

Abstract
A key property possessed by the mammalian cochlea is its ability to dynamically alter its own sensitivity. Because hair cells and ganglion cells are prone to damage following exposure to loud sound, extant mechanisms limiting cochlear damage include modulation involving both the mechanical (via outer hair cell motility) and neural signaling (via inner hair cell-ganglion cell synapses) steps of peripheral auditory processing. Feedback systems such as that embodied by the olivocochlear system can alter sensitivity, but respond only after stimulus encoding, allowing potentially damaging sounds to impact the inner ear before sensitivity is adjusted. Less well characterized are potential cellular signaling systems involved in protection against metabolic stress and resultant damage. Although pharmacological manipulation of the olivocochlear system may hold some promise for attenuating cochlear damage, targeting this system may still allow damage to occur that does not depend on a fully functional feedback loop for its mitigation. Thus, understanding endogenous cell signaling systems involved in cochlear protection may lead to new strategies and therapies for prevention of cochlear damage and consequent hearing loss. We have recently discovered a novel cochlear signaling system that is molecularly equivalent to the classic hypothalamic-pituitary-adrenal (HPA) axis. This cochlear HPA-equivalent system functions to balance auditory sensitivity and susceptibility to noise-induced hearing loss, and also protects against cellular metabolic insults resulting from exposures to ototoxic drugs. This system may represent a local cellular response system designed to mitigate damage arising from various types of insult.
Keywords: cochlea, corticotropin releasing factor, HPA axis, noise-induced hearing loss (NIHL), hair cell
INTRODUCTION
Peripheral processing of auditory stimuli is a complex, active computational process that depends not only on the function of numerous classes of cells, but also on feedback that can fine-tune sensitivity. Sensitive hearing is critical for success, of a species, and therefore the ability to establish the most sensitive hearing possible, either by controlling the computational elements of the inner ear, or generating filtering strategies useful for extracting information from a noisy background, should be under heavy genetic selection through evolution. Yet, hair cells are prone to damage and loss following exposure to constant moderate level sounds via metabolic insults (free radical formation, etc.), impact-like high intensity sounds, and numerous ototoxic compounds and natural organisms such as bacteria and viruses. This is an especially significant problem for mammals because, unlike the case for birds and reptiles, cochlear hair cells do not regenerate. Thus, the cochlea faces a significant biological problem- it must balance maximal sensitivity with susceptibility to damage that can result in permanent loss of frequency representation and a decreased probability of survival success. Feedback to the cochlea therefore plays a critical role in overall cochlear function and maintenance. Current ideas of feedback to the mammalian cochlea are mostly centered on the ability to adjust the cochlear amplifier via activity involving modulation of the outer hair cells, thereby dynamically controlling auditory sensitivity via physical mechanisms. By establishing dynamic control over its own sensitivity, the cochlea ensures that loud sounds are encoded at an operating point that is less than its peak of sensitivity, thereby minimizing metabolic and physical insults, and preserving cochlear structure and function. Yet, this classic olivocochlear efferent feedback mechanism is a reactive servitor dependent on encoding and transmitting information into the brain that potentially harmful sound has been encountered before the system is modulated. Delays between encountering a stimulus and final olivocochlear-induced modulation of cochlear activity can prove detrimental to tissue homeostasis. Additionally, it is questionable whether the olivocochlear system is fully functional over the full frequency spectrum. A local signaling system expressed in the cochlea and based on hypothalamic and pituitary signaling systems has recently been described that may provide for faster and more inclusive types of homeostatic control beyond the physical control that is generated by olivocochlear activity. This review will cover these new insights and describe how this system may be involved in setting cochlear sensitivity and balancing that sensitivity with protection from acoustic trauma and metabolic insults initiated by exposure to ototoxic compounds.
VARIOUS BIOLOGICAL SIGNALING MECHANISMS DYNAMICALLY CONTROL COCHLEA FUNCTION
It is self-evident that the cochlea is a complex physical and functional system. It is characterized by a large number of elements, with many interactions (at various levels) occurring between such elements. The organization of the cochlea gives rise to non-linear and stochastic results (neural outputs). Viewing the cochlea as a complex system, it should not be surprising that feedback loops with varying time delays are an integral component of cochlear function. At the most basic level, feedback can take the form of extrinsic or intrinsic influences. While both exist in the cochlea, and examples are briefly outlined below, the extrinsic feedback loop is to date the feedback loop that figures most prominently in our ideas of control over cochlear function.
Extrinsic feedback- control originating outside the cochlea: the olivocochlear system
An elucidation of the mechanisms allowing the cochlea to control it’s own activity has been the target of experimental investigations for many years, and much has been learned about cochlear control systems. The system that has figured most prominently in the history of such investigation has been the olivocochlear system. This neural feedback system between the lower brainstem and the hair cells and ganglion cells has been recognized since the 1940’s (Rasmussen, 1942; Rasmussen, 1955). Several studies implicate the olivocochlear efferent system in prevention of noise-induced hearing loss (NIHL). While a full review of the olivocochlear system is beyond the scope of this review, it should be noted that studies demonstrate increased resistance to noise damage in mice that either over-express (Maison et al., 2002) the alpha 9 subunit of the nicotinic acetylcholine receptor (nAChR), which is expressed by hair cells (Elgoyhen et al., 1994) and has been shown to be intimately involved in cholinergic efferent neurotransmission (Vetter et al., 1999), or that express a point mutation in the alpha 9 subunit that renders the nAChR more active due to hypersensitivity to acetylcholine and slow desensitization (Taranda et al., 2009). Thus individual variation in the activity of the medial olivocochlear (MOC) system could contribute to the observed variability in noise susceptibility, as indicated by a study in guinea pigs where the strength of the MOC reflex to incoming sound was a good indicator of resistance to NIHL (Maison and Liberman, 2000). However, the olivocochlear system is by its very organization a reactive system. Thus, the cochlea witnesses a delay between its exposure to potentially destructive stimuli and the return signal from the brain following this over-exposure before the efferent system comes to the rescue by decreasing the physical activity of the cochlea. The olivocochlear system also probably functions over a relatively short time frame, and once its activity is terminated, the cochlea is left in its original state of sensitivity.
Intrinsic feedback- control originating within the cochlea: the purinergic signaling system
While the olivocochlear system may be sufficient to protect against NIHL if one can escape the overly loud environment, or when the loud sound is transient, it may not function optimally under prolonged exposures or at certain frequencies of auditory stimulation. Other mechanisms involving release of small active compounds, such as ATP, within the cochlea are also known to alter cochlear function (Housley et al., 1999; Housley et al., 2002; Housley et al., 2006), and likely represent a faster solution to adjusting sensitivity. An attractive property of such signaling is that since purinergic transmission can include G-protein coupled receptors, signaling may leave the cochlea resistant to damage even after the local signaling episode at the receptor is terminated, thereby imparting resistivity to near-future events. While knowledge regarding cochlear regulation by this system remains relatively incomplete, it may include not only regulation via neural processes, but also via mechanisms that regulate ion homeostasis (Housley et al., 2006). Thus, this system may be more complicated but at the same time offer a more broad-spectrum modulation of cochlear function, i.e. via its actions on numerous signaling systems.
Despite the long history of experimental investigation into cochlear modulating systems, significant questions remain open pertaining to how the cochlea dynamically modulates its own sensitivity: what are the complete molecular signaling systems involved in generating and modulating cochlear sensitivity, and are there other signaling systems expressed wholly within the cochlea (i.e. that can minimize response delay) that be activated by auditory environments and respond in some manner to protect against hearing loss? Such a defense system should work constitutively to modulate acoustic sensitivity and reactivity based on past and present experience rather than functioning as a delayed reactive feedback mechanism that only engages upon intense, potentially dangerous, stimulation. Indeed, sound conditioning experiments, which demonstrate toughening of the cochlea against noise insult following previous exposure to more moderate (non-destructive) sound stimuli, reveal that such an integrative defense system exists and, intriguingly, that systemic stress hormones appear to play an important role. Though progress has been made in identifying various manipulations and pharmacological treatments that confer at least partial protection against noise-induced hearing loss (Pirvola et al., 2000; McFadden et al., 2001; Darrat et al., 2007; Monge Naldi et al., 2009), the molecular mechanisms and associated cell biology involved in protecting the cochlea against its daily metabolic and physical stress remain less clear. Since small 2009), it is likely that the cochlea expresses an endogenous protective signaling system within its own cellular ensemble. An investigation into cochlear signaling systems involved in maintaining homeostasis and combating cellular stress seems warranted. Understanding such endogenous sources of auditory protection may also lead to novel targeted therapeutic strategies for prevention of cochlear damage and consequent hearing loss. The question is, where do we look for models that may fill the requirements for a truly endogenous signaling system involved in an integrative feedback control of the cochlea?
THE HYPOTHALAMIC-PITUITARY-ADRENAL (HPA) AXIS
A major integrator/feedback system for biological signaling at the organismal level is the system collectively known as the HPA axis (Fig. 1). This diverse system of tissues, cells and molecular signals participates in a hierarchical control system that integrates standard bodily information for normal function of such things as gonads, growth of the organism, pregnancy and development, food and water intake, etc. It performs its function by using several key neuroendocrine tissues. Nuclei within the hypothalamus serve as integrating centers for both intrinsic (CNS-based) and extrinsic (originating outside of the nervous system) stimuli. Neurosecretory cells within the hypothalamus synthesize peptides and catecholamines. These substances are either released into the posterior pituitary directly, or into the hypophyseal portal circulatory system to ultimately enter either lobe of the pituitary and affect the release of hormones into the systemic circulation to affect distant target tissues. Corticotropin-releasing factor (CRF) is a major signaling molecule released by the hypothalamus into the portal circulation. Modulation of CRF release can be brought about by either intrinsic signals such as those derived from the CNS (e.g. stress) or extrinsic signals such as those derived from outside the body (e.g. diurnal rhythms). CRF stimulates CRF receptor 1 (CRFR1, one of two mammalian CRF receptors), in the anterior pituitary to stimulate production of pro-opiomelanocortin (POMC) and release of its major cleavage products, chief among them being adrenocorticotropic hormone (ACTH). ACTH is released into the systemic circulation and travels to the adrenal cortex where it binds to its receptor, the melanocortin 2 receptor (MC2R), and stimulates synthesis and release of the adrenocortical hormones cortisol, corticosterone, and aldosterone (Stevens and White, 2010), collectively also known as glucocorticoid hormones, or glucocorticoids. One major role for cortisol (in humans; corticosterone in rodents) is to feedback onto the CRF signaling system to turn off the stimulus leading to ACTH release (Fig. 1). Thus, a hierarchical feedback control is established to limit the activity of the signaling system. Glucocorticoids exert three major effects: 1) they stimulate release of adrenaline from adrenal chromaffin cells; 2) they stimulate gluconeogenesis to supply cellular fuel for a ‘fight or flight’ response; and 3) they suppress immune response and inflammation.
Figure 1. Organization of the classic hypothalamic-pituitary-adrenal (HPA) axis.

The HPA axis is the major stress response system of the body. During HPA signaling, corticotropin-releasing factor (CRF) is released from the hypothalamus and travels via blood circulation to the pituitary where it binds to its receptor, CRFR1. Activation of CRFR1 stimulates proteolytic cleavage of pro-opiomelanocortin (POMC) to generate (among others) adrenocorticotropic hormone (ACTH). ACTH is then secreted into the systemic blood circulation and travels to the adrenal glands, where it binds to its receptor, melanocortin receptor 2 (MCR2) to finally stimulate the production and release of glucocorticoids (primarily cortisol in humans, and corticosterone in rodents). This system is hierarchical in its nature of signaling, and is under inhibitory feedback by the secreted glucocorticoids, allowing it to be switched off and thereby avoiding the deleterious effects of prolonged heightened glucocorticoid signaling.
