

Abstract
Walker-Warburg syndrome (WWS) is clinically defined as congenital muscular dystrophy accompanied by a variety of brain and eye malformations. It represents the most severe clinical phenotype in a spectrum of alpha-dystroglycan posttranslational processing abnormalities, which share a defect in laminin binding glycan synthesis1. Although six WWS causing genes have been described, only half of all patients can currently be diagnosed genetically2. A cell fusion complementation assay using fibroblasts from undiagnosed WWS individuals identified five novel complementation groups. Further evaluation of one group by linkage analysis and targeted sequencing identified recessive mutations in the isoprenoid synthase domain containing (ISPD) gene. Confirmation of the pathogenicity of the identified ISPD mutations was demonstrated by complementation of fibroblasts with wild-type ISPD. Finally, we show that recessive mutations in ISPD abolish the initial step in laminin binding glycan synthesis by disrupting dystroglycan O-mannosylation. This establishes a novel mechanism for WWS pathophysiology.
The hallmark of dystroglycanopathies - and the common pathogenic denominator in all WWS (MIM# 236670) patients - is loss of functional glycosylation of α-dystroglycan (α-DG)3. Lack of proper α-DG glycosylation reduces binding to extracellular matrix proteins since the ligand binding is mediated through the α-DG sugar moiety3,4. All six of the causative genes previously identified in WWS patients demonstrate autosomal recessive inheritance and code for known or putative glycosyltransferases; mutations therein result in abnormal α-DG glycosylation5. Nevertheless, approximately half of the WWS patient population has no mutations in these known genes2, emphasizing the need for WWS gene discovery.
We established a complementation assay to enable us to identify additional genes that contribute to WWS pathology. This assay was developed based on a panel of fibroblasts from six genetically defined, but heterogeneous dystroglycanopathy cases (Supplementary Table 1), for which western blotting revealed a lack of functional glycosylation (defined as immunoreactivity to IIH6, a monoclonal antibody specific for requisite sugar moiety) and an inability to bind laminin (Fig. 1a). The results are consistent with previously published data obtained from various dystroglycanopathy patient tissues3,6 and cells7–9. Notably, the degrees of α-DG hypoglycosylation varied by the gene mutated, and the molecular weight of α-DG produced in each case is hypothesized to reflect an abnormality at different steps in α-DG glycan biosynthesis (Fig. 1a). For each patient population the α-DG glycosylation defect was rescued by transferring a wild-type copy of the mutant gene. For example, in WWS fibroblasts with known POMT1 mutations, α-DG functional glycosylation was restored by adenovirus-mediated gene transfer of POMT1, but not by expression of the other known WWS genes (Fig. 1b). This complementation assay was adapted to On-Cell western blotting, and rescue of α-DG functional glycosylation was demonstrated for all known WWS genes (Fig. 1c). Previously, it was shown that forced overexpression of the putative glycosyltransferase LARGE can induce α-DG hyperglycosylation in control cells and bypass the α-DG glycosylation defect in dystroglycanopathy patient cells8,10. However, we now demonstrate that the ability of LARGE to hyperglycosylate α-DG is dependent on the availability of O-mannosyl phosphate acceptor sites and correlates with the severity of the clinical phenotype (Fig. 1d).
Fig. 1. α-DG glycosylation status in dystroglycanopathy patient dermal fibroblasts.
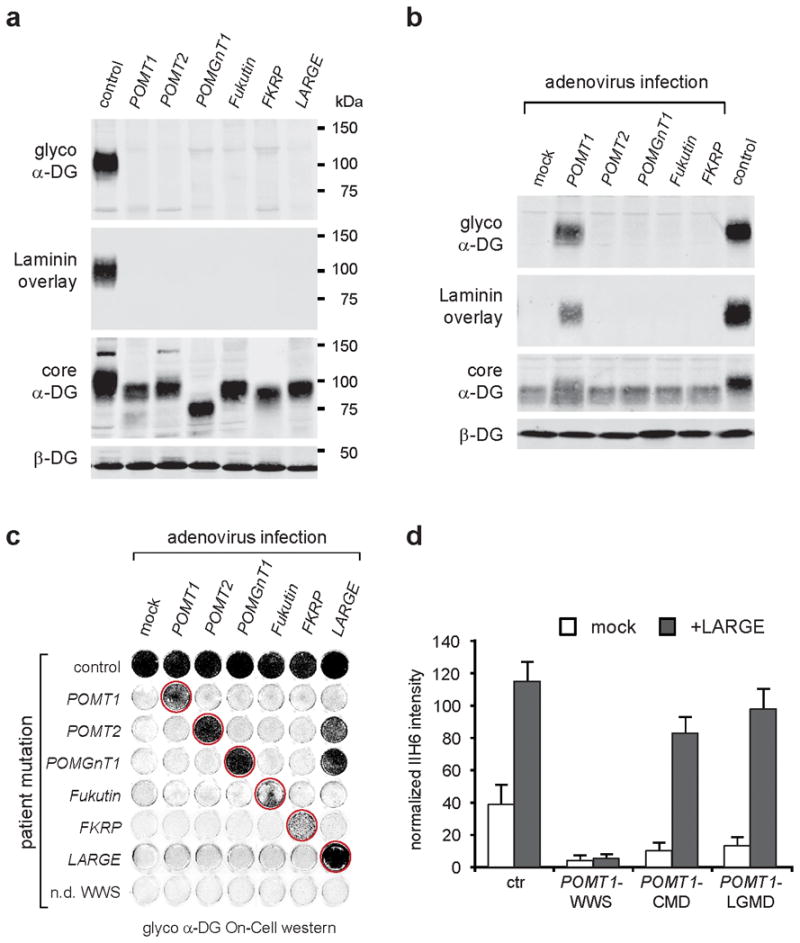
WGA-enriched cell lysates from control and patient fibroblasts (samples with defects in six different genes, Supplementary Table 1) were subjected to biochemical analysis. (a) α-DG glycosylation status as assessed by western blotting using antibodies against the glycosylated form of α-DG (IIH6), core α-DG (G6317), and by laminin overlay assay. An antibody against β-DG (AP83) was used to assess loading. Apparent molecular masses are indicated. (b) Complementation assay. Immunoblot of POMT1 deficient -WWS cells infected with a panel of adenoviruses expressing WWS candidate genes; only adenovirus mediated gene transfer with a POMT1wild-type copy rescues the defect. (c) On-Cell western analysis of fibroblasts from