residence and systemic absorption of lung-administered AMPs. Further, using cell biological and biophysical approaches, we explored the previously reported phenomenon that pegylation of AMPs has beneficial effects on the selectivity of interaction between prokaryotic and eukaryotic cell membranes (20). Using both circular dichroism (CD) and fluorescence spectroscopy, the biophysical interactions of the AMPs with artificial lipid models of eukaryotic and prokaryotic membranes were examined to explore the issues of AMP conformation and membrane selectivity.
MATERIALS AND METHODS
Tissue culture plastics were from Corning (High Wycombe, United Kingdom). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPG) were purchased from Avanti Polar Lipids (Alabaster, AL) as dry powders. All other reagents and general lab consumables were purchased from Fisher (Loughborough, United Kingdom) or Sigma-Aldrich (Poole, United Kingdom).
Bacterial strains, growth conditions, and media.
Staphylococcus aureus 29213, Escherichia coli 25922, and Bacillus anthracis UM23-CL2 were obtained from the culture collection at the Defence Science and Technology Laboratory (DSTL) (Porton Down, Salisbury, United Kingdom). All strains were handled under Advisory Committee on Dangerous Pathogens (ACDP) level II conditions and were maintained on lysogeny broth (LB) or LB agar. All strains were cultured at 37°C.
Preparation of B. anthracis spores.
B. anthracis UM23-CL2 was cultured on new sporulation medium (3.0 g Difco tryptone, 6.0 g Oxoid bacteriological peptone, 3.0 g Oxoid yeast extract, 1.5 g Oxoid Lab Lemco, 1 ml 0.1% MnCl2 · 4H2O, and 25 g Difco Bacto agar made up to 1 liter with H2O) in Roux bottles at 37°C until the cultures contained more than 95% phase-bright spores, as measured by phase-contrast microscopy, typically between 7 and 10 days. The spores were harvested by centrifugation at 10,000 × g for 10 min and then washed 10 times with ice-cold distilled water to remove any vegetative cells and cell debris. The spore preparation was heated at 70°C for 1 h to kill any remaining vegetative cells, and then the final spore concentration was determined by serial dilution. Spore preparations were stored at −20°C until they were required.
Animals.
Male specific-pathogen-free Sprague-Dawley rats (250 to 350 g; Harlan, United Kingdom) were used throughout and housed in rooms maintained at 20 to 25°C and 40 to 70% relative humidity. The animals had free access to food and water during acclimatization for ≥2 days prior to experimentation. All animal experiments and the protocols described below adhered to the Animal (Scientific Procedures) Act 1986 (United Kingdom).
Peptide synthesis, characterization, and quantitation.
The peptides listed in Table 1 were synthesized by Alta Biosciences (Birmingham, United Kingdom) at a purity of ≥90%. Subscript numbers in PEG2-CaLL and PEG3-CaLL denote the number of 22-atom PEG units attached sequentially to the N terminus of CaLL to form a linear PEG chain.
Table 1.
Sequence data of peptides used in this study
| Peptide | Sequencea | Molecular mass (Da) |
|
|---|---|---|---|
| Theoretical | Measured | ||
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 4,493.3 | 4,493.0 |
| Cecropin A | KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK | 4,004.8 | 4,006.4 |
| CaLL | KWKLFKKIFKRIVQRIKDFLR | 2,791.5 | 2,791.6 |
| PEG2-CaLL | (PEG)2-KWKLFKKIFKRIVQRIKDFLR | 3,462.3 | 3,463.0 |
| PEG3-CaLL | (PEG)3-KWKLFKKIFKRIVQRIKDFLR | 3,797.7 | 3,798.9 |
Underlined amino acids identify those sequences combined to make CaLL.
HPLC.
Peptide purity was assessed by reversed-phase high-performance liquid chromatography (HPLC) using a Luna C18 column (4.6 by 150 mm; Phenomenex, United Kingdom) attached to a Thermo Finnigan HPLC system. HPLC was performed using a binary gradient: 0 to 5 min, 95% A; 5 to 35 min, linear gradient from 95 to 0% A; 35 to 45 min, hold at 0% A; 45 to 50 min, linear gradient from 0 to 95% A, where mobile phase A was 0.1% (vol/vol) trifluoroacetic acid (TFA) in H2O and B was 0.1% (vol/vol) TFA in acetonitrile (MeCN). Peptide elution was monitored simultaneously at 220 and 254 nm. Cecropin, CaLL, and PEG2-CaLL samples were also monitored using fluorescence detection (λex, 280 nm; λem, 348 nm).
LL-37 ELISA.
LL-37 was detected in isolated perfused rat lung (IPRL) perfusate samples, bronchoalveolar lavage fluid (BALF), and tissue homogenates using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Hycult Biotech, Frontstraat, Netherlands). Kits were used according to the manufacturer's instructions.
Mass spectrometry.
Peptide molecular masses were measured by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry using α-cyano-4-hydroxycinnamic acid as a matrix. Spectra were recorded on a Waters MicroMass MX spectrometer (Waters, MA) in reflectron mode.
In vitro assessment of antimicrobial activity.
The antimicrobial activities of AMPs were determined as a 50% inhibitory concentration (IC50), i.e., the lowest peptide concentrations at which bacterial growth was inhibited by 50%, using the modified microtiter broth dilution method (41). For these assays, spores or bacterial cells cultured to early exponential phase were used at 105 to 107 (typically ∼106) CFU/ml and tested against AMP concentration ranges of 62.5 μg/liter to 128 mg/liter in 2-fold dilution steps. Microtiter plates were prepared immediately prior to use, with peptide concentrations, arranged in triplicate, suspended in 100 μl 0.02% acetic acid and 0.4% bovine serum albumin (BSA/AA buffer; Sigma-Aldrich, United Kingdom). A suspension containing 100 μl of diluted bacteria was added to each well of a 96-well microtiter plate, except for the negative controls, where just broth was added. Plates were incubated at 37°C for 16 h. Bacterial growth was determined by measuring the absorbance at 620 nm with a Bio-Tek EL800 microplate reader (Bio-Tek Industries, Bedfordshire, United Kingdom). IC50s were determined for each AMP as the concentration necessary to inhibit bacterial growth by 50% or more compared to the growth observed in control wells.
In vitro cell viability assay in primary rat lung alveolar epithelial cells.
The isolation and primary culture of type II rat alveolar epithelial (ATII) cells was undertaken according to procedures reported previously (2). Over 6 to 8 days of culture, ATII cells have been shown to adopt a phenotype more characteristic of alveolar epithelial type-I-like (ATI-like) cells (9) whereas day 2 cultures retain the features of the ATII cell phenotype. AMPs prepared as ∼2-mg/ml stock solutions in sterile phosphate-buffered saline (PBS) were serially diluted in full culture medium (containing the necessary volume of PBS vehicle as a control). Primary cultures were incubated with AMP solutions at 37°C for 8 h in a cell culture incubator, and cell viability was determined using the MTT substrate as described previously (38).
Assessment of pulmonary barrier toxicity and AMP pulmonary absorption in an IPRL model.
An IPRL preparation employing a forced intratracheal solution instillation technique was used as previously described (8, 27, 33). Briefly, a rat lung was surgically removed and housed horizontally in a humidified artificial glass thorax maintained at 37°C. The lung was perfused (15 ml/min) through the pulmonary artery with perfusate comprising Krebs-Henseleit buffer supplemented with 4% (wt/vol) bovine serum albumin (BSA). The lungs were ventilated under positive pressure with atmospheric air at a tidal volume of 2.5 ml, 20 cycles per min.