NOVEL BIOLOGICAL SYSTEMS UNDERLYING DYNAMIC CONTROL OF COCHLEAR FUNCTION
Because the classic HPA system can integrate a wide variety of stimuli and respond with an equally wide array of actions, it should not be too surprising to find that HPA activity impacts auditory processing.
Non-neural extrinsic feedback- The systemic HPA axis-induced stress response and auditory protection
The impact of the systemic stress response on auditory function and protection has been appreciated for decades. The systemic stress response involves a chain of events collectively coordinated via the HPA axis as described above that end with release of glucocorticoids, steroid hormones with pleiotropic affects to various tissues.
Previous clinical investigations and experiments have shown that glucocorticoids have significant roles in auditory function, and may help protect the system from damage. It was shown in the 1960s that patients with adrenocorticosteroid deficiency possessed greater auditory sensitivity compared to the normal population (Henkin et al., 1967). Treatment of these patients with prednisone, a synthetic corticosteroid, re-established normal hearing thresholds, demonstrating that the observed hypersensitivity was related to levels of circulating corticosteroids. Meniere’s disease patients exhibit low levels of circulating corticosteroids. Administration of adrenal cortex extract improved auditory function in these patients (Powers, 1972). Steroids are also in widespread use for the treatment of idiopathic sudden sensorineural hearing loss (Kuhn et al., 2011; Rauch et al., 2011). Synthetic glucocorticoids also protect the cochlea against damage induced by ototoxic drugs, acoustic trauma, and ischemia/reperfusion injury (Himeno et al., 2002; Takemura et al., 2004; Tabuchi et al., 2006). Given the modulatory role of glucocorticoid receptors over transcription, several molecular changes likely underlie the observed protection. In particular, experiments point to enhanced biosynthesis of glutathione, reduced secretion of tumor necrosis factor induced cytokines, and altered expression of apoptotic genes as some of the changes likely to combat the free radical damage and apoptosis associated with noise- and chemically-induced cochlear damage (Maeda et al., 2005; Nagashima and Ogita, 2006; Hoang Dinh et al., 2009).
Investigations into the role of the systemic stress axis in sound conditioning have revealed the ability of glucocorticoids to confer protection against sound levels normally capable of damaging the auditory system. The experimental paradigm involved in sound conditioning consists of pre-exposure to sound stimuli that toughen ears against subsequent noise trauma. Interestingly, a high-intensity conditioning stimulus is not required to produce subsequent auditory protection (Canlon et al., 1988; Canlon and Fransson, 1995; Yoshida and Liberman, 2000). Exposure to moderate or low level sound, even of short duration, confers protection against future acoustic insult. Thus, toughening is the result of an adaptive processes induced by a more basic response to sound.
It has been acknowledged for years that sound can activate a systemic stress response (Henkin and Knigge, 1963), and it has been suggested that sound-induced systemic stress underlies some of the maladaptive consequences of constant noise exposure in the workplace, including elevated blood pressure and heart rate (Lusk et al., 2002). Given the connection between sound and stress-response, it is possible that activation of the systemic stress axis (the HPA axis) contributes to sound conditioning-mediated protection. If general HPA activation is involved, one may postulate that non-auditory induction of HPA activity should also result in changes to the auditory system. Following this logic, it was shown that mice undergoing a fifteen minute heat stress exhibited greater resistance to auditory threshold shifts following acoustic insult than did non-stressed mice (Yoshida et al., 1999). Using similar logic, restraint stress was also shown to generate auditory protection, and this was directly correlated to levels of circulating corticosterone (Wang and Liberman, 2002). If the traumatizing auditory stimulus was presented after corticosterone levels returned to normal, protection was not observed. Thus, systemic corticosterone appeared to be an important component of acquired resistance to NIHL. A causal link was established by experiments that showed sound conditioning no longer yielded protection if HPA activation was disrupted via adrenalectomy or administration of glucocorticoid synthesis inhibitors and receptor antagonists (Tahera et al., 2007). Most recently, a corticosteroid-responsive transcription factor, promyelocytic leukemia zinc-finger protein (PLZF), was shown to mediate cochlear protection induced by acoustic conditioning stimuli and restraint stress (Peppi et al., 2011). In PLZF null mice, auditory protection was not generated by typical cochlear conditioning paradigms. Thus, these studies all implicated HPA activation, and more specifically, circulating glucocorticoids, as an endogenous source of cochlear protection, particularly the adaptations leading to acquired resistance against NIHL.
While HPA signaling seems firmly established as being capable of modulating cochlear processing, classic HPA activity related to auditory function still suffers from a potential lag between detection of a harmful stimulus and the response it ultimately delivers. For example, while CRF release along the classic HPA axis can be induced in less than a minute following a stressor, it can take 10-30 minutes before glucocorticoids are finally secreted in response to the same stressor (Sapolsky et al., 2000), owing largely to the requirement of attaining a critical concentration of ACTH in the bloodstream. In addition, adding a greater number of biological elements between the ear and the final classic HPA-induced response that can then feed back to alter inner ear function introduces numerous modulators that may not be in place in a completely local signaling system. For example, age plays a major role in stress response in fish, with juveniles attaining peak responses to handling between 5 and 30 minutes and adults being more resistant, attaining peak responses after 60 minutes (Koakoski et al., 2012). It is also well known that genetic background can play a major role in susceptibility to stress response, and at least some of these differences seem to involve lower susceptibility to activation of other systems such as an immune response. This seems especially prominent in C57Bl/6 versus BALB/c mice (Flint and Tinkle, 2001). Moving from an extrinsically based modulatory system to an intrinsic feedback system functioning in a paracrine-like manner that still maintains the diversity of responses that an HPA-like signaling system should possess, and that can be more responsive along the temporal dimension may be worth searching for.
Non-neural intrinsic feedback- Preliminary evidence for a local HPA axis-induced stress response and auditory protection
While previous results described above suggested that the systemic (central) stress axis likely plays a significant role in protecting hearing, challenges emerged over whether systemic HPA activation could be regarded as the sole mechanism involved in acquired (condition-induced) resistance. For example, an experimental design used to discriminate systemic and local contributions revealed that animals undergoing sound conditioning with one ear plugged and the other left open to the sound stimuli produced unilateral protection. Only the ear left open to the preconditioning stimuli acquired resistance against sound-induced auditory threshold elevation (Yamasoba et al., 1999), suggesting that systemic responses could not account for conditioning-mediated protection. If systemic responses were involved, both ears should have been protected even if acoustic exposure was limited to one ear. Local adaptations therefore seemed responsible for the acquired resistance. Could local adaptations within the cochlea share aspects of cell—cell signaling with classic HPA activation? Two questions in particular arise: is there any precedent for the same hormones involved in HPA axis-induced stress response being expressed outside of the classic HPA axis, and could these signaling molecules be expressed within the cochlea to provide a local stress response system?
On a systems integration level, the cochlea is much like the skin. Both function as a barrier between the environment and the internal biological milieu. Both function as sensory integrators/processors, and send critical information into the CNS. Because of their interaction with the external environment, both are potentially exposed to a variety of potentially harmful factors capable of damaging the tissue, for example UV irradiation in the case of skin, and intense sound in the case of the cochlea. Finally, both systems face a similar problem concerning the need for immediate signaling to occur. In the face of potentially traumatizing challenges, signal delay inherent in waiting for feedback from afar increases the risks for cellular/tissue damage. Numerous processes involved in homeostatic balance of each system would be better served by local, rather than remote, signaling that could bring about a return to homeostatic balance in the face of challenge to homeostasis. Thus, it has been shown that the skin actually expresses a local signaling system involved in homeostatic balance, and the system is a local replication of the classic HPA axis, complete with HPA signaling molecules that function in the same way as they do in the classic HPA axis (down to inducing release of glucocorticoids) but within the local confines of the skin (Slominski et al., 1996; Slominski et al., 1999; Slominski et al., 2000; Slominski, 2005). We therefore asked, could the cochlea also contain a local HPA-like signaling system that might help it maintain homeostatic balance from both a functional (sensitivity) and molecular (protein expression changes in the face of cytotoxic challenges) perspective in a manner similar to that of skin.
THE COCHLEA EXPRESSES AN HPA-EQUIVALENT SIGNALING SYSTEM
The cochlea expresses glucocorticoid receptors (Shimazaki et al., 2002; Terakado et al., 2011). Additionally, systemic HPA activation influences hearing (Wang and Liberman, 2002; Canlon et al., 2007), presumably via delivery of systemic glucocorticoids through the circulation. CRF receptors (discussed more below) were also localized in the cochlea via in situ hybridization (Vetter et al., 2002) and immunolocalization (Graham et al., 2010; Graham and Vetter, 2011) (Fig. 2). Additionally, CRF expression (Fig. 3) has recently been described in the cochlea (Graham and Vetter, 2011), indicating that both the start point (CRF) and end point (glucocorticoid receptors) of the systemic HPA axis signaling are expressed in the cochlea along with CRF’s requisite receptors for initiating CRF-based cell signaling. In addition to its obvious role as the peripheral auditory organ, we wondered: can the cochlea also be considered a neuroendocrine organ, responsible for its own feedback modulation via local signaling? Despite the expression of some aspects of HPA-like signaling in the cochlea, a large gap between signaling molecules (CRF and CRFRs) and effectors (glucocorticoid receptors) seemed to exist. Yet, the presence of many different cellular elements within the cochlea and the complexity of their interactions, not all of which are understood, may be an indication that unknown signaling systems are still to be discovered.
Figure 2. CRFR1 and CRFR2 expression in the mouse cochlea.

CRFR1 expression overlaps and juxtaposes sites of CRF expression suggesting paracrine and juxtacrine signaling. (A) Immunofluorescent detection of GFP driven by the CRFR1 promoter demonstrates expression in the inner sulcus (IS) and support cells lateral to the organ of Corti (OC). Intense immunoreactivity is also observed in the organ of Corti (boxed), shown at higher magnification in C. (B) Double label with CRF reveals regions of overlapping expression (inner sulcus and lateral support cells) suggesting the possibility of paracrine/autocrine signaling. (C) At higher magnification intense CRFR1-GFP immunofluorescence is observed in the Deiter’s cells and in the border cell (BC). (D) Overlay with CRF reveals that these CRFR1-positive cells juxtapose cells expressing CRF, including the inner hair cell (IHC) and outer hair cells (OHCs, arrows). (E) CRFR2 immunolocalization was carried out using antibodies labeled with the green fluorphore, while calbindin, a marker for hair cells, was co-localized using antibodies labeled with a red fluorphore. The tunnel of Corti (ToC) spans the area between the inner and outer hair cells, labeled in red. CRFR2 expression is localized to cells lining the lumen of the endolymphatic space. These include the inner sulcus cells (IS) the lateral support cells (asterisks) up to the spiral prominence (SP), and marginal cells of the stria vascularis (arrows). Cells within the spiral ligament (SLg) behind the stria vascularis, and cells within the spiral limbus (SLm) are also immunopositive. TM, tectorial membrane; SGN, spiral ganglion neurons; SpLim, spiral limbus; OS, outer sulcus; SpLig, spiral ligament; RM, Reissner’s membrane; ToC, tunnel of Corti. Scale bar in C represents 60μm; scale bar in D represents 10μm; scale bar in E represents 50μm. (A-D reprinted from (Graham and Vetter, 2011) with permission, E reprinted from (Graham et al., 2010) with permission)
Figure 3. Immunolocalization of CRF in the mouse cochlea.

