

Abstract
The failure of antibiotics to inactivate in vivo pathogens organized in biofilms has been shown to trigger chronic infections. In addition to mechanisms involving specific genetic or physiological cell properties, antibiotic sorption and/or reaction with biofilm components may lessen the antibiotic bioavailability and consequently decrease their efficiency. To assess locally and accurately the antibiotic diffusion-reaction, we used for the first time a set of advanced fluorescence microscopic tools (fluorescence recovery after photobleaching, fluorescence correlation spectroscopy, and fluorescence lifetime imaging) that offer a spatiotemporal resolution not available with the commonly used time-lapse confocal imaging method. This set of techniques was used to characterize the dynamics of fluorescently labeled vancomycin in biofilms formed by two Staphylococcus aureus human isolates. We demonstrate that, at therapeutic concentrations of vancomycin, the biofilm matrix was not an obstacle to the diffusion-reaction of the antibiotic that can reach all cells through the biostructure.
INTRODUCTION
Biofilms are composed of microorganisms entrapped in a hydrated matrix of organic exopolymeric substances (EPS) produced by the cells themselves. EPS are mainly composed of polysaccharides, proteins, nucleic acids, lipids, and heteropolymers that are interlaced by a vast number of weak interactions (hydrogen bonds, hydrophobic, electrostatic, van der Waals interactions, divalent cation bridges, etc.) giving the three-dimensional assemblage a viscoelastic nature (9). Such a reactive molecular environment pointed to the proposal that this biomass could act as a diffusion barrier that lessens the antimicrobial bioavailability and thus be a contributing factor to biofilm antibiotic tolerance. This is the case of tobramycin, a polycationic aminoglycoside that interacts electrostatically with the polyanionic alginate matrix of Pseudomonas aeruginosa biofilms (22). In addition, antibiotic sorption may be highly dependent on microenvironment heterogeneity (dense clusters of polysaccharides, DNA resulting from cell lysis, etc.) that can also induce heterogeneity of the antibiotic irrigation. The cells are thus locally exposed to various concentrations of antibiotics, leading to some subinhibitory concentrations of the antibiotic through the biofilms.
Fluorescence microscopy represents an essential and noninvasive method to assess antimicrobial dynamics and reactivity in biofilms, contributing to our understanding of what makes bacteria resistant in these biostructures (3, 5, 8, 29). In this respect, confocal laser scanning microscopy (CLSM) has been shown to be a powerful tool to analyze antimicrobial actions indirectly by a time course (hour-to-day scale) visualization of live and dead cells through the biofilm structure (4, 17, 29, 34). However, this approach is unlikely to allow distinguishing survivor cells that were not reached by the active molecules (diffusion-reaction limitation through the matrix) from survivors with resistant physiology (biofilm “phenotype”).
More recently, the emergence of fluorescently tagged antimicrobials, including antibiotics, allowed their direct tracking (second-to-minute scale) through biofilms by time-lapse microscopy (18, 28). The available studies highlight various behaviors depending on the biofilm models and/or the antibiotics considered. For example, it was observed that tetracycline and daptomycin quickly (i.e., within a few minutes) make their way into Escherichia coli and Staphylococcus epidermidis biofilms, respectively (27, 28), whereas it takes about 1 h for vancomycin to diffuse through a mucoid model of Staphylococcus aureus biofilm (18). From these studies, it is evident that no consensus has been reached yet on the role of the EPS matrix of biofilms as a barrier to the diffusion-reaction of antibiotics.
Although time-lapse imaging represents an essential versatile tool, visualizing in real time the penetration of the antibiotic through the biofilm depth, the method is limited in its temporal resolution giving only access to initial diffusive rates. No further measurements are available when the antimicrobials have reached equilibrium inside the biomatrix, although it is important to follow their reactivity on the time scale of antibiotic exposure (several hours to days). To overcome this limitation, we took advantage of the large potential offered by advanced CLSM methods, including fluorescence recovery after photobleaching (FRAP) (7, 32), fluorescence correlation spectroscopy (FCS) (2, 13, 14), and fluorescence lifetime imaging (FLIM) (5, 13, 21). These methods have not only allowed significant advances owing to their performance in terms of spatial resolution (submicrometric) and sensitivity (nanomolar-to-micromolar therapeutic antibiotic concentrations), but they have also provided noninvasive conditions of observations consistent with studies on biofilms after several hours of contact with the antibiotics. They allow discriminating free diffusion from constrained diffusion of the antibiotics and hence quantifying their interaction with biofilm components.
The purpose of the present study was to highlight the strength of such correlative time-resolved fluorescence microscopy approaches in order to assess and understand the diffusion-reaction of fluorescently labeled vancomycin within S. aureus biofilms of two different human isolates.
MATERIALS AND METHODS
Bacterial strain, media, and antibiotics.
Two S. aureus strains (ATCC 6538 and ATCC 27217) and one P. aeruginosa strain (ATCC 15442) were used in the present study. The stock cultures were kept at −80°C in tryptic soy broth (TSB; BD-Difco, France) containing 20% (vol/vol) glycerol. Prior to each experiment, the frozen cells were subcultured twice in TSB. Bacterial growth and experiments were both conducted at 30°C. The fluorescently labeled antibiotic BODIPY-vancomycin and free BODIPY (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid) were purchased from Invitrogen (Cergy-Pontoise, France). The lyophilysates were dissolved in sterile Milli-Q water for BODIPY-vancomycin and in dimethyl sulfoxide for BODIPY to obtain stock solutions of 69 and 34.5 mM, respectively. These stock solutions, kept at −20°C before use, were then diluted to working concentrations: 345 nM for FCS, 6.9 μM for FLIM, and 69 μM and 34.5 mM for FRAP.
Biofilm preparation.
For the preparation of S. aureus biofilms, 500-μl portions of an overnight subculture adjusted to an optical density at 600 nm of 0.03 (corresponding to ∼107 CFU/ml) were added to 4-well sterile microscopic chambers (Lab-Tek; Nunc/Thermo Scientific, France), or 250-μl portions were added to 96-well microplates (μClear; Greiner Bio-One, France) under humidified air conditions. After a 1.5-h adhesion period at 30°C, the wells were rinsed with 150 mM NaCl in order to eliminate nonadherent cells, refilled with sterile TSB, and then incubated for 24 h at 30°C to allow biofilm growth. Since TSB contains traces of fluorescent compounds, the biofilms were rinsed with 150 mM NaCl and refilled with nonfluorescent RPMI 1640 medium (Gibco/Invitrogen, France). Three-day-old P. aeruginosa biofilms were prepared according to S. aureus biofilm protocol but using M9 minimal growth medium (Difco, France) that was replaced with fresh medium every day. Examples of the distribution of cells, EPS, and interstitial voids after a 24-h biofilm growth period are provided in Fig. S1 in the supplemental material.