AMP dosing solutions (0.1 ml) were prepared containing 100 μg of the respective AMP dissolved in 20 mM PBS, pH 7.4. Dosing solutions also contained a nominal dose of ∼0.2 μCi [14C] mannitol and 200 μg 4-kDa fluorescein isothiocyanate (FITC) dextran (FD4000) serving as transport probes to monitor pulmonary membrane integrity. Dosing solutions were administered into the airways of the IPRL as a coarse spray through a single actuation of the pressurized metered dose inhaler, delivering 5.5 cm3 propellant vapor. Serial aliquots were sampled from the recirculating perfusate at predetermined times and centrifuged (13,000 × g, 10 min, 4°C) to remove trace blood cells, and the supernatant was stored at −20°C until assay. To quantify AMP content, BALF and perfusate samples (0.5 ml) were treated with 1 ml MeCN and incubated for 5 min on ice and then centrifuged at 12,000 × g to sediment the protein pellet. Supernatants were transferred to clean tubes and stored at −20°C. Lung tissue was homogenized and extracted into 5 ml of 60% MeCN plus 1% trifluoroacetic acid and mixed end over end at 4°C. This was followed by centrifugation at 12,000 × g to sediment insoluble material, and then the homogenate was syringe filtered (0.2-μm-pore-size filter) into a 10-ml round-bottom flask before all solvent was removed under reduced pressure. Extracted CaLL, PEG-CaLL, and cecropin were redissolved in 2 ml H2O plus 0.1% TFA prior to HPLC analysis. LL-37 was resuspended in PBS prior to quantification by ELISA. Quantitation of the permeability probe [14C]mannitol was by liquid scintillation counting using a Tricarb 2900TR (Perkin Elmer, Shelton, CT). The permeability probe FD4000 was quantified by microplate fluorescence spectroscopy (λex, 485 nm; λem, 520 nm; FLUOstar; BMG, Aylesbury, United Kingdom) using calibration curves constructed in perfusate solution. Cumulative mass transfer of analyte was determined by multiplying the sample concentration by the total volume of perfusate, correcting for mass removed during sampling, as described previously (27).
Proteolytic stability of AMPs in BALF.
BALF was harvested from rats that had been euthanized by a pentobarbital overdose and cervical dislocation. The trachea was cannulated with a 23G intravenous cannula, and then the lungs were removed from the thoracic cavity en bloc. Lungs were lavaged by the introduction under gravity of three 5-ml aliquots of normal saline. BALF harvested from three animals was pooled and transferred to sterile 12-well plates that had been blocked overnight with 0.5% (wt/vol) BSA in PBS to prevent nonspecific adsorption. AMPs were incubated in BALF or normal saline (control) at a concentration of 0.4 mg/ml for up to 12 h. Samples (0.3 ml) were taken at predetermined time intervals and immediately centrifuged at 12,000 × g for 5 min to pellet luminal cells. BALF supernatants were mixed with two volumes of MeCN to precipitate proteins and then applied to an ultrafiltration plate with a molecular weight cutoff of 10,000 (Millipore, Watford, United Kingdom) and centrifuged at 4,235 × g at 4°C for 90 min (Rotanta 460R; Hettich Lab Technology, Tuttlingen, Germany). Ultrafiltrate samples were stored at −20°C before analysis by HPLC. To ensure correct interpretation of our HPLC data, the concentration of each BALF-incubated sample was normalized to the recovery of peptide from the saline-only control at each time point, thus accounting for nonspecific loss through peptide adsorption to the plasticware and filters. The recovery of each peptide was calculated from the time zero sample and found to be >90% for all analytes. The intra-assay analytical precision was determined to be ∼5% relative standard deviation (RSD), as determined from separate injections of 100-μg/ml standard peptide solutions.
Antimicrobial activity of AMPs in the presence of BALF.
The antimicrobial efficacy of AMPs against B. anthracis spores was determined in the presence of BALF at an initial AMP concentration of 100 μg/ml (22 μM LL-37, 35 μM CaLL, and 30 μM PEG2-CaLL). Briefly, 180 μl of BALF was mixed with 40 μl of a 10× concentrate of CaLL, PEG2-CaLL, or LL-37 (1 mg/ml in BSA/AA buffer) and then mixed with 180 μl of B. anthracis spore suspension (∼106 spores/ml). Samples were removed at 0, 0.5, 1, 2, 3, and 4 h and serially diluted in PBS. Viable bacteria were enumerated on agar plates. To control for possible effects of BALF on bacterial growth, additional samples were prepared where 40 μl of the AMP solution was replaced with BSA/AA buffer.
CD spectroscopy.
Spectra were recorded on an Aviv 215 CD spectrophotometer (Aviv Biomedical Inc., Lakewood, NJ; 1-nm bandwidth, 0.2-nm steps, 3 to 5 s per point accumulation) equipped with a thermostatted cell holder. Samples were prepared in 100 mM NaF–20 mM KH2PO4-NaOH, pH 7.2 (CD buffer). NaF was used to avoid the chloride absorbance of NaCl at a λ of <195 nm. Spectra were recorded in a 0.1-cm quartz cell at peptide concentrations of 0.1 to 0.2 mg/ml determined from the optical density at 280 nm (OD280) assuming absorption coefficients calculated from the amino acid composition (30) or, in the case of LL-37, the peptide bond absorbance (36). Buffer baselines were subtracted, and data were smoothed and normalized to mean residue ellipticities with the equation [θ]MRW = θobs × MRW/(l × c), where θobs is the observed ellipticity (mdeg), MRW the mean residue weight, l the path length (mm), and c the concentration (mg/ml). The instrument was calibrated with camphorsulfonic acid (10). α-Helical content fH was estimated from [ϴ]222 as fH = ([ϴ]222−[ϴ]c)/([ϴ]h−[ϴ]c) with [ϴ]c = 2,200−53 × T and [ϴ]h = (250 × T −44,000)/(1−3/n), where T is the temperature (°C) and n the number of residues (25). Induction of α-helix formation by 2,2,2-trifluoroethanol (TFE) was monitored at 222 nm, and data were fitted assuming a one-site binding mechanism fH = fhmax × c/(KD + c), where fhmax and KD are the maximum helix fraction achievable and apparent dissociation constant, respectively.
Small unilamellar vesicles (SUV).
Thin lipid films were produced by rotary evaporation of lipid solutions (10 mg/ml in chloroform) and further drying overnight under vacuum. Films were hydrated at a lipid concentration of 10 mg/ml in either CD buffer or PBS. Lipid suspensions were subjected to 10 freeze-thaw cycles (2 min in liquid N2, 5 min in a 37°C water bath, 2 min vortex mixing) and left overnight at room temperature (RT). Vesicle suspensions were extruded 10 times at RT through two stacked 100-nm or 50-nm Nuclepore membranes (Whatman, United Kingdom) for fluorescence and CD experiments, respectively. Vesicles were stored at RT and used within 24 h.
Fluorescence spectroscopy.