(A) CRF is localized in numerous regions of the cochlea. The highest immunofluorescence intensities are located in the inner hair cell region (IHC), the spiral ganglion cells (SGC) and the associated afferent fibers, and in the lateral support cells along the basilar membrane (arrowheads) extending up the lateral wall to the spiral prominence (SP). Also, the stria vascularis (SV) is moderately labeled for CRF. (B) In the organ of Corti, inner hair cells (IHC) and outer hair cells (OHCs) are immunolabeled for CRF. The Deiter’s cells are also immunopositive, but are less intensely labeled. (reprinted from (Graham et al., 2010) with permission)
In order to understand the role and complexity of potential CRF-like signaling for the cochlea, we sought to fill the gap between expression of known HPA-like signaling elements expressed in the cochlea (such as CRFRs and glucocorticoid receptors) and the rest of the classic systemic HPA axis signaling molecules by examining whether expression of these other molecules occurred in the cochlea as well. Classic HPA signaling involves CRF release from the hypothalamus. As previously described at greater length (Graham et al., 2011), CRF binds to CRFR1 expressed by pituitary corticotropes and stimulates cleavage of POMC to produce ACTH, which then travels via blood circulation to the adrenal cortex where it binds melanocortin 2 receptor (MC2R, also known as the ACTH receptor), inducing production and release of glucocorticoids. Therefore, immunofluorescence was used to ascertain whether and where POMC, ACTH, and MC2R expression occurs in the cochlea (Graham and Vetter, 2011). All of these key HPA-related molecules were detected in the mouse cochlea (Fig. 4, Table I), and a common site of expression for CRF, POMC, ACTH, and MC2R was the spiral ganglion cells. Otherwise, both POMC and ACTH were observed in support cells lining the inner sulcus and the lateral support cells (Claudius cells, Boetcher cells). Support cells immediately surrounding the organ of Corti also express CRF, CRFR1, and CRFR2 (Fig. 2). However, despite this overlap, mismatches were also observed in the localization of POMC, ACTH, and CRFR1. For instance, the inner hair cell contained ACTH but little to no POMC and no CRFR1. The Deiter’s cells were highly CRFR1-positive and also expressed abundant POMC and ACTH, but MC2R, the ACTH receptor was only expressed in cells situated at a distance from this source. ACTH was abundantly expressed in Tectal cells and Hensen’s cells, with little expression of POMC. Finally, while the spiral ganglion cells expressed POMC, ACTH, and MC2R, they did not express CRFR1. Such mismatches in expression of HPA components suggest that HPA-like signaling within the cochlea is not as straightforward as CRF binding to CRFR1 on a cell to stimulate breakdown of POMC and production of ACTH in that cell. Instead, the cochlear HPA equivalent signaling system likely represents a dynamic signaling system in which the components interact across the various cell types of the cochlea to orchestrate a local response between cell populations to cochlear stress.
Figure 4. The cochlea expresses an HPA equivalent signaling system.

Immunofluorescent labeling of POMC, ACTH, and MC2R reveals expression of classic HPA components in the cochlea. (A, D, G) POMC and ACTH are expressed in inner and outer sulcus cells lining the cochlear duct (IS and OS, respectively). MC2 is expressed in these regions to a lesser extent (C). All components are expressed in the spiral ganglion cells (SG). The organ of Corti region is boxed in A and is the region from which the higher magnification illustrations were produced. (B) Higher magnification of the organ of Corti region reveals intense POMC labeling in the Deiter’s Cells (DCs) and this expression overlaps with CRFR1-GFP label (C). (E, F) ACTH shows less immunolabeling in Deiter’s cells compared to POMC, but an intense labeling of the inner hair cell (IHC). (H, I) Finally, in the organ of Corti region, MC2R shows an intense and specific labeling for the IHC and a lack of Deiter’s cell labeling. Bottom panels- Expression of CRF, CRFR1, and HPA components in the cochlea is mapped for clarity. CRF signaling molecules are expressed in the cochlea as defined in the immunostaining panels, and are re-created here in schematic fashion. Cells of the inner sulcus (IS) express CRF, CRFR1, and CRFR2. CRF alone is expressed in the inner and outer hair cells (IHC, OHC respectively) of the cochlea, shown in green. These CRF-positive sensory cells are juxtaposed by support cells such as the border cell (BdC, shown in red), which expresses CRFR1, and the Deiter’s cells (DC, cells directly below the outer hair cells and shown in dark yellow) that express CRF, CRFR1, and CRFR2. In addition to the Deiter’s cells, Tectal cells (TC) and Lateral Tunnel Cells (LTC) are directly apposed to the outer hair cells and Deiter’s cells, but do not express any CRF signaling components. Support cells located more laterally include the Hensen’s cells (HC), which flank the Tectal cells and Lateral Tunnel Cells laterally and express CRF, CRFR1, and the Claudius cells (CC) and Boettcher cells (BoC), which express CRF and CRFR2. Thus there is a potential for juxtacrine interaction between hair cells and support cells in their immediate vicinity. The inner sulcus cells medial to the border cell and support cells lateral to the organ of Corti express CRF, CRFR1, and CRFR2, suggesting autocrine and paracrine communications in these peripheral support cells that could also involve the hair cell populations. Finally, molecules of the classic HPA signaling system are expressed in the cochlea as defined in the immunostaining panels, and are schematically mapped for clarity. POMC, ACTH, and to a lesser extent, MC2R are expressed in inner sulcus cells, border cell near the IHC, and the lateral-most support cells which include Claudius cells and Boettcher cells (blue). Deiter’s cells express POMC and ACTH (depicted in yellow), but with little to no expression of MC2R. ACTH and its receptor, MC2R, are expressed in the IHC (pink), suggesting a convergence of HPA signaling on the afferent auditory transducer. ACTH alone seems to be expressed in the Hensen’s and Tectal cells (purple), with no discernable POMC expression found to date. Localization of previously described glucocorticoid receptors within the organ of Corti (based on (Terakado et al., 2011) is indicated by red nuclei, and demonstrates spatial proximity between cells expressing ACTH and MC2R and cells expressing glucocorticoid receptors. Scale bar in A represents 60μm and pertains to A, D, G. Scale bar in B represents 10μm and pertains to all other panels. HC = Hensen’s cell. CRFR1 designates CRFR1-GFP immunolabeling. (all figures reprinted from (Graham and Vetter, 2011) with permission. With permission, schematic figures were re-drawn and annotated from an original provided by Dr. M. Charles Liberman, Mass Eye and Ear Infirmary, Boston, MA)
Table 1. CRF signaling molecule expression within the cochlea.
| CRF alone |
CRFR1 alone |
CRFR2 alone |
CRF+ CRFR2 |
CRF+ CRFR1 +CRFR2 |
|
|---|---|---|---|---|---|
| Organ of Corti | |||||
| Inner phlangeal cell | X | ||||
| IHCs | X | ||||
| Border cell | ? | ||||
| Pillar cells | |||||
| OHCs | X | ||||
| Deiter’s cells | X | ||||
| Other support cells | |||||
| Interdental cells | X | ||||
| Inner sulcus cells | X | ||||
| Tectal cells | |||||
| Hensen’s cells | X | ||||
| Lateral support cells (Claudius and Boettcher’s cells) |
X | ||||
|
Other cells lining scala media |
|||||
| Reissner’s membrane | X | ||||
| Spiral prominence | X | ||||
| Lateral Wall | |||||
| Type I fibrocytes | X | ||||
| Type II fibrocytes | X | ||||
| Type III fibrocytes | X | ||||
| Type IV fibrocytes | X | X | |||
| Stria Vascularis | |||||
| Marginal cells | X | ||||
| Intermediate cells | X | ||||
| Basal cells | X | ||||
|
Spiral Ganglion neurons |
X |
The mapping of HPA-associated molecules within the cochlea has revealed that ACTH and POMC are not necessarily co-localized. For example, this is found with regards to the Hensen’s cells, and in general is somewhat unexpected given that ACTH is a breakdown product of POMC. One way to reconcile this apparently discordant localization of expression is to recognize that the actual levels of POMC (protein) expression can reflect the extent of POMC cleavage. Thus, cells expressing ACTH while expressing little to no POMC may contain more of the enzyme (PC1) that converts POMC to ACTH. Cells containing POMC with little to no ACTH may simply exhibit less POMC proteolytic activity at the time of the assay, or they may express PC2, the enzyme that converts ACTH to alpha-melanocyte stimulating hormone (α-MSH). This has been shown to occur in the brain and skin (reviewed in (Stevens and White, 2010). POMC is also constitutively secreted by cells of the pituitary, medial hypothalamus and skin without first undergoing proteolytic cleavage (Stevens and White, 2010). Since POMC has been shown to directly stimulate melanocortin receptors, one cannot yet rule out directed secretion and physiological activity of POMC in the cochlea. Finally, experiments indicate that POMC can be processed extracellularly to produce its cleavage products, including ACTH-like peptides (Konig et al., 2006). Direct signaling activity of POMC coupled with its constitutive transcription and secretion suggest that this molecule can act independently and eliminates the necessity for co-localization with either CRFR1 or ACTH. Separately or together, these possibilities may explain mismatches in expression of CRFR1, POMC and ACTH, and serve to highlight the extraordinary diversity of signaling implicit in cellular signaling based on “pre-pro-hormone” systems.
THE QUESTION OF LOCAL GLUCOCORTICOID PRODUCTION IN THE COCHLEA
Glucocorticoids and other bioactive compounds are released as the final product of classic HPA signaling. Glucocorticoids promote survive during stressful events by modifying the glucose utilization of organs, and are also involved in regulation of the immune response, among other mechanisms of action. Thus, one may suggest, or even expect, that a complete HPA-equivalent signaling system within the cochlea should involve local production and release of glucocorticoids. However, it is still a matter of debate whether cells within the cochlea are capable of synthesizing glucocorticoids such as corticosterone, and releasing it locally. At least one study suggests that glucocorticoid-synthesizing enzymes are not expressed in the cochlea (Lecain et al., 2003). However, based upon localization of key steroidogenic enzymes within the cochlea, other data indicate that local glucocorticoid production may take place (ten Cate et al., 1994; Terakado et al., 2011). The initial step in steroid biosynthesis involves conversion of cholesterol to pregnenolone via cholesterol side chain cleavage enzyme. Pregnenolone is then converted to progesterone, which can be processed in two separate pathways, one producing sex steroids and one producing glucocorticoids. Corticosterone is created from its precursor 11-deoxycorticosterone via activity of steroid 11-beta-hydroxyalse (cytochrome P450 11B1, mitochondrial). Aldosterone, a mineralocorticoid, has been identified in the cochlea, and is created from the same precursor in a two-step reaction that produces corticosterone as an intermediate. The enzyme aldosterone synthase accomplishes this two-step conversion. Interestingly, experiments investigating cochlear expression of steroidogenic enzymes localized enzymes responsible for the early phases of steroid synthesis and aldosterone synthase, but failed to detect 11-β-hydroxylase (Lecain et al., 2003). These results suggested that sex steroids and mineralocorticoids are produced in the cochlea, but not glucocorticoids. Yet, it has been demonstrated that 11-beta-hydroxysteriod dehydrogenase (11-HSD) isoforms are expressed in the cochlea (ten Cate et al., 1994; Terakado et al., 2011). 11-HSD is responsible for converting cortisol (with activity at glucocorticoid receptors) to cortisone (with little to no activity at the glucocorticoid receptors). While it remains an open question whether corticosterone is produced locally, it must be acknowledged that the likelihood of local production is high. This is suggested given that corticosterone is a necessary intermediate for mineralocorticoid synthesis, that molecules involved in earlier steps in the synthesis process for glucocorticoids are present in the cochlea, and that molecules involved in glucocorticoid degradation are located in the cochlea. Nonetheless, it is possible that aldosterone synthase does not act on local precursors but on systemic corticosterone delivered through the blood supply.
