

Abstract
Myxococcus xanthus undergoes a starvation-induced multicellular developmental program during which cells partition into three known fates: (i) aggregation into fruiting bodies followed by differentiation into spores, (ii) lysis, or (iii) differentiation into nonaggregating persister-like cells, termed peripheral rods. As a first step to characterize cell fate segregation, we enumerated total, aggregating, and nonaggregating cells throughout the developmental program. We demonstrate that both cell lysis and cell aggregation begin with similar timing at approximately 24 h after induction of development. Examination of several known regulatory proteins in the separated aggregated and nonaggregated cell fractions revealed previously unknown heterogeneity in the accumulation patterns of proteins involved in type IV pilus (T4P)-mediated motility (PilC and PilA) and regulation of development (MrpC, FruA, and C-signal). As part of our characterization of the cell lysis fate, we set out to investigate the unorthodox MazF-MrpC toxin-antitoxin system which was previously proposed to induce programmed cell death (PCD). We demonstrate that deletion of mazF in two different wild-type M. xanthus laboratory strains does not significantly reduce developmental cell lysis, suggesting that MazF's role in promoting PCD is an adaption to the mutant background strain used previously.
INTRODUCTION
The myxobacteria are excellent model systems for multicellular bacterial behavior. Myxococcus xanthus, the best studied of this group of bacteria, exhibits a complex life cycle. Under nutrient-replete conditions, cells swarm over surfaces, obtaining nutrients cooperatively by secreting sufficient degradative enzymes to digest organic macromolecules (47). When nutrients become limited, the population enters a complex developmental program consisting of at least three distinct cell fates: sporulation within multicellular fruiting bodies (reviewed in reference 18), differentiation into persister-like cells termed peripheral rods (39–41), and cell lysis (36, 64).
Fruiting bodies, the best-characterized cell fate, are produced when some cells are induced to aggregate into mounds of approximately 105 cells prior to induction of sporulation. The roles of several developmental regulators in promoting fruiting body formation have been well defined. MrpC, a transcriptional regulator of the cyclic AMP (cAMP) receptor protein (CRP) family, is necessary for both the aggregation and sporulation programs (34, 35, 56, 57). MrpC is present at low levels in vegetative cells, but mrpC is transcriptionally upregulated after initiation of development (37, 38, 56, 57). MrpC is subject to complex posttranslational regulation, which appears to control its affinity for identified target promoters. Under vegetative conditions, MrpC is thought to be phosphorylated (MrpC-P), which reduces its affinity for target sequences. Under developmental conditions, MrpC is no longer phosphorylated and is thought to be processed into MrpC2, an isoform which lacks approximately 25 amino acids from the amino terminus (38). MrpC2 shows increased affinity for target sequences in vitro (37, 38) and is proposed to more efficiently induce their transcription. An important target of MrpC2 is the developmental transcriptional regulator gene, fruA (60). FruA is proposed to be activated in response to C-signaling (12, 43), a cell contact-dependent signal transmission pathway. C-signal (p17) is generated by proteolytic processing of the surface-exposed CsgA (p25) protein (24, 46). It is proposed that C-signal binds to an unidentified receptor on a neighboring cell, which initiates an unknown signaling pathway that results in activation of FruA. Low levels of activated FruA are proposed to stimulate the methylation state of the methyl-accepting chemosensory protein (MCP), FrzCD. An increase in FrzCD methylation induces cell aggregation (6, 29, 30, 51, 54), which leads to increased cell contact and therefore higher levels of C-signaling and activated FruA. Increased activation of FruA is proposed to induce FruA-dependent transcription of several target genes. In many cases, FruA acts in combination with MrpC2 (22, 33, 34, 55). One important target of FruA/MrpC2 is the dev locus (62), which is expressed at high levels in aggregation centers (16), is necessary for efficient sporulation (59), and additionally promotes upregulation of fruA, inducing a positive-feedback loop (7). Thus, MrpC/MrpC2, C-signal, FruA, and FrzCD constitute core components necessary for induction and coordination of fruiting body formation.
Two additional cell fates, differentiation into peripheral rods and cell lysis, are wired into the developmental program. Peripheral rods represent a minor proportion of the developing population that neither aggregate nor sporulate and may have evolved to take advantage of low levels of nutrients insufficient to induce germination of fruiting body spores (40). These cells do not divide but can be restored to vegetative growth if transferred to rich medium (40). This cell type was initially characterized using a differential centrifugation assay; cells in aggregation centers pellet during a low-speed centrifugation assay while peripheral rods remain in the supernatant. These assays demonstrated that peripheral rods display total protein accumulation patterns distinct from both vegetative and sporulating cells (39, 41). It was also previously observed that CsgA (p25) is produced at lower levels in peripheral rods (16) and that dev and nfs loci, which represent fruiting body- and sporulation-specific loci, respectively, are not expressed in peripheral rods (16, 35). However, little is known about the regulatory details which induce certain cells to form peripheral rods. It has been debated whether the final cell fate, lysis, is wired into the developmental program (14, 48, 64), is an artifact of manipulating fragile cells (42), or is a strain-specific phenomenon (1). Most recently, lysis was attributed to induction of programmed cell death (PCD) by an atypical toxin-antitoxin system comprised of MazF and MrpC (36). Deletion of the orphan mazF gene, which encodes an endoribonuclease, reduced developmental cell lysis, produced a severe delay in aggregation, and reduced sporulation. MrpC interacts both in vivo and in vitro with MazF, and this interaction interferes with MazF activity. Thus, it is proposed that MrpC acts as an antitoxin to MazF, but it is unknown how MazF is released to induce PCD.
As part of our interest in identifying the regulatory mechanisms which control cell fate segregation in developing M. xanthus cells, we first examined the timing of cell fate production by enumerating total cells, cells which can be sedimented in aggregation centers (41), and cells which are not tightly associated in large groups. We observed a robust pattern of subpopulation production in which at between 24 and 30 h of development there is both a burst of cell lysis and a rapid increase of cells in aggregation centers. Comparison of several developmental and structural proteins in the separated subpopulations revealed previously unrecognized heterogeneity in the accumulation of developmental regulatory proteins and structural proteins. Furthermore, investigation of the developmental cell lysis revealed that the endoribonuclease MazF does not play a significant role in developmental cell lysis of M. xanthus wild-type strains.
MATERIALS AND METHODS
Strains and growth conditions.
Bacterial strains and plasmids used are listed in Table 1. M. xanthus strains were grown vegetatively at 32°C on CYE agar plates [1% Casitone, 0.5% yeast extract, 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.6, 4 mM MgSO4, 1.5% agar] or in CYE broth (CYE lacking agar). Plates were supplemented with 100 μg ml−1 kanamycin, where necessary. Escherichia coli was grown under standard laboratory conditions in LB medium (27) unless otherwise described. Plates were supplemented with 50 μg ml−1 kanamycin, where necessary.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or characteristic(s) | Reference or source |
|---|---|---|
| M. xanthus strains | ||
| DZ2 | Wild type | 9 |
| DK1622 | Wild type | 63 |
| DK101 | Wild type (sgl1) | 11 |
| PH1021 | DZ2 ΔmazF | This study |
| PH1022 | DZ2 Mxan_1658::pPH165 | This study |
| PH1023 | DK1622 ΔmazF | This study |
| PH1024 | DK101 ΔmazF | This study |
| PH1025 | DZ2 ΔmrpC | This study |
| PH1014 | DZ2 csgA::Tn5 lacΩLS205 | 13 |
| E. coli strains | ||
| TOP10 | Host for cloning [F−mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 deoR recA1 arsD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG] | Invitrogen |
| GJ1158 | proUp-T7 RNAP | 3 |
| Plasmids | ||
| pET28 | T7 promoter overexpression plasmid, Kmr | Novagen |
| pPH158 | pET28-mrpC | This study |
| pBJ114 | Backbone for in-frame deletions; galK Kmr | 16 |
| pPH165 | pBJ114 ΔmazF | This study |
| pPH164 | pBJ114 ΔmrpC | This study |
Plasmid and strain construction.