control and genetically heterogeneous WWS patients demonstrates that the complementation approach, using adenovirus mediated gene transfer, can successfully be applied to all known WWS genes. Restoration of the glycosylation defect is indicated with a red circle. Bottom: sample from a non-diagnosed WWS patient; lack of rescue suggests a novel genetic defect. Note, LARGE overexpression rescues and bypasses the α-DG hypoglycosylation in less severe WWS-MEB cells with hypothesized residual activity (POMT2 and POMGnT1), but not in the most severe loss-of-function WWS patient cells. The On-Cell western blot was probed with antibodies against the glycosylated form of α-DG (IIH6) and for signal enhancement the cells were co-infected with dystroglycan expressing adenovirus (Ad5CMV-DAG1). (d) Quantitative On-Cell western blot analysis of LARGE-induced α-DG hyperglycosylation in control and POMT1 deficient cells from three patients with different clinical severity. The ability of LARGE to increase the affinity of the cell surface for the IIH6 antibody and bypass the glycosylation defect in POMT1 deficient patient cells correlates with the residual activity of the mutant gene product and the severity of the clinical manifestation. IIH6 On-Cell quantitative data were normalized with DRAQ5 cell DNA dye (n=3). Error bars represent s.d.
We next applied the On-Cell complementation assay to fibroblasts from a cohort of 63 dystroglycanopathy patients (Supplementary Fig. 1), identifying 11 WWS individuals from 10 unrelated families, who we postulated to have mutations in novel genes. Our first step toward defining the genetic basis for WWS in these patients was to establish complementation groups; to this end we adapted a cell fusion approach that is commonly used in yeast research11 and has proven successful in mammalian cells12. Whereas fusion is achieved by mating in the case of yeast cells, polyethylene glycol (PEG)13 treatment is used for mammalian cells. We hypothesized that fusion between co-cultured cells from patients harboring recessive mutations in the same gene would not rescue the α-DG glycosylation defect, whereas fusion between cells from patients with independent genetic defects would rescue successfully. Immunofluorescence analysis revealed IIH6-positive fused cells (as indicated by the presence of multiple nuclei) only when two cell lines from genetically different patients were co-cultured; complementation between POMT1 and FKTN fibroblasts is shown in Fig. 2a. Two-way fusions of WWS cells with mutations in each of the known genes rescued α-DG glycosylation (data not shown). Application of the PEG fusion approach to all 11 genetically unidentified WWS patients led to the identification of five separate complementation groups (Fig. 2b,c), suggesting that five novel WWS genes are represented in this small cohort of patients. While four complementation groups were represented by a single WWS patient, one group consisted of seven WWS individuals. If mutations in a single novel gene are responsible for disease in all seven patients, this complementation group should represent a relative common cause of WWS.
Fig. 2. Cell fusion experiments reveal novel genetic complementation groups.
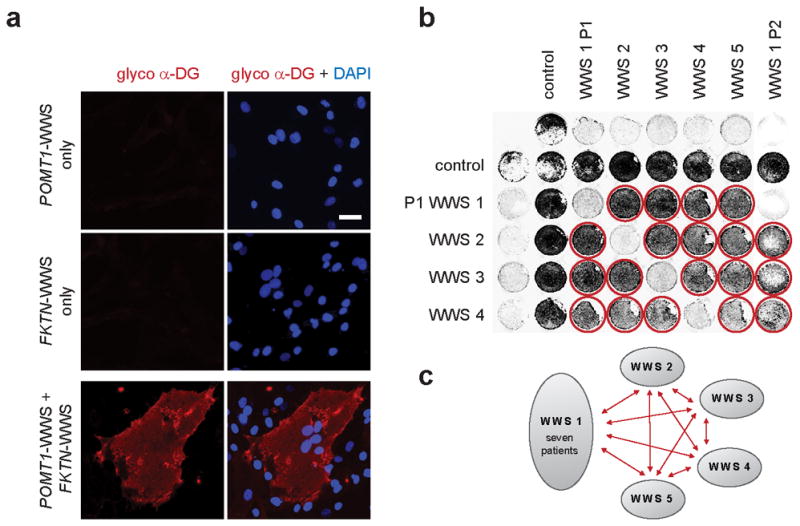
Cell fusion among co-cultured dermal fibroblasts induced with polyethylene glycol (PEG). (a) Immunofluorescence-based detection of restored α-DG functional glycosylation with glyco α-DG antibodies (IIH6) (scale bar 50μm). In contrast to cultures containing POMT1-WWS or FKTN-WWS patient cells only, co-cultures of these cells exhibited restored functional α-DG glycosylation in multinucleated cell fusions. Nuclei are stained with DAPI. (b) Fibroblasts from control and different WWS patients (five) with unknown genetic defects were co-cultured and subjected to cell-fusion complementation. Complementation was assessed as rescue of functional α-DG glycosylation, using glycosylation-specific α-DG antibodies (IIH6) by On-Cell western blotting. Rescue of the glycosylation defect is indicated by red circles. For signal enhancement the cells were co-infected with dystroglycan expressing adenovirus (Ad5CMV-DAG1). (c) Schematic representation summarizing our identification of five new complementation groups. Red arrows indicate successful fusion complementation. One group contains seven patients, whereas the remaining four groups currently only consist of a single patient.