GLUCOCORTICOID RELEASE MAY NOT BE THE FINAL RESULT OF COCHLEAR HPA-EQUIVALENT SIGNALING
Whether or not glucocorticoids are produced locally within the cochlea does not detract from the hypothesis that local HPA-like signaling is biologically relevant for cochlear processing. The cochlear HPA-equivalent system could be involved in other processes that do not require the classic endpoint of glucocorticoid production and release. Although classic HPA signaling involves ACTH binding to the MC2 receptor and the subsequent production and release of glucocorticoids, MC2R activity is not limited to promoting glucocorticoid synthesis alone. Indeed, MC2R is a G-protein-coupled receptor that couples to the cAMP-PKA pathway. Working through this pathway, ACTH activation of MC2R could potentially exert several effects on auditory processing beyond glucocorticoid production. In fact, the major site of MC2R expression, the inner hair cell, also hosts a potential target of ACTH-induced PKA signaling. Inner hair cells express the large-conductance potassium channel known as the BK channel (Brunton et al., 2007). BK activity is directly and intimately involved in hair cell physiological responses (Skinner et al., 2003; Beurg et al., 2005). ACTH has been shown to alter splicing of the BK channel and inhibit BK activity via cAMP-PKA signaling cascades (Shipston et al., 1996; Tian et al., 2001; Lai and McCobb, 2002) in other tissues. Thus local HPA-like signaling within the cochlea can have effects on cochlear function that may be unrelated to glucocorticoid synthesis.
CRF RECEPTOR SIGNALING IN THE COCHLEA
The classic HPA system is activated by CRF release from synaptic terminals innervating the pituitary and emanating from hypothalamic nuclei. Thus, if HPA-equivalent signaling occurs in the cochlea, CRF signaling within the cochlea should exist and modulate basic cochlear processes. CRF receptors are 7-transmembrane G-protein coupled receptors that modulate cellular function via second messenger signaling cascades. Two receptors exist in mammals and are encoded by the CRFR1 and CRFR2 genes. Pituitary (corticotrope) cells express CRFR1, and therefore CRFR1 activation is considered to be the initiator of the classic systemic stress-response. While CRFR1 activity initiates HPA response, CRFR2 has been shown to provide regulation/modulation over elements of HPA activity. For example, CRFR2 null mice exhibit early termination of the ACTH release following CRFR1 stimulation (Bale et al., 2000). CRFR1 and CRFR2 are also expressed in numerous other tissues, including the brain, and therefore are also thought to participate in non-hormonal signaling systems including neurotransmission. As described above, both CRF receptors are expressed within the mouse cochlea (Fig. 2), as is CRF itself (Fig. 3) (Vetter et al., 2002; Graham et al., 2010; Graham and Vetter, 2011). In order to begin unraveling the function(s) of the cochlear HPA-like signaling system, mice carrying null alleles for each CRF receptor have been used to examine in vivo the structural and functional phenotypes induced by constitutive loss of each CRF receptor gene (Graham et al., 2010; Graham and Vetter, 2011), thereby generating data on the functions involved for CRF signaling through these separate receptor systems. Additionally, using pharmacological activation of CRFR2 in an in vitro model of cochlear response to cytotoxic compounds, a role for CRF in protection metabolic insult is emerging (Basappa et al., 2010). While refinements to these approaches must still be made, these initial experiments begin to reveal roles for CRF activity at either of its receptors that is complementary to the other, further suggesting a homeostatic, sensitivity-modulating and metabolic buffering role for CRF signaling in the cochlea.
CRFR1 Function in the Cochlea
Decreased auditory sensitivity following loss of CRFR1 expression
Auditory brainstem response (ABR) measures in CRFR1 null mice revealed a 20-30dB deficit in auditory sensitivity across all frequencies (Fig. 5), along with slightly impaired cochlear mechanics indicated by a 5-10 dB elevation of distortion product otoacoustic emissions thresholds (Graham and Vetter, 2011). These results suggest a mixed, but predominantly inner hair cell based etiology. A number of mechanisms may induce the altered sensitivity observed, including defects in afferent neurotransmission and/or afferent innervation. In order to better understand some of the processes involved in CRFR1’s role in cochlear function, and the mechanisms by which loss of CRFR1 expression resulted in a decrease in sensitivity, experiments were carried out to examine whether biochemical and/or structural defects associated with afferent function occurred in the CRFR1 null mice.
Figure 5. ABR thresholds of CRFR1 and CRFR2 null mice compared to wild type controls.

Auditory brainstem response (ABR) thresholds were measured in wild type and CRFR1 null (top) and CRFR2 null (bottom) mice and plotted against ABRs from line-specific wild type controls. Symbols mark the average threshold observed at each frequency tested (5.66, 8, 16, 22.65, 32, 45.25 kHz) ± SEM. CRFR1 null exhibited a 20-30 decibel (dB) increase in ABR thresholds across all frequencies tested. CRFR2 null and wild type mice were born and raised in an acoustic attenuation chamber in cages on standard wire rack shelving. At approximately two months of age, baseline ABR thresholds were obtained and plotted as a function of stimulus frequency (solid lines). Mice were then exposed within 24hrs to the 8-16kHz 100dB sound for 2hrs. Two weeks post-exposure, mice were again tested for ABR thresholds, and post-trauma results (dashed lines) were plotted over baseline results obtained prior to exposure (solid lines). The average ABR threshold shift induced by noise exposure was computed, and a repeated measure ANOVA used to calculate statistical significance. On average, wild type mice underwent a 9dB threshold shift, while the CRFR2 null mice underwent an 18dB threshold shift (p<0.01). (top panel reprinted from (Graham and Vetter, 2011) with permission; bottom panel reprinted from (Graham et al., 2010) with permission)
Elimination of CRFR1 alters afferent fiber targeting, suggesting a role for CRF signaling in the development of dendrites of spiral ganglion cells
CRFR1 null mice exhibit an abnormal distribution of afferent synapses along the inner hair cells (Fig. 6). Both presynaptic ribbons and postsynaptic ganglion cell dendrites tend to cluster on the modiolar side of the inner hair cell (Graham and Vetter, 2011). CRF and CRFR1 have previously been linked to dendritic differentiation (Chen et al., 2004) and dendritic spine maintenance (Chen et al., 2008) in the hippocampus. Postsynaptic activity of ganglion cell fibers has been shown to correlate well with the face (modiolar or pillar) of the hair cell with which they make synaptic contact. For example, in the cat cochlea, synapses localized on the modiolar side of the hair cell exhibit low spontaneous rates and high response thresholds, while synapses localized on the pillar side exhibit high spontaneous rates and low response thresholds (Liberman, 1980; Liberman, 1982). Similar properties have been found relating fiber innervation of pillar/modiolar inner hair cell face and their physiological responses in the mouse cochlea (Taberner and Liberman, 2005). Therefore, the modiolar bias of synaptic distribution observed in CRFR1 null mice could indicate loss of low threshold (pillar side) fibers, with sparing of the high threshold (modiolar) fibers. This could then explain the high ABR thresholds observed in the CRFR1 null mice.
Figure 6. Elimination of CRFR1 leads to abnormal afferent fiber innervation to inner hair cells (IHC).

Afferent fibers and their most distal endings were visualized using antibodies directed against sodium-potassium ATPase α3 subunit (NKA, green). IHCs were immunolabeled using antibodies against myosin VI (red). Whole-mount sections of matched cochlear middle turns were imaged in CRFR1 wild type (top) and CRFR1 null (bottom) mice. Significant changes to hair cell width (modiolar to pillar along the y axis) are evident following loss of CRFR1 expression. In the CRFR1 null mice, afferent fibers (star) were found on all sides of the IHCs, and were decorated with immunoreactive puncta that outlined the termination of the fibers (double arrow in top panel). These fibers appeared stunted in CRFR1 null mice (bottom panel), with few endings reaching toward the pillar side of the inner hair cell, and none exhibiting the immunoreactive puncta observed in wild type mice. Additionally, moderate disorganization of the efferent fibers crossing the tunnel of Corti (arrows in top and bottom panels) and efferent innervation to the OHCs (single arrowhead) was also observed in the CRFR1 null mice. (reprinted from (Graham and Vetter, 2011) with permission)
Inner hair cells of the CRFR1 null mice were significantly smaller than those from homologous cochlear regions of wild type mice. While CRFR1 null mice possessed a normal number of synaptic ribbons in inner hair cells, the smaller soma induced a tighter packing of the ribbons. A smaller inner hair cell could lead to two main changes that could affect afferent physiology. The smaller size of the inner hair cell could alter the calcium micro-domains around the ribbons (Brandt et al., 2003; Moser et al., 2006a; Moser et al., 2006b). These microdomains could be so close to each other in the CRFR1 null inner hair cells that they interfere with function of neighboring ribbons, e.g. via changes in local calcium buffering around the ribbon, thereby perturbing the tight coupling between activity at a single ribbon, synchronous synaptic vesicle release (Graydon et al., 2011), and normal firing of a single ganglion cell dendrite. Since the ABR is the sum of synchronized responses of numerous ganglion cells synapsing with an individual inner hair cell, lack of sufficient spatial segregation of ribbons may result in abnormal recruitment of ribbon activity in response to inner hair cell depolarization, loss of postsynaptic response synchrony (Buran et al., 2010; Frank et al., 2010), and therefore decreased thresholds measured with ABR techniques. Secondly, with a decrease in inner hair cell size, the capacitance of the inner hair cell will certainly be altered, leading to changes in basic physiological behavior of the inner hair cells of the CRFR1 null mice.
Loss of CRFR1 expression results in decreased levels of glutamine synthetase expression
CRFR1 is expressed in border cells, a support cell population immediately adjacent to the medial face of inner hair cells. Thus, CRFR1 is well positioned to regulate communication between these support cells and the inner hair cells, potentially exerting an indirect influence over auditory afferent transduction. It has been proposed that various cochlear support cells interact with hair cells in a manner similar to astrocyte interactions with neurons in the central nervous system, and one potential interaction involves glutamate recycling via the glutamate-glutamine cycle (Ottersen et al., 1998; Rio et al., 2002). In the cochlear version of this cycle, glutamate is cleared from the synaptic cleft via the glutamate transporter GLAST, which is expressed by the border cell. Glutamate is then broken down to glutamine via the enzyme glutamine synthetase (GS). Glutamine is then shuttled back to the inner hair cell where it is used to synthesize glutamate. In this manner, excitotoxicity is avoided by sending the precursor of glutamate back to the IHC to ultimately replenish the glutamate neurotransmitter pool. There is a 50% reduction of cochlear GS levels in CRFR1 null mice compared to wild type mice, suggesting a deficiency of glutamate-glutamine cycling and potentially reflecting a reduced ability to convert glutamate taken up from the synapse into glutamine. In turn this deficiency may lead to decreased glutamate recycling in the inner hair cell and thus a rundown of neurotransmitter supply in the face of frequent stimulation. It is possible that such a rundown contributes the hearing deficiency observed in CRFR1 null mice. It should be noted, however, that GS expression was examined in whole cochlear lysates, and therefore may not reflect processes occurring specifically at the afferent synapse. Recent work demonstrated activity of the excitatory amino acid transporter GLAST in fibrocytes lining the lateral wall and suggests a role for these cells in regulating glutamate homeostasis (Furness et al., 2009). It follows that glutamine synthetase may also be expressed in these cells as well to regulate glutamate recycling. If so, the GS deficiency observed in CRFR1 null mice might reflect problems that extend beyond the local confines of the inner hair cell- ganglion cell afferent synapse, representing a more global role in cochlear glutamate homeostasis.