Strain PH1021 (DZ2 ΔmazF) was generated using pPH165 (pBJ114 ΔmazF) and the kanamycin resistance (Kanr)-galK galactose selection/counterselection method (61) as previously described in detail (21). Briefly, strain DZ2 was transformed with pPH165, and integration of the plasmid (via homologous recombination) into the region surrounding mazF was selected by kanamycin resistance, generating strain PH1022 (DZ2 Mxan_1658::pPH165). pPH165 contains a 1,043-bp region surrounding a mazF deletion (deletion of codons 10 to 111) cloned into the EcoRI and BamHI sites of pBJ114; this insert was generated by overlap PCR using the primers listed in Table S1 in the supplemental material as previously described in detail (21). To generate an in-frame deletion of mazF, PH1022 was plated in the presence of 2.5% galactose to screen for loss of the plasmid via a second homologous recombination event. Resulting Galr Kans colonies were screened by PCR for the wild-type or ΔmazF genotype. Strains PH1023 (DK1622 ΔmazF) and PH1024 (DK101 ΔmazF) were generated by transforming DK1622 and DK101, respectively, with genomic DNA from PH1022 and selecting for double homologous recombination into the mazF region by kanamycin resistance. The mazF deletion was generated in the resulting backgrounds by selecting for growth on galactose as described above. For all deletion strains, the developmental phenotype was confirmed for three independent clones. Strain PH1025 (DZ2 ΔmrpC) was generated using construct pPH164 (pBJ114 ΔmrpC) as described above. pPH164 contains a 1,105-bp fragment surrounding a mrpC deletion mutant (ΔmrpC; deletion of codons 14 to 228) cloned into the EcoRI and BamHI sites of pBJ114. Plasmid pPH158 (pET28-mrpC) used for overproduction of recombinant MrpC containing a six-histidine affinity tag at the amino terminus (H6-MrpC) was constructed by PCR amplifying mrpC from DZ2 genomic DNA using the primers indicated in Table S1; the resulting insert was cloned into the BamHI and XhoI sites of pET28 (Novagen), and the inserted region was sequenced to confirm the absence of PCR-generated sequence errors.
Developmental subpopulation analysis and cell enumeration.
Cells were induced to develop under submerged culture in 90-cm petri plates as follows. Cells from an overnight culture in CYE broth were diluted to an optical density at 550 nm (OD550) of 0.035 in fresh CYE medium. Sixteen milliliters of diluted cells was added to 90-cm petri plates, followed by incubation at 32°C for 24 h. To initiate the developmental program, CYE medium was replaced by an equivalent volume of sterile MMC starvation medium (10 mM MOPS, pH 7.6, 2 mM CaCl2, 4 mM MgSO4), and plates were incubated at 32°C for the times indicated in the respective figures. Developmental phenotypes were recorded at the times indicated with a Leica MZ8 stereomicroscope and attached Leica DFC320 camera.
For subpopulation segregation analysis, the developing cells were harvested by using a 20-ml pipette to flush the cell mat off the surface of the petri dish. The entire 16-ml volume was then transferred to a 50-ml Falcon tube, and the mats were dispersed by vigorously pipetting up and down 30 times. Cells in aggregates were then sedimented by centrifugation at 50 × g (Heraeus Multifuge 1 S-R centrifuge and 75002002 G swinging bucket rotor) for 5 min at room temperature (RT). The supernatant fraction was carefully removed and transferred to a 50-ml Falcon tube, and the pellet (sedimented cell fraction) was resuspended in an equivalent volume (16 ml) of MMC starvation medium. Cells in the supernatant fraction were present as single cells or in clusters of 1 to 4 cells, whereas resuspended cells in the sedimented fraction were detected as very large clusters of cells (data not shown). Both fractions were dispersed at least three times at 5 m s−1 for 45 s in a FastPrep 24 cell and tissue homogenizer (MP Biomedicals) at 4°C. After dispersal, both the supernatant and sedimented cell fractions were detected as single cells (data not shown). Both fractions were enumerated using a Helber bacterial counting chamber (Hawksley, United Kingdom). The total population was calculated as the sum of the supernatant and sedimented cell fractions.
Relative quantification of EPS by trypan blue binding.
Relative extracellular polymeric substance (EPS) production was assayed using a modification of a trypan blue binding assay (4). Wild-type DZ2 cells were developed under submerged culture and harvested and separated into supernatant and sedimented fractions by centrifugation for 5 min at 50 × g as described above. The supernatant fraction was pelleted for 10 min at 4,700 × g. This pellet and the sedimented cell fraction pellet were each resuspended in 1 ml of 50 mM MOPS, pH 7.6, transferred to a 2-ml screw-cap tube, and dispersed in a FastPrep tissue homogenizer as described above. A total of 1 × 109 cells from each fraction were diluted into 3.5 ml of 10 mM MOPS (pH 7.2)–1 mM CaCl2. Trypan blue was added to 5 μg ml−1 in triplicate 1-ml samples, followed by incubation in the dark for 30 min at RT. Cells were pelleted for 5 min at maximum speed in a microcentrifuge, and the absorbance of the supernatant was measured at 585 nm. To calculate the dye remaining, the average absorbance value was subtracted from that of a cell-free control and recorded as the fraction of the cell-free control. Data shown are the average of two independent biological experiments.
Immunoblot analysis.
To prepare protein lysates for immunoblot analysis, cell fractions were separated by centrifugation as above; the pellet fraction was immediately stored at −20°C, and the supernatant fraction was pelleted at 4,700 × g for 10 min and stored at −20°C as a cell pellet. Duplicate cultures were used to enumerate the cells in each fraction as described in the cell enumeration section above. The frozen cell pellets were resuspended to 3.6 × 106 cells μl−1 in protein sample buffer (19) containing a 1:20 dilution of mammalian protease inhibitor cocktail (Sigma) and heated at 99°C for 10 min. Samples (200 μl) were then disrupted by bead-beating in 2-ml screw-cap tubes containing 0.2 g of 0.1-mm zirconia-silica beads six times for 45 s at 6.5 m s−1 in a FastPrep 24 cell homogenizer and stored at −20°C. Samples were heated at 99°C prior to use, and 10-μl samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For analysis of CsgA p25 and p17, samples were instead resolved on Tris-Tricine gels as described previously (50). Proteins were transferred to polyvinylidene difluoride (PVDF) membrane using a semidry transfer apparatus (Hoeffer). Western blot analyses were performed using the following antibody dilutions: anti-protein S at 1:5,000 (41), anti-protein C polyclonal antibodies (pAb) (31) (which recognize FibA [20]) at 1:5,000, anti-PilC pAb at 1:10,000 (8), anti-PilA pAb at 1:10,000 (65), anti-MrpC pAb at 1:1,000 (below), anti-FruA pAb at 1:5,000 (24), anti-CsgA C-terminal pAb (24) at 1:2,500, and anti-FrzCD pAb (32) at 1:5,000. Secondary anti-rabbit IgG-horseradish peroxidase (HRP) antibody (Pierce) was used at 1:20,000, and signals were detected with enhanced chemiluminescence substrate (Pierce), followed by detection in an LAS-4000 luminescent image analyzer (Fujifilm). The PVDF membrane was subsequently stained using Coomassie stain (50% [vol/vol] methanol, 7% [vol/vol], acetic acid, 0.1 Coomassie blue R-250) to determine that the total proteins detected in each fraction were equivalent and to confirm that the lysates were prepared from equivalent numbers of cells. Representative immunoblot patterns are shown, but similar patterns were obtained from at least two independent biological replicates.
Anti-MrpC rabbit polyclonal antiserum was generated by Eurogentec (Belgium) using a 28-day Super Speedy polyclonal antibody protocol. Rabbits were immunized with recombinant MrpC containing an amino-terminal six-histidine affinity purification tag (H6-MrpC). To overproduce H6-MrpC, the salt-inducible T7 RNA polymerase strain (GJ1158) was freshly transformed with the plasmid pPH158 (pET28-mrpC) and plated on LB medium lacking NaCl (LBNS, for LB no salt) containing kanamycin. Several colonies were inoculated into LBNS medium plus kanamycin, grown overnight at 37°C, subcultured 1:100 into fresh LBNS medium plus kanamycin, and grown at 37°C until an optical density at 600 nm (OD600) of 0.6. The culture was transferred to a shaking incubator at 18°C for 1 h and then induced with 0.3 M NaCl (final concentration) overnight. Soluble H6-MrpC was purified under native conditions using nickel affinity resin (Qiagen) and gravity flow according to the manufacturer's instructions.
Analysis of the effect of the ΔmazF mutant on development.