All seven patients within this complementation group met the classic diagnostic criteria for WWS (Supplementary Table 2). Two of the cases, P56,14 and P615, were previously published and described as WWS. In the case of P1, brain MRI examinations performed at 3 days and 5 months of age, showed hydrocephalus, cobblestone lissencephaly of the cerebral cortex, severe brainstem hypoplasia with a kink at the isthmus, and severe hypoplasia of the cerebellum (Fig. 3a). This patient also displayed evidence of severe muscular dystrophy (Fig. 3b), bilateral microophthalmia with cataracts and arrested retinal development. Immunofluorescence and western blot analysis of a skeletal muscle biopsy from this patient showed that this new complementation group of WWS patients manifests the typical α-DG glycosylation defect in skeletal muscle with loss of both functional glycosylation and receptor function (Fig. 3b,c). Comparative analysis of the α-DG glycosylation status in fibroblasts from five different WWS cases in the same complementation group confirmed that all samples share a defect in α-DG processing, with complete loss of functional glycosylation and laminin binding (Supplementary Fig. 2). Moreover, the loss of post-translational modification and a subsequent shift to lower molecular weight was comparable in all samples, consistent with the hypothesis that they share a common genetic defect.
Fig. 3. Clinical presentation and α-DG glycosylation defect in ISPD-WWS patient P1.
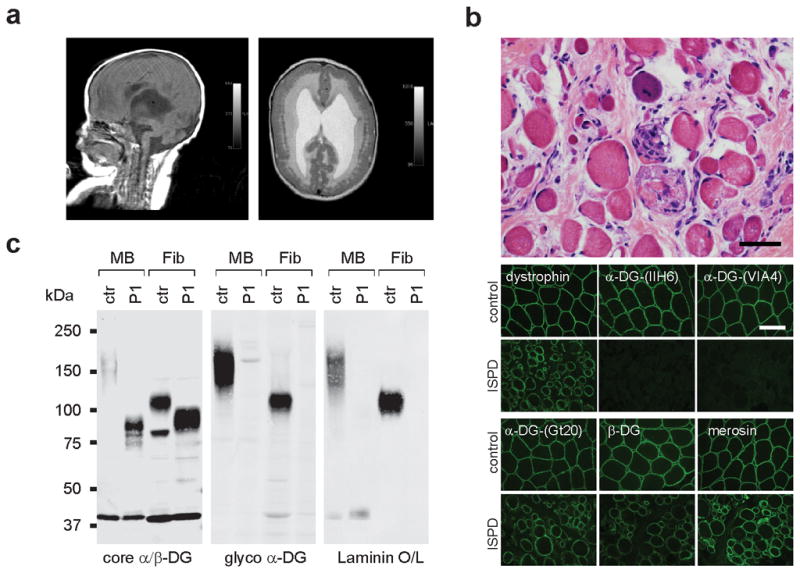
(a) Sagittal and axial T1 MRI brain images at 5 months of age showed severe ventriculomegaly, agyria and a significantly malformed (Z-shaped) hypoplastic brainstem, as well as severely hypoplastic cerebellar vermis. In addition, the axial image reveals subcortical heterotopia. (b) Histologic staining of frozen cross-sections from a skeletal muscle biopsy, showing severe dystrophic histopathology with muscle fiber necrosis and regeneration, as well as endomysial fibrosis (top panel: H&E stain, scale bar 50 μm). The immunofluorescence with two antibodies against glycosylated α-DG reveals a complete loss of functional α-DG glycosylation. Antibodies against dystrophin, α-DG core protein, β-DG and laminin α2 show mildly reduced to normal staining (scale bar 100 μm). (c) Western blot of skeletal muscle biopsy (MB) and skin fibroblasts (Fib) of control and ISPD-WWS P1. Both patient samples reveal core α-DG hypoglycosylation and loss of α-DG functional glycosylation. Glycoproteins were WGA-enriched from muscle or cell lysates (MB: 250μg protein/lane, Fib: 1000μg/lane). The immunoblot was probed with antibodies against the glycosylated form of α-DG (IIH6) and core α-DG (G6317), as well as withβ-DG (AP83) as a loading control.
