

Abstract
Oxidation products of 1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphatidylcholine (PAPC), referred to as OxPAPC, and an active component, 1-palmitoyl-2-(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphatidylcholine (PEIPC), accumulate in atherosclerotic lesions and regulate over 1,000 genes in human aortic endothelial cells (HAEC). We previously demonstrated that OxPNB, a biotinylated analog of OxPAPC, covalently binds to a number of proteins in HAEC. The goal of these studies was to gain insight into the binding mechanism and determine whether binding regulates activity. In whole cells, N-acetylcysteine inhibited gene regulation by OxPAPC, and blocking cell cysteines with N-ethylmaleimide strongly inhibited the binding of OxPNB to HAEC proteins. Using MS, we demonstrate that most of the binding of OxPAPC to cysteine is mediated by PEIPC. We also show that OxPNB and PEIPE-NB, the analog of PEIPC, bound to a model protein, H-Ras, at cysteines previously shown to regulate activity in response to 15-deoxy-Δ12,14-prostaglandin J2 (15dPGJ2). This binding was observed with recombinant protein and in cells overexpressing H-Ras. OxPAPC and PEIPC compete with OxPNB for binding to H-Ras. 15dPGJ2 and OxPAPC increased H-Ras activity at comparable concentrations. Using microarray analysis, we demonstrate a considerable overlap of gene regulation by OxPAPC, PEIPC, and 15dPGJ2 in HAEC, suggesting that some effects attributed to 15dPGJ2 may also be regulated by PEIPC because both molecules accumulate in inflammatory sites. Overall, we provide evidence for the importance of OxPAPC-cysteine interactions in regulating HAEC function.
Keywords: Oxidized phospholipid, endothelial cells, gene regulation, atherosclerosis, inflammation, oxidative stress, covalent binding, microarray, mass spectrometry
Minimally modified LDL (mm-LDL) was demonstrated by our group to stimulate endothelial cells, resulting in the recruitment of monocytes to the vascular wall, an important initial event in the early stages of atherogenesis (1). We have subsequently shown that several biologically active components of mm-LDL are the oxidation products of 1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphatidylcholine (PAPC), a naturally occurring phospholipid found in cell membranes and lipoproteins (2). OxPAPC was also shown to regulate over 1,000 genes in human aortic endothelial cells (HAEC) (3). Although our group and others have demonstrated that OxPAPC activates several signaling pathways in HAECs, the primary event in OxPAPC signaling has not been determined. The goal of this study was to gain insight into this primary event.
In addition to our studies showing strong activity of OxPAPC in regulating gene expression in endothelial cells, we have shown that a biotinylated analog of OxPAPC, OxPAPE-N-biotin (OxPNB), covalently binds to a group of endothelial cell proteins and contains similar oxidation products (4). We hypothesized that this covalent binding plays a role in some aspects of OxPAPC action. For these studies, we developed methods to synthesize and oxidize PAPE-N-biotin to produce OxPNB. We subsequently demonstrated that OxPNB regulates a number of genes that are regulated by OxPAPC, suggesting that OxPNB is an appropriate analog of OxPAPC. We demonstrated covalent binding of OxPNB to a group of proteins in HAECs (4). However, we previously had not determined the mechanism of the covalent binding by OxPAPC, including which lipid is responsible for this binding and which amino acid is bound by OxPAPC. We hypothesize that a primary mechanism of this binding is the interaction of electrophilic OxPAPC components, such as 1-palmitoyl-2-(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphatidylcholine (PEIPC), with available cysteines on endothelial proteins, and we address this mechanism in this article.
PEIPC is the most bioactive component in OxPAPC with respect to gene regulation in HAECs, and it is active at the lowest concentration of the eight OxPAPC lipids that we have tested in several assays (3). This lipid is formed by the free radical oxidation and cyclization of the arachidonic acid group of PAPC, resulting in a 5,6-expoxyisoprostane. PEIPC has an electrophilic α,β-unsaturated enone group in the sn-2 position, which is capable of Michael addition with nucleophilic amino acid residues like cysteine and lysine (Fig. 1). We hypothesize that PEIPC interacts with cysteines on proteins, and that this interaction of PEIPC is an important mechanism in the action of OxPAPC on the endothelium. Although the structure of PEIPC suggests that its interaction with cysteines is important, there are several reasons why this may not be true. In a recent publication by Gao et al., it is reported that for two other bioactive, α,β-unsaturated enone-containing oxidized phos-pholipids, KOdiaPC and KDdiaPC, the α,β-unsaturation does not play a major role in the interaction of these phospholipids with CD36 (5). Furthermore, we previously demonstrated that the epoxide group of PEIPC bound strongly to a model amine-containing compound (used as a surrogate compound for peptides containing lysine) (6), and we had not previously confirmed the binding of PEIPC to cysteine.
Fig. 1.

Molecular structures of (A) PEIPC and (B) 15-deoxy-Δ12,14-prostaglandin J2. The α, β carbons are shown, which are important in covalent binding.
To address the possibility of OxPAPC-cysteine interactions playing a role in OxPAPC action, we employed OxPNB. We examined the binding of OxPNB and its constituents to total HAEC protein and to a model protein, H-Ras. Ras is a central protein in cell signaling in inflammatory pathways, including the activation of MAPK/ERK and Akt pathways, which have both been reported by our group and others to be induced by OxPAPC (7, 8). Furthermore, our group has reported that H-Ras has a role in the recruitment of monocytes to endothelial cells, an important initial event in atherogenesis (9–11). We demonstrated that the balance between H-Ras and R-Ras is part of a signaling pathway that leads to deposition of CS-1 containing fibronectin on the endothelial cell surface. Monocyte α4β1 then binds to the CS-1 fibronectin. H-Ras also serves as a good model because there has been considerable study of the role played by specific cysteines in Ras activation (12–15). The current study examines the mechanism by which OxPNB interacts with H-Ras. We demonstrate that this interaction is specific by developing methods to identify OxPNB binding sites on the protein. We also demonstrate that OxPAPC activates H-Ras.
PEIPC has striking similarities in structure to 15dPGJ2, including an electrophilic enone group and a cyclopentenone component, prompting our speculation of similar chemical activity (Fig. 1). For these studies we use 15dPGJ2 as a model compound to elucidate the mechanism by which OxPAPC acts on HAEC. The current study compares the binding of 15dPGJ2, PEIPC, and OxPNB to H-Ras, and it examines H-Ras activation by OxPAPC and 15dPGJ2. Finally, this article explores similarities in gene regulation by OxPAPC, PEIPC, and 15dPGJ2 using microarray analysis. Overall, we demonstrate the importance of cysteines in OxPNB, Ox-PAPC, and PEIPC binding to endothelial cells and regulation of gene expression.