For analysis of the ΔmazF strain developmental phenotypes under submerged culture, the mutant and corresponding DZ2 background strain were induced to develop as described for the cell enumeration analyses. For analysis of total cell number, cells at each time point were harvested into a 50-ml Falcon tube, dispersed as described above, and enumerated in a cell counter (Beckman Coulter Multisizer 3) with a 20-μm-aperture tube. Data from three independent biological replicates were used to calculate the average number of cells at each time point. Isolation and enumeration of heat- and sonication-resistant spores were performed as described previously (21). Briefly, cells from each time point were resuspended in MMC buffer, heated at 50°C for 60 min, sonicated, and enumerated using a Helber bacteria counting chamber (Hawksley, Lancing, United Kingdom).
For analysis of the ΔmazF strain developmental phenotype on clone fruiting (CF) agar plates, 1 ml of CF agar [0.15% Casitone, 0.2% sodium citrate, 0.1% sodium pyruvate, 0.02% (NH4)2SO4, 10 mM MOPS (pH 7.6), 8 mM MgSO4, 1 mM KH2PO4, 1.5% agar] was added to each well of a 24-well tissue culture plate (Sarstedt) and allowed to solidify overnight at RT. To mimic the high-density developmental conditions of Nariya and Inouye (36), cells were grown to mid-log phase in CYE broth, and the OD550 was measured. Five milliliters of each culture was pelleted for 20 min at 4,600 × g, washed, and resuspended to a calculated OD550 of 35 in MMC starvation medium. Five microliters of cells was spotted onto the CF agar in one well. Developmental phenotypes were recorded as described above. For analysis of total cell number at each time point, cells from triplicate wells were scraped from the agar, resuspended into 0.5 ml of MMC starvation buffer in a 2-ml screw cap tube, dispersed as described above, and enumerated in a cell counter as described above. Data from two independent biological replicates of duplicate samples were used to calculate the average number of cells at each time point. Enumeration of heat- and sonication-resistant spores was performed as described above.
RESULTS
The timeline of developmental cell fate production.
Detailed regulatory mechanisms controlling segregation of developing M. xanthus cells into different cell fates (lysis, formation of fruiting bodies, or differentiation of peripheral rods) are unknown. As a first step to characterization, we set out to define the timing of cell fate production by enumerating total, aggregated, and nonaggregated cells throughout development. For these assays, cells were developed under submerged culture: cells were grown on the surface of petri dishes in rich medium (time [T] zero), and development was induced by replacing the medium with starvation buffer. Under these conditions, cells began to form visible translucent aggregates by 30 h of development, and by 48 h they formed darkened fruiting bodies as the cells differentiated into spores (Fig. 1A). Cells were harvested at several periods during development by repeated pipetting and subjected to low-speed centrifugation to separate cells in groups (pellet) from single cells or cells in small groups (supernatant) (41). Cells in each fraction were then dispersed, enumerated, and plotted individually or summed as a function of time during development (Fig. 1B). During development of this assay, we ascertained that (i) neither the harvesting nor dispersal steps significantly affected the total cell count, (ii) the dispersal procedure resulted in single or small and countable groups of cells, and (iii) the total number of cells counted without the separation step was not appreciably different from the sum of the two populations of separated cells (data not shown).
Fig 1.
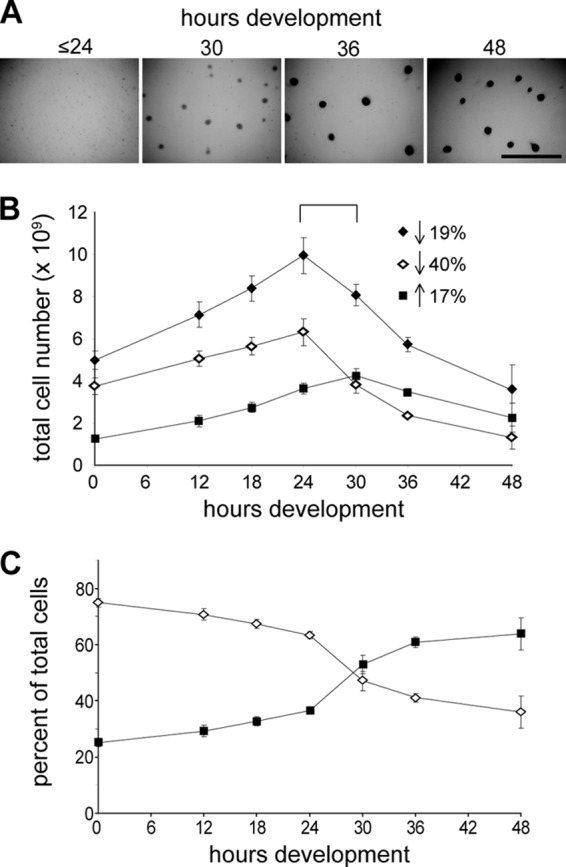
Enumeration of supernatant and sedimented cell fractions during development. (A) Fruiting body formation of wild-type DZ2 developing under submerged culture. Visible aggregation centers can be identified by 30 h of development which enlarge by 36 h and darken with the onset of sporulation (between 36 and 48 h). Scale bar, 2 mm. (B) The number of cells at the indicated hours of development in the sedimented (black squares) or supernatant (white diamonds) cell fractions after centrifugation at 50 × g for 5 min. The total cell number (black diamonds) is the sum of the cells enumerated in the sedimented and supernatant cell fractions. Data are shown as the average and associated standard deviations of three independent biological replicates. Arrows indicate the percent increase (up) or decrease (down) of the number of total, supernatant, or sedimented cells from 24 to 30 h of development. (C) Percentage of the total number of cells enumerated at each time point located in the sedimented (black squares) and supernatant (white diamonds) cell fractions.
The total cell number enumeration indicated that the population doubled over the first 24 h of development and then dramatically decreased such that between 24 and 48 h ∼64% of the cells had lysed (Fig. 1B). Surprisingly, even prior to initiation of development (0 h), 25% ± 1% of the total cells in the population could be isolated in the pellet (sedimented fraction) (Fig. 1B and C). However, between 24 and 36 h, concurrent with the formation of visible aggregation centers, the sedimented cell population sharply increased from 37% ± 1% to 61% ± 2% (Fig. 1) and essentially remained at this level until the final time point assayed at 48 h. The number of spores was not enumerated in these experiments, but in similar experiments (pre)spores (i.e., spherical cells which were not tested for heat and sonication resistance) were enumerated as follows: none detected, ∼4 × 108, ∼5 × 108, and ∼9 × 108 at ≤ 36, 48, 72, and 144 h of development, respectively. The percentage of cells in the supernatant fraction decreased corresponding to the increase in sedimented cells and was maintained at essentially 64% ± 6% after 36 h of development (Fig. 1B and C). These cells were likely predominantly peripheral rods (41).
These data suggest the time period between 24 and 30 h as the onset of both cell lysis and aggregation. During this time period, the total cell population decreased by 19%, the supernatant fraction decreased by 40%, and the cells in the sedimented fraction increased by 17% (Fig. 1B). Therefore, the 40% decrease in number of cells in the supernatant subpopulation is most likely due to approximately equal numbers of cells undergoing lysis and being stimulated to aggregate. The onset of peripheral rod production is less clear but was likely approximately 36 h of development.
Aggregating cell fractions display distinct cellular properties.
We next sought to confirm that the sedimented and supernatant cell fractions displayed distinct characteristics. The production of extracellular polymeric substances (EPS) was first examined by monitoring binding of the dye trypan blue to the cells in each fraction. Dye binding was normalized to the number of cells in the respective fractions at each time point. Under vegetative conditions (T = 0), cells in the sedimented fraction bound slightly more (∼5-fold) trypan blue than the cells in the supernatant fraction (Fig. 2A), suggesting that they produce more EPS (5). Between 24 and 30 h, cells in the sedimented fraction began to bind more trypan blue such that by 48 h these cells bound approximately 12 times more trypan blue than was observed in equivalent numbers of vegetative cells. In contrast, equivalent numbers of cells in the supernatant did not appear to bind significantly more trypan blue (Fig. 2A). These results suggest that EPS is upregulated specifically in the cells which are induced to aggregate into fruiting bodies.
Fig 2.