Our first step toward identifying the underlying genetic defect in the large complementation cohort, was to perform linkage analysis to identify candidate regions. As reliable family history regarding consanguinity was not available for all cases, regions of homozygosity-by-descent were identified using high-resolution SNP arrays. Besides the sibling pair P2 and P3, four of the five unrelated patients showed multiple long (>10cM) stretches of homozygosity, suggesting some degree of consanguinity (Supplementary Fig. 3). We searched for regions where P2 and P3 are identical on both alleles and all or a subset of the four suspected consanguineous samples are homozygous. All 7,113 coding exons across 14 overlapping intervals were subjected to targeted sequencing (Supplementary Table 3). All seven samples were bar-coded, pooled, captured by one custom-designed capture array and sequenced on a lane of an Illumina HiSeq2000 flowcell as a 50bp paired-end run. The sequence data were processed through a custom-built analysis pipeline, and after variant filter strategies were applied, six genes were identified for which at least two independent protein-damaging variants passed the hard-filtration (Supplementary Table 4). Based on genetic evidence, ISPD was the most likely candidate gene. After manually examining the variants that did not meet filter criteria, as well as augmentation of the dataset with Sanger sequencing of P5 which was poorly covered, a total of four heterozygous variants and four homozygous variants were found in ISPD which identified multiple rare variants in all six independent cases (Table 1, Supplementary Fig. 4). All mutations were predicted to damage or abolish protein function, as expected in individuals with a severe form of dystroglycanopathy such as WWS7. In addition, ISPD was localized to chromosome 7p21.2, a region in which three of the four suspected consanguineous patients had intervals of homozygosity longer than 10cM and P2 and P3 shared both parental alleles (Fig. 4a,b). A schematic representation of all ISPD mutations identified in our patient cohort is shown in Fig. 4c.
Table 1.
A summary of pathogenic ISPD mutations detected in this study
| Patients | Zygosity | Chr | Genomic position (b37) | Nucleotide variant | Amino acid | Comment |
|---|---|---|---|---|---|---|
| P1 | heterozygous | 7 | 16,415,758 | c.643C>T | p.Gln215* | nonsense mutation |
| heterozygous | 7 | g.(16,107,358–16,115,680)_(16,289,931–16,297,326)del | exon 9–10 deletion | |||
|
| ||||||
| P2 and P3 (siblings) | heterozygous | 7 | 16,348,146 | c.789+2T>G | IVS4+2T>G | ivs4 splice-site mutation, ΔExon4 |
| heterozygous | 7 | 16,445,940 | c.277–279del ATT | p.Ile93del | single amino-acid (aa) deletion | |
|
| ||||||
| P4 | homozygous | 7 | 16,131,322 | c.1354T>A | p.*452Arg | mutation of original stop-codon, next stop codon 27 aa downstream |
|
| ||||||
| P5 | homozygous | 7 | 16,255,823 | c.1120-1G>T | IVS8-1G>T | ivs8 splice site mutation, ΔExon9 |
|
| ||||||
| P6 | homozygous | 7 | g.(16,401,191–16,406,273)_(16,409,318–16,431,594)del | in frame deletion of Exon 3 | ||
|
| ||||||
| P7 | homozygous | 7 | 16,415,851 | c.550C>T | p.Arg184* | nonsense mutation |
Fig. 4. Identification and validation of ISPD as disease gene in WWS patients.

(a) Alignment of identical-by-descent (IBD) and homozygosity-by-descent (HBD) intervals among ISPD patients on chromosome 7 is shown with the genomic position in hg19 coordinates on the top and chromosome bands at the bottom. The minimal region of overlap between the three out of four suspected consanguineous samples were homozygous where P2 and P3 share both parental alleles is highlighted by a red box and (b) zoomed in to show the genes within the region. (c) Schematic representation (not drawn to scale) of the ISPD exon-intron gene structure. Human ISPD cDNA (NM_001101426, 5,524 bp) contains 10 coding exons spread across 333,796 bp genomic DNA. All identified pathogenic ISPD protein changes are indicated, as are regions with gene deletions and splice-site mutations. Coding exons are indicated by black boxes, and untranslated regions (UTR) by open boxes. (d) On-Cell western-based complementation assay of control and ISPD-WWS patient fibroblasts after nucleofection with a wild-type or mutant ISPD expression construct. Rescue of α-DG functional glycosylation was detected with α-DG glyco (IIH6) antibodies. (e) Adenovirus-mediated ISPD gene transfer rescues α-DG glycosylation defect in ISPD-WWS P2 patient cells. WGA-enriched cell lysates from fibroblasts were subjected to immunoblotting with α-DG glyco (IIH6), α-DG core (G6317), anti-myc (4A6), and β-DG (AP83) and by laminin overlay. Infection with ISPD-myc expressing adenovirus restored functional glycosylation in ISPD-WWS P2 patient cells, but did not significantly alter α-DG functional glycosylation in control cells.
To confirm the pathogenicity of the identified ISPD mutations, we conducted complementation assays on fibroblasts derived from the ISPD-WWS patients. In the patient cells, expression of wild-type ISPD, but not that of a mutant isoform (P6, ISPD-ΔE3) restored functional glycosylation (Fig. 4d); in control cells, such overexpression did not significantly alter functional α-DG glycosylation (Fig. 4e). Functional rescue of patient cells confirmed that the identified ISPD mutations have pathogenic relevance, and indicated that severe mutations in ISPD can cause WWS.