EXPERIMENTAL PROCEDURES
Reagents
1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphatidylcholine (PAPC) and 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (PAPE) were purchased from Avanti Lipids. Recombinant H-Ras was purchased from EMD Biosciences or Abgent, the H-Ras activation kit from Pierce Biotechnologies, anti-HA resin and monoclonal anti-HA antibody from Roche, polyclonal H-Ras antibody from Santa Cruz, strepavidin-HRP from RD Biosciences, and biotin, dimethylaminopyridine (DMAP), and dicyclohexylcarbodiimide (DCC) from Sigma-Aldrich.
PAPE-N-biotin synthesis, production of OxPNB, and isolation of PEIPE-NB
PAPE was biotinylated and oxidized as described previously (4). PEIPE-NB was isolated from OxPNB with semipreparative, normal Phase LC-MS using an isocratic mobile phase of 77:15:8 acteonitrile:water:methanol, as previously published for PEIPC isolation (16).
Human aortic endothelial cell culture
HAECs were isolated as described previously (17, 18). HAECs were cultured in VEC complete media (VEC Technologies), and media was changed to 10% FBS (Thermo Scientific) in M199 media (Mediatech) overnight before use in experiments.
Measurement of lipid binding to recombinant H-Ras
Human recombinant H-Ras (hr-H-Ras) (1 μg) was treated with 50 μg/ml (52 μM) OxPNB or 12 μM PEIPE-NB at 37°C for 30 min. In some experiments, hr-H-Ras was pretreated with N-ethylmaleimide (NEM; 1 mM) or 50 μg/ml (158 μM) 15dPGJ2 for 30 min. Additional competition studies were performed to determine similarity in binding of OxPAPC, PEIPC, and OxPNB. In these studies, 100 ng H-Ras was incubated with 10 μM OxPNB for 15 min, with or without pre- and cotreatment with 100 μM OxPAPC or 10 μM PEIPC for 60 min. After lipid incubation, protein was analyzed with Western blotting using streptavidin-HRP (RD Systems). Western blots were imaged on a Bio-Rad Versadoc™ 4000 system. Densitometry was performed using Quantity One software (Bio-Rad).
Measurement of N-acetylcysteine effect on OxPAPC regulation of genes
HAECs were cultured and pretreated in 3 mM N-acetylcysteine (NAC) in M199 media containing 1% FBS for 1 h. 50 μg/ml (64 μM) OxPAPC was then added to the media, and the cells were cotreated with NAC (3 mM) and OxPAPC for an additional 4 h. Cells were washed with PBS and lysed, and then RNA was extracted and qPCR analysis was performed, measuring GAPDH, IL-8, ATF-3, and HO-1 mRNA levels. GAPDH levels showed minimal change. IL-8, ATF-3, and HO-1 levels were normalized to GAPDH levels. The primer sequences used for qPCR were GAPDH: Forward: 5′-CCT CAA GAT CAT CAG CAA TGC CTC CT-3′, Reverse: 5′-GGT CAT GAG TCC TTC CAC GAT ACC AA-3′ HO-1: Forward: 5′-ATA GAT GTG GTA CAG GGA GGC CAT CA-3′, Reverse: 5′-GGC AGA GAA TGC TGA GTT CAT GAG GA-3′ IL-8: Forward: 5′-ACC ACA CTG CGC CAA CAC AGA AAT-3′, Reverse: 5′-TCC AGA CAG AGC TCT CTT CCA TCA GA-3′ ATF-3: Forward: 5′-TTG CAG AGC TAA GCA GTC GTG GTA-3′, Reverse: 5′-ATG GTT CTC TGC TGC TGG GAT TCT-3′.
LC/MS detection of NAC adduct(s) with OxPAPC components
N-acetylcysteine (1 μg total, 50 μg/ml) was incubated with 50 μg/ml (64 μM) OxPAPC in PBS at 37°C for 4 h. Analytical LC/MS was then performed on a 1.0 × 150 mm Zorbax 300SB-C18 column (Agilent) using a flow rate of 50 μl/min and a gradient of 5% acetonitrile to 100% acetonitrile with 1 mM formic acid as an additive in positive ion mode over 75 min, and then held at 100% acetonitrile for an additional 30 min. PC-containing species were identified using MS/MS on an ABI Sciex 4000 Qtrap instrument, searching for ions in the full mass spectra with a daughter ion fragment of 184 Da representing the phosphatidylcholine headgroup.
LC/MS screening for OxPNB binding sites on H-Ras
Hr-H-Ras was incubated with or without OxPNB at a molar ratio of 1:10 (hr-H-Ras:OxPNB) at 37°C for 30 min. 20 μg H-Ras was used for each MS/MS analysis. Hr-H-Ras was denatured with 1 mM DTT, followed by alkylation with 100 mM iodoacetamide. The protein was acetone-precipitated, pelleted by centrifugation, and resuspended in pH 7.8 50 mM NH4HCO3. Trypsin was added according to manufacturer specifications and incubated at 37°C overnight for complete digestion. Samples were desalted with C18 ZipTip (Millipore) for MS analysis. Analytical LC/MS of digested H-Ras peptides was performed on a 1.0 × 150 mm Zorbax 300SB-C18 column (Agilent) using a flow rate of 50 μl/min and a gradient of 5% acetonitrile to 100% acetonitrile with 1 mM formic acid as an additive in positive ion mode over 75 min, and then held at 100% acetonitrile for an additional 30 min. During analysis of tryptic digests, full MS scans were performed followed by zoom scans and MS/MS scans of the four most abundant ions in the full scan.
MS-MS database search for peptides
Database searching for peptides was performed using the web-based Global Proteome Machine (GPM) Organization to match MS/MS data to known peptides, using modifications of 57 Da at cysteines to account for carbamidomethyl modifications from iodoacetimide treatment and other default settings. The X! Tandem algorithm and a cutoff of peptides with a log(e) value of less than −2 were used as options in the GPM analysis.
Site-directed mutagenesis
The pEGFP-C3 vector containing human H-Ras (h-H-Ras) ORF was a kind gift from Dr. Junji Yamauchi (Tokyo, Japan). The plasmid encoded GFP-HA-H-Ras fusion protein, which contains GFP, 1xHA, and H-Ras, sequentially. To produce C181S and C184S point mutations, we used a long-range PCR amplification procedure employing appropriate primers with point mutations. Finally, the GFP sequence was removed, as the GFP tag contains cysteines and may bind OxPNB and PEIPE-NB. The integrity of plasmids was confirmed at the UCLA Sequencing Core Facility.
Transfection of HEK293 cells
Overexpression of H-Ras in HEK293 cells was performed using a modified plasmid. Our group modified the plasmid by removing the GFP tag and performing the aforementioned site-directed mutagenesis on selected cysteine sites. Transfection of HEK293 cells was done with Lipofectamine 2000 according to manufacturer specifications. Cells were grown to 85–95% confluence before transfection and harvested the day after transfection. Our lab achieves 90–100% transfection for HEK293 cells using these conditions.