Fluorescence spectra were recorded on an Aminco-Bowman Series 2 luminescence spectrometer (SLM-Aminco Spectroscopic Instruments, Rochester, NY). A 200-μl portion of CaLL or PEG2-CaLL (4 μM) was mixed 1:1 (vol/vol) with lipid vesicle suspension to give a final lipid concentration between 0 and 50 μM. Tryptophan was excited at 280 nm and the emission spectrum scanned between 300 and 450 nm at 1-nm increments over 75 s. Emission curves were fitted using a Gaussian nonlinear regression analysis (GraphPad Prism v4.0) to determine individual λmax values for each scan.
RESULTS
In vitro AMP activity.
To determine whether the attachment of small PEG chains to the N terminus of CaLL effected a change in antimicrobial potency, IC50s were determined against a select range of bacteria. Table 2 shows the IC50s for CaLL and two pegylated derivatives against Gram-positive B. anthracis (both vegetative and spore forms) and against the Gram-positive S. aureus and the Gram-negative E. coli. Compared to the parental LL-37 peptide, CaLL was more potent against germinating B. anthracis spores by almost 5-fold and at least 11-fold more potent against the vegetative form.
Table 2.
Antibacterial activity of AMPs
| Bacterial type | Strain | IC50 (μM)a |
|||
|---|---|---|---|---|---|
| CaLL | PEG2-CaLL | PEG3-CaLL | LL-37 | ||
| Gram positive | B. anthracis spores | 2.8 | 4.5 | 8.2 | 13.9 |
| B. anthracis vegetative | 5.6 | 9.0 | 16.5 | >65 | |
| S. aureus | 2.8 | 18.1 | 16.5 | ND | |
| Gram negative | E. coli | 0.7 | 2.3 | 8.2 | ND |
IC50 values (n = 3). ND = not determined.
The pegylated CaLL derivatives retained significant antimicrobial activity, with the IC50s remaining within an order of magnitude of that of CaLL. The PEG2 conjugate retained the greater potency compared to the PEG3 conjugate, with the former displaying only a 2-fold- to 3-fold-higher IC50 compared to CaLL toward B. anthracis and E. coli.
Lung epithelial toxicity of AMPs.
The potential for AMPs to induce membrane damage to lung epithelial cells over an 8-h period was investigated using the MTT assay with primary cultures of rat lung alveolar epithelial (AE) cells. Both the ATII and the ATI-like cultures showed decreases in cell viability with increases in AMP concentration. At the highest AMP concentration tested (90 μM) the cell viability of the ATII cells (Fig. 1A) was reduced by more than 60% by CaLL and by 40% and 30% following exposure to PEG2-CaLL and PEG3-CaLL, respectively. The loss of cell viability was more profound in the attenuated ATI-like cells (Fig. 1B) with a >70% loss of viability associated with CaLL and the PEG2- and PEG3-CaLL derivatives at 90 μM. The response of the ATII and ATI-like cells to cecropin A and LL-37 peptides was less consistent (Fig. 1C and D). Even at the highest concentration tested (90 μM) the LL-37 peptide did not affect the turnover of the MTT dye in the ATII cells (Fig. 1C) although it did elicit an approximate 60% loss in MTT turnover in the ATI-like cells (Fig. 1D). In contrast, cecropin A (90 μM) appeared to have no adverse effect in ATI-like cells (Fig. 1D) but reduced the viability of ATII cells by 60% (Fig. 1C). The effects of AMPs were comparable when cells were incubated with AMPs in medium without serum (data not shown).
Fig 1.

In vitro biocompatibility of AMPs. ATII primary cultures of rat AE cells were incubated for 8 h with increasing concentrations of CaLL or pegylated CaLL derivatives (A) or cecropin A and LL-3 (C). ATI-like primary cultures of rat AE cells were incubated with increasing concentrations of CaLL or pegylated CaLL (B) or cecropin A and LL-37 (D). Data shown are means ± standard errors of the means (SEM) (n ≥ 6 from three different primary cultures). AMPs were applied in full culture medium including 10% serum.
We next assessed in the IPRL model the integrity of the intact pulmonary barrier over a 90-min period following administration of the AMPs into the airways. This was achieved by coinstillation (simultaneously with the AMP) of the hydrophilic paracellular permeability probe [14C]mannitol or FD4000. The pulmonary absorption half-life (t1/2) for mannitol administered alone (i.e., without AMP) was 19 min (Table 3). However, upon coinstillation with 30 nmol CaLL, the mannitol absorption t1/2 shortened considerably (P < 0.05), to 3 min, indicating that the permeability of the IPRL epithelium to mannitol had greatly increased. Further, at the 30-nmol dose of CaLL there was significant visual evidence of pulmonary edema developing soon after dosing. In contrast, instillation of an equivalent molar dose of PEG2-CaLL (33 nmol) failed to cause any visible evidence of edema and no barrier perturbation, as evidenced by the mannitol absorption t1/2 remaining similar (P > 0.05) to the control value. Reducing the dose of CaLL by 10-fold (3 nmol CaLL) also prevented any gross morphological evidence of pulmonary barrier alteration or quantitative changes in mannitol absorption (Table 3). This observation indicates that pegylation of CaLL had a profound beneficial impact to reduce CaLL toxicity in the IPRL model. With respect to the parental peptides, no change (P > 0.05) in the pulmonary absorption t1/2 of mannitol was evident upon coinstillation with LL-37 (12 nmol). However, a surprising and as-yet-unexplained lengthening (P < 0.05) in mannitol absorption t1/2 was observed when coadministered with cecropin A (deposited dose, 21 nmol), which was indicative of a retardation in the mannitol absorption process or a reduced availability of mannitol for absorption. We also studied the effect of the AMPs upon the IPRL permeability to coinstilled FD4000, a larger 4-kDa hydrophilic permeability probe. Here, the only AMP coinstillation treatment to alter FD4000 permeability in the IPRL model was again high-dose (30-nmol) CaLL, which significantly (P < 0.05) shortened the absorption half-life (t1/2) of FD4000, indicating acute barrier perturbation (data not shown).
Table 3.
Permeability of pulmonary epithelium to mannitol and pulmonary absorption of AMPs in the IPRL modela
| Treatment | AMP deposited dose (nmol) | Extent of AMP absorptionb | Mannitol absorption t1/2 (min)c |
|---|---|---|---|
| Control | 18.9 ± 8.4 | ||
| CaLL | 30 ± 0.7 | <3 | 2.6 ± 1.3† |
| CaLL | 3 ± 0.4 | 18.3 ± 1.1 | |
| PEG2-CaLL | 33 ± 5.1 | <3 | 16.1 ± 5.9 |
| LL-37 | 12 ± 0.2 | 0.5 ± 0.1 | 17.8 ± 1.0 |
| Cecropin A | 21 ± 0.2 | <3 | 31.6 ± 1.2† |
Data shown are mean ± standard deviation (SD) (n ≥ 3).
Extent of AMP absorption was determined at 90 min from the IPRL perfusate concentration and the lung-deposited dose.
Mannitol absorption t1/2 was determined by nonlinear regression of the cumulative transport curves. †, P < 0.05 compared to control by one-way analysis of variance (ANOVA) and Dunnett's post hoc test.
Pulmonary absorption of AMPs in the IPRL model.