Notably, ISPD has not been characterized in mammals. Quantitative reverse transcriptase PCR (qRT-PCR) revealed that ISPD is ubiquitously expressed in all tissues analyzed, with expression highest in brain (Supplementary Fig. 5). ISPD belongs to the family of 4-diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) synthases (also known as 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferases), that are conserved from bacteria to mammals (Supplementary Fig. 6). In E.coli, IspD activity contributes to the methylerythritol pathway (MEP) in its synthesis of isoprenoid precursors16,17, which give rise to the polyisoprenoid alcohols (e.g., dolichols and polyprenols) found in all living organisms18 (Supplementary Fig. 7). However, the MEP pathway is used only by eubacteria, green algae and the chloroplasts of higher plants19. Eukaryotes, archaebacteria, and the cytosol of higher plants are thought to use an alternative, ISPD-independent mevalonate (MVA) pathway for isoprenoid synthesis20. As the MEP pathway involving ISPD is postulated to be absent from animals, the specific role of ISPD in humans, especially in regards to α-DG glycosylation, is unclear.
To investigate the role of ISPD in α-DG glycosylation, we tested ISPD-WWS patients for changes in any known steps in laminin binding glycan synthesis. Notably, protein O-mannosylation, the initiating step, was markedly reduced in fibroblasts lacking functional ISPD, as were downstream events like O-mannosyl phosphorylation and LARGE-induced hyperglycosylation (Fig. 5a–c). These findings suggest that ISPD function is crucial for efficient POMT-dependent O-mannosylation and subsequent glycosylation of α-DG (Supplementary Fig. 8).
Fig. 5. ISPD loss of function causes α-DG O-mannosylation defect.

(a) POMT activity in control cells and patient-derived dermal fibroblasts, as assayed by the rate of radioactive [3H]-mannose transfer from Dol-P-[3H]-Man (125,000 dpm/pmol) to a GST-α-DG fusion protein. POMT1-WWS and ISPD-WWS patient cells show comparable defects in POMT enzyme activity. Specific POMT activity in control cells was determined as 536.7 pmol/g/h. The diagram shows relative POMT activity in reference to control cells (n=3). Error bars represent s.d. (b) [32P]-orthophosphate labeling of DGFc5-expressing cultured cells from control and ISPD P2 cells. After O-mannosyl residues are transferred, the radioactive [32P]-orthophosphate is incorporated to form a phosphorylated O-linked mannose glycan21. The ISPD sample shows markedly reduced [32P] labeling due to reduced number of O-mannosyl acceptor sites. (c) Quantitative On-Cell western analysis of LARGE-induced α-DG hyperglycosylation. The glycosyltransferase LARGE participates in a post-phosphoryl modification transferring the laminin-binding glycan. Forced expression of LARGE increases the affinity of the cell surface for the IIH6 antibody in control cells, but not in POMT1 and ISPD deficient WWS cells, confirming that the mutant cells lack the O-mannosyl acceptors of the post-phosphoryl modification. IIH6 On-Cell quantitative data were normalized with DRAQ5 cell DNA dye (n=3). Error bars represent s.d.
In this study, we have identified a novel WWS disease gene in patient fibroblasts by using a complementation assay in combination with targeted sequencing. This approach provided conclusive genetic and biochemical evidence that recessive mutations in ISPD lead to impaired α-DG O-mannosylation, establishing a novel WWS pathomechanism. Further studies are needed to determine how defects in ISPD influence protein O-mannosylation, as this is the first WWS gene without proposed glycosyltransferase activity and direct role in α-DG glycosylation.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Supplementary Material
Acknowledgments
We thank the Gene Transfer Vector Core (UI, supported by NIH/NIDDK P30 DK 54759) for generating adenoviruses; Traci Toy at the UCLA Genome Sequencing Center (see URLs) for assisting with construction of the sequencing libraries, and Dr. Suhua Feng at the UCLA Broad Stem Cell Research Center (BSCRC) for assisting in running HiSeq2000, and Bret Harry for maintaining the data analysis pipeline. Sequencing and sequence analysis were supported by the Genomics and Bioinformatics Core of the UCLA Muscular Dystrophy Core Center (P30). This study used fibroblast samples from the NINDS Cell Line Repository (see URLs) and the Miami Brain and Tissue Bank for Developmental Disorders, funded by NICHD. We thank members of the Campbell laboratory and Colleen A. Campbell for fruitful discussions; Alex Dietz, Andrew Crimmins, Greg Morgensen, Jamie Eskuri, Pascale Guicheney and Hans v. Bokhoven for technical support; Christine Blaumueller for critical reading of the manuscript. This work was supported in part by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant (1U54NS053672, K.P.C., K.D.M., S.A.M. and T.W.) and an ARRA Go Grant (1 RC2 NS069521-01, K.P.C. and T.W.). The Muscular Dystrophy Campaign Grant to F.M. is also gratefully acknowledged. S.C. and F.M. are investigators of, and supported by, the European FP7 NMD-Chip and Bio-NMD projects. F.M. is supported by the Great Ormond Street Hospital Children’s Charity. K.P.C. is an investigator of the Howard Hughes Medical Institute.
ONLINE METHODS
Subjects and samples
We obtained and tested all tissues and patient cells in agreement with the guidelines set out by the Human Subjects Institutional Review Board of the University of Iowa; informed consent was obtained from all subjects or their legal guardians. More detailed information on fibroblasts from control and affected dystroglycanopathy individuals with known genetic defects is summarized in Supplementary Table 1.