Measurement of OxPNB binding to H-Ras expressed in cells
To study binding of OxPNB to transfected HEK293 cells, cells were pretreated in PBS (with Ca and Mg) with or without NEM (1 mM) for 60 min or 50 μg/ml (158 μM) 15dPGJ2 for 30 min at 37°C. Then 50 μg/ml (52 μM) OxPNB was added, and cells were incubated for an additional 15 min in the presence of NEM or 30 min in the presence of 15dPGJ2. Cells were scraped into 3 ml cold PBS, centrifuged, washed with an additional 10 ml PBS, and centrifuged again. Cells were then lysed in RIPA buffer (Sigma) containing PMSF, phosphate inhibitors (Sigma), and protease inhibitors (Sigma). Lysate (1 ml), from one 100 mm plate of transfected HEK293 cells was incubated with 60 μl anti-HA resin (Roche) at 4°C, rotating overnight, to immunoprecipitate H-Ras. We centrifuged and washed the beads three times in cold PBS, and then boiled for 5 min in 45 μl Laemmeli sample buffer with 5% β-mercaptoethanol to elute H-Ras. Western blotting was then performed, detecting HA and biotin.
Ras activation assay
H-Ras activity was tested using an Active Ras Pull Down and detection kit (Pierce) according to manufacturer specifications. HAECs were cultured and washed with PBS and treated with 50 μg/ml (64 μM) OxPAPC or 20 μg/ml (64 μM) 15dPGJ2 in PBS and incubated for 60 min. After incubation, cells were scraped into PBS, centrifuged, washed with PBS, and lysed with 700 μl RIPA buffer (Sigma). The kit directions were then followed.
Microarray analysis
Duplicate wells of HAECs were treated with 1% M199 media or media containing 64 μM OxPAPC, 5 μM PEIPC, or 5 μM 15dPGJ2 for 4 h, and then RNA was extracted. RNA was prepared for hybridization to Illumina arrays, measuring 45,000 probes, using a standard protocol described previously (3). Data were analyzed using the Genome Studio software package. Data were analyzed using background subtraction, quantile normalization, and the content descriptor file “HumanHT-12_V4_0_R1_15002873_B.bgx” in the Genome Studio files. The data were filtered for results with either the detection P value less than 0.001 for the PEIPC/15dPGJ2/OxPAPC RNA measurement or for the control measurement. The fold changes in probes were calculated as the average probe level for each treatment divided by the average probe level for control treatment and converted to log 2 values to analyze for gene regulation. Up/downregulated genes were identified as those which changed at least 1.25-fold from control values in the presence of lipids.
Gene functional annotation analysis on regulated genes was performed using the NIH DAVID Bioinformatics Resources 6.7 website platform. The Illumina IDs of probes that were identified as up/downregulated from the initial Genome Studio analysis were sent to the DAVID server, and the genes were analyzed for enrichment of gene ontology (GO) annotation categories using the medium stringency setting. We used a P value cutoff of 1e−6 or lower for significant GO category enrichment.
Furthermore, we used the Ingenuity software database to search for common pathways regulated by OxPAPC and 15dPGJ2. For this analysis, we used a detection P value cutoff of 0.05 and generated an Illumina probe list of those with a fold change of 1.5 or higher.
Array data are available in Gene Expression Omnibus accession GSE35709 and in the supplementary tables.
RESULTS
Evidence for a general role of cysteine in OxPAPC action in HAECs
We first tested the hypothesis that cysteines play an important role in OxPAPC action. Treatment of HAECs with 50 μg/ml (64 μM) OxPAPC for 4 h increased the expression of IL-8, HO-1, and ATF-3, hub genes representing three major functions regulated by OxPAPC. Pre- and cotreatment with 3 mM NAC strongly inhibited OxPAPC induction of these genes, as measured by qPCR (Fig. 2A). We next examined the role of cysteine in covalent binding of OxPAPC to HAEC proteins using the biotinylated analog OxPNB. HAECs were cultured in VEC media and then incubated in serum-free M199 media for 15 min with 2.5 μg/ml (2.6 μM) OxPNB (Fig. 2B, lane 1) with or without pre- and cotreatment with 1 mM N-ethylmaleimide, a simple cysteine-binding compound. The cells were then lysed, run on a gel, and probed by Western blotting with streptavidin-HRP (SA-HRP) to detect biotinylated, modified protein. Treatment of HAECs with NEM strongly inhibits binding of OxPNB to several HAEC proteins (Fig. 2B, lane 2). The large band halfway down the blot corresponds to intrinsic nonspecific biotin binding protein, present in HAECs (4). The bands that appear most clearly diminished are at about 45, 90, 110, 200, and 220 kDa. These bands are marked by arrows on the left of the figure. These studies demonstrate an important role for cysteine in OxPAPC action and covalent binding to HAEC proteins.
Fig. 2.

PEIPC and OxPAPC regulate genes via interaction with cysteines. A: HAECs were incubated for 4 h in M199 or M199 with 50 μg/ml (64 μM) OxPAPC with or without pre- and cotreatment with 3 mM N-acetylcysteine (NAC). IL-8, ATF-3, and HO-1 mRNA levels were measured by qPCR and normalized to GAPDH levels. *P < 0.05 compared with the OxPAPC-treated sample, based on Student t-test. B: HAEC cells were pretreated with or without 1 mM N-ethylmaleimide (NEM) for 1 h, and then cotreated with 2.5 μg/ml (2.6 μM) OxPNB for 15 min. Western blot analysis was performed with streptavidin-HRP to detect biotinylated proteins. Molecular weight ladder and corresponding weights are shown on right. Arrows on the left identify bands that are substantially decreased in intensity with NEM treatment, indicating that NEM competes with OxPNB binding to these proteins. QPCR and binding experiments were performed at least three times, showing similar results. A representative experiment is shown.
Determination that PEIPC accounts for most of the covalent binding of OxPAPC to cysteine
We next screened for OxPAPC lipids that covalently bind cysteines to confirm our hypothesis that PEIPC accounts for a significant amount of this binding. In this experiment, we incubated NAC (1 μg total, 50 μg/ml), a simple cysteine-containing compound, with 50 μg/ml (64 μM) OxPAPC for 4 h at 37°C and analyzed the sample with reverse-phase LC-MS/MS, screening for compounds with a daughter fragment of 184 Da representing the phosphatidylcholine headgroup. A sharp peak was shown to elute at 40.96 min followed by subsequent peaks, corresponding to unbound OxPAPC lipid components (Fig. 3A). This peak contained a molecule of MW 991 corresponding to PEIPC bound to NAC. Although OxPAPC contains many lipids, NAC adducts with other OxPAPC components were either undetectable or minimally detectable over several experiments. This experiment demonstrates that PEIPC is the primary OxPAPC component that covalently binds N-acetylcysteine. On the basis of this observation, we tested the ability of biotinylated PEIPC (PEIPE-NB) to bind to HAEC proteins. We demonstrated that PEIPE-NB also has strong binding to many HAEC proteins (Fig. 3C).
Fig. 3.