The IPRL model was also used to examine the pulmonary disposition of each of the AMPs. Table 3 also shows the lung-deposited dose for each of the peptides administered into the IPRL and the extent of AMP dose absorption across the lung epithelial barrier into the perfusate (i.e., vascular compartment of the IPRL) by 90 min. The concentrations of cecropin A, CaLL, and PEG2-CaLL in pulmonary perfusate samples at the end of each 90-min IPRL experiment were below the lower limit of detection (LLD) (fluorescence HPLC assay LLD of ∼50 ng/ml). This indicates a low extent of absorption from the airways. Specifically, based on the LLD and an average final IPRL perfusate volume of approximately 50 ml, the theoretical maximal possible extent of pulmonary absorption for cecropin A, CaLL, and PEG2-CaLL could not be greater than 3% of the AMP dose, and in all probability it was considerably lower than this. This estimation was reinforced by the absorption data for LL-37, which was quantified using an ELISA (limit of quantification, ≤0.1 ng/ml). For LL-37 we determined only 0.5% ± 0.1% of the lung-deposited LL-37 dose to be absorbed over the 90-min IPRL experiment. The mass balance recovery for the AMPs from the combined perfusate, lung tissue, and lung ELF, the latter obtained with significant dilution by bronchoalveolar lavage, was consistently high, within the range of 83 to 94% of the deposited AMP dose. However, a mass balance assessment for the PEG2-CaLL peptide conjugate was not possible, as it was undetectable in BALF and lung tissue although confirmed to be fully intact in the IPRL dosing solution.
Proteolytic stability and bactericidal kinetics of AMPs in the presence of BALF.
To investigate further the disposition of AMPs in the airway luminal environment, each of the AMPs was incubated in fresh rat BALF and the kinetics of AMP degradation was determined (Fig. 2). LL-37 showed the greatest proteolytic stability, evidenced by >80% intact peptide remaining at 12 h. Some 50% of cecropin A was recovered intact after 12 h of in vitro incubation in BALF. The stability of CaLL was intermediate between the two parental peptides, while the stability of the PEG2-CaLL derivative was considerably lower (P < 0.05) than all other AMPs tested; i.e., >50% of PEG2-CaLL peptide conjugate was degraded within 1 h. This degradation involved breakdown of the peptide component, as quantitative analysis could not detect the presence of free intact CaLL in the in vitro BALF medium (data not shown). Of note, in control studies with incubations of the AMPs at 37°C in sterile saline, all of the peptides (including the pegylated derivatives) displayed minimal degradation, with each AMP showing >80% stability over the 12 h of the incubation period (data not shown).
Fig 2.

In vitro AMP stability in fresh rat BALF. AMPs (0.3 mg/ml) were added to BALF and incubated at 37°C for up to 12 h. Samples were removed and the amount of peptide present quantified by HPLC. Data shown are mean ± standard deviation (SD) (n ≥ 4). Only negative SD bars are shown for clarity.
Acknowledging the potential for lung components to attenuate the potency of AMPs delivered into the pulmonary tract, the bactericidal kinetics of AMPs were determined in the presence of rat lung BALF. Figure 3 shows the AMP-mediated killing of germinating B. anthracis spores in the standard bacterial culture medium (Fig. 3A) and in culture medium containing 50% (vol/vol) rat lung BALF (Fig. 3B). In the absence of BALF, CaLL killed approximately 99% of B. anthracis spores within 1 h of spore germination commencement (represented as time zero in Fig. 3); the rate of PEG2-CaLL-mediated kill was similar, i.e., ∼95% bacterial kill within 1 h (Fig. 3). The rates of B. anthracis kill by all AMPs tested were not significantly slowed (P < 0.05) by the presence of rat BALF. Under both experimental conditions CaLL and PEG2-CaLL displayed greater activity against germinating spores than LL-37.
Fig 3.

Bactericidal kinetics of AMPs. Kill kinetics of AMPs against germinating B. anthracis are shown. A 100-μg/ml concentration of LL-37, CaLL, or PEG2-CaLL was incubated with spores in the absence (A) or presence (B) of 50% (vol/vol) rat lung BALF. Data shown are means ± standard errors of the means (SEM) (n ≥ 6). Error bars are within the area of the symbols.
Solution conformation of AMPs.
The solution conformations of peptides cecropin A, LL-37, and CaLL with and without PEG chains attached were evaluated by CD spectroscopy. Cecropin A shows a minimum at 200 nm and a shoulder around 220 nm indicative of a mainly unordered secondary structure (Fig. 4A), which is in agreement with former studies on porcine cecropin P1 (37). The LL-37 spectrum, with minima at 222 and 208 nm and a maximum at 192 nm, is characteristic for an α-helical conformation at the given conditions, which agrees with published data (22). The CaLL spectrum, with extrema at 192 and 205 nm, qualitatively represents an additive effect of the conformations of its parent peptides, with a 2:1 contribution of cecropin A to LL-37, suggesting that the two parts behave structurally independently. PEG2-CaLL spectra exhibit the same characteristics as the unpegylated peptide, though the CD amplitude is slightly increased, which might indicate that the PEG chains preferentially stabilize the ordered conformations of CaLL.
Fig 4.
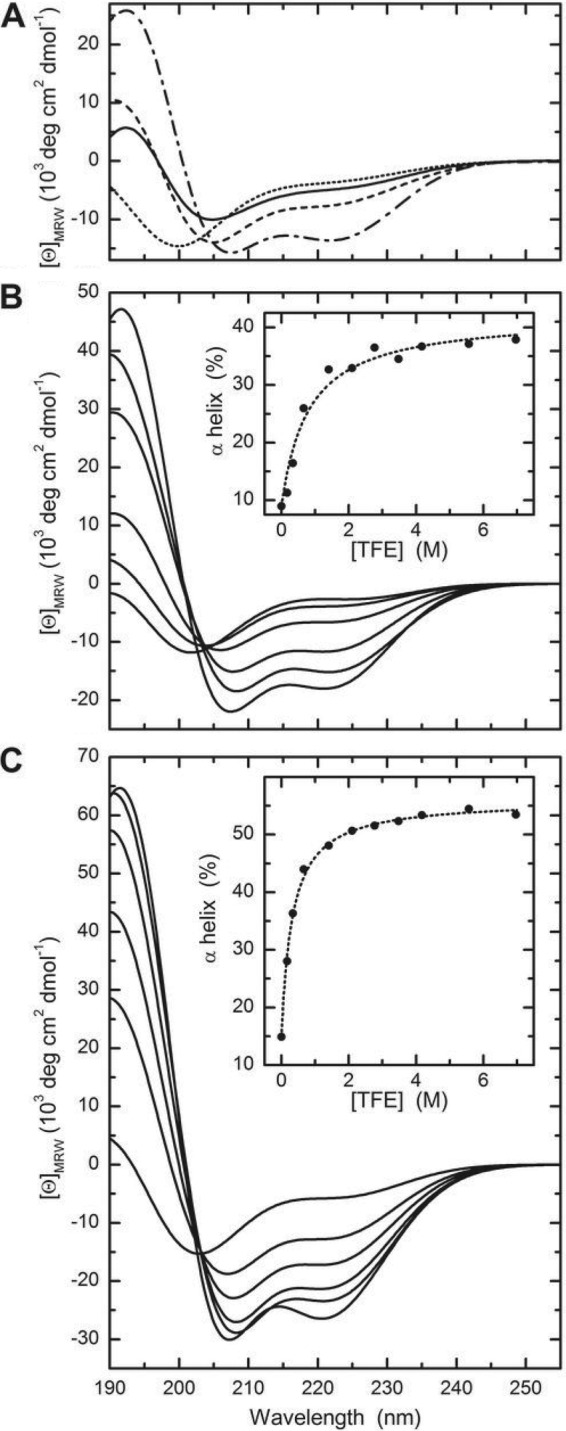
CD spectra of AMPs in buffer. (A) CD spectra were recorded for cecropin A (dotted line), LL-37 (dashed-dotted line), CaLL (solid line), and PEG2-CaLL (dashed line) in CD buffer at 37°C. CaLL (B) and PEG2-CaLL (C) spectra were recorded in the presence of TFE. Spectra shown correspond to TFE concentrations of (from top to bottom at 220 nm) 0, 1, 2, 5, 10, and 50%. Insets show the apparent α-helical content as calculated according to reference 25. Data were fitted to a one-site binding model resulting in fHmax = 0.42 (CaLL) and 0.56 (PEG2-CaLL) with apparent dissociation constants of 800 and 320 nM, respectively.