Cell cultures
Cells were maintained at 37 °C and 5% CO2 in DMEM medium plus 20% fetal bovine serum and 0.5% penicillin-streptomycin (Invitrogen, Carlsbad, CA).
Biochemical analyses
The monoclonal antibodies to the fully glycosylated form of α-DG (IIH6 and VIA4)22 and β-dystroglycan (AP83)23 have been characterized previously. Gt20 (coreαDG) from goat antiserum was raised against the dystrophin-glycoprotein complex (DGC) in its entirety and purified against a hypoglycosylated full-length α-DG–human IgGFc fusion protein3,24. Additionally, antibodies against the protein backbone of alpha-dystroglycan were generated by injecting a keyhole limpet hemocyanin (KLH)-conjugated synthetic peptide consisting of amino acids 485–514 of human dystroglycan (Swiss-Prot Q14118) into rabbits (G6317, Genemed Biosynthesis). The anti-core-α-dystroglycan antibody was affinity purified using BSA-conjugated C-terminal peptide25. Mouse monoclonal anti-myc tag (clone 4A6) antibodies were purchased from Millipore (Billerica, MA).
Assay for protein O-mannosyltransferase activity
Protein O-mannosyltransferase activity was determined based on the amount of [3H]-mannose transferred from Dol-P-[3H]-mannose to an α-DG glutathione-S-transferase fusion, as described elsewhere7,26.
[32P] orthophosphate labeling of cells
Phosphorylation of α-DG in patient fibroblasts was determined based on the incorporation of [32P] into a secreted Fc-tagged α-DG recombinant protein, as described elsewhere21.
Adenovirus generation and gene transfer
E1-deficient recombinant adenoviruses (Ad5CMV-DAG1, Ad5CMV-DGFc5, Ad5CMV-POMT1/RSVeGFP, Ad5CMV-POMT2/RSVeGFP, Ad5CMV-POMGnT1/RSVeGFP, Ad5CMV-FKTN/RSVeGFP, Ad5CMV-FKRP/RSVeGFP and Ad5CMV-LARGE/RSVeGFP) were generated by the University of Iowa Gene Transfer Vector Core and described previously 8,27. Similarly, Ad5CMV-ISPD-myc/RSVeGFP was generated by cloning a C-terminal myc-tagged open reading frame corresponding to human ISPD (NM_001101426) into the polylinker region of pAd5CMVK-NpA. Cell cultures were infected with viral vector for 12 h, at an MOI of 400. We examined cultures 3–5 d after treatment. We also used nucleofection, as nonviral method for gene transfer into cells. Nucleofection of fibroblasts was performed using the Human Dermal Fibroblast Nucleofector kit, according to an optimized protocol provided by the manufacturer (Amaxa Biosystems, Germany).
ISPD expression vector construction
cDNA was generated from control and ISPD-WWS P6 (in frame ΔE3, 50aa deletion) fibroblasts. The C-terminal myc-tagged coding region of the human wild-type ISPD (1,395bp) and mutant ISPD (1,245bp) gene were amplified from cDNA by PCR, using primer pair 7717/7718 (Supplementary Table 5). The amplified PCR product was subcloned using the pcDNA™3.1/V5-His TOPO® TA Expression Kit (Invitrogen).
Immunohistochemical analysis
Cryosections of the skeletal muscle biopsy (10μm) were processed for immunofluorescence as described23. Mouse monoclonal anti-laminin α2 (anti-merosin, clone 5H2) antibodies were purchased from Millipore (Billerica, MA). Cultured cells were fixed in 4% paraformaldehyde for 10 min, and permeabilized with PBS/0.1% TX-100 for 10 min, before being blocked and incubated with primary antibodies. Slides were observed with an Axio Imager.M1 microscope (Zeiss).
Glycoprotein enrichment and biochemical analysis
WGA-enriched glycoproteins of frozen samples and cultured cells were processed as described3. Immunoblots were done on polyvinylidene difluoride membranes as described3. Blots were developed with IR-conjugated secondary antibodies (Pierce) and scanned with an Odyssey infrared imaging system (LI-COR Bioscience, Lincoln, NE). Laminin overlay assays were performed as previously described3.
On-Cell complementation assay of patient fibroblasts
2×105 cells were seeded into a 48-well dish. The next day the cells were co-infected with 200 MOI of Ad5CMV-DAG1 for signal enhancement, and 200 MOI of the Ad5 complementation construct (except Ad5CMV-FKRP/RSVeGFP was infected with 40 MOI), in growth medium. Four days later, the cells were washed in TBS and fixed with 4% paraformaldehyde/TBS for 10min. After blocking with 3% dry milk in TBS + 0.1% Tween (TBS-T), the cells were incubated with primary antibody (glyco α-DG, IIH6) in blocking buffer overnight. For developing the On-Cell westerns we conjugated goat anti-mouse IgM (Millipore) with IR800CW dye (LI-COR), subjected the sample to gel filtration, and isolated the labeled antibody fraction. After staining with IR800CW secondary antibody in blocking buffer, we washed the cells in TBS and scanned the 48-well plate with an Odyssey infrared imaging system (LI-COR Bioscience, Lincoln, NE). For cell normalization DRAQ5 (Biostatus Limited) cell DNA dye was added to the secondary antibody incubation.