Visualization of antibiotic cell wall binding.
The antibiotic cell binding was visualized as described by Gilbert et al. (11). Briefly, BODIPY-vancomycin was mixed with an equal amount of unlabeled vancomycin, giving a final concentration of 3 μg/ml, and the mixture was added to S. aureus cells from exponentially growing cultures. After a 30-min incubation period at 30°C, the cells were deposited on a polylysine-coated slide (Kindler GmbH; VWR, France) and then visualized by fluorescence microscopy. In addition, the visualization of the nascent peptidoglycan was obtained according to the protocol of Pinho and Errington (23).
Visualization of antibiotic penetration.
BODIPY-vancomycin diffusive penetration through our biofilm models was measured by time-lapse microscopy using a Leica SP2 AOBS confocal laser scanning microscope (Leica Microsystems, France), implemented at the MIMA2 microscopy platform (Massy). For these experiments, the labeled antibiotic was excited with a continuous argon laser line at 488 nm through a ×63 oil immersion objective, and the emitted fluorescence was recorded within the range 500 to 650 nm. The focal plane inside the biofilms (thickness, ∼30 μm) was set ∼5 μm above the glass surface. An x-y time series was then initiated in which an image of 512 × 128 pixels corresponding to ∼40 × 10 μm2 was collected every 3 s. The time series was recorded for 10 min. Images were analyzed using Leica software (Lite; Leica Microsystems). BODIPY and BODIPY-vancomycin solutions were added very gently to the medium over the biofilm; transmission imaging ascertains that no structural alteration of the biofilm occurred during this process.
The corresponding diffusive penetration coefficients (Dp) through the biofilms were determined according to the relationship described by Stewart for flat structures (26):
| (1) |
where L is the biofilm thickness and t90 is the time required to attain 90% of the equilibrium staining intensity at the deeper layers of the biofilm.
Time-resolved fluorescence microscopy.
All time-resolved fluorescence measurements were obtained on the same confocal Leica SP5 microscope (Leica Microsystems; implemented at the Centre de Photonique Biomédicale of Orsay) equipped with a ×63 high numerical aperture (1.4) oil immersion objective and coupled with either continuous lasers for FCS and FRAP acquisitions or a femtosecond titanium-sapphire laser (Chameleon-XR; Coherent, USA) running at a 80-MHz repetition rate and delivering pulses of 150 fs for fluorescence decay time and FLIM measurements.
FRAP.
The principle of FRAP is based on a brief excitation of fluorescent molecules by a high-intensity laser pulse spatially confined to the volume defined by the confocal microscope objective (for single-spot FRAP) or, in a user-defined region, to quench irreversibly their fluorescence (photobleaching). Fluorescence recovery in the photobleached area is then observed as a function of time.
In the present study, an image-based FRAP protocol was applied that can be readily applied by anyone familiar with a confocal laser scanning microscope and is well adapted to study in situ local molecular diffusion with accuracy. This protocol includes (i) the image acquisition of photobleached areas and (ii) a kymogram representation to control the bacterial movement that may lead to an incorrect estimation of the molecular diffusion coefficient (32). Kymograms are two-dimensional graphs showing fluorescence intensity fluctuations over time along a chosen trajectory. Such representation is essential in order to discard distorted acquisitions.
The image-based FRAP protocol has been explained in detail elsewhere (7, 32). Briefly, the fluorescence intensity image size was fixed to 512 × 128 pixels with a 80-nm pixel size in order to ensure usable spatial information on the biofilm (∼40 × 10 μm2) and recorded using 16-bit resolution to improve the image analysis. The line scan rate frequency was fixed at 1,400 Hz, which corresponds to a total time between frames of ∼265 ms. The full widths at half-maximum in the x and y directions and the z direction (i.e., along the optical axis) of the bleached profiles were 0.8 and 14 μm, respectively, as determined previously (32).
Each FRAP experiment started with the acquisition of 50 image scans at 3% of laser maximum intensity (which was measured to be ∼7 μW at the object level) and followed by a single bleached spot of 100 ms at 100% laser intensity. A series of 300 single-section images were then collected with the laser power attenuated to its initial value (3% of the bleach intensity); the first image was recorded 365 ms after the beginning of bleaching. Under these image acquisition conditions, FRAP measurements could be acquired in 74 s, and this ensured bacterial viability as controlled with a live-dead staining.
The fluorescence recovery curves here reported were analyzed using the adapted mathematical model of Braga et al. (1) as previously detailed (32). It should be noted that a two-dimensional diffusion was considered in view of the axial (14-μm) and lateral (0.8-μm) extents of the photobleaching pattern; diffusion along the axial/vertical axis can be neglected.
FCS.
Fluorescence correlation spectroscopy (FCS) is based on monitoring the emission intensity fluctuations due to a small number of fluorophores (until probes concentrations of few hundreds nanomolar) passing through the confocal excitation volume. These fluctuations can be quantified in their amplitudes and durations by temporally autocorrelating the recorded intensity signals. In the absence of chemical reaction or other dynamic processes, these temporal fluctuations in intensity can be attributed solely to the translational diffusion of the fluorescent probes.
Our FCS measurements were performed point by point using the 488-nm line of a continuous Ar laser. The emission output was focused on a PicoHarp 300 module (PicoQuant, Germany) equipped with a single-photon avalanche diode detector (time resolution of 250 ps). The emitted fluorescence was recorded within a range of 500 to 650 nm. Data acquisition was performed using SymPhoTime software (PicoQuant), which computes online the correlation function of fluorescence fluctuations. To measure the translational diffusion time with 10 to 20% accuracy, fluorescence temporal signals in water were collected in runs of 60 s (number of iterations, 5 to 10). In the biofilms, acquisition time was increased to 120 s, and each experiment was repeated 10 times at ∼15 randomly selected locations (to account for biofilm heterogeneity). The data were analyzed using both SymPhoTime software (PicoQuant) and an in-house Matlab routine, which enabled (i) the selection and averaging of chosen successive acquisitions, (ii) the adaptation of different diffusion models to analyze the autocorrelation function, and (iii) the calculation of associated standard deviations. The incident laser energy was attenuated to less than 1 mW at the sample in order to reduce the risk of photobleaching.