To estimate the possible structure formation in an environment with reduced water activity mimicking the hydrophobic core of membranes (for a review, see reference 7), CD spectra of CaLL (Fig. 4B) and PEG2-CaLL (Fig. 4C) were recorded at increasing TFE concentrations. Even very low TFE concentrations had a substantial effect, resulting in an increase of the CD amplitudes. At higher concentrations, a clear minimum at 222 nm appeared resulting in spectra similar to LL-37 in benign solvent. Fitting [θ]222 as a function of TFE concentration assuming a one-binding-site model, and relating the values to the degree of α-helix content, results in maximal helicities of 42 and 56% for CaLL and PEG2-CaLL with apparent dissociation equilibrium constants of 800 and 320 mM−1 TFE, respectively (insets in Fig. 4B and C).
AMP conformational changes upon lipid membrane interaction.
Given the substantial effect observed for TFE on the conformation of CaLL and PEG2-CaLL, we examined whether a similar helical change could be induced by interaction with SUVs. All CD experiments were conducted with 50-nm-diameter vesicles in order to reduce light-scattering effects. Whereas no obvious changes of the CD spectrum were seen when POPC (i.e., zwitterionic) vesicles were mixed with cecropin A (Fig. 5A), LL-37 showed a large increase in the CD amplitude (Fig. 5B), and the signal observed for the hybrid peptides CaLL (Fig. 5C) and PEG2-CaLL (Fig. 5D) indicated a transition from a mostly unordered state to one containing a substantial degree of α-helix, e.g., an increase by a factor of ∼2.5 in the cases of CaLL and PEG2-CaLL (Table 4). POPG (i.e., anionic) vesicles showed even larger effects, with an ∼3-fold increase of helical content for cecropin A, CaLL, and PEG2-CaLL. Indeed, the α-helical contents of CaLL and PEG2-CaLL of 41 and 54% in the presence of POPG SUVs are in close agreement with the values of 42 and 56%, respectively, extrapolated from exposure of the peptides to TFE (Fig. 4B and C).
Fig 5.
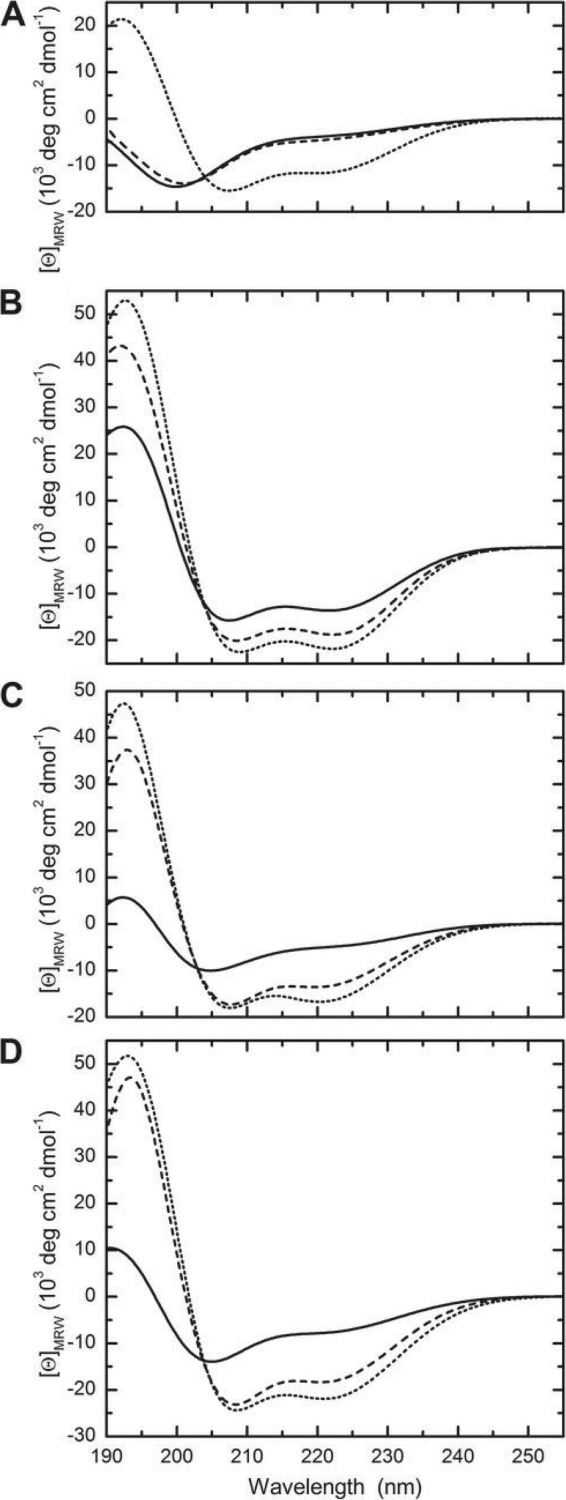
CD spectra of AMPs in the presence of SUVs. Spectra were recorded for cecropin A (A), LL-37 (B), CaLL (C), and PEG2-CaLL (D) at 37°C (solid lines) and in the same buffer in the presence of POPC (dashed line) or POPG (dotted line) SUVs (lipid concentration: 1 mg/ml).
Table 4.
AMP α-helical content in the absence and presence of POPC and POPG SUVs
| Peptide | Lipid | α helix (%)a |
|---|---|---|
| Cecropin A | None | 10 |
| POPC | 12 | |
| POPG | 31 | |
| LL-37 | None | 36 |
| POPC | 50 | |
| POPG | 58 | |
| CaLL | None | 13 |
| POPC | 33 | |
| POPG | 41 | |
| PEG2-CaLL | None | 19 |
| POPC | 45 | |
| POPG | 54 |
Helix content derived from [θ]222 at 37°C according to reference 24.
Fluorescence spectroscopy investigation of AMP lipid membrane binding.
Fluorescence spectroscopy was used to further explore the lipid membrane-binding interactions of CaLL and PEG2-CaLL with experiments conducted at peptide concentrations approximating their IC50s (Table 1). Broadly, in these experiments membrane binding of the peptide was indicated by a shift (Δλmax) in emission maximum to shorter wavelengths while an increase in fluorescence intensity (ΔFl) inferred greater sequestration of the tryptophan indole group into an environment of lower polarity (i.e., the lipid membrane). Mixing CaLL with increasing concentrations of POPG SUVs caused increases in Δλmax and ΔFI (Fig. 6A and C) that were greater than the respective increases observed when CaLL was mixed with POPC vesicles (Fig. 6A and C). This is consistent with the CD data for CaLL. In contrast, PEG2-CaLL showed a more marked preferential interaction with POPG SUVs. For example, a 6-fold-greater Δλmax (Fig. 6B) and a 1.7-fold-greater ΔFI (Fig. 6D) were observed in the presence of 50 μM POPG vesicles than in the interactions measured with an equivalent 50 μM concentration of POPC vesicles (Fig. 6B and D). Studies with the tryptophan-containing parental peptide cecropin A also showed selectivity in lipid-membrane interactions toward the POPG vesicles, although the magnitude of the difference between the anionic and zwitterionic membranes was not as great as those recorded for PEG2-CaLL (data not shown).