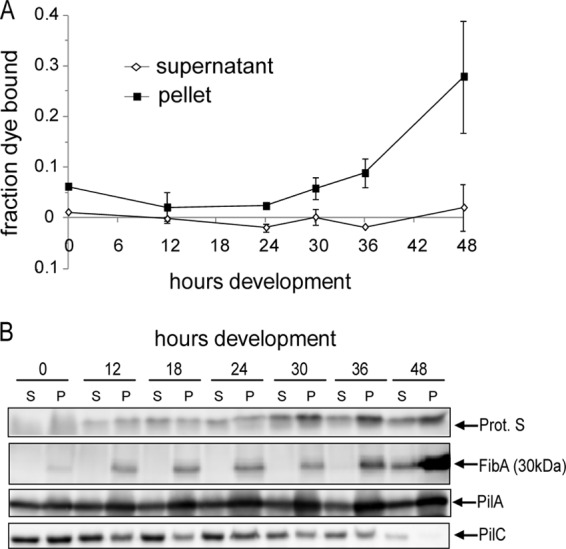
Cells in the pellet and supernatant fractions display distinct characteristics. (A) Assay for production of extracellular polymeric substances (EPS). Approximately 2.8 × 108 cells from the dispersed supernatant (white diamonds) or sedimented (black squares) cell fractions harvested at the indicated hours were incubated with trypan blue. The fraction of dye bound to the cells was calculated by measuring the absorbance of the dye remaining in solution relative to a cell-free control. Data shown are the average and associated standard deviation of two independent biological experiments. (B) Immunoblot analysis of cells from the supernatant (S) and pellet (P; sedimented) fractions using antisera specific for protein S, the 30-kDa fragment of FibA, and the PilA and PilC components of the type IV pilus motility machinery. Each lane contains total cell lysate prepared from 4 × 107 cells.
We next prepared protein lysates from equivalent numbers of cells in each fraction at each time point and subjected them to immunoblot analysis using antisera to several M. xanthus proteins, including protein S (a spore coat protein which has been examined in peripheral rod and prespore fractions previously [41]) and FibA, which we have previously shown is specific for the aggregated-cell fraction (20). Similar to what was described previously, protein S was produced in both nonaggregated and aggregated fractions of the population but by 48 h was upregulated at least 2-fold in the sedimented fraction (Fig. 2B). In contrast, as we had observed previously (20), FibA was specific to the sedimented fraction from 0 to 36 h of development but by 48 h was also observed in the cells remaining in the supernatant.
Two proteins of the type IV pilus (T4P)-mediated motility machinery (reviewed in reference 68), PilA and PilC, which are equally represented in the two cell fractions in vegetative cells (20), displayed heterogeneous accumulation patterns as development progressed (Fig. 2B). Interestingly, these analyses revealed that PilA gradually accumulated during development in the sedimented cell fraction but was maintained at nearly constant levels in the supernatant cell fraction such that after 18 h of development, the sedimented cell fraction was overrepresented in PilA. In contrast, PilC decreased in both fractions during development but decreased more rapidly in the sedimented-cell fraction. Thus, the relative PilA/PilC ratio increases during development, particularly in the sedimented-cell fraction. Together, these results confirm that even early during the developmental program, the differential centrifugation assay can differentiate two apparently distinct populations of cells.
Developmental regulator proteins display distinct accumulation patterns in the aggregating and nonaggregating fractions.
Having established that the sedimented and supernatant fractions of a low-speed centrifugation represent distinct subpopulations of cells, we next examined each fraction for the accumulation of proteins known to regulate the developmental program. We first examined MrpC, a transcriptional regulator required for induction of aggregation and sporulation (36, 56, 57) which has also been implicated in control of cell lysis (36). MrpC can be detected in at least two isoforms by immunoblotting: full-length MrpC, which migrates at ∼25 kDa, and the smaller MrpC2 form, which migrates at ∼23 kDa (38). In cells grown on a surface under vegetative conditions (T = 0), MrpC was represented equally in both the supernatant and pellet cell fractions (Fig. 3A). By 12 h of development, the MrpC signal had decreased 1.2-fold in the supernatant and 2.4-fold in the pellet fraction, suggesting that the protein is turned over in at least some of the cells in both populations (Fig. 3A). Between 12 and 24 h of development, MrpC accumulated dramatically in the supernatant cell fraction, but between 24 and 48 h of development it decreased such that by 48 h of development, MrpC was essentially absent in this cell fraction. In contrast, in the pellet fraction, MrpC accumulated linearly, but more slowly, from 12 to 48 h of development. MrpC2 was observed to increase between 18 and 24 h of development in the supernatant fraction (Fig. 3A, dashed lines), approximately 6 h later than MrpC. After 24 h of development, as observed with MrpC, MrpC2 began to decrease in the supernatant fraction. In the sedimented fraction, MrpC2 increased 3-fold between 24 and 36 h, after which it was essentially cleared. Thus, the two MrpC isoforms displayed significantly different accumulation patterns from each other and within the two populations of developing cells. Together, these analyses revealed previously unrecognized fluctuations and heterogeneity in the accumulation pattern of MrpC and/or MrpC2.
Fig 3.
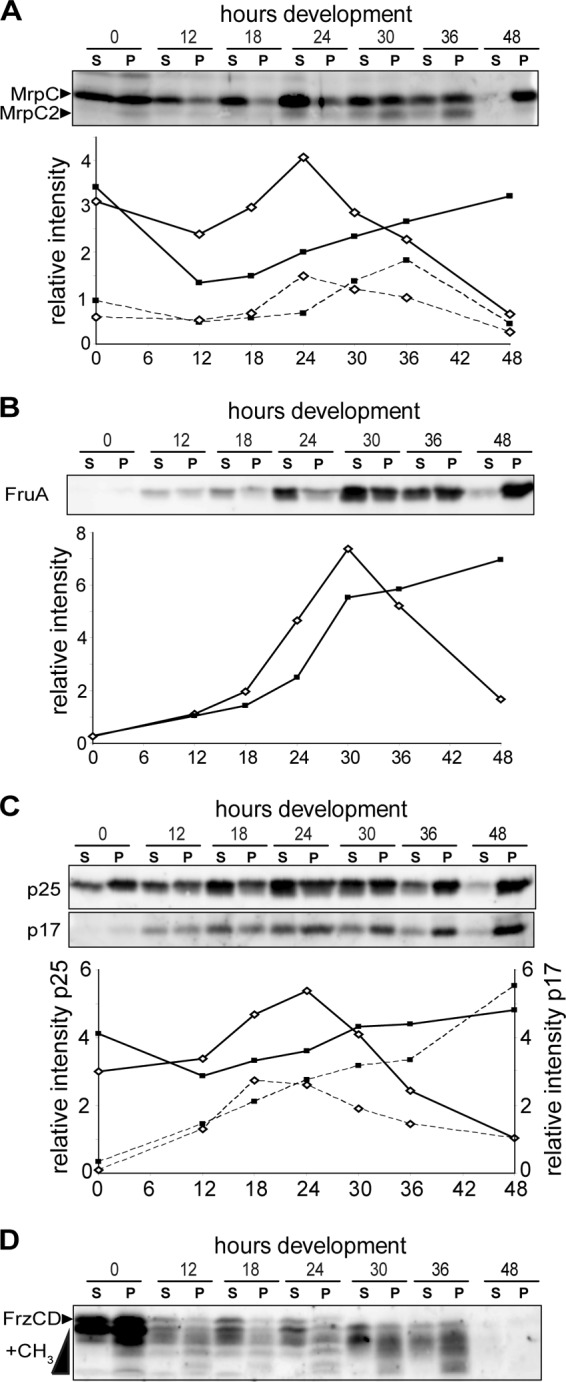
Cells in the pellet and supernatant fractions display distinct patterns of developmental regulator accumulation. Panels show immunoblot analysis of cells from the supernatant (S) and pellet (P; sedimented) fractions using antisera specific for MrpC (A), FruA (B), CsgA (C), and FrzCD (D). Each lane contains cell lysates generated from 4 × 107 cells. For panels A to C, the respective relative intensity measurements for each band from the supernatant (white diamonds) and pellet (black squares) are shown underneath each blot. Representative intensity plots are shown, but similar patterns were observed in at least two biological replicates. Solid lines, MrpC (panel A) and CsgA p25 (panel C); dashed lines, MrpC2 (panel A) and C-signal (p17) (panel C). The relative signal intensities of p25 and p17 were quantified from different exposure times. (D) The methylated forms (+CH3) of FrzCD are the more rapidly migrating bands.
We next examined the accumulation patterns of FruA, a transcriptional target of MrpC2 (60) which is necessary for induction of aggregation and sporulation. We observed that by 12 h of development, FruA began to accumulate in both cell populations, but between 18 and 30 h it dramatically increased in the supernatant fraction and then dramatically decreased between 30 and 48 h of development (Fig. 3B). In the sedimented fraction, FruA accumulated between 24 and 30 h of development and remained at high levels until 48 h of development. Thus, like MrpC and MrpC2, FruA accumulated first in the supernatant fraction and then simultaneously decreased in the supernatant and increased in the sedimented fractions.