In these experiments, we assumed that the excitation intensity profile could be approximated using a three-dimensional Gaussian distribution and that no additional “blinking” dynamics of the fluorophore occurred other than its diffusion through the excitation volume; then each fluorescence autocorrelation [g(τ)] curve was fitted using a two-component free Brownian motion model:
| (2) |
where N is the number of fluorescent molecules in the excitation volume; α and “(1 − α)” are the fractions of the molar concentrations of the two fluorescent diffusive species, τ1 and τ2 are their respective translational diffusion times, and ω0 and z0 are the radial and axial radii of the confocal volume. These were determined by previous calibration experiments (ω0 = 0.25 ± 0.04 μm and z0 = 0.84 ± 0.1 μm) and fixed throughout the fittings. In the present study, this two-component model allowed us to discriminate the fast-diffusing fluorescent molecules of antibiotics from the slower ones, which are either autoaggregates or complexed with the biofilm components.
The diffusion coefficient (D) could be related to the translational diffusion time τi using the following equation:
| (3) |
All of the FCS data reported here are averages of at least 10 independent sample preparations.
Fluorescence lifetime and FLIM measurements.
Fluorescence intensity is dependent on fluorophore concentration and on environmental conditions and thus can be difficult to understand or interpret. Fluorescence decay time measurements allowed us to address this problem. Fluorescence decay time corresponds to the average time a fluorophore remains in the excited state after excitation. The fluorescence lifetime is an intrinsic characteristic of the fluorophore, independent on its concentration, but may locally vary depending on the reactivity of the fluorophore with the biological environment. It is thus possible to build fluorescence lifetime images that allow assessment of the reactivity of a fluorophore at the molecular level throughout a three-dimensional biological structure such as a biofilm.
In the present study, the BODIPY-labeled samples were biphotonically excited (two-photon excitation) at 950 nm. The fluorescence signal was collected by using a PicoHarp 300 device based on time-correlated single-photon counting method. An 800-nm short-pass emission filter was used to remove any residual laser light, and the emitted fluorescence was recorded within a range of 500 to 650 nm.
The observed time-resolved decays were deconvoluted with the instrumental response function obtained on a picric acid solution. It was possible here to fit all of the fluorescence decays, with a single exponential giving the fluorescence lifetime of the sample with a 100-ps time resolution (30). To obtain two-dimensional FLIM, the laser beam was scanned on the cell surface at 400 Hz, which gave an acquisition mean time of ∼10 min to cover a field of view of 50 by 50 μm2 (64 × 64 pixels).
RESULTS
Assessing BODIPY-vancomycin interaction with planktonic S. aureus bacteria.
The specific binding of BODIPY-vancomycin to planktonic S. aureus bacteria was determined by using both FCS and time-resolved fluorescence emission.
Before carrying out studies in the presence of bacteria, FCS measurements were performed using the free BODIPY and BODIPY-vancomycin probes in aqueous solution (water viscosity η = 0.96 cF at 295 K). Typical experimental fluorescence correlation curves are presented in Fig. 1a, showing a reproducible shift of BODIPY-vancomycin curve compared to that of free BODIPY. The diffusion times obtained by fitting the experimental curves (equation 2) and the corresponding diffusion coefficient values calculated using equation 3 are summarized in Table 1. The slight difference between the major diffusion coefficient (D1) of BODIPY-vancomycin and the one of free BODIPY in the data measured was in line with the influence of the probe size on mobility. The minor diffusion coefficient D2 of BODIPY-vancomycin likely corresponds to molecular aggregates.
Fig 1.
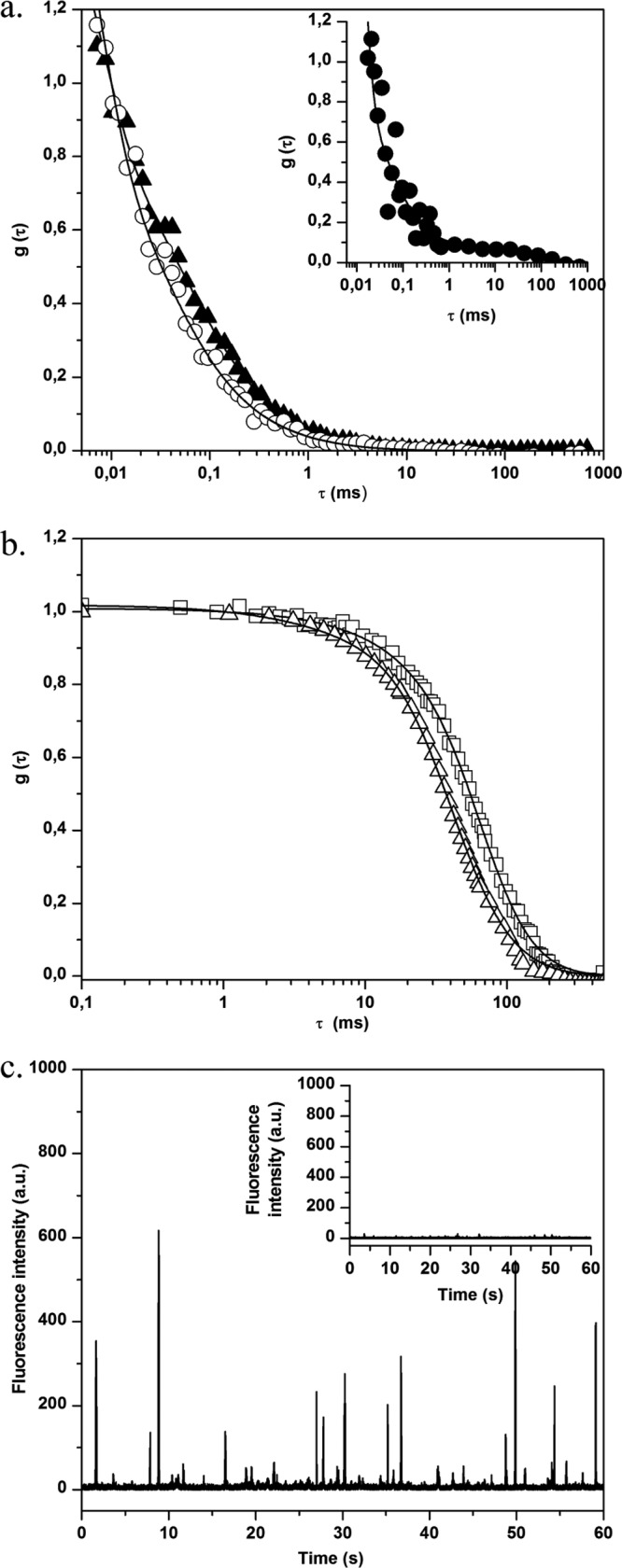
(a) Normalized fluorescence autocorrelation curves g(τ) for BODIPY (○) and BODIPY-vancomycin (▲) in solution. The inset shows the correlation curve g(τ) for BODIPY (●) in the presence of S. aureus planktonic cells. (b) Normalized fluorescence autocorrelation curves g(τ) for S. aureus planktonic cells without (□) or with (△) BODIPY-vancomycin revealing interaction of vancomycin with S. aureus cell wall. (c) Fluorescence time traces for S. aureus planktonic cells without (inset) and with BODIPY-vancomycin. In the presence of labeled vancomycin, the fluorescence intensity (given in arbitrary units [a.u.]) is much higher, confirming the presence of the antibiotic in the excited volume.