Fig 6.

Fluorescence spectroscopy investigation of AMP lipid membrane interactions. Tryptophan fluorescence blue shifts (Δλmax) (A and B) and increases in fluorescence emission intensity (ΔFI) (C and D) were recorded in the presence of SUVs. Data for 2 μM CaLL (A and C) and 2 μM PEG2-CaLL (B and D) were recorded upon titration of increasing concentrations of 100-nm-diameter SUVs comprising either POPG (empty symbols) or POPC (filled symbols).
DISCUSSION
We previously reported upon the design (15) and antimicrobial activity (13) of CaLL, a synthetic AMP derived from selected sequence regions of the cecropin A and LL-37 peptides. While CaLL possesses significantly greater antimicrobial effectiveness compared to its parental AMPs, it also exhibits significant toxicity as determined by increased CaLL-induced hemolysis. Recognizing that improved outcomes can be gained by delivery of inhaled AMPs for the local treatment of pulmonary infections, we examined if pegylation of CaLL provided an advantage in terms of minimizing pulmonary lung tissue toxicity and in the pulmonary pharmacokinetics of the AMP while nevertheless maintaining antimicrobial activity. Further, using spectroscopic techniques we explored the hypothesis that pegylation of CaLL would lead to selectivity of interaction with prokaryotic cell membranes. Enhanced AMP potency through improved selectivity for bacterial membranes might therefore reduce the requisite dose of costly peptide drugs while concurrently improving biocompatibility.
Extending previous studies of the antimicrobial activity of CaLL (13), here we investigated activity against the pulmonary pathogen B. anthracis (in spore and vegetative forms) and also against laboratory strains of E. coli and S. aureus, both of which are potential pulmonary pathogens. We found CaLL to be significantly more potent against all bacterial organisms tested than were its parent peptides; the IC50s of CaLL observed against B. anthracis were among the lowest reported for AMPs tested against the Sterne strain of B. anthracis (12, 24). We also noted that CaLL displayed slightly higher potency against the germinating spore form of B. anthracis, substantiating further investigations into the use of CaLL in the prevention of pulmonary infection following exposure to B. anthracis spores. Previous reports of AMP pegylation, employed for the purposes of increasing peptide solubility or improving proteolytic stability, have documented decreases in antimicrobial activities of between 4- and 30-fold against non-spore-forming bacteria (16, 18, 19, 46). We found N-terminal pegylation of CaLL only slightly diminished its antimicrobial activity, with the PEG2 conjugate of CaLL retaining greater antimicrobial activity than the larger PEG3 conjugate. The additional 22-atom PEG unit present in the latter may impart a conformation whereby the PEG chain interferes with insertion of the modified CaLL into bacterial membranes. The diminished activity of the PEG3 conjugate led subsequent experiments to focus on the biological and mechanistic aspects of PEG2-CaLL.
Unfolded AMPs interact with membranes principally via electrostatic and/or hydrophobic interactions with the lipid bilayer surface. Subsequent insertion of peptide nonpolar side chains into the interfacial membrane region is believed to accompany the structural transition of the AMP to an α-helical conformation which disrupts the cell membrane and causes microbial death. This conformational transition can be mimicked by exposing the peptide to fluorinated solvents or artificial lipid membranes (7, 35). Using both CD and fluorescence spectroscopy, we examined the induction of α-helicity in the AMPs and their selectivity of membrane interactions using lipid models of anionic (POPG) and zwitterionic (POPC) model membranes.
In the in silico modeling and design of CaLL the aim was to retain the critical ability of CaLL to form an α-helical conformation (15). Our CD data confirmed this and that this content increased in the presence of the fluorinated solvent TFE or lipid SUVs. It is of note that a differential responsiveness toward lipid vesicles was apparent between the parenteral cecropin A and CaLL. Specifically, while the α-helical content of the former was not augmented by exposure to POPC vesicles, that of CaLL was almost equally amplified by POPC or POPG SUVs. Our observations contrast with some reports showing pegylation of AMPs to inhibit α-helix formation, for example, that of Imura et al. (18), who attached a 5-kDa PEG to the N terminus of magainin-2 peptide. We found that pegylation of CaLL did not have a detrimental impact upon α-helical content in any of the environments we studied. Indeed, the data indicated that the PEG chains may have preferentially stabilized the ordered conformations of CaLL.
Fluorescence spectroscopy was exploited to extend our mechanistic understanding of CaLL activity and, in particular, the interactions of CaLL and its pegylated derivative with anionic and zwitterionic SUVs. Unlike the near-equivalent interactions of CaLL with POPC and POPG SUVs, PEG2-CaLL showed marked preferential interactions with the anionic POPG SUVs. CaLL is composed of the first eight N-terminal residues from cecropin A, a fragment enriched in positively charged amino acids. The remainder of CaLL is comprised of 13 residues (residues 17 to 29) from LL-37. Given the array of structural data available for cecropin (37) and LL-37 (22, 40), an appropriate speculation for CaLL interactions with a membrane would include initial membrane anchoring of CaLL mediated through electrostatic interactions of the cationic N-terminal residues from cecropin with the propensity for CaLL to assume an α-helical conformation being attributable to the 13 residues from LL-37. Therefore, we could propose that the presence of the PEG2 chain at the N terminus of CaLL is sufficient to disrupt, to some extent, anchoring and subsequent membrane insertion of the peptide with zwitterionic membranes, while the PEG2 chain has less impact on the peptide interactions with the more anion-rich POPG membranes.
With the aim of defining the pulmonary disposition and acute biocompatibility of CaLL and pegylated CaLL derivatives in normal, uninfected lung models, we performed a series of experiments that employed our unique combination of in vitro primary rat AE cell cultures and the ex vivo IPRL model. These primary cultures are a particularly valuable tool for the examination of cellular toxicity; the ATII cell, which covers only 5% of the alveolar surface area, is accepted as the sole in vivo progenitor of the ATI cell (42), which represents the remaining 95% of the surface area. As might be expected from a highly cationic peptide (8+ charge at pH 7), CaLL reduced the viability of ATII and ATI-like cell cultures over an 8-h incubation period, albeit most profoundly at high concentrations on the order of 100 μM. Notwithstanding this, a modest reduction in membrane damage was afforded by the attachment to CaLL of the PEG2 and PEG3 chains, an observation supportive of the role of PEG to shield the detrimental interactions of CaLL with epithelial cells. CaLL and pegylated CaLL reduced the cell viability of ATII as well as ATI cells, which indicates that the role of the ATII cell in restoring alveolar epithelial confluence after injury to ATI cells may be compromised. These in vitro models present simple single-cell phenotype monolayer cultures and contrast markedly to the multicell phenotype and complex architecture of the intact lung. Our MTT assays provide valuable information about the nature of epithelial toxicity that may be observed at the alveolar epithelial surface when AMPs accumulate at the alveolar epithelial surface due to slow absorption into the pulmonary vasculature.