FruA is proposed to induce aggregation and then sporulation in proportion to the degree of activation by C-signal, a 17-kDa proteolytic fragment of the CsgA protein (12, 24, 43, 46). We therefore examined the accumulation of C-signal (and its precursor, CsgA) in the supernatant and sedimented fractions. We observed that in the supernatant fractions, CsgA (p25) accumulated ∼2-fold between 12 and 24 h, followed by a rapid decrease between 24 and 48 h (Fig. 3C, upper panel and solid lines). In the sedimented fraction, CsgA exhibited slow and essentially linear accumulation between 12 and 48 h of development. Thus, the pattern of CsgA accumulation was similar to that of MrpC and FruA. In contrast, C-signal (p17) accumulated to essentially similar levels in the supernatant and sedimented fractions from 0 to 18 h (Fig. 3C, lower panel, dashed lines). After 18 h, C-signal decreased in the supernatant fraction, whereas accumulation continued in the pellet fraction through to 48 h. These data are consistent with the model in which C-signal is produced in response to cell-cell contact, which is predominant in the aggregating cells (15).
One of the proposed outputs of activated FruA is an increase in the methylation state of the FrzCD MCP which is thought to stimulate aggregation (53). Therefore, we next examined the methylation state of FrzCD, which can be recognized by more quickly migrating FrzCD species by anti-FrzCD immunoblotting (29). In both cell fractions, the total FrzCD levels decreased dramatically between the vegetative state (T = 0) and 12 h of development, remained relatively constant between 12 and 36, and then disappeared between 36 and 48 h (Fig. 3D). In all sedimented fractions, FrzCD was observed with a more pronounced methylation state, consistent with cells in close contact (30). After 30 h, concurrent with formation of visible aggregation centers (Fig. 1A), the most slowly migrating FrzCD band is absent from both fractions, and the intensity of the lower bands increases.
Together, the heterogeneous accumulation patterns of the major developmental regulators are consistent with the hypothesis that the sedimented cell fraction and the supernatant cell fraction represent at least two developmental subpopulations that are following different regulatory programs.
Developmental cell lysis is independent of MazF in wild-type M. xanthus.
It has been previously proposed that cell lysis is controlled by the MazF-MrpC toxin-antitoxin system (36). Based on our observation that MrpC displays heterogeneous accumulation patterns, we next set out to examine the accumulation pattern of MazF with the goal to determine whether we could identify when cells may segregate into the lysis fate. Since the role of MazF in mediating programmed cell lysis was investigated in an alternate M. xanthus strain, we first generated a deletion of the mazF gene in our wild-type DZ2 strain and assayed the developmental phenotype. Unexpectedly, when the total cell number was assayed during development, the ΔmazF mutant displayed a distinct decrease in cell number similar to the wild-type parent (Fig. 4A). Furthermore, the ΔmazF mutant displayed only a slight (4-h) delay in development and no significant reduction in sporulation efficiency compared to the wild type (Fig. 4B). As controls, we also assayed the total cell number of strains bearing a deletion in mrpC (ΔmrpC strain) and a disruption in csgA (csgA::Tn5-132 ΩLS205) which have been previously observed to fail to increase in cell number and decrease in cell number, respectively (14, 36). As expected in the ΔmrpC strain, the cell number did not significantly increase by 24 h of development, and the csgA strain cell number did not decrease after 24 h of development. Also consistently, neither the ΔmrpC nor the csgA strain was able to form fruiting bodies (Fig. 4B). Thus, only the deletion of mazF produced a phenotype inconsistent with previously published results.
Fig 4.
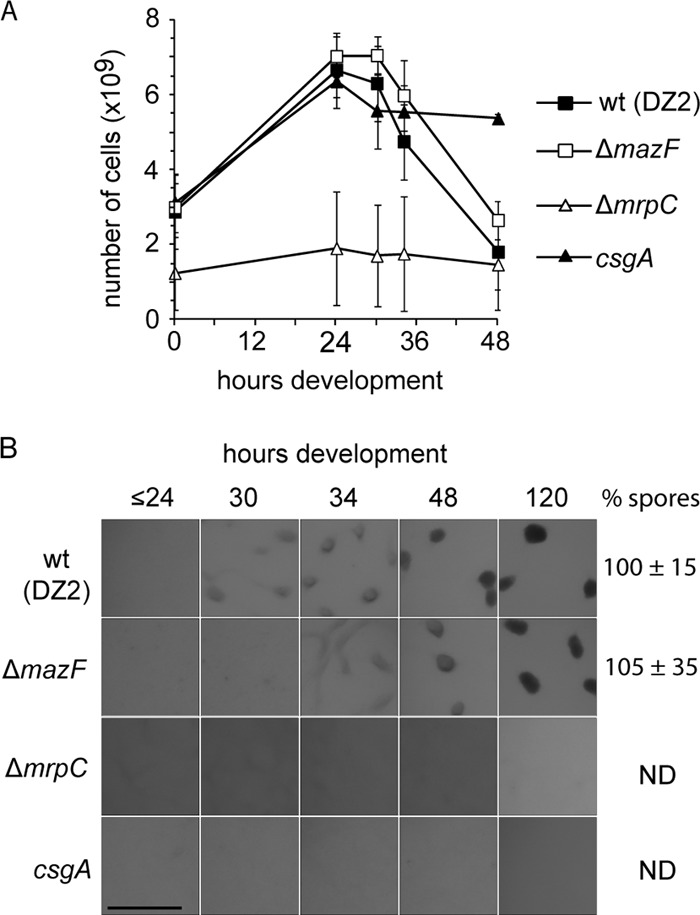
Deletion of mazF does not prevent developmental cell lysis in M. xanthus wild-type DZ2. (A) Developmental phenotype of the DZ2 wild-type (wt) and ΔmazF (PH1021), ΔmrpC (PH1025), csgA (PH1014) strains induced to develop under submerged culture for the indicated hours. The number of heat- and sonication-resistant spores was enumerated from cells harvested at 120 h of development and recorded as the percentage of wild-type spores (% spores). Numbers represent the average and associated standard deviation of three independent biological replicates. ND, not determined. Scale bar, 0.5 mm. (B) Enumeration of the total cell number during development. DZ2 wild-type, ΔmazF, ΔmrpC, and csgA cells from panel A were dispersed and enumerated at the indicated time points using an impedance cell counter. Data are the average and associated standard deviations from three independent biological experiments.
Previous studies indicating MazF played a major role in M. xanthus PCD were performed in a different genetic background (strain DZF1, also known as DK101 and FB [49, 63]), using a different mazF deletion construct, and development was assayed at high density on nutrient-limited agar plates rather than submerged culture (36). To determine if the difference in mazF phenotype was due to any of these factors, we additionally generated our mazF deletion in the DK101 as well as the commonly employed DK1622 M. xanthus backgrounds and assayed all strains under the developmental conditions described by Nariya and Inouye (36). Consistent with the previously described phenotype, the DK101 ΔmazF (PH1024) mutant exhibited no striking decrease in cell number during development, a delayed developmental phenotype, and a reduction in sporulation efficiency to 34% (Fig. 5A). Similar to what was observed under submerged culture development, the DZ2 ΔmazF mutant (PH1021) displayed a significant decrease in cell number during development, produced an essentially wild-type developmental phenotype, and had only a slight reduction in sporulation efficiency (Fig. 5B). Finally, the DK1622 ΔmazF mutant (PH1023) had a decrease in cell number indistinguishable from the DK1622 parent, no significant developmental defect, and only a slight reduction in sporulation efficiency (Fig. 5C). Under these developmental conditions, we did not observe an initial increase in cell number in either DK1622 or PH1023, likely because these cells develop more rapidly.
Fig 5.

MazF is not necessary for developmental cell lysis in wild-type M. xanthus strains DK1622 and DZ2. (A to C) Total cell enumeration (left panels) and developmental phenotype (right panels) of the DK101 wild-type and DK101 ΔmazF (PH1024) (A), DZ2 wild-type and DZ2 ΔmazF (PH1021) (B), and DK1622 wild-type and DK1622 ΔmazF (PH1023) (C) strains. Left panels show total cell numbers of the developmental time course represented in the right panels. Data are the average and associated standard deviation of two independent biological experiments of duplicate samples. Right panels show developmental phenotypes of the indicated strains induced to develop on nutrient-limited CF agar. Pictures were recorded at the indicated times of development. Scale bar, 0.5 mm. The number of heat- and sonication-resistant spores harvested at 120 h of development was recorded as a percentage of wild-type spores. Numbers represent the average and associated standard deviation from two independent biological replicates of duplicate samples.