PEIPC is the major component of OxPAPC that binds N-acetylcysteine. NAC (1 μg total, 50 μg/ml) was incubated for 4 h with 50 μg/ml (64 μM) OxPAPC and analyzed with reverse phase LC-MS/MS for PC-containing compounds by screening for compounds that yield a fragment daughter ion of 184 Da, molecular weight of the phosphatidylcholine headgroup. A: The chromatogram represents the total ion current of species eluting from the column. The sharp peak at 40.96 min represents the NAC-PEIPC adduct eluting from the column. The peaks that elute at later retention times are unbound residual OxPAPC species. B: The MS spectra of the peak eluting at 40.96, with a molecular weight of 991.7 Da, representing the NAC-PEIPC adduct (structure shown in inset); C: HAEC cells were lysed and either run directly or treated with 12 μM PEIPE-NB. Western blot analysis was performed with streptavidin-HRP to detect biotinylated proteins.
A role for cysteines in the covalent binding of OxPNB and PEIPE-NB to H-Ras
Next, we tested the hypothesis that OxPAPC and PEIPC bind to cysteines in H-Ras by using the biotinylated analogs OxPNB and PEIPE-NB. We performed experiments to determine whether OxPNB and PEIPE-NB bind hr-H-Ras and H-Ras expressed in cells. H-Ras was treated with lipid, run on a gel, and probed with Western blotting with SA-HRP to detect modified protein. We demonstrated that oxidized PAPE-N-biotin (OxPNB), but not unoxidized PAPE-N-biotin (PNB), covalently modifies commercially available, purified hr-H-Ras (Fig. 4A). Covalent binding of H-Ras in cells by OxPNB was also examined. HEK293 cells were transfected with HA-tagged H-Ras. Cells were treated with 50 μg/ml (52 μM) OxPNB or 50 μg/ml (52 μM) PNB for 30 min, lysed, and then H-Ras was immunoprecipitated with anti-HA beads, followed by Western blotting with anti-H-Ras and SA-HRP. Similar to results with hr-H-Ras, endogenous H-Ras in transfected HEK293 cells was covalently modified by OxPNB but not by PNB (Fig. 4B).
Fig. 4.

Oxidized PAPE-N-biotin binds to recombinant H-Ras and H-Ras in cells, whereas unoxidized PAPE-N-biotin does not bind. A: Human recombinant H-Ras (hr-H-Ras) was incubated with no lipid, 50 μg/ml (52 μM) unoxidized PAPE-N-biotin (PNB), or 50 μg/ml (52 μM) oxidized PAPE-N-biotin (OxPNB) for 30 min. Western blot analysis was performed with SA-HRP to detect biotinylated (lipid-modified) H-Ras. B: HEK293 cells were transfected with HA-tagged wild-type H-Ras. Cells were treated with PBS or 50 μg/ml (52 μM) of unoxidized or oxidized PAPE-N-biotin for 30 min. Lysates were immunoprecipitated with anti-HA beads, and samples were analyzed by Western blot for HA and streptavidin (lipid-modified H-Ras). Densitometry shows the ratio of lipid-modified H-Ras (SA-HRP) to total H-Ras (anti-HA); These experiments were performed at least three times, showing similar results. C: 100 ng hr-H-Ras was incubated with no lipid, 100 μM OxPAPC, or 10 μM PEIPC for 60 min, followed by incubation with no lipid or 10 μM OxPNB for an additional 15 min. Western blot analysis was performed as in panel A. Densitometry shows the ratio of lipid-modified H-Ras normalized to H-Ras treated only with OxPNB.
Previous studies have shown that headgroup can play an important role in the interaction of oxidized phospholipids with cells (5). To provide further evidence that OxPNB is a good surrogate for OxPAPC and its component PEIPC, we performed competition studies, examining the ability of these molecules to compete with OxPNB for H-Ras binding. Hr-H-Ras (100 ng) was incubated in PBS with or without 100 μM OxPAPC or 10 μM PEIPC. Then 10 μM OxPNB was added, and the sample was further incubated. The samples were then examined with Western blotting using SA-HRP to detect modified H-Ras (Fig. 4C). These results show strong competition of OxPAPC and PEIPC for OxPNB binding to H-Ras. We also tested the ability of OxPAPC to compete with OxPNB for binding to H-Ras overexpressed in HEK293 cells. A 70% inhibition was observed (data not shown). These studies demonstrate that headgroup differences have a minimal effect on binding to H-Ras.
We next examined the effect of NEM on covalent binding of OxPNB and PEIPE-NB to human recombinant H-Ras. Hr-H-Ras (1 μg) was incubated in PBS with or without 1 mM NEM. Then 1 μg OxPNB or 0.5 μg PEIPE-NB was added, and the samples were further incubated and examined with Western blotting for H-Ras modification (Fig. 5A). We demonstrate that OxPNB and PEIPE-NB binding to hr-H-Ras was strongly competed by NEM. We also tested the ability of NEM to inhibit the binding of OxPNB to H-Ras that is overexpressed in cells. HEK293 cells overexpressing HA-tagged wild-type H-Ras were pretreated with or without 1 mM NEM for 60 min and then cotreated with 10 μg/ml (10.4 μM) OxPNB for 15 min. The cells were lysed, H-Ras was immunoprecipitated with anti-HA resin, and samples were analyzed for modified H-Ras with Western blotting (Fig. 5B). The results show that in cells the binding of OxPNB to H-Ras was inhibited by pre- and cotreatment with NEM. This set of experiments demonstrated that both OxPNB and PEIPE-NB covalently modify H-Ras and that NEM blocks this binding, showing that cysteines are important in this interaction.
Fig. 5.

N-ethylmaleimide competes for binding of OxPAPE-N-biotin and PEIPE-N-biotin to H-Ras. A: 1 μg hr-H-Ras was incubated with no lipid, 0.2 mM NEM, 0.5 μg PEIPE-NB, 0.5 μg PEIPE-NB and 0.2 mM NEM, 0.5 μg OxPNB, or 0.5 μg OxPNB and 0.2 μg NEM for 30 min, corresponding to 10:1 NEM or lipid:H-Ras molar ratios. Western blot analysis was performed as in Fig. 3A. B: HEK293 cells were transfected with HA-tagged wild-type H-Ras and treated with PBS or 1 mM NEM for 60 min, then with 10 μg/ml (10.4 μM) OxPNB for 15 min. Lysates were prepared for Western blotting as in Fig. 3B. Densitometry shows the ratio of lipid-modified H-Ras (SA-HRP) to total H-Ras (anti-HA). Competition experiments were performed at least three times, showing similar results.