Polyethylene glycol (PEG)-induced fusion of dermal fibroblasts
Cell fusion assays were performed as published previously28. In brief, cells were seeded into a 48-well dish or on a tissue culture chamber slide. When the cells reached 90–100% confluency, the culture medium was removed and cell fusion was induced by adding 55% PEG1500 (w/v) in DMEM to the cell monolayer, and incubating for 1min at room temperature. The fusion solution was then discarded and the cell monolayer was rapidly rinsed four times with complete growth medium at room temperature, and then returned to the 37 °C tissue-culture incubator. The next day, fusion-induced cells were infected with 200 MOI of Ad5CMV-DAG1 for signal enhancement. After four days, cells were fixed and stained (see method for On-Cell complementation assay and immunohistochemical analysis).
Linkage analysis
Genomic DNA samples were genotyped on an Illumina Omni-1 Quad BeadChip. Identical-by-descent (IBD) and Homozygosity-by-descent (HBD) analyses were performed using a custom Mathematica script (Wolfram Research; B. Merriman, available on request), which compares genomes for genotype identity and identifies long intervals of homozgosity29.
Targeted next generation sequencing
Regions over 10cM in length where the sibling pair P2 and P3 is IBD for both alleles were selected as were regions that were homozygous for over 2cM in one or more of the four suspected consanguineous samples (P4, P5, P6, P7), and used to create a custom-capture array. In total, 7,113 coding exons across 14 overlapping intervals were used to design a 244K CGH array, using Agilent eArray (Supplementary Table 3). Repeat regions were excluded. 3μg of genomic DNA was used to generate each sequencing library, using the Agilent SureSelect Target Enrichment System for Illumina Paired-End Sequencing Library Protocol (version 2.0.1); the only exception was that rather than the commercial adapter, 7 different custom-made bar-coded adapters were used (sequence available on request). After amplification, samples were pooled at equal molar concentration, captured on one array following in-house protocol30 and sequenced on the Illumina HiSeq2000 as 50bp paired-end reads.
Sequence Read Alignment
The sequence reads were de-barcoded and aligned to the Human reference genome b37 (hg19) using Novobarcode and Novoalign from Novocraft Short Read Alignment Package (see URLs) . Data were processed with SAMtools (see URLs) ,31 version 0.1.15, and potential PCR duplicates were removed using Picard (see URLs) . The mean coverage achieved within the intended captured region was 35.9X per base, with 82.2% of the targeted base positions covered at ≥10X. Local realignment was performed using the GATK32,33 ‘IndelRealigner’ tool.
Variant Calling
Variants were called using the GATK ‘Unified Genotyper’ tool simultaneously for all 7 samples. Small indels were called with the ‘-glm DINDEL’ option. Variants with a phred-scale Qscore of 50.0 or greater were reported as PASSed calls and those with an Qscore of 10.0 or greater and of less than 50.0 were reported as ‘Low Qual’ calls. Using the GATK ‘VariantFiltrationWalker’ tool, both the SNPs and INDELs were hard-filtered to eliminate low-quality variants. The following filtering parameters were used, as suggested as standard by GATK: 1) clusterWindowSize 10; 2) MAPQ0 (mapping quality of zero) >40; 3) QD (Quality-by-depth) < 5.0; 4) SB (Strand Bias) > −0.10. The GATK ‘VariantEval’ tool was used to collect the statistics for the PASSed variants (Supplementary Table 6A).
Variant Annotation
The PASSed variants that are not found at dbSNP132 positions were annotated using SeattleSeqAnnotation version 6.16 (see URLs) ; SNVs and INDELs were annotated separately. Variants present in the 1000 Genomes database (March 2010 release) or dbSNP131, as well as those that result in coding-synonymous changes or that are found outside the coding region, were removed from further analysis (Supplementary Table 6B). The remaining 84 variants were prioritized based on frequency of coexistence within the same gene.
Copy Number Variant Detection
To identify exonic deletions within the ISPD gene, LRR (log R ratio), Covi,j/Covi,P4 was calculated for each exon i, in each sample j, using ExomeCNV with sample P4 as control31.
Sanger sequencing
Genomic DNA was extracted from dermal fibroblasts or transformed lymphoblastoid cell lines using standard methods (Qiagen DNeasy Blood & Tissue Kit). The coding regions (10 exons) and exon-intron boundaries of ISPD were amplified using PCR (Primers sequences and PCR conditions are available upon request). Primer sets for PCR were designed using the web-based design tool ExonPrimer. After PCR amplification the purified products were evaluated by Sanger sequencing, using standard protocols.
Footnotes
URLs.
UCLA Genome Sequencing Center (http://gsc.ucla.edu/)
NINDS Cell Line Repository (http://ccr.coriell.org/ninds)
Novocraft Short Read Alignment Package (http://www.novocraft.com/index.html)
SAMtools (http://samtools.sourceforge.net/)
Picard (http://picard.sourceforge.net/)
SeattleSeqAnnotation (http://snp.gs.washington.edu/SeattleSeqAnnotation131/)
Author Contributions T.W., H.L., S.F.N., and K.P.C. designed the research. T.W. performed the research and analyzed the data. H.L. and S.F.N. performed SNP analysis, next generation sequencing and data filtering. M.L. and S.S. carried out POMT enzyme activity assays. T.Y.-M. performed α-DG orthophosphate cell labeling experiments. D.B. performed qPCR expression analysis. D.V. carried out antibody affinity purifications and labelings. T.L.W. carried out Sanger sequencing of known WWS genes. S.A.M. performed the muscle histology and clinical data interpretation. H.S., J.V., S.C., F.M., T.V., A.S.L., W.B.D. and K.D.M. provided clinical data and/or patient fibroblast samples. K.P.C. supervised and mentored the project; T.W. and K.P.C. wrote the initial manuscript and all authors approved and commented on the manuscript.