The IPRL model afforded investigation of whole-lung biocompatibility of the AMPs through monitoring of the absorption of coadministered hydrophilic probes, i.e., 182-Da mannitol and 4-kDa dextran. The improved lung biocompatibility arising from pegylation of CaLL was clearly evidenced by PEG2-CaLL administration not leading to pulmonary barrier solute transport perturbation or the marked pulmonary edema seen with CaLL administration. Although the IPRL experiment represented an acute (90-min) exposure to AMPs, it nevertheless exposed the lung tissue to high concentrations of AMPs predicted to have reached close to 100-fold greater than the respective antibacterial IC50s measured in earlier experiments. For example, based on an accepted ELF volume of 250 to 300 μl in a 250-g rat, a 30-nmol dose of PEG2-CaLL would be predicted to give an in situ lung ELF concentration, immediately after administration, of approximately 100 to 150 μM peptide.
The administration of the AMPs into the airways of the IPRL also provided the opportunity to collect information on the pulmonary absorption and stability of these peptides. Over the 90-min duration of the IPRL experiment the extent of absorption of the 2.8- to 4.5-kDa AMPs into the pulmonary perfusate was very low (<3%). It is reasonable to predict from this that the AMPs likely reside in the airway lumen for a prolonged period postadministration, which would be beneficial for antimicrobial efficacy within the lung lumen itself. Nevertheless, the low extent of absorption was unexpected, as the lung is recognized as one of the more permeable epithelial barriers to the systemic absorption of macromolecules, including the display of significant pulmonary absorption of polypeptides and proteins (17). We (27) and others (32) have previously reported on the relatively high bioavailability of macromolecules administered via the lung, including for example that of 4-kDa dextran, which displays an extent of pulmonary absorption of approximately 30% over a 90-min IPRL experiment, an observation which is consistent with 4-kDa dextran absorption in vivo (28). The AMPs utilized in this work are highly cationic, with 22% to 43% of their respective peptide chains comprising basic amino acid residues. This is not unlike the cationic peptides defined as cell-penetrating peptides (CPPs) (14), whose systemic disposition is characterized by high tissue binding and accumulation in well-perfused tissue beds, including that of the lung (1, 34). Indeed, it is generally recognized that cationic character leads to a propensity for exogenously administered molecules, even low-molecular-weight entities, to accumulate in lung tissue (4). A variety of endogenous intraluminal molecules, including lipids and proteins, have been associated with the pulmonary sequestration and prolonged luminal retention of exogenously administered polypeptides (26). A number of individual ELF components, including surfactant proteins (45), apolipoprotein A1 (44), and albumin (39), have been shown to bind to AMPs. Indeed, Bergsson and colleagues (5) recently reported that LL-37 undergoes electrostatic binding interactions with cell membrane and matrix glycosaminoglycans (GAGs), a binding interaction previously documented for a number of CPPs (48).
We studied the stability of the AMPs to rat lung BALF to determine their potential as anti-infective agents for pulmonary delivery. While the PEG2-CaLL conjugate showed good stability in saline, in contrast its stability in BALF was relatively poor (t1/2, ∼1 h) and not simply a reflection of removal of the PEG moiety but instead involved breakdown of the peptide component. While it is possible to conjecture that the lack of toxicity of PEG2-CaLL in the IPRL was simply due to its degradation by what will be a more enriched in vivo ELF, this argument is not wholly sustainable when one considers the almost instantaneous (within 1 min postadministration) pulmonary edema induced by CaLL in the IPRL biocompatibility experiments and the avoidance of this hyperacute toxicity when PEG2-CaLL was administered. The pegylation of the CaLL peptide may decrease CaLL stability through disrupting the capacity of CaLL to self-associate. For example, a growing body of evidence supports the enzymatic stability of LL-37 through its ability to undergo concentration-dependent self-association of LL-37 monomers (29). The reduced stability of PEG2-CaLL in the presence of BALF may initially appear at variance with its bactericidal actions tested in this work in the presence of BALF. In particular, against germinating B. anthracis spores we showed the actions of pegylated CaLL to be at least as effective as CaLL itself and indeed to be unaffected by the coincubation with BALF. Critically, the bactericidal actions in this assay reflect predominantly the first hour following spore germination, where concentrations of PEG2-CaLL will have remained at levels much greater than the IC50. Repeated dosing of cationic AMPs displaying prolonged residence in the lung lumen may cause deleterious effects on lung integrity. The ability to control the rate of proteolytic cleavage of such AMPs through pegylation, however, may offer an opportunity to avoid potential harmful AMP accumulation in the lung at the same time as providing efficacious antimicrobial cover.
In conclusion, we have demonstrated that attachment of a short N-terminal PEG chain to CaLL offers improvements in the acute lung biocompatibility of this AMP without interfering with the critical membrane binding and peptide conformational changes necessary for sustained antimicrobial potency. This study provides evidence that pegylation may find utility in the development of inhaled AMP therapeutics for pulmonary infection.
ACKNOWLEDGMENTS
This work was performed with funding from the United Kingdom Ministry of Defence. Purchase of the CD instrument was partially funded by BBSRC grant 75/REI18433.