OxPNB binding to H-Ras involves cysteines 181 and/or 184
To determine which cysteines bound H-Ras, we employed both recombinant and overexpressed H-Ras. Hr-H-Ras (20 μg) was incubated with or without OxPNB at 10:1 OxPNB molar excess for 30 min at 37°C in PBS. The H-Ras was then treated with DTT, followed by iodoacetimide, and digested with trypsin overnight. The tryptic digests of H-Ras treated with OxPNB, along with an untreated control, were analyzed with ESI-LC-MS/MS, and peptide database searching was performed with the Global Proteome Database (www.thegpm.org), using modifications of 57 Da at cysteines to account for carbamidomethyl modifications from iodoacetimide treatment, as mentioned in Experimental Procedures. The H-Ras fragments were identified using MS/MS data. Database searching of the MS/MS data from untreated H-Ras tryptic digests identified 88% of the predicted peptides, whereas database searching of the OxPNB-treated H-Ras tryptic digest identified only 71% of the predicted H-Ras fragments, suggesting that some of the protein had been modified by OxPNB. One peptide eluting at 16.21 min was found only in untreated H-Ras and not in OxPNB-treated H-Ras (Fig. 6). The inset in Fig. 6 shows the MS/MS spectra of the peptide eluting at 16.2 min with the sequence KLNPPDESGPGCMSCK. Peaks used for matching the MS/MS peaks to the peptide in the database are marked with asterisks. The observed m/z for this fragment is 888.87, representing a doubly charged ion of molecular weight 1,775. This data demonstrated that the binding of OxPNB to H-Ras involved C181 and/or C184.
Fig. 6.

LC/MS shows that OxPAPE-N-biotin binding to H-Ras involves cysteines 181 and 184. Hr-H-Ras was incubated with or without OxPNB at a molar ratio of 1:10 (hr-H-Ras:OxPNB) at 37°C for 30 min, digested with trypsin, and analyzed with ESI-LC/MS/MS, followed by identification of H-Ras fragments. One peptide, eluting at 16.21 min, was found only in untreated H-Ras and not in OxPNB-treated H-Ras. The inset shows the MS/MS spectra of the peptide eluting at 16.2 min with the sequence KLNPPDESGPGCMSCK. Asterisks indicate peaks used for matching the MS/MS peaks to the peptide in the database. The observed m/z for this fragment is 888.87, representing a doubly charged ion of molecular weight 1775. This data shows that the binding of OxPNB to H-Ras involves C181 and/or C184.
Unambiguous determination of whether OxPNB primarily binds to C181 and/or C184 in H-Ras was achieved using plasmids containing HA-tagged H-Ras with mutations from cysteine → serine at cysteine 181 or cysteine 184 (C181S and C184S H-Ras, respectively). HEK293 cells were transfected with empty vector or HA-tagged wild-type, C181S, or C184S H-Ras. The cells were then treated with 50 μg/ml (52 μM) OxPNB and analyzed by Western blotting with anti-HA and SA-HRP probes. The data show less binding to the C181S- and C184S-mutant H-Ras than to wild-type H-Ras (Fig. 7C). Over a series of experiments, there was no significant difference between the binding to C181 and binding to C184. These results suggest that both C181 and C184 are covalently bound by OxPNB.
Fig. 7.

Mutant H-Ras and 15dPGJ2 provide evidence that binding of Ox-PAPE-N-Biotin to H-Ras involves Cysteines 181 and 184. A: Ability of 15dPGJ2 to compete with OxPNB for binding to recombinant H-Ras. Human recombinant H-Ras was preincubated with either PBS or 50 μg/ml (158 μM) 15dPGJ2 for 30 min, and then coincubated with 50 μg/ml (52 μM) OxPNB for 30 min. Western blotting was performed as in Fig. 3A. B: Ability of 15dPGJ2 to compete with OxPNB for binding to H-Ras in cells. HEK293 cells were transfected with HA-tagged wild-type H-Ras and treated with PBS or 50 μg/ml (158 μM) 15dPGJ2 for 30 min, and then 50 μg/ml (52 μM) OxPNB for 30 min. Lysates were prepared for Western blotting as in Fig. 3B. Densitometry shows the ratio of lipid-modified H-Ras (SA-HRP) to total H-Ras (anti-HA). C: HEK293 cells were transfected with empty vector, HA-tagged wild-type H-Ras, C181S-mutant H-Ras, or C184S-mutant H-Ras. Cells were treated with 50 μg/ml (52 μM) OxPNB for 30 min. Lysates were immunoprecipitated with anti-HA resin, and Western blotting was carried out as in Fig. 3A. D: HEK293 cells were transfected with empty vector, HA-tagged wild-type H-Ras, or HA-tagged C181S-mutant H-Ras. Cells were treated with 10 μg/ml (10.4 μM) OxPNB for 30 min. The last lane was pretreated with 50 μg/ml (158 μM) 15dPGJ2 for 15 min, and then cotreated with 10 μg/ml (10.4 μM) OxPNB for 30 min. Cells were immunoprecipitated, and Western blotting was carried out as in Fig. 3A. Mutant and competition H-Ras experiments were performed at least three times, showing similar results.
Similarity in interaction of 15dPGJ2 and OxPNB with H-Ras
Previous studies had demonstrated that 15dPGJ2 binds to C181 and C184 (15). Because of the similarity in structure of PEIPC and 15dPGJ2 and because both bind to cysteines 181 and 184 in H-Ras, we examined the ability of 15dPGJ2 to inhibit binding of OxPNB to H-Ras. Human recombinant H-Ras (1 μg) was incubated with 50 μg/ml (52 μM OxPNB) with or without pre- and cotreatment of 50 μg/ml (158 μM) 15dPGJ2. OxPNB binding to recombinant, purified H-Ras was strongly competed by 15dPGJ2 (Fig. 7A). In addition, we examined the ability of 15dPGJ2 to inhibit binding of OxPNB to H-Ras expressed in cells. HEK293 cells overexpressing HA-tagged wild-type H-Ras were exposed to 50 μg/ml (52 μM) OxPNB with or without pre- and cotreatment of 50 μg/ml (158 μM) 15dPGJ2 (Fig. 7B). These results show that the binding of OxPNB to H-Ras in cells is inhibited by 50% after pre- and cotreatment with 15dPGJ2. Both the studies with recombinant H-Ras and overexpressed H-Ras support our hypothesis that OxPNB binds to cysteines with a similar mechanism to that of 15dPGJ2.
It has been previously reported that 15dPGJ2 binds mainly to cysteine 184 (12). To further support the hypothesis that OxPNB is only binding to C181 and C184 in H-Ras, we transfected HEK293 cells with either wild-type H-Ras or C181S-mutant H-Ras and pre- and cotreated the C181S-transfected cells with or without 50 μg/ml (158 μM) 15dPGJ2; we then treated with OxPNB. We reasoned that 15dPGJ2 should compete more strongly with OxPNB binding to C181S-mutant H-Ras (Fig. 7D). The Western blotting demonstrates that 15dPGJ2 strongly competes with OxPNB binding to C181S-mutant H-Ras, almost eliminating binding and supporting our hypothesis that OxPNB binds to C181 and C184 in H-Ras with very little or no binding to other cysteines.