Table 1.
Diffusion times, their corresponding calculated diffusion coefficients, and fluorescence lifetimes for BODIPY and BODIPY-vancomycin in water and in the presence of S. aureus planktonic cellsa
| Method | Mean ± SEM or rangeb |
|||
|---|---|---|---|---|
| In water |
With S. aureus planktonic cells |
|||
| BODIPY | BODIPY-vancomycin | BODIPY | BODIPY-vancomycin | |
| FCS | ||||
| τ1 (μs) | 38 ± 6 (100) | 52 ± 11 (92) | 54 ± 9 (87) | 30,000–100,000 (100) |
| D1 (μm2/s) | 395 ± 60 | 290 ± 60 | 278 ± 46 | 0.15–0.50 |
| τ2 (ms) | 1.26 ± 0.62 (8) | 30–100 (13) | ||
| D2 (μm2/s) | 12 ± 6 | 0.15–0.50 | ||
| FLIM (ns) | 5.7 ± 0.2 | 5.7 ± 0.2 | 5.7 ± 0.2 | 4.5 ± 0.2 |
Diffusion times (τ1 and τ2) obtained from fitting with the equation from the fluorescent correlation curves, their corresponding calculated diffusion coefficients (D1 and D2), and the fluorescence lifetimes for BODIPY and BODIPY-vancomycin in water and in the presence of S. aureus plankton cells were determined. The percentages given in parentheses correspond to the ratio of molecules diffusing with the indicated time.
Values are expressed as means ± the SEM or as a range, where applicable. Percentages are indicated in parentheses where applicable.
We checked that the remaining autofluorescence of planktonic S. aureus bacteria was high enough to create an additional correlation signal as illustrated in Fig. 1b. The corresponding time range of bacteria diffusion was ca. 30 to 100 ms, depending on the presence of cell aggregates. In the presence of free BODIPY, the fluorescence correlation curve analysis using equation 2 corresponds to a combination of the diffusion coefficients for BODIPY and planktonic bacteria (Fig. 1a, inset, and Table 1). Because of this bacterial contribution, some dispersion in the fast BODIPY diffusion time was observed compared to water. However, it can be ascertained that the probe diffusion remains unchanged in the presence of bacteria.
By contrast, the correlation curves measured after adding BODIPY-vancomycin to a bacterial suspension could be superposed on those obtained with planktonic bacteria in solution (Fig. 1b and Table 1). Furthermore, a much more intense fluorescence count rate burst was recorded in the presence of BODIPY-vancomycin (Fig. 1c) compared to planktonic bacteria that indicates the presence of the antibiotic in the bacterial suspension. Together, these results demonstrate that BODIPY-vancomycin interacts with planktonic bacteria from the vancomycin part of the molecule, which is confirmed by time-resolved fluorescence emission.
In the absence of bacteria, the fluorescence decay of BODIPY-vancomycin could be superposed on that of free BODIPY. It is monoexponential, corresponding to a fluorescence lifetime of 5.7 ± 0.2 ns (Table 1 and Fig. 2a), which is in good agreement with a previous report (19). The presence of bacteria does not affect the dynamic fluorescence properties of BODIPY. Combining this result with FCS data suggests the absence of strong interaction between the fluorescent probe and the bacterial cell wall. In contrast, the BODIPY-vancomycin fluorescence lifetime is shortened to 4.5 ± 0.2 ns in the presence of bacteria (Table 1 and Fig. 2). Such a variation in the fluorescence decay time reflects a change in the vicinity of the fluorophore, which is consistent with an interaction of the vancomycin part of the labeled antibiotic with the bacterial surface.
Fig 2.

(a) Fluorescence lifetime decay for BODIPY-vancomycin in solution (solid triangles), with S. aureus planktonic cells (gray triangles), and in biofilms (open circles). (b) FLIM representation (in an x-y plane) of BODIPY-vancomycin inside S. aureus biofilms (50 by 50 μm2). The white scale bar represents 10 μm.
In the light of these data, we examined the possibility of visualizing the cellular binding pattern of the fluorescent conjugated antibiotic by fluorescence microscopy using the protocol described in Materials and Methods. Figure 3a shows that BODIPY-vancomycin was not distributed evenly around the planktonic cell wall but preferentially to the division septum sites upon cell division (Fig. 3b). This is in good agreement with the reported specificity and affinity of the antibiotic to bind preferentially to the terminal d-Ala-d-Ala of nascent peptidoglycan at the cell septum but also to the dipeptide present on the surface of the bacteria (11, 23). Similar fluorescence imaging was also performed in the presence of free BODIPY: neither bacterial nor BODIPY fluorescence was detected, confirming that the fluorophore did not interact with the S. aureus cell wall.
Fig 3.

Fluorescence imaging of BODIPY-vancomycin interacting with S. aureus planktonic cells (a) and at the single-cell level (b), allowing peptidoglycan synthesis to be visualized.
BODIPY-vancomycin diffusion-reaction inside S. aureus biofilms. (i) Time-lapse imaging.