Footnotes
Published ahead of print 19 March 2012
REFERENCES
- 1. Aguilera TA, Olson ES, Timmers MM, Jiang T, Tsien RY. 2009. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr. Biol. (Camb.) 1:371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barar J, et al. 2007. Cell selective glucocorticoid induction of caveolin-1 and caveolae in differentiating pulmonary alveolar epithelial cell cultures. Biochem. Biophys. Res. Commun. 359:360–366 [DOI] [PubMed] [Google Scholar]
- 3. Bartlett KH, McCray PB, Jr, Thorne PS. 2003. Novispirin G10-induced lung toxicity in a Klebsiella pneumoniae infection model. Antimicrob. Agents Chemother. 47:3901–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bend JR, Serabjit-Singh CJ, Philpot RM. 1985. The pulmonary uptake, accumulation, and metabolism of xenobiotics. Annu. Rev. Pharmacol. Toxicol. 25:97–125 [DOI] [PubMed] [Google Scholar]
- 5. Bergsson G, et al. 2009. LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J. Immunol. 183:543–551 [DOI] [PubMed] [Google Scholar]
- 6. Brogden KA, et al. 2001. The ovine cathelicidin SMAP29 kills ovine respiratory pathogens in vitro and in an ovine model of pulmonary infection. Antimicrob. Agents Chemother. 45:331–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buck M. 1998. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 31:297–355 [DOI] [PubMed] [Google Scholar]
- 8. Byron PR, Roberts NS, Clark AR. 1986. An isolated perfused rat lung preparation for the study of aerosolized drug deposition and absorption. J. Pharm. Sci. 75:168–171 [DOI] [PubMed] [Google Scholar]
- 9. Campbell L, et al. 1999. Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung alveolar epithelial primary cultures. Biochem. Biophys. Res. Commun. 262:744–751 [DOI] [PubMed] [Google Scholar]
- 10. Chen GC, Yang JT. 1977. Two-point calibration of circular dichrometer with d-10-camphorsulfonic acid. Anal. Lett. 10:1195–1207 [Google Scholar]
- 11. Cudic M, et al. 2002. Development of novel antibacterial peptides that kill resistant isolates. Peptides 23:2071–2083 [DOI] [PubMed] [Google Scholar]
- 12. Dawson RM, McAllister J, Liu CQ. 2010. Characterisation and evaluation of synthetic antimicrobial peptides against Bacillus globigii, Bacillus anthracis and Burkholderia thailandensis. Int. J. Antimicrob. Agents 36:359–363 [DOI] [PubMed] [Google Scholar]
- 13. Dean RE, et al. 2010. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 31:1966–1972 [DOI] [PubMed] [Google Scholar]
- 14. Fonseca SB, Pereira MP, Kelley SO. 2009. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 61:953–964 [DOI] [PubMed] [Google Scholar]
- 15. Fox MA, Thwaite JE, Ulaeto DO, Atkins TP, Atkins HS. 2012. Design and characterization of novel hybrid antimicrobial peptides based on cecropin A, LL-37 and magainin II. Peptides 33:197–205 [DOI] [PubMed] [Google Scholar]
- 16. Guiotto A, et al. 2003. PEGylation of the antimicrobial peptide nisin A: problems and perspectives. Farmaco 58:45–50 [DOI] [PubMed] [Google Scholar]
- 17. Gumbleton M. 2001. Caveolae as potential macromolecule trafficking compartments within alveolar epithelium. Adv. Drug Deliv. Rev. 49:281–300 [DOI] [PubMed] [Google Scholar]
- 18. Imura Y, Nishida M, Matsuzaki K. 2007. Action mechanism of PEGylated magainin 2 analogue peptide. Biochim. Biophys. Acta 1768:2578–2585 [DOI] [PubMed] [Google Scholar]
- 19. Imura Y, Nishida M, Ogawa Y, Takakura Y, Matsuzaki K. 2007. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta 1768:1160–1169 [DOI] [PubMed] [Google Scholar]
- 20. Jacob MK, Leena S, Kumar KS. 2008. Peptide-polymer biotherapeutic synthesis on novel cross-linked beads with “spatially tunable” and “isolated” functional sites. Biopolymers 90:512–525 [DOI] [PubMed] [Google Scholar]
- 21. Jenssen H, Hamill P, Hancock RE. 2006. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19:491–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. 1998. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 273:3718–3724 [DOI] [PubMed] [Google Scholar]
- 23. Lisanby MW, et al. 2008. Cathelicidin administration protects mice from Bacillus anthracis spore challenge. J. Immunol. 181:4989–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loose C, Jensen K, Rigoutsos I, Stephanopoulos G. 2006. A linguistic model for the rational design of antimicrobial peptides. Nature 443:867–869 [DOI] [PubMed] [Google Scholar]
- 25. Luo P, Baldwin RL. 1997. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 36:8413–8421 [DOI] [PubMed] [Google Scholar]
- 26. McAllister SM, Alpar HO, Teitelbaum Z, Bennett DB. 1996. Do interactions with phospholipids contribute to the prolonged retention of polypeptides within the lung? Adv. Drug Deliv. Rev. 19:89–110 [Google Scholar]
- 27. Morris CJ, Smith MW, Griffiths PC, McKeown NB, Gumbleton M. 2011. Enhanced pulmonary absorption of a macromolecule through coupling to a sequence-specific phage display-derived peptide. J. Control. Release 151:83–94 [DOI] [PubMed] [Google Scholar]
- 28. Ohtani T, Murakami M, Yamamoto A, Takada K, Muranishi S. 1991. Effect of absorption enhancers on pulmonary absorption of fluorescein isothiocyanate dextrans with various molecular weights. Int. J. Pharm. 77:141–150 [Google Scholar]
- 29. Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y. 1999. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 341(Pt 3):501–513 [PMC free article] [PubMed] [Google Scholar]
- 30. Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryan G, Mukhopadhyay S, Singh M. 2000. Nebulised anti-pseudomonal antibiotics for cystic fibrosis. Cochrane Database Syst. Rev. CD001021 doi:10.1002/14651858.CD001021 [DOI] [PubMed] [Google Scholar]
- 32. Sakagami M, Byron PR, Rypacek F. 2002. Biochemical evidence for transcytotic absorption of polyaspartamide from the rat lung: effects of temperature and metabolic inhibitors. J. Pharm. Sci. 91:1958–1968 [DOI] [PubMed] [Google Scholar]
- 33. Sakagami M, et al. 2006. Expression and transport functionality of FcRn within rat alveolar epithelium: a study in primary cell culture and in the isolated perfused lung. Pharm. Res. 23:270–279 [DOI] [PubMed] [Google Scholar]
- 34. Sarko D, et al. 2010. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 7:2224–2231 [DOI] [PubMed] [Google Scholar]
- 35. Sato H, Feix JB. 2006. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 1758:1245–1256 [DOI] [PubMed] [Google Scholar]
- 36. Scopes RK. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59:277–282 [DOI] [PubMed] [Google Scholar]
- 37. Sipos D, Andersson M, Ehrenberg A. 1992. The structure of the mammalian antibacterial peptide cecropin P1 in solution, determined by proton-NMR. Eur. J. Biochem. 209:163–169 [DOI] [PubMed] [Google Scholar]
- 38. Smith MW, et al. 2007. Phage display identification of functional binding peptides against 4-acetamidophenol (Paracetamol): an exemplified approach to target low molecular weight organic molecules. Biochem. Biophys. Res. Commun. 358:285–291 [DOI] [PubMed] [Google Scholar]
- 39. Svenson J, Brandsdal BO, Stensen W, Svendsen JS. 2007. Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J. Med. Chem. 50:3334–3339 [DOI] [PubMed] [Google Scholar]
- 40. Thennarasu S, et al. 2010. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 98:248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thwaite JE, Hibbs S, Titball RW, Atkins TP. 2006. Proteolytic degradation of human antimicrobial peptide LL-37 by Bacillus anthracis may contribute to virulence. Antimicrob. Agents Chemother. 50:2316–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uhal BD. 1997. Cell cycle kinetics in the alveolar epithelium. Am. J. Physiol. 272:L1031–L1045 [DOI] [PubMed] [Google Scholar]
- 43. Veronese FM, Harris JM. 2002. Introduction and overview of peptide and protein pegylation. Adv. Drug Deliv. Rev. 54:453–456 [DOI] [PubMed] [Google Scholar]
- 44. Wang Y, Agerberth B, Lothgren A, Almstedt A, Johansson J. 1998. Apolipoprotein A-I binds and inhibits the human antibacterial/cytotoxic peptide LL-37. J. Biol. Chem. 273:33115–33118 [DOI] [PubMed] [Google Scholar]
- 45. Wang Y, Walter G, Herting E, Agerberth B, Johansson J. 2004. Antibacterial activities of the cathelicidins prophenin (residues 62 to 79) and LL-37 in the presence of a lung surfactant preparation. Antimicrob. Agents Chemother. 48:2097–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang G, Han B, Lin X, Wu X, Yan H. 2008. Modification of antimicrobial peptide with low molar mass poly(ethylene glycol). J. Biochem. 144:781–788 [DOI] [PubMed] [Google Scholar]
- 47. Zhang L, et al. 2005. Antimicrobial peptide therapeutics for cystic fibrosis. Antimicrob. Agents Chemother. 49:2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ziegler A. 2008. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 60:580–597 [DOI] [PubMed] [Google Scholar]