CRFR1 null mice are deficient in circulating corticosterone levels due to atrophy of their adrenal cortex (Smith et al., 1998). Therefore any glucocorticoid-dependent processes altered in the cochlea of CRFR1 null mice could result from either local or systemic glucocorticoid depletion. A glucocorticoid-response element is present on the promoter of the glutamine synthetase gene, suggesting that the reduced levels of GS observed in the CRFR1 null mice could result from deficient glucocorticoid activity (Vardimon et al., 1999). To distinguish whether or not the decrease in GS was a glucocorticoid-dependent effect, GS levels were evaluated in wild type and CRFR1 null mice following corticosterone presentation via drinking water. Glucocorticoid treatment beginning in utero via administration to pregnant dams rescued GS expression in CRFR1 null offspring (Graham and Vetter, 2011), indicating that CRFR1-mediated changes in cochlear GS expression depend on signaling through glucocorticoids. However, the endogenous source of the glucocorticoids (local or systemic) acting on the cochlear GS gene remains elusive. Most importantly, however, glucocorticoid administration did not rescue the auditory deficits generated by the loss of CRFR1 expression (Graham and Vetter, 2011). This data indicates that there is a distinction between what is controlled by steroid hormones (whether they be locally derived or products of the classic HPA system), and what is controlled by locally expressed ( cochlear) HPA-like signaling systems that may by-pass glucocorticoid synthesis/release, and rather may include more neural-like processes as the final output of system activity.
CRFR2 Function in the Cochlea
Following loss of CRFR2 expression, auditory sensitivity and susceptibility to noise-induced hearing loss are both enhanced
The ABR thresholds of CRFR2 null mice are 20-25dB lower than age matched wild type mice (Fig. 5), and distortion product otoacoustic emission (DP) thresholds are 10-15dB lower than wild types (Graham et al., 2010). The lower DP thresholds suggest enhanced cochlear amplification, contributed by outer hair cell activity, occurs in the CRFR2 null mice. However, since the degree of afferent hypersensitivity is almost twice that of the enhanced DPs, at least some portion of the hypersensitivity must originate from changes in afferent transduction and/or neurotransmission. Additionally, noise exposure was found to induce approximately twice the degree of permanent threshold shift compared to wild type mice (Graham et al., 2010). Since the ABR threshold shift following noise exposure is not accompanied by changes in distortion product threshold, a predominantly afferent mechanism most likely underlies the noise-induced hearing loss observed in the CRFR2 null mice. Remarkably, noise-induced threshold shifts (i.e. loss of sensitivity) in CRFR2 null mice occurred with sound intensities as low as 50dB (Graham et al., 2010), the intensity of quiet to moderate intensity human speech. Similar to the case with the CRFR1 null mice, data derived from the CRFR2 null mice suggested defects in afferent signaling. We therefore examined various components involved with afferent neurotransmission in the CRFR2 null mice, especially centered on the composition of the afferent glutamatergic neurotransmitter receptor.
Auditory environment alters GluR expression in CRFR2 null mice
Using immunofluorescence techniques, CRFR2 was localized to the afferent synaptic zone of inner hair cells (Graham et al., 2010). Afferent transmission in the cochlea is mediated by glutamate released from the inner hair cells binding to AMPA type glutamate receptors (GluR) expressed on the postsynaptic surface of spiral ganglion cell dendrites (Puel, 1995). With respect to AMPA class glutamate receptors, the mature cochlea expresses the GluR2, 3, and 4 receptor subunits (Eybalin et al., 2004). CRFR2 activity has been shown to alter GluR subunit expression and membrane insertion (Gounko et al., 2005), CRFR2 null mice express 50% less GluR2/3 than wild type mice but similar levels of GluR4 when reared under quiet conditions (sound attenuation chamber environments). When raised in an environment exposed to constant low to moderate level noise, CRFR2 null mice express similar levels of GluR2/3 as wild type mice but 80% more GluR4 (Graham et al., 2010).
Implications of Glutamate receptor 2 subunit expression changes in CRFR2 null mice
Little is known regarding the contribution of each type of AMPA receptor subunit to overall glutamatergic receptor function and afferent transmission/sensitivity in the cochlea. The majority of GluR2 subunits expressed in the mature rat cochlea is of the edited form and therefore, when incorporated into AMPA receptors, renders the GluR complex calcium impermeable (Eybalin et al., 2004). In other systems (such as the CNS), inclusion of edited GluR2 reduces conductance and slows kinetics of AMPA receptors (Swanson et al., 1997; Isaac et al., 2007). Reduced levels of GluR2/3 expressed by CRFR2 null mice under quiet conditions might reflect deficient expression of the GluR2 subunit (but this has yet to be directly tested). If this is the case, an increased pool of calcium-permeable AMPA receptors may reside within the spiral ganglion cell surface membrane. Such an enrichment of calcium permeable glutamatergic receptors can lead to the observed increase in afferent sensitivity. GluR2 trafficking is under the control of various signaling mechanisms. In particular, PKC-mediated signaling plays an integral role in activity-dependent recruitment of calcium-impermeable GluR2-containing AMPA receptors to the postsynaptic surface (reviewed in (Isaac et al., 2007). CRFR2 stimulates PKC signaling, and therefore a decrease in PKC signaling induced by the loss of CRFR2 activity could lead to deficient GluR2 recruitment to the postsynaptic membrane.
Changes in GluR expression in CRFR2 null mice may not stem from altered CRFR2 activity in spiral ganglion neurons. GluR2/3 expression changes could also represent a compensatory reaction. Given the similarities in GluR4 expression levels under quiet conditions between CRFR2 null and wild type mice, one may argue that loss of GluR2/3 expression represents a loss of AMPA receptors at the cell surface as compensation for increased presynaptic activity. Previous work has demonstrated a decrease in cell surface GluR2 expression on the spiral ganglion cells in response to excess sound in vivo or excess glutamate in vitro (Chen et al., 2007), presumably reflecting a decrease of surface AMPA receptors. If loss of cell surface expression is prevented through genetic hindrance of endocytosis, spiral ganglion cells are more susceptible to excitotoxic stress (Chen et al., 2009). Thus, the observed decrease in GluRs may represent a compensatory adjustment to over-activity occurring elsewhere in the afferent transduction chain. Alternatively, if decreased GluR2/3 levels reflect decreases specifically in GluR2, this reduction may result from an excitotoxic response that has already occurred (rather than an attempt to avoid such a response). Although paradoxical, ischemic or excitotoxic insult can repress GluR2 expression via activation of REST. This leads to expression of more calcium-permeable AMPA receptors (reviewed in (Isaac et al., 2007). If a similar phenomenon occurs in the cochlea, down regulation of GluR2/3 under quiet conditions may indicate an early stage of excitotoxic stress in CRFR2 null mice, and may also explain the greater susceptibility to loss of cochlear sensitivity following exposures to even moderate level sound intensities. Spiral ganglion cell counts in CRFR2 null mouse cochleae revealed no overt loss of postsynaptic cells ten days following noise exposure (Graham et al., 2010). However, afferent cell damage may initially be subtle, involving only loss of synaptic contacts that is followed by loss of ganglion cells over a period of months to years (Kujawa and Liberman, 2009), and this has not been examined thus far in the CRFR2 null mice.
Implications of Glutamate receptor 4 subunit expression changes in CRFR2 null mice
Wild type mice exhibit an increase in GluR4 expression (albeit, slight) under noise conditions that is concurrent with a dramatic drop in GluR2/3 levels. Under the same noise conditions, GluR4 expression increases 80% in CRFR2 null mice. The data from CRFR2 null mice suggests that the majority of AMPA receptors expressed under noise conditions contain GluR4, and may indicate the presence of GluR4 homomeric complexes. Although GluR4 homomeric receptors seem to be relatively rare, tonically active synapses between photoreceptors and second order cells in the salamander retina employ postsynaptic AMPA receptors composed only of GluR4 (Pang et al., 2008). GluR4 homomeric receptors undergo fast desensitization, but also are quick to recover following glutamatergic stimulation. Enrichment of surface membrane associated GluR4 subunits may be key for normal synaptic activity involving tonic stimulation. Like the retinal synapses, the afferent synapse in the cochlea is spontaneously active at all times. Perhaps the fast and sensitive desensitization kinetics of GluR4-enriched AMPA receptors limit postsynaptic current in the face of constant sound-associated neurotransmission and allows the cochlea to maintain a dynamic signaling range with tonic stimulation. By extension, the exaggerated increase in GluR4 observed in CRFR2 null mice under noise conditions could account for the observed hearing loss compared to wild type mice due to the desensitization kinetics of the receptor. It should be noted, however, this proposal contradicts work from other systems suggesting that GluR4 enhances postsynaptic currents and is therefore recruited for synaptic strengthening. In the early postnatal hippocampus, recruitment of GluR4 to silent synapses precedes recruitment of the other GluR subtypes and leads to a sustained increase in neuronal activity (Zhu et al., 2000). Similarly, studies comparing response properties of corticothalamic targets reveal that the neurons in the reticular nucleus exhibit a 2.6 fold larger post synaptic response to corticothalamic stimulation than relay neurons and this increase in activity correlates with enriched GluR4 subunit expression in the reticular nucleus (Golshani et al., 2001). These findings would suggest that the exaggerated elevation of GluR4 levels in CRFR2 null mice should lead to increased afferent activity under noise conditions. To reconcile these ideas, one may hypothesize an acute excitotoxic damage of the afferent synapses that would render them nonfunctional. Spiral ganglion cell counts obtained from CRFR2 null mice (Graham et al., 2010 ) have not revealed a loss of neurons, but the synaptic microstructure, including ribbons and postsynaptic contacts, was not examined. Thus, the exact consequence of CRFR2 control over GluR4 expression remains to be more completely described.
CRFR2 SIGNALING MAY REPRESENT A CELL STRESS-RESPONSE MECHANISM INVOLVED IN COCHLEAR PROTECTION
Data suggest that abnormal expression of CRFR1 and CRFR2 lead to altered auditory sensitivity and, following loss of CRFR2 activity, significantly greater susceptibility to noise-induced hearing loss. However, other kinds of insults, such as metabolic stresses, can also negatively impact cochlear function. If the cochlear HPA-equivalent signaling system is involved in homeostatic maintenance, then it should not only be involved in neural processes taking place in the cochlea, but also be involved with metabolic signaling to protect against the irretrievable loss of hair cells. Oxidative stress caused by accumulation of reactive oxygen species within cochlear tissue is considered to be a major mechanism underlying noise-induced damage to the cochlea that results in hearing loss. CRFR2 has an important anti-oxidative role in OCK-3 cells, an immortalized cochlear cell line. Cells treated with urocortin 2, a CRFR2-specific ligand, prior to incubation with H2O2 or the ototoxic aminoglycoside class antibiotic gentamicin (Fig. 7), underwent significantly less ROS production compared to control cells. The effect of urocortin 2 was blocked by CRFR2 antagonists (Basappa et al., 2010 ). Therefore, CRFR2 activity may prevent oxidative stress in cochlear cells and, as described in other systems, modulation of connexin expression may contribute to this antioxidant effect. Related to this, it has been shown that CRFR2 null mice express less connexin 26 and connexin 30 under both quiet and noise conditions than do wild type mice. Connexins are the protein subunits of gap junctions, and the avascular organ of Corti depends on exchange of nutrients and biochemical messengers through these gap junctions for normal function (Zhang et al., 2005; Chang et al., 2008). For example, Cx30 null mice exhibit impaired glucose transfer between support cells, leading to an increase in production of free radicals within the organ of Corti (Chang et al., 2008). Thus, deficient connexin expression in CRFR2 null mice may cause free radical 21 production due to insufficient nutrient exchange and it may also impair the ability to cope with these free radicals. This suggests one mechanism by which the greater susceptibility to noise-induced hearing loss, even at very mild noise levels, occurs in the CRFR2 null mice.