OxPAPC and 15dPGJ2 activate H-Ras at similar concentrations
Previous publications demonstrated that H-Ras is activated by the binding of 15dPGJ2 to C181 and C184 on H-Ras (12). We compared the ability of OxPAPC and 15dPGJ2 to activate H-Ras, using 15dPGJ2 as a positive control for H-Ras activation. HAECs were treated with serum-free M199, M199 with 50 μg/ml (64 μM) OxPAPC, or M199 with 20 μg/ml (64 μM) 15dPGJ2, and activation was determined using a kit that involves binding to Raf and detection of the amount of activated Ras on Western blots. We demonstrated equivalent activation at 60 min treatment (Fig. 8). Furthermore, the fold change measured by this activation kit is comparable to change already reported in other publications (19, 20). This experiment demonstrates that OxPAPC treatment activates H-Ras in HAECs. The similarity of activation again confirms a similar mechanism of action of OxPAPC and 15dPGJ2.
Fig. 8.
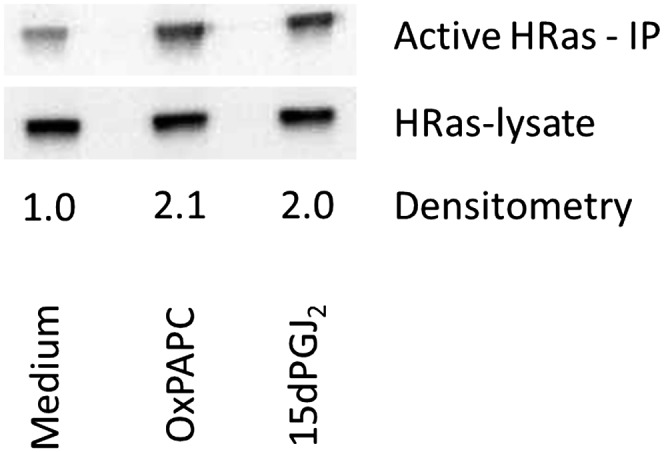
H-Ras in HAECs is activated by both OxPAPC and 15dPGJ2. One hundred millimeter dishes of HAECs were incubated in M199 or M199 with 64 μM OxPAPC or 15dPGJ2 for 60 min. Activated H-Ras was isolated as per Ras pulldown kit manufacturer instructions (Pierce Biotechnologies). Densitometry shows the ratio of active H-Ras (after kit immunoprecipitation) to total H-Ras (before immunoprecipitation); Densitometry was performed normalized to lysate and to M199 signals. Activation data were replicated three times, showing consistent activation by OxPAPC and 15dPGJ2 at 60 min.
PEIPC, 15dPGJ2, and OxPAPC regulate many of the same genes and pathways
Because of the evidence of similarity in the effect of OxPAPC, PEIPC, and 15dPGJ2 on H-Ras and because of the common structural features of 15dPGJ2 and PEIPC, we hypothesized that 15dPGJ2 might inhibit the binding of PEIPC to multiple proteins. Whole-cell lysates of HAEC were incubated with 12 μM PEIPE-NB, for 30 min at 37°C, with or without 120 μM 15dPGJ2 and analyzed with Western blotting. PEIPE-NB binding to HAEC proteins was almost completely inhibited by incubation with 15dPGJ2 (Fig. 9A).
Fig. 9.

Evidence for an overlap in binding and regulation of gene expression in HAEC by PEIPC, OxPAPC, and 15dPGJ2. A: HAEC cells were lysed, and lysates were treated with 12 μM PEIPE-NB or cotreated with 120 μM 15dPGJ2 and 12 μM PEIPE-NB. Western blotting was then performed. This experiment was performed at least three times with similar results. B: Duplicate wells were treated with media or media containing 50 μg/ml (64 μM) OxPAPC or equimolar amounts of either PEIPC or 15dPGJ2 for 4 h, and then RNA was extracted and analyzed with microarray on an Illumina chip. Data were filtered for P ≤ 0.001 and fold change of 1.25 during data analysis with Genome Studio software; Data are represented in Venn diagrams of PEIPC versus 15dPGJ2 downregulated and upregulated genes and OxPAPC versus 15dPGJ2 downregulated and upregulated genes.
The results shown in Fig. 7, in addition to previous studies showing similar pathways activated by OxPAPC and 15dPGJ2, suggested that OxPAPC, PEIPC, and 15dPGJ2 might also show similar regulation of gene expression. To more extensively compare gene regulation of the two molecules, we performed microarray analyses comparing the mRNA levels of PEIPC-, OxPAPC-, and 15dPGJ2-treated HAECs. HAECs were treated in M199 media containing 1% FBS with no additions, 5 μM PEIPC, 5 μM 15dPGJ2, or 64 μM OxPAPC for 4 h, and RNA was collected and hybridized to Illumina microarrays for analysis. Fold changes in probe levels were analyzed for genes up- or downregulated by at least 1.25-fold. The regulated genes were then categorized into those of similar regulation between 15dPGJ2 and OxPAPC or between 15dPGJ2 and PEIPC (Fig. 9B). Approximately 50% of the genes regulated by PEIPC were also regulated by 15dPGJ2 and slightly more overlap was observed with OxPAPC. There was also a significant overlap in regulation between PEIPC and 15dPGJ2 when analyzing genes up- or downregulated by at least 1.5-fold (supplementary Fig. I).
We also performed functional annotations on the genes regulated by PEIPC, 15dPGJ2, and OxPAPC in HAECs using the DAVID enrichment tool. We used a P value cutoff of 1e−6 or lower to identify significant enrichment in the GO categories (supplementary tables). We then separated the categories into those up- or downregulated by both 15dPGJ2 and PEIPC and those GO categories that were exclusively regulated by 15dPGJ2 or PEIPC. There are several overlaps in 15dPGJ2- and PEIPC-regulated categories. Stress response, response to protein stimulus, and unfolded protein response GO categories are upregulated by both PEIPC and 15dPGJ2. DNA binding, DNA-dependent regulation of transcription, nucleus category genes, zinc finger-related genes, and regulation of RNA metabolic process categories are downregulated by both PEIPC and 15dPGJ2. Although PEIPC and 15dPGJ2 shared most biological activities, some of the GO annotations were different in the DAVID analysis. Only PEIPC showed upregulation of cytokines and cytokine receptor interaction, negative regulation of apoptosis, and negative regulation of cell death, whereas 15dPGJ2 showed more upregulation of heat shock proteins.