Lower cell numbers at T = 0 were observed for DK101 and PH1024 (Fig. 5A) because cells in the DK101 background do not pellet well (see Discussion). We can exclude this lower cell density as the basis for the strong mazF phenotype in DK101 because the mazF deletion in either the DZ2 or DK1622 background does not display a strong phenotype when cells are plated at lower density (data not shown), and Nariya and Inouye observed a similar strong mazF phenotype when the cells were plated at higher cell densities (36). We concluded that the absence of the toxin MazF perturbs developmental cell lysis only in the mutant DK101 background, and MazF did not represent a marker for cell lysis in our strain. Consistently, preliminary immunoblot analysis with anti-MazF immunoserum did not detect accumulation patterns consistent with a marker for PCD (data not shown).
DISCUSSION
In this study, our main goal was to determine the timing of M. xanthus developmental cell fate segregation and to correlate this segregation to accumulation patterns of known developmental regulators. To identify the period during development in which different cell fates could be observed, we adapted a sedimentation assay originally employed by O'Conner and Zusman to separate peripheral rods from aggregated cells (41). However, our assay differed in certain points. First, we harvested cells developing under submerged culture for ease of harvesting developing cells and to reduce the variation in development which we sometimes observe on agar plates. Second, prior to the sedimentation step, the developing cell mat was harvested by repeatedly pipetting the entire 16 ml of solution with a 20-ml pipette. Thus, cells which were not tightly associated in aggregates, such as the cells which move in and out of aggregation centers (52), were in the supernatant. Third, we examined cells from vegetative and preaggregating developmental time points in addition to the later stages of development.
One surprising outcome of this study was the observation that ∼25% of the vegetative cells can be isolated in tightly associated groups even after the vigorous cell harvesting procedure. This population displayed distinct characteristics, including slightly increased EPS production (Fig. 2A), increased methylation of the chemosensory protein, FrzCD (Fig. 3D), and the exclusive production of the detected fragments of FibA, an extracellular metalloprotease (20) (Fig. 2B). We propose that this population be termed “cell clusters” to distinguish them from cells which are induced to aggregate into mounds during the developmental program (between 24 and 36 h). Although it has been recognized that cells in groups may have different molecular characteristics (2, 28, 51), most studies addressing the function of proteins involved in EPS production (e.g., Dif, FrzCD, or FibA) have been explored in isolated single-cell analyses or on the entire population of cells. Addressing these regulatory systems in cell groups is important for further understanding these regulatory systems.
Our analysis revealed that the onset of both aggregation and lysis is the time period between 24 and 30 h of development (Fig. 1B). It has been previously suggested that lysis occurs after the formation of aggregation centers and is specifically necessary for spore production (64). Under our conditions, lysis immediately precedes, or is concurrent with, induction of aggregation but may also continue within the aggregation centers. Aggregation is essentially complete by 36 h of development although a small proportion of cells may continue to join the aggregation centers after this time. The timing of production of peripheral rods is less clear but likely at, or prior to, approximately 36 h of development. We suggest that by 48 h the supernatant population consists primarily of peripheral rods because (i) the proportion of cells in the supernatant is maintained at ∼36% from at least 48 h (Fig. 1) to 120 h of development (data not shown) and (ii) most of the proteins which are necessary for aggregation (MrpC, FruA, C-signal, and FrzCD) are at very low levels in this population by 48 h of development. However, at these later time points, single-cell analyses are necessary to confirm whether cells continue to enter aggregates or whether peripheral rods transiently associate with aggregation centers.
We next set out to determine whether differences in accumulation patterns of developmental regulators could be correlated with segregation of cell fates. Cell lysates were prepared from cells isolated in the sedimented or supernatant subpopulations, and equal numbers of cells in each fraction at each time point were examined by immunoblotting against several developmental regulators. These analyses revealed significant previously unrecognized heterogeneity in the accumulation of many of the examined proteins. For instance, we observed for the first time that MrpC is likely turned over after induction of starvation because the average per cell level in both populations of vegetative cells was higher than the average per cell level at 12 h of development. This pattern was likely not recognized previously because the MrpC accumulation patterns were examined using lysates loaded to equal protein concentrations (13, 38), which are influenced both by decreases in total protein content after induction of starvation and by the changes in cell numbers during development.
By separating aggregating and nonaggregated cells, we observed here that accumulation of the known developmental regulators MrpC, FruA, CsgA p25, and C-signal (p17) is heterogeneous within the population. Between 12 and 24 h, prior to aggregation or lysis, most of these proteins accumulate more rapidly in the nonaggregated (supernatant) subpopulation than in the sedimented population, which may contain primarily cell clusters (Fig. 3A to C). After 24 h, MrpC, MrpC2, CsgA p25, and C-signal begin to decrease in the supernatant fraction, whereas FruA continues to increase until 30 h of development. All of these regulatory proteins continue to increase in the aggregated cell fraction after their respective decrease in the supernatant fraction. The simplest interpretation of this observation is that accumulation of these proteins in some cells in the supernatant fraction stimulates aggregation and therefore causes a transition of these cells into the sedimented population (Fig. 6). Once in the aggregation centers, these cells undergo increased C-signaling leading to increased production of at least FruA via a DevT-dependent positive-feedback loop (7). From our results we cannot ascertain whether the cell clusters remain as a distinct population or also accumulate the regulatory proteins as part of the aggregation centers. With respect to formation of peripheral rods, it is tempting to speculate that certain cells in the supernatant do not accumulate threshold levels of MrpC, FruA, and C-signal and thus remain in the supernatant as peripheral rods (Fig. 6). Consistently, these proteins are present at low levels in the supernatant fraction at least by the 36-h time point, and we propose this population consists primarily of peripheral rods. We are currently examining the single-cell accumulation of the known regulatory proteins to address this hypothesis.
Fig 6.
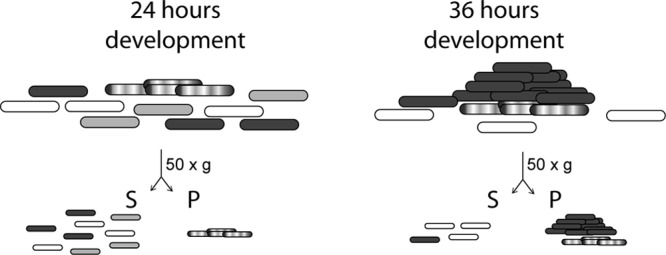
Regulation of developmental heterogeneity—a working model. Cells growing on surfaces in nutrient-rich medium or under starvation conditions can be can be segregated by centrifugation at 50 × g. The supernatant fraction (S) contains single or small groups of cells. The sedimented cell fraction (pellet, P) contains large cell groups. During early stages of development, prior to the formation of visible aggregation centers (e.g., 24 h of development), the sedimented fraction contains cell clusters (hatched cells) which accumulate no or low levels of the developmental regulators MrpC and FruA. The supernatant fraction may contain a mixed population of cells destined to follow different cell fates. Cells which accumulate at least MrpC and FruA (dark gray) are destined to aggregate into fruiting bodies, and cells which do not (white) are destined to become peripheral rods. The remaining cells (light gray) destined to lyse in a process that may involve MrpC but does not require MazF. Later during development (e.g., ≥36 h of development), aggregated cells (dark gray cell mounds) are in the sedimented (P) population. Cell clusters (hatched cells) may remain as a distinct population. Peripheral rods (white cells) remain in the supernatant, and lysed cells are absent.