Figure 7. UcnII protects against gentamicin-induced ROS formation.
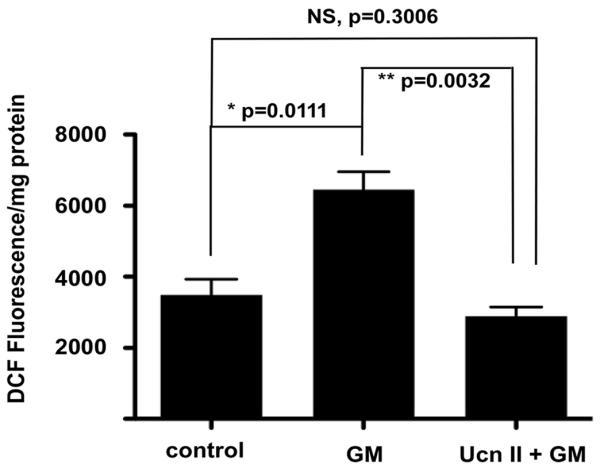
Gentamicin induces ROS generation (measured as DCF fluorescence intensity in the ROS assay) significantly greater than background (second bar). Pretreatment for one hour with UcnII, which specifically activates CRFR2, protects cells against gentamicin-induced ROS formation (third bar), and normalizes the ROS levels to control values. Reprinted from Basappa et al., 2010 with permission)
In addition to the role CRFR2 signaling plays in handling ototoxic drug-induced ROS accumulation, results have also shown that CRFR2 activity prevents aminoglycoside-induced caspase-3 activity (Basappa et al., 2010). Activity of typical cell death pathways therefore also seems to be inhibited by CRFR2 activity. Using iTRAQ-based mass spectrometry, a quantitative differential mass spectrometry approach, we further revealed the consequences following CRFR2 activation in OCK3 cochlear cells (Basappa et al., 2010). Protein expression was compared between gentamicin stimulated cells and CRFR2 pre-stimulated cells that were then challenged with a gentamicin application. In general, apoptosis-related proteins were up-regulated following exposure to gentamicin, but were down-regulated in cells that were first pre-treated to activate CRFR2 that were then treated with the same dose of gentamicin. Proteins involved in cell survival (for example, growth factors and proteins involved in cell-cell adhesion) and proteasome elements involved in protein degradation (presumably clearing misfolded proteins resulting from cellular stress) were up-regulated under CRFR2 pre-treatment followed by gentamicin challenge, while these proteins were down-regulated under gentamicin only exposure.
Various critical components of the AKT/PKB signaling pathway are altered following loss of CRFR2 expression (Graham et al., 2010). Basal AKT1 (protein) expression is approximately 60% lower in CRFR2 null mice in both quiet and noise conditions compared to wild type mice. AKT/PKB signaling is involved in a wide range of cellular functions and disease states (Hers et al., 2011), including control over apoptotic events (Zhang et al., 2011) and cellular protein synthesis (Shi et al., 2011) pathways. Thus, CRFR2 signaling is probably involved in modulating very basic cellular functions that have an impact on survival in the face of environmental and ototoxic drug challenges. CRF signaling in general may therefore represent a physiologically significant endogenous cellular stress-response system responsible for cochlear homeostasis.
In total, the data described above demonstrate the importance of recognizing the multiple mechanisms by which the cochlear HPA-equivalent signaling system can influence peripheral auditory processing and mechanisms involved in general homeostasis of the inner ear tissue. The data thus far collected suggest impacts on both neural aspects (via GluR expression as one example) of cochlear function and, potentially via local glucocorticoid release, response to cellular stress events important for cell survival.
TOWARD A MODEL OF CRF/HPA-LIKE SIGNALING IN THE COCHLEA
As touched upon briefly above, the skin locally expresses an ensemble of molecules previously recognized only along the classic HPA axis. The molecular signaling elements participate (and locally replicate) HPA-like signaling to maintain homeostasis via numerous mechanisms. We have recently discovered that the same molecules also exist in the cochlea. Although the state of research into the role of cochlear HPA-like signaling is only beginning, numerous results point to its complexity and its potential over-arching roles in maintaining the cochlea at a homeostatic balance. If the hypothesis is correct and the cochlear HPA-like signaling system represents a local response system untethered to (but perhaps still responsive to) the classic HPA axis, one nagging question, among many, remains to be resolved- what is the initiator for activation of the cochlear HPA-like system? Data from the CRFR2 nulls suggest that exposure to intense sound may somehow initiate CRF signaling through the CRFR2. But initiators of CRF release and CRFR1 activation have yet to be defined. Examining the skin for hints, it is clear that the environment can initiate CRF release and CRFR1 activation in that system. For the cochlea, this would again implicate sound-induced cellular stress, either in the form of metabolic or structural stress, as initiation signals. However, it is also well known that the immune response, via cytokine signaling, is also capable of initiating HPA-like responses (both in skin and in the classic HPA axis). Thus, while currently untested, it is possible that cytokine signaling brought about by immune reactions in the inner ear could play a role in inducing cochlear HPA-like signaling. These are all topics for future research, and carry with them the possibility of localized, rather than systemic, treatments for numerous inner ear maladies.
Neural signaling
The evidence for CRF-mediated neuromodulation in the cochlea builds upon a robust literature describing CRF as a neuromodulator in the central nervous system. In particular, CRF signaling exerts powerful affects on glutamatergic potentiation in the amygdala and hippocampus, leading to behaviors such as drug addiction, anxiety, and fear-associated learning and memory. There are three major avenues by which CRF signaling can influence afferent transduction and neural processing in the cochlea. First, CRFR2 expressed in the postsynaptic afferent dendrites could be stimulated by input from CRF expressed in and released from the inner hair cell as well as via autocrine signaling from the spiral ganglion cell fibers, and finally from urocortin expressed in and released from the lateral efferent terminals synapsing on the afferent dendrites. The net effect of CRFR2 activation, as demonstrated by results from the CRFR2 null mice, is to modulate GluR expression and/or membrane insertion in the postsynaptic ganglion cell. Under normal conditions, this ultimately leads to a decrease in acoustic sensitivity and a resistance to noise-induced hearing loss. Second, CRFR1 expressed in the border cell next to the presynaptic inner hair cell is in position to receive input from CRF expressed in and released from the inner hair cell and the spiral ganglion cells (and possibly urocortin from the lateral efferents). CRFR1 activation in the border cells can lead to increased expression of glutamine synthetase, thereby sustaining the glutamate-glutamine cycle between inner hair cells and their neighboring support cells. Based on the phenotype resulting from loss of CRFR1 expression, the net effect of normal CRFR1 activation is an increase in acoustic sensitivity. Third, MC2 receptors expressed by the inner hair cell could receive autocrine input from ACTH expressed in the inner hair cell as well as paracrine input from ACTH expressed in more distant support cells. ACTH can exert a variety of effects on the inner hair cell by signaling through MC2R and the cAMP pathway. One potential effect is modulation of BK potassium channel activity. BK channels are a major regulator of presynaptic afferent transduction by the inner hair cells. This potassium channel is important for inducing membrane potential hyperpolarization in response to calcium and voltage increases, and therefore helps to repolarize cells following excitation. In the cochlea, BK activity is crucial for enabling repetitive firing in response to sustained stimuli (Fettiplace and Fuchs, 1999). Intriguingly, experiments demonstrate a reciprocal activity between BK and HPA signaling involving both transcriptional and post-translational modifications in BK channel activity in other tissues. In pituitary corticotropes, CRF and ACTH signal through PKA to inhibit BK activity, thereby enhancing cell excitability and consequent ACTH release (Shipston et al., 1996; Brunton et al., 2007). Conversely, glucocorticoids counter this inhibition via fast, non-genomic mechanisms to hold cells in a hyperpolarized state and prevent ACTH secretion. In theory, activation of MC2R also leads to glucocorticoid synthesis, and the resulting glucocorticoids can then stimulate numerous targets within the cochlea, particularly the spiral ganglion cells and the inner border cell. Synergistically, ACTH released from the inner hair cell could bind MC2R in spiral ganglion cells and exert effects there including local glucocorticoid activity.
Support cell signaling
The evidence for CRF signaling in cochlear support cells add to the otherwise limited knowledge of the roles in auditory function and protection of this large, heterogeneous cell population. In particular, evidence points to a molecular mechanism for maintenance of cochlear homeostasis via support cell function, and their role in protecting against cellular stress. This finding compliments earlier descriptions of calcium waves that extend through support cell networks in response to hair cell damage. It has been suggested that these calcium waves represent a cell-stress signal that is communicated across the region of injury (Gale et al., 2004; Piazza et al., 2007). Given that CRF represents the quintessential stress signal of the body, the demonstration of a CRF based HPA-equivalent signaling system in cochlear support cells advances ideas concerning mechanisms of action related to the major role these cells have in responding to stress. Understanding the nature of this stress response may provide useful knowledge of endogenous mechanisms conferring resistance to NIHL, and may therefore lead to novel methods and therapeutic strategies for prevention of hearing loss.
There are two major ways in which CRF signaling can influence support cell activity. First, CRF signaling modulates connexin expression, potentially leading to changes in ATP release through connexin hemi-channels, and in potassium recycling from perilymphatic compartments back to the scala media, which contains the potassium rich endolymph. In the adult cochlea, CRFR2-induced changes in Cx26 and Cx30 expression (Graham et al., 2010) may be important for suppressing endocochlear potential, thereby dulling sensitivity to sound, and also in mounting a response against oxidative stress. Second, CRF signaling may regulate HPA components expressed in support cells. In particular, POMC and ACTH co-localize with CRFR1 in both the inner sulcus cells and the lateral support cells (Fig. 4). The complete role(s) of this HPA-equivalent signaling system in the cochlea remain to be fully elucidated; however we propose that it represents a local stress response system similar to that observed in the skin (Slominski et al., 1999; Slominski et al., 2000). The localization of the ACTH receptor, MC2R, to the inner hair cell and spiral ganglion cell suggests that the majority of support cell HPA signaling converges on the afferent system to influence activity there.
CONCLUSION
The revelation of a CRF-based, HPA-like signaling system expression in the cochlea opens new frontiers in describing mechanisms involved in normal and pathological auditory processing. The experiments recently presented demonstrate the existence of numerous previously unrecognized molecules in the cochlea that are positioned in a way that suggests their involvement with many cochlear processes, and may help define new cellular interactions not previously suggested or imagined. The work also highlights the need for developing new methodologies to be employed in further investigating the role of the cochlear HPA-equivalent system. Such methods need to define the intracellular signaling pathways of specific cochlear cells influenced by the cochlear HPA system, and robust quantitative methods to further define changes not only to general protein expression levels at both the organ and individual cell/cell population, but also to phosphorylation state changes as we have begun to outline above. Revelation of an HPA-equivalent signaling system expressed in the cochlea forces a re-evaluation of the mechanisms whereby the cochlea acquires and maintains homeostatic balance in the face of constant stimulation. These mechanisms likely include both autonomous functions in the face of cellular stress, as well as feedback from higher neural centers. Finally, discovery of a cochlear HPA-equivalent signaling system suggests the need for a reinterpretation of the cochlea as a complex neuroendocrine system capable of controlling a wide range of processes that may alter cochlear function over relatively long timeframes, thereby proactively protecting the delicate structures of the inner ear not only from present dangers, but, given the kinds of signaling involved, also from potential cell stress events in the near future. We propose that the cochlear HPA-equivalent signaling system represents a dynamically active, locally distributed neuroendocrine response system allowing the cochlea to monitor, coordinate and calibrate cellular responses to acoustic stimuli, thereby maintaining a homeostatic norm.