Visualization of both BODIPY and BODIPY-vancomycin penetration through S. aureus biofilm depths (thickness, ∼30 μm) was performed. The fluorescence signals at the deeper layers of the biofilm (biofilm slide adjacent to the coverslip) were measured within 2 to 3 min for S. aureus ATCC 6538 and within ∼8 min for S. aureus ATCC 27217 (Fig. 4 and see Movie S1 in the supplemental material). The fluorescence intensity reached a plateau that remained steady. The equilibrium distribution of the labeled antibiotic through each S. aureus strain is illustrated in Fig. 4c and d. We observed that the antibiotic labeled preferentially bacterial cell walls through all depths of the biofilms. By comparison, maximal fluorescence of free BODIPY occurred within the time taken to obtain an x-y plane image (<3 s). The corresponding Dp values obtained from equation 1 are listed in Table 2. Diffusion of free BODIPY was too fast to be quantified by time-lapse confocal imaging.
Fig 4.

Time course of BODIPY-vancomycin penetration measured at the bottom of S. aureus ATCC 6538 (a) and ATCC 27217 (b) biofilms. The relative intensities correspond to a normalization of the maximum intensity value to 1. (c and d) Corresponding fluorescence intensity images taken at the bottom of the biofilms (in an x-y plane) showing BODIPY-vancomycin penetration in the whole structure of biofilms at 400 s (c) and 1,000 s (d), when the antibiotic equilibrium is reached. The images are sectioned along two vertical and orthogonal x-z and y-z planes: the substratum-biofilm interface is the outside part of the rectangle, and the biofilm-medium interface is the one close to the square fluorescence intensity image. Scale bar, 20 μm.
Table 2.
Diffusion coefficients of BODIPY and BODIPY-vancomycin measured in ATCC 6538 and ATCC 27217 S. aureus biofilms using different time-resolved fluorescence techniques
| Method | Variable | Mean ± SEMa |
||
|---|---|---|---|---|
| BODIPY | BODIPY-vancomycin |
|||
| ATCC 6538 strain | ATCC 27217 strain | |||
| Time-lapse | Dp (μm2/s) | <140 | 2.0 ± 0.7 | 0.8 ± 0.3 |
| FRAP | D (μm2/s) | 180 ± 60 | 0.5 ± 0.2 | 0.2 ± 0.1 |
| FCS | D (μm2/s) | 242 ± 36 | Biofilm motion | |
Values are indicated as means ± the SEM where applicable.
(ii) FRAP imaging.
Our group has recently developed an image-based FRAP protocol to improve the accuracy of FRAP measurements inside biofilms (7, 32). In particular, we have introduced a kymogram representation (illustrated in Fig. 5a and d) to check biofilm stability during fluorescence recovery curve acquisitions and to validate the data.
Fig 5.

FRAP acquisitions for BODIPY (a to c) and BODIPY-vancomycin (d to f) inside S. aureus ATCC 27217 biofilms. (a and d) Kymogram representation (xt) of FRAP acquisitions (representation of fluorescence intensity along a horizontal line passing through the photobleached area as a function of time t) from t = 0 to 75 s. (b and e) Sequence of fluorescence intensity images starting just before photobleaching and showing the beginning of the recovery phase. The time interval between two images is 265 ms. Scale bar, 5 μm. (c and f) Typical fluorescence recovery curves recorded in three different zones of the biofilms showing a total fluorescence recovery for BODIPY and a partial fluorescence recovery for BODIPY-vancomycin. The fluorescence intensity is expressed in arbitrary units (a.u.).
FRAP experiments (image acquisition, fluorescence recovery curve) of BODIPY and BODIPY-vancomycin in each type and different parts of S. aureus biofilms were performed (Fig. 5b and e). Over the time range of our observation (74 s), complete fluorescence recovery of BODIPY was observed (Fig. 5c). In contrast, only partial recovery of BODIPY-vancomycin was measured inside each strain of S. aureus biofilm corresponding to a mobile fraction, respectively, of ca. 60% ± 10% in S. aureus ATCC 6538 biofilm and ranging from 20 to 60% in S. aureus ATCC 27217 biofilm (Fig. 5f) depending on the local EPS nature: FRAP measurements acquired in extracellular DNA clusters (corresponding to the red zones in Fig. S1 in the supplemental material) reveal much lower mobile fractions. A control experiment showed that without the photobleaching phase, the fluorescence signal is stable during the acquisition time. The local diffusion coefficients at equilibrium (D) determined using the adapted mathematical model of Braga described in Materials and Methods are summarized in Table 2. They are of the same order of magnitude as Dp estimated from time-lapse confocal imaging analysis.
Otherwise, the image sequences acquired during FRAP measurements (Fig. 5b and e) reinforce the privileged location of the antibiotic on the bacterial cell surface, whereas free BODIPY localized in the intercellular space.
(iii) FLIM measurements.
A typical fluorescence lifetime image of BODIPY-vancomycin in S. aureus biofilm is reported in Fig. 2b. On an x-y plane, the fluorescence lifetime appears homogeneous over the entire surface. The corresponding fluorescence decay times acquired by collecting all of the photons in an x-y plane gave an average lifetime τf of 5.0 ± 0.2 ns. That can be correlated with 50% of BODIPY-vancomycin molecules in their free form (τf = 5.7 ns) and 50% in their bound form to S. aureus biofilm components, as determined in the presence of planktonic bacteria (τf = 4.5 ns).
(iv) FCS measurements.
FCS measurements were also performed in different areas of S. aureus biofilms (at the biofilm base and surface and in the bulk biofilm). In the absence of BODIPY-vancomycin, only small fluorescence intensity fluctuations around a low mean value are observed (Fig. 6a, inset). The analysis of corresponding autocorrelation curves leads to a diffusion time of a few hundred milliseconds (Fig. 6b). This value, which was higher than that obtained for planktonic cells, is consistent with the constricted movement of bacteria embedded in an exopolymeric matrix, which is more viscous than a culture medium (1). We also checked to determine whether free BODIPY has similar diffusion properties inside the biofilm as in an aqueous environment, confirming that no strong interaction occurs between the fluorophore and bacteria and/or the EPS matrix (Fig. 6b, insert).
Fig 6.