Our initial plan was to address developmental cell lysis by examining the accumulation pattern of the toxin MazF, which was proposed to induce programmed cell death (PCD) via the unusual MazF toxin-MrpC antitoxin system (36). Importantly, however, we demonstrated here that deletion of mazF in either our wild-type DZ2 or the alternate DK1622 wild-type background did not significantly reduce cell lysis or produce a dramatic developmental phenotype. Since we could reproduce the strong mazF phenotype observed by Nariya and Inouye (36) in the DK101 (also known as DZF1) background, we suggest that MazF-dependent cell lysis appears to be an adaption of this particular strain background. DK101 contains a known sgl (pilQ1) mutation resulting in a partial social motility defect; it is additionally the progenitor of the DK1622 strain in which the pilQ1 mutation was repaired (63). DZ2 has an independent derivation (9). As was previously reported, in contrast to both DZ2 and DK1622, DK101 does not develop by submerged culture (data not shown), likely due to the pilQ1-associated loss of cohesiveness (63). In addition, unlike the wild-type strains, DK101 cells were difficult to pellet (Fig. 5). Thus, we suggest that the DZF1 (DK101)-specific MazF role in cell lysis may be an adaption to the pilQ1 mutation and that MazF is not solely responsible for developmentally induced programmed cell death. Perhaps in the DZ2 and DK1622 backgrounds, MazF's function is redundant; many other organisms contain multiple toxin-antitoxin systems (17, 26, 45) which show additive phenotypes when deleted (25). Alternatively, MazF's role may not normally be related to lysis but, rather, as has been proposed for a number of RNase-toxin modules in other organisms (25, 44, 58), to the promotion of growth arrest under stress conditions.
Although we have shown that MazF does not appear to be solely necessary for developmental cell lysis, our results are still consistent with developmental induction of lysis, rather than lysis as an artifact of handling fragile cells (42). First, we observe cell lysis at 24 to 30 h of development, prior to visible mound formation (32 to 36 h) and well before the transition into the fragile prespores (36 to 48 h). Second, we can observe that the total number of cells isolated after 5 days of undisturbed development is ∼20% of the initial population (unpublished observations). Third, consistent with previous results (14), we observed that a DZ2 csgA mutant did not decrease in cell number during development, suggesting that lysis is not simply a result of starvation and is related to induction of a specific signaling mechanism involving CsgA or C-signal.
Apart from the analysis of developmental regulators, we also analyzed the accumulation patterns of proteins involved in social motility and EPS production: PilA, PilC, and FibA. Consistent with our previous results (20), we observed that FibA accumulates specifically in the aggregated cell fraction. The late increase is consistent with the observation that the ∼31-kDa form of FibA, which represents primarily the two PPC repeats (10), was additionally isolated as the major spore coat protein C (20, 31). PilA accumulation is known to increase during development (65), but our analyses indicate that PilA increases specifically in the aggregated cell fraction and remains relatively constant in the supernatant cell fraction. In contrast, PilC, the inner membrane component of the T4P machinery, decreases in both cell populations but considerably more rapidly in the aggregated cell fraction. Thus, the relative ratio of PilA to PilC observed in vegetative cells is strongly increased in the aggregated cell fraction. Since biogenesis of the pilus requires PilC (65, 66), it is tempting to speculate that the concomitant accumulation of PilA and reduction of PilC could trap PilA either in the inner membrane pool or as pili at the cell surface. PilA trapped in the inner membrane reduces EPS production and prevents fruiting body formation (67). Since the sedimented cell fraction displays significantly more EPS (Fig. 2A), we speculate that PilA is instead trapped as cell surface type IV pili. An attractive model arises in which extruded T4P attach to the EPS of neighboring cells (23) and cement the aggregated cells together, providing the basis for compact fruiting body formation. The observation that EPS is upregulated specifically in the sedimented cell fraction after formation of visible aggregation centers (Fig. 2A and 1A) is consistent with nonretractable T4P stimulating EPS production via activation of the Dif chemosensory system (4). It should be noted, however, that it is not known whether M. xanthus PilC is necessary for pilus retraction in addition to extension.
In summary, this study provides the framework for characterization of M. xanthus population heterogeneity and developmental cell fate segregation. Our data raise the testable hypothesis that differences in the accumulation of key developmental regulators may lead to segregation of cells induced to aggregate or remain as peripheral rods. We are currently testing this hypothesis by single-cell analyses of developmental regulator accumulation. Importantly, since we demonstrated that MazF does not exclusively mediate PCD in wild-type M. xanthus strains, it is clear that the mechanism mediating developmental PCD must be reinvestigated. The identification of the time period during which PCD can be first observed will help pinpoint the factors involved. Finally, this study demonstrates previously unrecognized population heterogeneity in vegetative and early developing cells and in the accumulation patterns of proteins involved in type IV pilus function.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by the Max Planck Society (P.I.H., B.L., and C.H.), a fellowship from the International Max Planck Research School for Environmental, Cellular and Molecular Microbiology (C.H.), and the German Research Federation (DFG Tr429/4-2) (A.T.-L.).
We gratefully acknowledge the labs of Lotte Søgaard-Andersen, David Zusman, and Dale Kaiser for sharing antibodies. We additionally thank past and present members of the Higgs lab for helpful discussions and Stuart Huntley for critical reading of the manuscript.
Footnotes
Published ahead of print 6 April 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Berleman JE, Chumley T, Cheung P, Kirby JR. 2006. Rippling is a predatory behavior in Myxococcus xanthus. J. Bacteriol. 188:5888–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berleman JE, et al. 2011. FrzS regulates social motility in Myxococcus xanthus by controlling exopolysaccharide production. PLoS One 6:e23920 doi:10.1371/journal.pone.0023920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhandari P, Gowrishankar J. 1997. An Escherichia coli host strain useful for efficient overproduction of cloned gene products with NaCl as the inducer. J. Bacteriol. 179:4403–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Black WP, Xu Q, Yang Z. 2006. Type IV pili function upstream of the Dif chemotaxis pathway in Myxococcus xanthus EPS regulation. Mol. Microbiol. 61:447–456 [DOI] [PubMed] [Google Scholar]
- 5. Black WP, Yang Z. 2004. Myxococcus xanthus chemotaxis homologs DifD and DifG negatively regulate fibril polysaccharide production. J. Bacteriol. 186:1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blackhart BD, Zusman DR. 1985. “Frizzy” genes of Myxococcus xanthus are involved in control of frequency of reversal of gliding motility. Proc. Natl. Acad. Sci. U. S. A. 82:8767–8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boysen A, Ellehauge E, Julien B, Sogaard-Andersen L. 2002. The DevT protein stimulates synthesis of FruA, a signal transduction protein required for fruiting body morphogenesis in Myxococcus xanthus. J. Bacteriol. 184:1540–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bulyha I, et al. 2009. Regulation of the type IV pili molecular machine by dynamic localization of two motor proteins. Mol. Microbiol. 74:691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campos JM, Zusman DR. 1975. Regulation of development in Myxococcus xanthus: effect of 3′:5′-cyclic AMP, ADP, and nutrition. Proc. Natl. Acad. Sci. U. S. A. 72:518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Curtis PD, Atwood J, 3rd, Orlando R, Shimkets LJ. 2007. Proteins associated with the Myxococcus xanthus extracellular matrix. J. Bacteriol. 189:7634–7642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dworkin M. 1962. Nutritional requirements for vegetative growth of Myxococcus xanthus. J. Bacteriol. 84:250–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ellehauge E, Norregaard-Madsen M, Sogaard-Andersen L. 1998. The FruA signal transduction protein provides a checkpoint for the temporal co-ordination of intercellular signals in Myxococcus xanthus development. Mol. Microbiol. 30:807–817 [DOI] [PubMed] [Google Scholar]
- 13. Higgs PI, Jagadeesan S, Mann P, Zusman DR. 2008. EspA, an orphan hybrid histidine protein kinase, regulates the timing of expression of key developmental proteins of Myxococcus xanthus. J. Bacteriol. 190:4416–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janssen GR, Dworkin M. 1985. Cell-cell interactions in developmental lysis of Myxococcus xanthus. Dev. Biol. 112:194–202 [DOI] [PubMed] [Google Scholar]
- 15. Jelsbak L, Sogaard-Andersen L. 2000. Pattern formation: fruiting body morphogenesis in Myxococcus xanthus. Curr. Opin. Microbiol. 3:637–642 [DOI] [PubMed] [Google Scholar]
- 16. Julien B, Kaiser AD, Garza A. 2000. Spatial control of cell differentiation in Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 97:9098–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim Y, et al. 2010. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 12:1105–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kroos L. 2007. The Bacillus and Myxococcus developmental networks and their transcriptional regulators. Annu. Rev. Genet. 41:13–39 [DOI] [PubMed] [Google Scholar]