DISCUSSION
These data highlight the importance of cysteine in the interaction of OxPAPC and its component PEIPC with HAEC. We postulated an important role for cysteine in OxPAPC gene regulation and protein interactions because the most active lipid in OxPAPC, PEIPC, contains an electrophilic group that can readily undergo Michael addition to cysteine. We now present evidence that treatment of OxPAPC with a cysteine-containing compound (NAC) inhibited its ability to activate genes important in the OxPAPC response (Fig. 2A). We previously demonstrated that OxPNB, a biotinylated analog of OxPAPC, can covalently bind to proteins and that it regulates several of the same pathways as OxPAPC in HAECs (4). We now demonstrate that NEM, a molecule that binds to cysteines, inhibited the binding of OxPNB to total cell proteins (Fig. 2B). We also determined that OxPNB and PEIPC binding to one protein, H-Ras, was strongly inhibited by NEM (Fig. 5). It is apparent that there is a difference in the extent of OxPNB binding in different experiments (Figs. 2B, 3C, and 9AFigs. 2B, 3C, and 9A). This is most likely due to a small difference in the relative amounts of PEIPE-NB in OxPNB preps used or different levels of OxPNB used in these experiments. We previously demonstrated that there is an 80% overlap in the genes regulated by Ox-PAPC and PEIPC (3). We now show with LC-MS/MS that PEIPC accounts for almost all of the binding of OxPAPC to cysteine (Fig. 3). Thus, we present strong evidence that both gene regulation and covalent binding of OxPAPC involve interactions with cysteine and that this interaction is primarily from PEIPC.
To examine the specificity of OxPNB binding to cysteine, we focused on interaction with H-Ras. We now demonstrate by MS that OxPNB binds specifically to two of the six free cysteines in H-Ras, C181 and 184 (Fig. 7). This is a well-studied protein in which specific cysteines (C184 and C181) have been shown to play a critical role in activation (12). Ras activation involves a process in which Ras, which normally moves from the Golgi to the cell membrane, becomes palmitoylated at C181 and is retained at the plasma membrane. Ras is also palmitoylated at C184, which allows for segregation between lipid raft and nonlipid raft domains to allow for Raf binding to Ras, resulting in activation of several pathways (13). We now demonstrate that OxPAPC activates H-Ras to the same extent as 15dPGJ2. We present evidence that binding to cysteines 181 and 184 accounts for at least some of the OxPAPC increase in activity by showing that OxPAPC and PEIPC compete for binding of OxPNB to H-Ras and that OxPNB binds to cysteines 181 and 184. Our studies strongly suggest that the increase in activity of H-Ras by Ox-PAPC is due to the binding of critical H-Ras cysteines. However, more experiments will be necessary to prove that this is the only mechanism.
There are several mechanisms by which OxPAPC might activate H-Ras, one involving the necessity for covalent binding and the other involving interaction without covalent binding. The two cysteines to which OxPAPC binds are particularly reactive cysteines in H-Ras, with palmitoylation of these cysteines required for movement of H-Ras to the plasma membrane, binding of H-Ras to Raf protein, and activation of downstream ERK/MAPK and Akt pathways. It is possible that the direct modification of these residues by OxPAPC results in activation of H-Ras, perhaps with the palmitoyl group on OxPAPC fulfilling the requirement for enzymatic palmitoylation. However, it is also possible that the interaction of OxPAPC with H-Ras catalyzes normal palmitoylation. Recent crystal structures of the peroxisome proliferator-activated receptor (PPAR)γ ligand binding domain complexed with 15dPGJ2 show distinct roles for 15dPGJ2 ligand, both noncovalently and covalently bound to PPARγ (21). Possibly both types of OxPAPC-H-Ras interactions are also important in cells.
In this study, we concentrated on OxPAPC and PEIPC interactions with cysteines, particularly with cysteines 181 and 184 due to their involvement in H-Ras activation. It is also possible that some OxPAPC components interact with lysine(s) on H-Ras to form reversible Schiff base adducts that would not be observed in this analysis.
Our studies document a strong similarity in the actions of OxPAPC and 15dPGJ2. The similarity of structure in these two molecules suggested that they might have similar interactions with cell proteins. Previous studies have shown similar effects of 15dPGJ2 to OxPAPC in several pathways (7, 9, 12, 14, 15, 22–27). We now demonstrate that OxPNB and 15dPGJ2 bind to the same cysteines in H-Ras (C181 and C184) and that 15dPGJ2 can inhibit the binding of OxPNB to H-Ras. The fact that 15dPGJ2 inhibits the binding of OxPNB to H-Ras in cells suggests that, even though 15dPGJ2 is a fatty acid and PEIPC a phospholipid, they can to some extent enter the same cell compartment. These similarities in activity and the similarity in binding by these two molecules to cysteine suggests that interaction with cysteines is important in gene regulation by both molecules in HAECs.
Our microarray analysis demonstrates that there is approximately 50% overlap of the genes up- or downregulated by PEIPC with the genes regulated by 15dPGJ2. Inflammation is well established as having a prominent role in progression of atherosclerosis, and the unfolded protein response is an important regulator of inflammatory genes in HAECs (28). Although 15dPGJ2 is known as an inhibitor of inflammation, major inflammatory genes IL-6 and IL-8 are strongly upregulated by both PEIPC and 15dPGJ2. However, an enrichment of chemokines and chemokine receptors is seen only with PEIPC. As shown in the DAVID GO category analysis, similarities in gene regulation are observed in stress response, unfolded protein response, transcriptional response, and zinc finger GO categories. Oxidative stress and production of reactive oxidative species have also been linked to atherosclerosis (1, 10, 11, 29–35). Both molecules cause an induction of Nrf2, which regulates genes protecting against oxidant stress. CYP26B1 was the most downregulated gene in PEIPC and OxPAPC arrays and one of the most downregulated genes in the 15dPGJ2 array. The metabolism of all trans-retinoic acid (atRA) has been reported to be regulated by this gene, and increased CRP26B1 activity has been reported to lead to decreased local atRA levels and increased stenosis (36, 37). SOX18, which is also reported to have a role in atherosclerosis (38), was among the most downregulated genes by OxPAPC, PEIPC, and 15dPGJ2. Both molecules also regulate the UPR response that protects against cell injury. Among the most upregulated genes by OxPAPC, PEIPC, and 15dPGJ2 were atherosclerosis marker HSPA1A (39) and UPR target gene DNAJB1 (28).
There are also a number of differences between the effects of 15dPGJ2 and PEIPC in regulating gene expression. As stated above, our microarray analysis also shows that PEIPC upregulates cytokines and cytokine receptors and downregulates apoptosis and cell death. However, these groups were not induced by 15dPGJ2. This result agrees with prior reports, as 15dPGJ2 has been reported to be anti-inflammatory and induce PPARγ-dependent endothelial cell apoptosis (40, 41). Even within GO categories, such as oxidative stress, which are stimulated by both agents, there are differences in the regulation of individual genes. Although both molecules activate Nrf2, downstream targets of Nrf2 are regulated differently. Interestingly, in our microarray studies in HAECs, Nrf2 and downstream targets NQO1, GCLC, GCLM, and HMOX1 are strongly upregulated by PEIPC. In 15dPGJ2, although Nrf2 and NQO1 are slightly downregulated, GCLM and HMOX1 are significantly upregulated. These differences could be important in the ability of HAECs to resist oxidative stress. Overall, the differences between 15dPGJ2 and PEIPC may be attributed to the differences in the interaction of a phospholipid and a fatty acid with cells. Elucidation of mechanisms by which the two molecules are transported may give insight into nonoverlapping regulation.