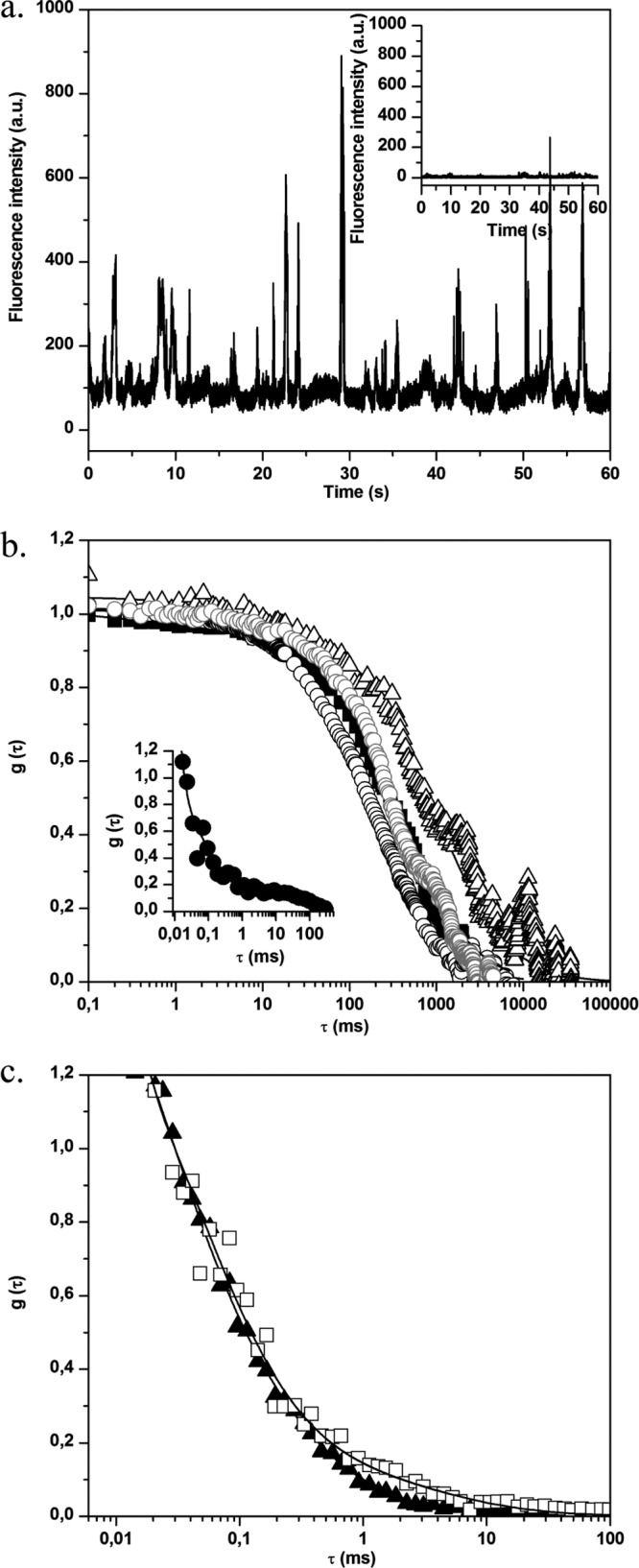
(a) Fluorescence time traces for S. aureus biofilm without (inset) and with BODIPY-vancomycin. In the presence of labeled vancomycin, the fluorescence intensity of biofilm cells increases, which corresponds to the motion of fluorescently cell aggregates in the excitation volume. (b) Normalized fluorescence autocorrelation curves g(τ) for BODIPY-vancomycin interaction with S. aureus cells inside biofilm. The measurements were obtained in different zones: ∼3 μm (△), ∼10 μm (■), and ∼25 μm (○) from the biofilm-coverslip interface. The fit of the curves (straight lines) was obtained using equation 2. The gray curve corresponds to the motion of S. aureus biofilm without BODIPY-vancomycin. (Inset) g(τ) for BODIPY diffusion inside S. aureus biofilm. (c) Fluorescence autocorrelation curves g(τ) for BODIPY-vancomycin diffusion with P. aeruginosa biofilm. The acquisitions were performed inside (□, zone 1) and outside (▲, zone 2) the mushroom structure of the biofilm (the zones are defined in Fig. S1c in the supplemental material). The fit of the curves (straight lines) was obtained using equation 2.
For S. aureus biofilms treated with BODIPY-vancomycin, the recorded fluorescence time trace (Fig. 6a) shows a significant increase in the fluorescence intensity fluctuations compared to the time trace obtained in the absence of the antibiotic. Whatever the area considered, diffusion times of the same order of magnitude as in untreated biofilms were obtained corresponding to that of the global movement of the biofilm (Fig. 6a). This suggests that, at the nanomolar concentration used in the FCS measurements, there was a total interaction of vancomycin with S. aureus biomass.
In order to test whether the inhibition of free antibiotic diffusion could be attributed to nonspecific interactions with the biomass, FCS experiments were extended to P. aeruginosa biofilm incubated with BODIPY-vancomycin. This Gram-negative bacterium is known to be insensitive to the antibiotic due to the absence of the d-Ala-d-Ala targets on the outer cell envelope. Figure 6c shows a typical autocorrelation curve, obtained over several horizontal scans and at different depths within low, dense zones of the biofilm matrix (see Fig. S1c in the supplemental material). The corresponding BODIPY-vancomycin diffusion time τ1 is 67 ± 25 μs (D = 224 ± 80 μm2 s−1), which is close to the value obtained for unreactive BODIPY in S. aureus biofilm). This shows that the labeled antibiotic freely diffuses through a biofilm composed of cells devoid of antibiotic targets.
DISCUSSION
Medicine today is faced with the consideration of infections involving biofilms on both the surface and the internal tissues of the host or on invasive devices such as catheters and implants (6). The presence of biofilms (versus planktonic cells) during infection is not diagnosed by current methods of medical bacteriology (except in few cases for superficial infections), and thus all infections are treated the same way. However, experimental evidence has accumulated showing that the biofilm can shield the action of antimicrobials and of the immune system (10). Hence, antibiotics with high performance against planktonic exponential-growth-phase bacteria in vitro may be less successful in clearing biofilm infections in vivo.
Understanding the mechanisms of biofilm resistance to antimicrobials, especially antibiotics, has become a challenge for the medical community. This is especially important at a time when bacterial strains resistant to any form of conventional antibiotherapy are emerging, as is the case for S. aureus (15).
It is now well established that horizontal gene transfers, and in particular those related to antibiotic resistance, occur at a much higher frequency between cells in a biofilm than between their planktonic counterparts (16). However, in addition to these acquired traits of genetic resistance, biofilm spatial organization may also be the cause of high antibiotic tolerance (16). The associated mechanisms may involve specific cell physiology and the matrix of the biofilm that could act as a barrier to the diffusion-reaction of the antibiotics by retarding its penetration and/or restricting its bioavailability.