- 19. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 20. Lee B, et al. 2011. The Myxococcus xanthus spore cuticula protein C is a fragment of FibA, an extracellular metalloprotease produced exclusively in aggregated cells. PLoS One 6:e28968 doi:10.1371/journal.pone.0028968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee B, Schramm A, Jagadeesan S, Higgs PI. 2010. Two-component systems and regulation of developmental progression in Myxococcus xanthus. Methods Enzymol. 471:253–278 [DOI] [PubMed] [Google Scholar]
- 22. Lee JS, Son B, Viswanathan P, Luethy PM, Kroos L. 2011. Combinatorial regulation of fmgD by MrpC2 and FruA during Myxococcus xanthus development. J. Bacteriol. 193:1681–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, et al. 2003. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 100:5443–5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lobedanz S, Sogaard-Andersen L. 2003. Identification of the C-signal, a contact-dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17:2151–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U. S. A. 108:13206–13211 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 4:19 doi:10.1186/1745-6150-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maniatis T, Fritsch EF, Sambrook J. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Mauriello EM, Astling DP, Sliusarenko O, Zusman DR. 2009. Localization of a bacterial cytoplasmic receptor is dynamic and changes with cell-cell contacts. Proc. Natl. Acad. Sci. U. S. A. 106:4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McBride MJ, Kohler T, Zusman DR. 1992. Methylation of FrzCD, a methyl-accepting taxis protein of Myxococcus xanthus, is correlated with factors affecting cell behavior. J. Bacteriol. 174:4246–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McBride MJ, Zusman DR. 1993. FrzCD, a methyl-accepting taxis protein from Myxococcus xanthus, shows modulated methylation during fruiting body formation. J. Bacteriol. 175:4936–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCleary WR, Esmon B, Zusman DR. 1991. Myxococcus xanthus protein C is a major spore surface protein. J. Bacteriol. 173:2141–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCleary WR, McBride MJ, Zusman DR. 1990. Developmental sensory transduction in Myxococcus xanthus involves methylation and demethylation of FrzCD. J. Bacteriol. 172:4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mittal S, Kroos L. 2009. A combination of unusual transcription factors binds cooperatively to control Myxococcus xanthus developmental gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:1965–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mittal S, Kroos L. 2009. Combinatorial regulation by a novel arrangement of FruA and MrpC2 transcription factors during Myxococcus xanthus development. J. Bacteriol. 191:2753–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Müller FD, Treuner-Lange A, Heider J, Huntley SM, Higgs PI. 2010. Global transcriptome analysis of spore formation in Myxococcus xanthus reveals a locus necessary for cell differentiation. BMC Genomics 11:264 doi:10.1186/1471-2164-11-264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nariya H, Inouye M. 2008. MazF, an mRNA interferase, mediates programmed cell death during multicellular Myxococcus development. Cell 132:55–66 [DOI] [PubMed] [Google Scholar]
- 37. Nariya H, Inouye S. 2005. Identification of a protein Ser/Thr kinase cascade that regulates essential transcriptional activators in Myxococcus xanthus development. Mol. Microbiol. 58:367–379 [DOI] [PubMed] [Google Scholar]
- 38. Nariya H, Inouye S. 2006. A protein Ser/Thr kinase cascade negatively regulates the DNA-binding activity of MrpC, a smaller form of which may be necessary for the Myxococcus xanthus development. Mol. Microbiol. 60:1205–1217 [DOI] [PubMed] [Google Scholar]
- 39. O'Connor KA, Zusman DR. 1991. Analysis of Myxococcus xanthus cell types by two-dimensional polyacrylamide gel electrophoresis. J. Bacteriol. 173:3334–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. O'Connor KA, Zusman DR. 1991. Behavior of peripheral rods and their role in the life cycle of Myxococcus xanthus. J. Bacteriol. 173:3342–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Connor KA, Zusman DR. 1991. Development in Myxococcus xanthus involves differentiation into two cell types, peripheral rods and spores. J. Bacteriol. 173:3318–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Connor KA, Zusman DR. 1988. Reexamination of the role of autolysis in the development of Myxococcus xanthus. J. Bacteriol. 170:4103–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ogawa M, Fujitani S, Mao X, Inouye S, Komano T. 1996. FruA, a putative transcription factor essential for the development of Myxococcus xanthus. Mol. Microbiol. 22:757–767 [DOI] [PubMed] [Google Scholar]
- 44. Pedersen K, Christensen SK, Gerdes K. 2002. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45:501–510 [DOI] [PubMed] [Google Scholar]
- 45. Ramage HR, Connolly LE, Cox JS. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767 doi:10.1371/journal.pgen.1000767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rolbetzki A, Ammon M, Jakovljevic V, Konovalova A, Sogaard-Andersen L. 2008. Regulated secretion of a protease activates intercellular signaling during fruiting body formation in M. xanthus. Dev. Cell 15:627–634 [DOI] [PubMed] [Google Scholar]
- 47. Rosenberg E, Keller KH, Dworkin M. 1977. Cell density-dependent growth of Myxococcus xanthus on casein. J. Bacteriol. 129:770–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosenbluh A, Nir R, Sahar E, Rosenberg E. 1989. Cell-density-dependent lysis and sporulation of Myxococcus xanthus in agarose microbeads. J. Bacteriol. 171:4923–4929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rudd K, Zusman DR. 1979. Rifampin-resistant mutants of Myxococcus xanthus defective in development. J. Bacteriol. 137:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schagger H, von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379 [DOI] [PubMed] [Google Scholar]
- 51. Shi W, Ngok FK, Zusman DR. 1996. Cell density regulates cellular reversal frequency in Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 93:4142–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sliusarenko O, Zusman DR, Oster G. 2007. Aggregation during fruiting body formation in Myxococcus xanthus is driven by reducing cell movement. J. Bacteriol. 189:611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sogaard-Andersen L. 2004. Cell polarity, intercellular signalling and morphogenetic cell movements in Myxococcus xanthus. Curr. Opin. Microbiol. 7:587–593 [DOI] [PubMed] [Google Scholar]
- 54. Sogaard-Andersen L, Kaiser D. 1996. C factor, a cell-surface-associated intercellular signaling protein, stimulates the cytoplasmic Frz signal transduction system in Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 93:2675–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Son B, Liu Y, Kroos L. 2011. Combinatorial regulation by MrpC2 and FruA involves three sites in the fmgE promoter region during Myxococcus xanthus development. J. Bacteriol. 193:2756–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun H, Shi W. 2001. Analyses of mrp genes during Myxococcus xanthus development. J. Bacteriol. 183:6733–6739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun H, Shi W. 2001. Genetic studies of mrp, a locus essential for cellular aggregation and sporulation of Myxococcus xanthus. J. Bacteriol. 183:4786–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Syed MA, et al. 2011. The chromosomal mazEF locus of Streptococcus mutans encodes a functional type II toxin-antitoxin addiction system. J. Bacteriol. 193:1122–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thony-Meyer L, Kaiser D. 1993. devRS, an autoregulated and essential genetic locus for fruiting body development in Myxococcus xanthus. J. Bacteriol. 175:7450–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ueki T, Inouye S. 2003. Identification of an activator protein required for the induction of fruA, a gene essential for fruiting body development in Myxococcus xanthus. Proc. Natl. Acad. Sci. U. S. A. 100:8782–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ueki T, Inouye S, Inouye M. 1996. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene 183:153–157 [DOI] [PubMed] [Google Scholar]
- 62. Viswanathan P, Ueki T, Inouye S, Kroos L. 2007. Combinatorial regulation of genes essential for Myxococcus xanthus development involves a response regulator and a LysR-type regulator. Proc. Natl. Acad. Sci. U. S. A. 104:7969–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wall D, Kolenbrander PE, Kaiser D. 1999. The Myxococcus xanthus pilQ (sglA) gene encodes a secretin homolog required for type IV pilus biogenesis, social motility, and development. J. Bacteriol. 181:24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wireman JW, Dworkin M. 1977. Developmentally induced autolysis during fruiting body formation by Myxococcus xanthus. J. Bacteriol. 129:798–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu SS, Kaiser D. 1997. Regulation of expression of the pilA gene in Myxococcus xanthus. J. Bacteriol. 179:7748–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu SS, Wu J, Kaiser D. 1997. The Myxococcus xanthus pilT locus is required for social gliding motility although pili are still produced. Mol. Microbiol. 23:109–121 [DOI] [PubMed] [Google Scholar]
- 67. Yang Z, Lux R, Hu W, Hu C, Shi W. 2010. PilA localization affects extracellular polysaccharide production and fruiting body formation in Myxococcus xanthus. Mol. Microbiol. 76:1500–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zusman DR, Scott AE, Yang Z, Kirby JR. 2007. Chemosensory pathways, motility and development in Myxococcus xanthus. Nat. Rev. Microbiol. 5:862–872 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.