Highlights.
hypothalamic-pituitary-adrenal axis-like signaling system defined in the mammalian cochlea
CRFR1 nulls exhibit increased ABR thresholds
CRFR2 nulls exhibit increased sensitivity (lower ABR thresholds)
CRFR2 nulls increased susceptibility to noise-induced hearing loss
Acknowledgements
We wish to acknowledge Dr. M.C. Liberman for his generous support in granting access to and use of his auditory physiology set-up for all of the studies described in this review, Dr. Paul Sawchenko for providing CRFR1-GFP transgenic mice, Dr. Carlos Arias for skillful assistance with the CRFR1-GFP mice, and Dr. Kuo-Fen Lee and the late Dr. Wylie Vale for providing initial CRFR1 and CRFR2 null lines used to generate the mice used in these studies. This work was funded by NIH R01DC006258 (to DEV), The Richard and Susan Smith Family Foundation (to DEV), and research funding from Univ. Mississippi Medical Center (to DEV). The reader is advised that some figures and portions of text have appeared in previous publications of the authors (Basappa et al., 2010; Graham et al., 2010; Graham et al., 2011; Graham and Vetter, 2011), and are reproduced in this invited review with permission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bale T, Contarino A, Smith G, Chan R, Gold L, Sawchenko P, Koob G, Vale W, Lee K. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- Basappa J, Turcan S, Vetter DE. Corticotropin-releasing factor-2 activation prevents gentamicin-induced oxidative stress in cells derived from the inner ear. J Neurosci Res. 2010;88:2976–2990. doi: 10.1002/jnr.22449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg M, Hafidi A, Skinner L, Ruel J, Nouvian R, Henaff M, Puel J, Aran J, Dulon D. Ryanodine receptors and BK channels act as a presynaptic depressor of neurotransmission in cochlear inner hair cells. The European journal of neuroscience. 2005;22:1109–1119. doi: 10.1111/j.1460-9568.2005.04310.x. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunton PJ, Sausbier M, Wietzorrek G, Sausbier U, Knaus HG, Russell JA, Ruth P, Shipston MJ. Hypothalamic-Pituitary-Adrenal Axis Hyporesponsiveness to Restraint Stress in Mice Deficient for Large-Conductance Calcium- and Voltage-Activated Potassium (BK) Channels. Endocrinology. 2007;148:5496–5506. doi: 10.1210/en.2007-0319. [DOI] [PubMed] [Google Scholar]
- Buran BN, Strenzke N, Neef A, Gundelfinger ED, Moser T, Liberman MC. Onset coding is degraded in auditory nerve fibers from mutant mice lacking synaptic ribbons. J Neurosci. 2010;30:7587–7597. doi: 10.1523/JNEUROSCI.0389-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canlon B, Fransson A. Morphological and functional preservation of the outer hair cells from noise trauma by sound conditioning. Hearing research. 1995;84:112–124. doi: 10.1016/0378-5955(95)00020-5. [DOI] [PubMed] [Google Scholar]
- Canlon B, Borg E, Flock A. Protection against noise trauma by pre-exposure to a low level acoustic stimulus. Hearing research. 1988;34:197–200. doi: 10.1016/0378-5955(88)90107-4. [DOI] [PubMed] [Google Scholar]
- Canlon B, Meltser I, Johansson P, Tahera Y. Glucocorticoid receptors modulate auditory sensitivity to acoustic trauma. Hearing research. 2007;226:61–69. doi: 10.1016/j.heares.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Chang Q, Tang W, Ahmad S, Zhou B, Lin X. Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PloS One. 2008;3:e4088. doi: 10.1371/journal.pone.0004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dube CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, Baram TZ. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci (USA) 2004;101:15782–15787. doi: 10.1073/pnas.0403975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Kujawa SG, Sewell WF. Auditory sensitivity regulation via rapid changes in expression of surface AMPA receptors. Nature neuroscience. 2007;10:1238–1240. doi: 10.1038/nn1974. [DOI] [PubMed] [Google Scholar]
- Chen Z, Peppi M, Kujawa SG, Sewell WF. Regulated expression of surface AMPA receptors reduces excitotoxicity in auditory neurons. J Neurophysiol. 2009;102:1152–1159. doi: 10.1152/jn.00288.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrat I, Ahmad N, Seidman K, Seidman MD. Auditory research involving antioxidants. Curr Opin Otolaryngol Head Neck Surg. 2007;15:358–363. doi: 10.1097/MOO.0b013e3282efa641. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–715. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- Eybalin M, Caicedo A, Renard N, Ruel J, Puel JL. Transient Ca2+-permeable AMPA receptors in postnatal rat primary auditory neurons. The European journal of neuroscience. 2004;20:2981–2989. doi: 10.1111/j.1460-9568.2004.03772.x. [DOI] [PubMed] [Google Scholar]
- Fettiplace R, Fuchs PA. Mechanisms of hair cell tuning. Annual review of physiology. 1999;61:809–834. doi: 10.1146/annurev.physiol.61.1.809. [DOI] [PubMed] [Google Scholar]
- Flint MS, Tinkle SS. C57BL/6 mice are resistant to acute restraint modulation of cutaneous hypersensitivity. Toxicol Sci. 2001;62:250–256. doi: 10.1093/toxsci/62.2.250. [DOI] [PubMed] [Google Scholar]
- Frank T, Rutherford MA, Strenzke N, Neef A, Pangrsic T, Khimich D, Fejtova A, Gundelfinger ED, Liberman MC, Harke B, Bryan KE, Lee A, Egner A, Riedel D, Moser T. Bassoon and the synaptic ribbon organize Ca(2)+ channels and vesicles to add release sites and promote refilling. Neuron. 2010;68:724–738. doi: 10.1016/j.neuron.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Lawton DM, Mahendrasingam S, Hodierne L, Jagger DJ. Quantitative analysis of the expression of the glutamate-aspartate transporter and identification of functional glutamate uptake reveal a role for cochlear fibrocytes in glutamate homeostasis. Neuroscience. 2009;162:1307–1321. doi: 10.1016/j.neuroscience.2009.05.036. [DOI] [PubMed] [Google Scholar]
- Gale JE, Piazza V, Ciubotaru CD, Mammano F. A mechanism for sensing noise damage in the inner ear. Curr Biol. 2004;14:526–529. doi: 10.1016/j.cub.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4- subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci (USA) 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounko N, Rybakin V, Kalicharan D, Siskova Z, Gramsbergen A, van der Want J. CRF and urocortin differentially modulate GluRdelta2 expression and distribution in parallel fiber-Purkinje cell synapses. Mol Cell Neurosci. 2005;30:513–522. doi: 10.1016/j.mcn.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Graham CE, Vetter DE. The mouse cochlea expresses a local hypothalamic-pituitary-adrenal equivalent signaling system and requires corticotropin-releasing factor receptor 1 to establish normal hair cell innervation and cochlear sensitivity. J Neurosci. 2011;31:1267–1278. doi: 10.1523/JNEUROSCI.4545-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham CE, Basappa J, Vetter DE. A corticotropin-releasing factor system expressed in the cochlea modulates hearing sensitivity and protects against noise-induced hearing loss. Neurobiol Dis. 2010;38:246–258. doi: 10.1016/j.nbd.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham CE, Basappa J, Turcan S, Vetter DE. The cochlear CRF signaling systems and their mechanisms of action in modulating cochlear sensitivity and protection against trauma. Mol Neurobiol. 2011;44:383–406. doi: 10.1007/s12035-011-8203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graydon CW, Cho S, Li GL, Kachar B, von Gersdorff H. Sharp Ca(2) nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:16637–16650. doi: 10.1523/JNEUROSCI.1866-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkin RI, Knigge KM. Effect of sound on the hypothalamic-pituitary-adrenal axis. Am J Physiol. 1963;204:701–704. doi: 10.1152/ajplegacy.1963.204.4.710. [DOI] [PubMed] [Google Scholar]
- Henkin RI, McGlone RE, Daly R, Bartter FC. Studies on auditory thresholds in normal man and in patients with adrenal cortical insufficiency: the role of adrenal cortical steroids. The Journal of clinical investigation. 1967;46:429–435. doi: 10.1172/JCI105544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Himeno C, Komeda M, Izumikawa M, Takemura K, Yagi M, Weiping Y, Doi T, Kuriyama H, Miller JM, Yamashita T. Intra-cochlear administration of dexamethasone attenuates aminoglycoside ototoxicity in the guinea pig. Hearing research. 2002;167:61–70. doi: 10.1016/s0378-5955(02)00345-3. [DOI] [PubMed] [Google Scholar]
- Hoang Dinh E, Ahmad S, Chang Q, Tang W, Stong B, Lin X. Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain Res. 2009;1277:52–69. doi: 10.1016/j.brainres.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley GD, Marcotti W, Navaratnam D, Yamoah EN. Hair cells--beyond the transducer. The Journal of membrane biology. 2006;209:89–118. doi: 10.1007/s00232-005-0835-7. [DOI] [PubMed] [Google Scholar]
- Housley GD, Jagger DJ, Greenwood D, Raybould NP, Salih SG, Jarlebark LE, Vlajkovic SM, Kanjhan R, Nikolic P, Munoz DJ, Thorne PR. Purinergic regulation of sound transduction and auditory neurotransmission. Audiol Neurootol. 2002;7:55–61. doi: 10.1159/000046865. [DOI] [PubMed] [Google Scholar]
- Housley GD, Kanjhan R, Raybould NP, Greenwood D, Salih SG, Jarlebark L, Burton LD, Setz VC, Cannell MB, Soeller C, Christie DL, Usami S, Matsubara A, Yoshie H, Ryan AF, Thorne PR. Expression of the P2X(2) receptor subunit of the ATP-gated ion channel in the cochlea: implications for sound transduction and auditory neurotransmission. J Neurosci. 1999;19:8377–8388. doi: 10.1523/JNEUROSCI.19-19-08377.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Koakoski G, Oliveira TA, da Rosa JG, Fagundes M, Kreutz LC, Barcellos LJ. Divergent time course of cortisol response to stress in fish of different ages. Physiology & behavior. 2012;106:129–132. doi: 10.1016/j.physbeh.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Konig S, Luger TA, Scholzen TE. Monitoring neuropeptide-specific proteases: processing of the proopiomelanocortin peptides adrenocorticotropin and alpha-melanocyte-stimulating hormone in the skin. Experimental dermatology. 2006;15:751–761. doi: 10.1111/j.1600-0625.2006.00472.x. [DOI] [PubMed] [Google Scholar]
- Kuhn M, Heman-Ackah SE, Shaikh JA, Roehm PC. Sudden Sensorineural Hearing Loss: A Review of Diagnosis, Treatment, and Prognosis. Trends Amplif. 2011 doi: 10.1177/1084713811408349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci. 2006;26:2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai GJ, McCobb DP. Opposing actions of adrenal androgens and glucocorticoids on alternative splicing of Slo potassium channels in bovine chromaffin cells. Proc Natl Acad Sci (USA) 2002;99:7722–7727. doi: 10.1073/pnas.112619799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecain E, Yang TH, Tran Ba Huy P. Steroidogenic enzyme expression in the rat cochlea. Acta oto-laryngologica. 2003;123:187–191. doi: 10.1080/0036554021000028106. [DOI] [PubMed] [Google Scholar]