The authors declare no conflicts of interest.
References cited in main text
- 1.Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet journal of rare diseases. 2006;1:29. doi: 10.1186/1750-1172-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Current opinion in genetics & development. 2011;21:278–85. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Michele DE, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–22. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 4.Cao W, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998;282:2079–81. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 5.Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 6.Satz JS, et al. Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:10567–75. doi: 10.1523/JNEUROSCI.2457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lommel M, et al. Correlation of enzyme activity and clinical phenotype in POMT1-associated dystroglycanopathies. Neurology. 2010;74:157–64. doi: 10.1212/WNL.0b013e3181c919d6. [DOI] [PubMed] [Google Scholar]
- 8.Barresi R, et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med. 2004;10:696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- 9.Clarke NF, et al. Congenital muscular dystrophy type 1D (MDC1D) due to a large intragenic insertion/deletion, involving intron 10 of the LARGE gene. European journal of human genetics : EJHG. 2011;19:452–7. doi: 10.1038/ejhg.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inamori K, et al. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–6. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masselot M, De Robichon-Szulmajster H. Methionine biosynthesis in Saccharomyces cerevisiae. I. Genetical analysis of auxotrophic mutants. Molecular & general genetics : MGG. 1975;139:121–32. doi: 10.1007/BF00264692. [DOI] [PubMed] [Google Scholar]
- 12.Stanley P. Membrane mutants of animal cells: rapid identification of those with a primary defect in glycosylation. Molecular and cellular biology. 1985;5:923–9. doi: 10.1128/mcb.5.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontecorvo G, Riddle PN, Hales A. Time and mode of fusion of human fibroblasts treated with polyethylene glycol (PEG) Nature. 1977;265:257–8. doi: 10.1038/265257a0. [DOI] [PubMed] [Google Scholar]
- 14.Kanoff RJ, et al. Walker-Warburg syndrome: neurologic features and muscle membrane structure. Pediatric neurology. 1998;18:76–80. doi: 10.1016/s0887-8994(97)00137-9. [DOI] [PubMed] [Google Scholar]
- 15.Vajsar J, Ackerley C, Chitayat D, Becker LE. Basal lamina abnormality in the skeletal muscle of Walker-Warburg syndrome. Pediatric neurology. 2000;22:139–43. doi: 10.1016/s0887-8994(99)00129-0. [DOI] [PubMed] [Google Scholar]
- 16.Skorupinska-Tudek K, et al. Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of dolichols in plants. The Journal of biological chemistry. 2008;283:21024–35. doi: 10.1074/jbc.M706069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard SB, et al. Structure of 4-diphosphocytidyl-2-C- methylerythritol synthetase involved in mevalonate- independent isoprenoid biosynthesis. Nature structural biology. 2001;8:641–8. doi: 10.1038/89691. [DOI] [PubMed] [Google Scholar]
- 18.Surmacz L, Swiezewska E. Polyisoprenoids - Secondary metabolites or physiologically important superlipids? Biochemical and biophysical research communications. 2011;407:627–32. doi: 10.1016/j.bbrc.2011.03.059. [DOI] [PubMed] [Google Scholar]
- 19.Kuzuyama T. Mevalonate and nonmevalonate pathways for the biosynthesis of isoprene units. Bioscience, biotechnology, and biochemistry. 2002;66:1619–27. doi: 10.1271/bbb.66.1619. [DOI] [PubMed] [Google Scholar]
- 20.Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Archives of biochemistry and biophysics. 2011;505:131–43. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida-Moriguchi T, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–31. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 23.Duclos F, et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. The Journal of cell biology. 1998;142:1461–71. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunz S, Sevilla N, McGavern DB, Campbell KP, Oldstone MB. Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]-dystroglycan. J Cell Biol. 2001;155:301–10. doi: 10.1083/jcb.200104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharp AH, Campbell KP. Characterization of the 1,4-dihydropyridine receptor using subunit-specific polyclonal antibodies. Evidence for a 32,000-Da subunit. The Journal of biological chemistry. 1989;264:2816–25. [PubMed] [Google Scholar]
- 26.Manya H, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci U S A. 2004;101:500–5. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Bernabe DB, et al. Loss of alpha-dystroglycan laminin binding in epithelium-derived cancers is caused by silencing of LARGE. The Journal of biological chemistry. 2009;284:11279–84. doi: 10.1074/jbc.C900007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidson RL, Gerald PS. Improved techniques for the induction of mammalian cell hybridization by polyethylene glycol. Somatic cell genetics. 1976;2:165–76. doi: 10.1007/BF01542629. [DOI] [PubMed] [Google Scholar]
- 29.Lee H, Jen JC, Cha YH, Nelson SF, Baloh RW. Phenotypic and genetic analysis of a large family with migraine-associated vertigo. Headache. 2008;48:1460–7. doi: 10.1111/j.1526-4610.2007.01002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, et al. Improving the efficiency of genomic loci capture using oligonucleotide arrays for high throughput resequencing. BMC Genomics. 2009;10:646. doi: 10.1186/1471-2164-10-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.