We sought here to dissect noninvasively the action of vancomycin on S. aureus biofilms using a set of advanced dynamic fluorescence imaging methods, including not only confocal time-lapse imaging, which is already widely used, but also FLIM, FRAP, and FCS methods.
Is the exopolymeric matrix of S. aureus biofilms an obstacle to the diffusion-reaction of vancomycin?
The only results available in the literature on the mobility of BODIPY-vancomycin inside S. aureus biofilms were reported by Jefferson et al. (18). These researchers observed a low rate of penetration of the antibiotic through their highly mucoid matrix (full biofilm irrigation with vancomycin can take >1 h). It was then hypothesized that a gradual cell exposure to the antibiotic could allow bacteria to undergo stress induced by metabolic or transcriptional changes that could increase their tolerance toward the antibiotic (18).
Our data obtained by time-lapse microscopy and fluorescence imaging demonstrated that BODIPY-vancomycin penetrates to the deepest layers (∼30 μm) of our S. aureus biofilms within minutes (2 to 3 min for ATCC 6538 biofilm and 8 min for ATCC 27217 biofilm) to reach preferentially the cell walls of the embedded bacteria (Fig. 4c and d). Remarkably, no measurable activity of the antibiotic on biofilm cell viability was recorded either by time-lapse imaging using the fluorescent live-dead staining or by conventional plating on agar. It was also shown that the BODIPY labeling of the antibiotics does not modify its antibacterial activity against both the S. aureus strains (data not shown). This fast irrigation by the antibiotic of the whole biofilm structure excludes bacterial stress response. The findings of Jefferson et al. may be related to the particular density of the EPS matrix due to poly-N-acetylglucosamine (PNAG) overproduction by the MN8m strain they used.
In the present study, we showed that fluorescently tagged vancomycin penetrates each S. aureus biofilm (time-lapse measurements) and further diffuses through the biomass (FRAP measurements) with similar diffusion coefficients. However, the relative values (i.e., the ratio between the diffusion coefficients measured in a biofilm and in water) of BODIPY-vancomycin in the S. aureus biofilms (ca. 0.01 to 0.03) do not range over the same scale as that commonly obtained for molecules of comparable molecular weight (0.1 to 0.35) (24). Furthermore, both FRAP and FLIM results reveal that at equilibrium (after a lag phase of about 30 min) approximately half of the antibiotic molecules are immobilized inside the biofilms.
The weak change in viscosity (ratio of ∼1.5) (2) between water and biofilm cannot be the only explanation for this diffusion limitation. Electrostatic interaction must also be considered due to the weak cationic charge of vancomycin (+0.7) (31), the globally negative charge of the cell surfaces at neutral pH (12, 20), and to a lesser extent the charged components of the EPS matrix. Indeed, it was previously reported by FCS measurements that electrostatic attraction between the probe and the biofilm resulted in a reduced diffusion of the probes (13, 33). Nonetheless, if such an electrostatic attractive effect governed the molecular mobility of the antibiotic, it would have also been observed in P. aeruginosa biofilm, which also has anionic charges on its cell walls and possibly on its extracellular polysaccharides (25); however, this was not the case.
More reasonably, the slow vancomycin diffusion through the S. aureus biofilms was likely due to specific adsorption of the antibiotic to the biofilm components. As mentioned above, the bacterial cell walls appeared as hot spots on fluorescence images, revealing the specific interaction of the antibiotic with its target. Nevertheless, some EPS components (proteins, polysaccharides, extracellular DNA, divalent ions, etc.) may also be expected to play a part in vancomycin immobilization inside the biomass. FRAP experiments in the extracellular DNA pockets of the ATCC 27217 biofilm matrix support this hypothesis. Indeed, in these specific zones, much higher immobilized fractions of the antibiotic are measured (60% versus ∼40%), a finding which is in good agreement with the reported ability of vancomycin to intercalate into DNA molecules (31).
Is vancomycin bioavailability an obstacle to its activity inside S. aureus biofilms?
Failure for some antibiotics to be active on cell viability in biofilms is often attributed to the failure of the drug to penetrate the biostructure. Our study is an additional contribution supporting that biofilm matrix is not an absolute physical barrier to the penetration of molecules at the size of antibiotics. However, sorption to, or reaction with, biofilm components may reduce antibiotic bioavailability and also be put forward to explain the decrease in antibiotic efficiency in biofilms.
To answer this question, two factors must be considered: (i) comparison between FRAP and FCS measurements and (ii) the difference in the concentration of antibiotics each method requires (a few hundred nanomoles for FCS and 3 to 10 times more than that for FRAP). With the low concentration range used for FCS experiments, we have obtained correlation curves corresponding to the biofilm motion due to a total interaction of BODIPY-vancomycin with the biomass. In the context of FRAP experiments, as mentioned above, a significant percentage of the antibiotic (50 to 60%) can freely diffuse in all parts of the biofilm, in agreement with an excess of antibiotic relatively to the concentration fixed on biofilm components. Thus, it seems unlikely that the lack of vancomycin bioavailability could explain the failure of the antibiotic activity in such biofilms. Shielding mechanisms of biofilms that derive from the specific physiology of embedded bacteria should be further studied.
In conclusion, we have demonstrated here that advanced fluorescence imaging methods (time-lapse, FLIM, FRAP, and FCS) represent versatile and nondestructive tools for studying the diffusion-reaction of molecules as small as antibiotics through the depth of biofilms. The time-lapse approach is accessible using current commercial confocal microscopes, and its advantage lies in the possibility of monitoring the penetration of fluorescent antibiotics over time in all parts of the biofilm. The FRAP, FLIM, and FCS methods require more experience in data analysis and/or additional instrumental acquisition (5). However, they allow diffusion-reaction measurements when the antimicrobials have reached equilibrium inside the biomatrix (within several hours). The use of these correlative time-resolved fluorescence microscopy methods has expanded our knowledge on the role of the biofilm structure on the local diffusion-reaction properties of antibiotics.
Supplementary Material
ACKNOWLEDGMENTS
S.D.O. was funded by the CNRS, France.
We thank Antoine Monsel, physician at the Pitié Salpêtrière Hospital (Paris, France), for valuable discussions. We thank Céline Merlin for English revision of the manuscript.