- Liberman M. Single-neuron labeling in the cat auditory nerve. Science. 1982;216:1239–1241. doi: 10.1126/science.7079757. New York, NY. [DOI] [PubMed] [Google Scholar]
- Liberman MC. Morphological differences among radial afferent fibers in the cat cochlea: an electron-microscopic study of serial sections. Hearing research. 1980;3:45–63. doi: 10.1016/0378-5955(80)90007-6. [DOI] [PubMed] [Google Scholar]
- Lusk SL, Hagerty BM, Gillespie B, Caruso CC. Chronic effects of workplace noise on blood pressure and heart rate. Arch Environ Health. 2002;57:273–281. doi: 10.1080/00039890209601410. [DOI] [PubMed] [Google Scholar]
- Maeda K, Yoshida K, Ichimiya I, Suzuki M. Dexamethasone inhibits tumor necrosis factor-alpha-induced cytokine secretion from spiral ligament fibrocytes. Hearing research. 2005;202:154–160. doi: 10.1016/j.heares.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Maison SF, Liberman MC. Predicting vulnerability to acoustic injury with a noninvasive assay of olivocochlear reflex strength. J Neurosci. 2000;20:4701–4707. doi: 10.1523/JNEUROSCI.20-12-04701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Luebke AE, Liberman MC, Zuo J. Efferent protection from acoustic injury is mediated via alpha9 nicotinic acetylcholine receptors on outer hair cells. J Neurosci. 2002;22:10838–10846. doi: 10.1523/JNEUROSCI.22-24-10838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden S, Ohlemiller K, Ding D, Shero M, Salvi R. The Influence of Superoxide Dismutase and Glutathione Peroxidase Deficiencies on Noise-Induced Hearing Loss in Mice. Noise Health. 2001;3:49–64. [PubMed] [Google Scholar]
- Monge Naldi A, Gassmann M, Bodmer D. Erythropoietin but not VEGF has a protective effect on auditory hair cells in the inner ear. Cell Mol Life Sci. 2009;66:3595–3599. doi: 10.1007/s00018-009-0144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Brandt A, Lysakowski A. Hair cell ribbon synapses. Cell Tissue Res. 2006a;326:347–359. doi: 10.1007/s00441-006-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Neef A, Khimich D. Mechanisms underlying the temporal precision of sound coding at the inner hair cell ribbon synapse. The Journal of physiology. 2006b;576:55–62. doi: 10.1113/jphysiol.2006.114835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima R, Ogita K. Enhanced biosynthesis of glutathione in the spiral ganglion of the cochlea after in vivo treatment with dexamethasone in mice. Brain Res. 2006;1117:101–108. doi: 10.1016/j.brainres.2006.07.113. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Takumi Y, Matsubara A, Landsend AS, Laake JH, Usami S. Molecular organization of a type of peripheral glutamate synapse: the afferent synapses of hair cells in the inner ear. Prog Neurobiol. 1998;54:127–148. doi: 10.1016/s0301-0082(97)00054-3. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Gao F, Barrow A, Jacoby RA, Wu SM. How do tonic glutamatergic synapses evade receptor desensitization? The Journal of physiology. 2008;586:2889–2902. doi: 10.1113/jphysiol.2008.151050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppi M, Kujawa SG, Sewell WF. A corticosteroid-responsive transcription factor, promyelocytic leukemia zinc finger protein, mediates protection of the cochlea from acoustic trauma. J Neurosci. 2011;31:735–741. doi: 10.1523/JNEUROSCI.3955-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza V, Ciubotaru CD, Gale JE, Mammano F. Purinergic signalling and intercellular Ca2+ wave propagation in the organ of Corti. Cell Calcium. 2007;41:77–86. doi: 10.1016/j.ceca.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WH. Metabolic aspects of Meniere’s disease. Laryngoscope. 1972;82:1716–1725. doi: 10.1288/00005537-197209000-00012. [DOI] [PubMed] [Google Scholar]
- Puel JL. Chemical synaptic transmission in the cochlea. Prog Neurobiol. 1995;47:449–476. doi: 10.1016/0301-0082(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Rasmussen GL. An efferent cochlear bundle. Anat Rec. 1942;82:441. [Google Scholar]
- Rasmussen GL. Descending, or “feed-back” connections of the auditory system of the cat. Am J Physiol. 1955;183:653. [Google Scholar]
- Rauch SD, Halpin CF, Antonelli PJ, Babu S, Carey JP, Gantz BJ, Goebel JA, Hammerschlag PE, Harris JP, Isaacson B, Lee D, Linstrom CJ, Parnes LS, Shi H, Slattery WH, Telian SA, Vrabec JT, Reda DJ. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. Jama. 2011;305:2071–2079. doi: 10.1001/jama.2011.679. [DOI] [PubMed] [Google Scholar]
- Rio C, Dikkes P, Liberman MC, Corfas G. Glial fibrillary acidic protein expression and promoter activity in the inner ear of developing and adult mice. The Journal of comparative neurology. 2002;442:156–162. doi: 10.1002/cne.10085. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine reviews. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Shi X, Li X, Wang Y, Zhang K, Zhou F, Chan L, Li D, Guan X. Glucagon-like peptide-2-stimulated protein synthesis through the PI 3-kinase-dependent Akt-mTOR signaling pathway. American journal of physiology Endocrinology and metabolism. 2011;300:E554–563. doi: 10.1152/ajpendo.00620.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki T, Ichimiya I, Suzuki M, Mogi G. Localization of glucocorticoid receptors in the murine inner ear. The Annals of otology, rhinology, and laryngology. 2002;111:1133–1138. doi: 10.1177/000348940211101213. [DOI] [PubMed] [Google Scholar]
- Shipston MJ, Kelly JS, Antoni FA. Glucocorticoids block protein kinase A inhibition of calcium-activated potassium channels. J Biol Chem. 1996;271:9197–9200. doi: 10.1074/jbc.271.16.9197. [DOI] [PubMed] [Google Scholar]
- Skinner L, Enée V, Beurg M, Jung H, Ryan A, Hafidi A, Aran J, Dulon D. Contribution of BK Ca2+-activated K+ channels to auditory neurotransmission in the Guinea pig cochlea. Journal of neurophysiology. 2003;90:320–332. doi: 10.1152/jn.01155.2002. [DOI] [PubMed] [Google Scholar]
- Slominski A. Neuroendocrine system of the skin. Dermatology. 2005;211:199–208. doi: 10.1159/000087012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski A, Baker J, Ermak G, Chakraborty A, Pawelek J. Ultraviolet B stimulates production of corticotropin releasing factor (CRF) by human melanocytes. FEBS Lett. 1996;399:175–176. doi: 10.1016/s0014-5793(96)01315-4. [DOI] [PubMed] [Google Scholar]
- Slominski A, Wortsman J, Luger T, Paus R, Solomon S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiological reviews. 2000;80:979–1020. doi: 10.1152/physrev.2000.80.3.979. [DOI] [PubMed] [Google Scholar]
- Slominski AT, Botchkarev V, Choudhry M, Fazal N, Fechner K, Furkert J, Krause E, Roloff B, Sayeed M, Wei E, Zbytek B, Zipper J, Wortsman J, Paus R. Cutaneous expression of CRH and CRH-R. Is there a “skin stress response system?”. Annals of the New York Academy of Sciences. 1999;885:287–311. doi: 10.1111/j.1749-6632.1999.tb08686.x. [DOI] [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee K-F. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Stevens A, White A. ACTH: cellular peptide hormone synthesis and secretory pathways. Results Probl Cell Differ. 2010;50:63–84. doi: 10.1007/400_2009_30. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner AM, Liberman MC. Response properties of single auditory nerve fibers in the mouse. Journal of neurophysiology. 2005;93:557–569. doi: 10.1152/jn.00574.2004. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Oikawa K, Murashita H, Hoshino T, Tsuji S, Hara A. Protective effects of glucocorticoids on ischemia-reperfusion injury of outer hair cells. Laryngoscope. 2006;116:627–629. doi: 10.1097/01.mlg.0000200963.69342.d7. [DOI] [PubMed] [Google Scholar]
- Tahera Y, Meltser I, Johansson P, Salman H, Canlon B. Sound conditioning protects hearing by activating the hypothalamic-pituitary-adrenal axis. Neurobiol Dis. 2007;25:189–197. doi: 10.1016/j.nbd.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Takemura K, Komeda M, Yagi M, Himeno C, Izumikawa M, Doi T, Kuriyama H, Miller JM, Yamashita T. Direct inner ear infusion of dexamethasone attenuates noise-induced trauma in guinea pig. Hearing research. 2004;196:58–68. doi: 10.1016/j.heares.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs PA, Elgoyhen AB. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol. 2009;7:e18. doi: 10.1371/journal.pbio.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Cate WJ, Monder C, Marandici A, Rarey KE. 11 beta-Hydroxysteroid dehydrogenase in the rat inner ear. Am J Physiol. 1994;266:E269–273. doi: 10.1152/ajpendo.1994.266.2.E269. [DOI] [PubMed] [Google Scholar]
- Terakado M, Kumagami H, Takahashi H. Distribution of glucocorticoid receptors and 11 beta-hydroxysteroid dehydrogenase isoforms in the rat inner ear. Hearing research. 2011 doi: 10.1016/j.heares.2011.05.006. in press. [DOI] [PubMed] [Google Scholar]
- Tian L, Duncan RR, Hammond MS, Coghill LS, Wen H, Rusinova R, Clark AG, Levitan IB, Shipston MJ. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem. 2001;276:7717–7720. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- Vardimon L, Ben-Dror I, Avisar N, Oren A, Shiftan L. Glucocorticoid control of glial gene expression. J Neurobiol. 1999;40:513–527. doi: 10.1002/(sici)1097-4695(19990915)40:4<513::aid-neu8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kolman J, Heinemann SF, Elgoyhen AB. Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron. 1999;23:93–103. doi: 10.1016/s0896-6273(00)80756-4. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Li C, Zhao L, Contarino A, Liberman MC, Smith GW, Marchuk Y, Koob GF, Heinemann SF, Vale W, Lee K-F. Urocortin-deficient mice show hearing impairment and increased anxiety-like behavior. Nat Genet. 2002;31:363–369. doi: 10.1038/ng914. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liberman MC. Restraint stress and protection from acoustic injury in mice. Hearing research. 2002;165:96–102. doi: 10.1016/s0378-5955(02)00289-7. [DOI] [PubMed] [Google Scholar]
- Yamasoba T, Dolan DF, Miller JM. Acquired resistance to acoustic trauma by sound conditioning is primarily mediated by changes restricted to the cochlea, not by systemic responses. Hearing research. 1999;127:31–40. doi: 10.1016/s0378-5955(98)00178-6. [DOI] [PubMed] [Google Scholar]
- Yoshida N, Liberman MC. Sound conditioning reduces noise-induced permanent threshold shift in mice. Hearing research. 2000;148:213–219. doi: 10.1016/s0378-5955(00)00161-1. [DOI] [PubMed] [Google Scholar]
- Yoshida N, Kristiansen A, Liberman MC. Heat stress and protection from permanent acoustic injury in mice. J Neurosci. 1999;19:10116–10124. doi: 10.1523/JNEUROSCI.19-22-10116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochimica et biophysica acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci (USA) 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nature neuroscience. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]