Footnotes
Published ahead of print 26 March 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Braga J, Desterro JMP, Carmo-Fonseca M. 2004. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol. Biol. Cell 15:4749–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Briandet R, et al. 2008. Fluorescence correlation spectroscopy to study diffusion and reaction of bacteriophages inside biofilms. Appl. Environ. Microbiol. 74:2135–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bridier A, Briandet R, Thomas V, Dubois-Brissonnet F. 2011. Resistance of bacterial biofilms to disinfectants: a review. Biofouling 27:1017–1032 [DOI] [PubMed] [Google Scholar]
- 4. Bridier A, Dubois-Brissonnet F, Greub G, Thomas V, Briandet R. 2011. Dynamics of the action of biocides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 55:2648–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bridier A, et al. 2011. Deciphering biofilm structure and reactivity by multiscale time-resolved fluorescence analysis. Adv. Exp. Med. Biol. 715:333–349 [DOI] [PubMed] [Google Scholar]
- 6. Bryers JD. 2008. Medical biofilms. Biotechnol. Bioeng. 100:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daddi Oubekka S, Briandet R, Wharate F, Fontaine-Aupart M-P, Steenkeste K. 2011. Image-based fluorescence recovery after photobleaching (FRAP) to dissect vancomycin diffusion-reaction processes in Staphylococcus aureus biofilms. SPIE-OSA Clin. Biomed. Spectrosc. Imaging II 8087 1I:1–8 [Google Scholar]
- 8. Davison WM, Pitts B, Stewart PS. 2010. Spatial and temporal patterns of biocide action against Staphylococcus epidermidis biofilms. Antimicrob. Agents Chemother. 54:2920–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 10. Fux CA, Costerton JW, Stewart PS, Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol. 13:34–40 [DOI] [PubMed] [Google Scholar]
- 11. Gilbert Y, et al. 2007. Single-molecule force spectroscopy and imaging of the vancomycin/d-Ala-d-Ala interaction. Nano. Lett. 7:796–801 [DOI] [PubMed] [Google Scholar]
- 12. Gross M, Cramton SE, Gotz F, Peschel A. 2001. Key role of teichoic acid net charge in Staphylococcus aureus colonization of artificial surfaces. Infect. Immun. 69:3423–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guiot E, et al. 2002. Heterogeneity of diffusion inside microbial biofilms determined by fluorescence correlation spectroscopy under two-photon excitation. Photochem. Photobiol. 75:570–578 [DOI] [PubMed] [Google Scholar]
- 14. Habimana O, et al. 2011. Diffusion of nanoparticles in biofilms is altered by bacterial cell wall hydrophobicity. Appl. Environ. Microbiol. 77:367–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hiramatsu K, Cui L, Kuroda M, Ito T. 2001. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 9:486–493 [DOI] [PubMed] [Google Scholar]
- 16. Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. 2010. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35:322–332 [DOI] [PubMed] [Google Scholar]
- 17. Hope CK, Wilson M. 2004. Analysis of the effects of chlorhexidine on oral biofilm vitality and structure based on viability profiling and an indicator of membrane integrity. Antimicrob. Agents Chemother. 48:1461–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jefferson KK, Goldmann DA, Pier GB. 2005. Use of confocal microscopy to analyze the rate of vancomycin penetration through Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 49:2467–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karolin J, Johansson LBA, Strandberg L, Ny T. 1994. Fluorescence and absorption spectroscopic properties of dipyrrometheneboron difluoride (BODIPY) derivatives in liquids, lipid membranes, and proteins. J. Am. Chem. Soc. 116:7801–7806 [Google Scholar]
- 20. Klodzinska E, et al. 2009. Differentiation of Staphylococcus aureus strains by CE, zeta potential and coagulase gene polymorphism. Electrophoresis 30:3086–3091 [DOI] [PubMed] [Google Scholar]
- 21. Neu TR, et al. 2010. Advanced imaging techniques for assessment of structure, composition and function in biofilm systems. FEMS Microbiol. Ecol. 72:1–21 [DOI] [PubMed] [Google Scholar]
- 22. Nichols WW, Dorrington SM, Slack MP, Walmsley HL. 1988. Inhibition of tobramycin diffusion by binding to alginate. Antimicrob. Agents Chemother. 32:518–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pinho MG, Errington J. 2003. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol. Microbiol. 50:871–881 [DOI] [PubMed] [Google Scholar]
- 24. Rani SA, Pitts B, Stewart PS. 2005. Rapid diffusion of fluorescent tracers into Staphylococcus epidermidis biofilms visualized by time lapse microscopy. Antimicrob. Agents Chemother. 49:728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shephard J, McQuillan AJ, Bremer PJ. 2008. Mechanisms of cation exchange by Pseudomonas aeruginosa PAO1 and PAO1 wbpL, a strain with a truncated lipopolysaccharide. Appl. Environ. Microbiol. 74:6980–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart PS. 2003. Diffusion in biofilms. J. Bacteriol. 185:1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stewart PS, Davison WM, Steenbergen JN. 2009. Daptomycin rapidly penetrates a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 53:3505–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stone G, Wood P, Dixon L, Keyhan M, Matin A. 2002. Tetracycline rapidly reaches all the constituent cells of uropathogenic Escherichia coli biofilms. Antimicrob. Agents Chemother. 46:2458–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takenaka S, Trivedi HM, Corbin A, Pitts B, Stewart PS. 2008. Direct visualization of spatial and temporal patterns of antimicrobial action within model oral biofilms. Appl. Environ. Microbiol. 74:1869–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valeur B. 2001. Molecular fluorescence: principles and applications, p 34–70 Wiley-VCH, New York, NY [Google Scholar]
- 31. Vijan LE. 2009. The interaction of vancomycin with DNA. Rev. Roum. Chem. 54:807–813 [Google Scholar]
- 32. Waharte F, Steenkeste K, Briandet R, Fontaine-Aupart MP. 2010. Diffusion measurements inside biofilms by image-based fluorescence recovery after photobleaching (FRAP) analysis with a commercial confocal laser scanning microscope. Appl. Environ. Microbiol. 76:5860–5869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z, Nadezhina E, Wilkinson KJ. 2010. Quantifying diffusion in a biofilm of Streptococcus mutans. Antimicrob. Agents Chemother. 55:1075–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Z, Stewart PS. 2004. Growth limitation of Staphylococcus epidermidis in biofilms contributes to rifampicin tolerance. Biofilms 1:31–35 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.