Although measured 15dPGJ2 levels are in the picomolar to nanomolar range (42, 43), PEIPC has been reported by our group in micromolar quantities in rabbit atherosclerotic lesions (44). The relative levels of these two molecules and activities may be important in the persistence of the inflammatory response in some disease states. Previous publications have questioned the significance of other studies with 15dPGJ2, arguing that most experimental studies measure effects at 15dPGJ2 concentrations that are several orders of magnitude higher than pathophysiological levels (42, 43). In contrast to the lower measured concentrations of 15dPGJ2, levels of OxPCs have been measured in total concentrations as high as 51 μM in human plasma (45), with several individual OxPC species being as high as several micromolar amounts in human plasma and atheromas (45, 46). Our experiments use concentrations that are comparable in magnitude to those reported in pathological environments. In the experiments presented, we use from 2.6 to 10.4 μM of OxPNB in most of our binding studies, and from 4 to 64 μM of OxPAPC in activity and microarray experiments. Furthermore, in experiments with OxPAPC (or OxPNB), PEIPC (or PEIPE-NB) only corresponds to at most 10% of the total OxPAPC moiety. We used 12 μM PEIPE-NB (Fig. 3C) in binding studies and 4 μM PEIPC in microarray studies. Both showed high regulation at this pathophysiological level (Fig. 9).
In summary, we have demonstrated that cysteine plays an important role in OxPAPC regulation of endothelial function. We show that PEIPC is the major cysteine-binding lipid in OxPAPC. OxPNB and 15dPGJ2 bind to the same cysteines in H-Ras, resulting in similar H-Ras activation. There is a strong overlap in protein binding and gene regulation by PEIPC, OxPAPC, and 15dPGJ2 in HAECs, including genes involved in inflammation, stress response, and atherosclerosis. Overall, this study strongly suggests that interaction with cysteines constitutes a major mechanism of action by PEIPC and OxPAPC in endothelial cells. Studies identifying reactive cysteines suggest that there is a strong correlation between hyperreactivity and functionality of cysteines (47). Thus, many of the proteins covalently binding OxPAPC in cells may play an important role in OxPAPC signaling.
Supplementary Material
Footnotes
Abbreviations:
- ATF3
- activating transcription factor 3
- HAEC
- human aortic endothelial cell
- HO-1
- heme oxygenase-1
- h-H-Ras
- human H-Ras
- hr-H-Ras
- human recombinant H-Ras
- IL-8
- interleukin-8
- mm-LDL
- minimally modified-LDL
- NAC
- N-acetylcysteine
- NEM
- N-ethylmaleimide
- OxPAPC
- autoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- OxPNB
- autoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl-(N-biotinylethanolamine)
- PEIPC
- 1-palmitoyl-2-(5,6)-epoxyisoprostane E2-sn-glycero-3-phosphocholine
- PEIPE-NB
- 1-palmitoyl-2-(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphatidyl-(N-biotinylethanolamine)
- PNB
- 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl-(N-biotinylethanolamine)
- 15dPGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
This work was supported by Ruth L. Kirschstein National Research Service Award T32-HL-69766 (J.R.S.) and National Institutes of Health Grants HL-30568 (J.R.S and J.A.B.), HL-064731 (J.R.S. and J.A.B.), and 1K99-HL-105577 (S.L.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one figure and 15 tables.
REFERENCES
- 1.Berliner J. A., Navab M., Fogelman A. M., Frank J. S., Demer L. L., Edwards P. A., Watson A. D., Lusis A. J. 1995. Atherosclerosis: basic mechanisms. oxidation, inflammation, and genetics. Circulation. 91: 2488–2496 [DOI] [PubMed] [Google Scholar]
- 2.Berliner J. A., Leitinger N., Tsimikas S. 2009. The role of oxidized phospholipids in atherosclerosis. J. Lipid Res. 50(Suppl): S207–S212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romanoski C. E., Che N., Yin F., Mai N., Pouldar D., Civelek M., Pan C., Lee S., Vakili L., Yang W. P., et al. 2011. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ. Res. 109: e27–e41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gugiu B. G., Mouillesseaux K., Duong V., Herzog T., Hekimian A., Koroniak L., Vondriska T. M., Watson A. D. 2008. Protein targets of oxidized phospholipids in endothelial cells. J. Lipid Res. 49: 510–520 [DOI] [PubMed] [Google Scholar]
- 5.Gao D., Ashraf M. Z., Kar N. S., Lin D., Sayre L. M., Podrez E. A. 2010. Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class B scavenger receptors CD36 and SR-BI. J. Biol. Chem. 285: 4447–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung M. E., Berliner J. A., Koroniak L., Gugiu B. G., Watson A. D. 2008. Improved synthesis of the epoxy isoprostane phospholipid PEIPC and its reactivity with amines. Org. Lett. 10: 4207–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimman A., Mouillesseaux K. P., Le T., Gharavi N. M., Ryvkin A., Graeber T. G., Chen T. T., Watson A. D., Berliner J. A. 2007. Vascular endothelial growth factor receptor 2 plays a role in the activation of aortic endothelial cells by oxidized phospholipids. Arterioscler. Thromb. Vasc. Biol. 27: 332–338 [DOI] [PubMed] [Google Scholar]
- 8.Birukov K. G., Leitinger N., Bochkov V. N., Garcia J. G. 2004. Signal transduction pathways activated in human pulmonary endothelial cells by OxPAPC, a bioactive component of oxidized lipoproteins. Microvasc. Res. 67: 18–28 [DOI] [PubMed] [Google Scholar]
- 9.Cole A. L., Subbanagounder G., Mukhopadhyay S., Berliner J. A., Vora D. K. 2003. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arterioscler. Thromb. Vasc. Biol. 23: 1384–1390 [DOI] [PubMed] [Google Scholar]
- 10.Fu P., Birukov K. G. 2009. Oxidized phospholipids in control of inflammation and endothelial barrier. Transl. Res. 153: 166–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leitinger N. 2005. Oxidized phospholipids as triggers of inflammation in atherosclerosis. Mol. Nutr. Food Res. 49: 1063–1071 [DOI] [PubMed] [Google Scholar]
- 12.Oliva J. L., Perez-Sala D., Castrillo A., Martinez N., Canada F. J., Bosca L., Rojas J. M. 2003. The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc. Natl. Acad. Sci. USA. 100: 4772–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy S., Plowman S., Rotblat B., Prior I. A., Muncke C., Grainger S., Parton R. G., Henis Y. I., Kloog Y., Hancock J. F. 2005. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol. Cell. Biol. 25: 6722–6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renedo M., Gayarre J., Garcia-Dominguez C. A., Perez-Rodriguez A., Prieto A., Canada F. J., Rojas J. M., Perez-Sala D. 2007. Modification and activation of Ras proteins by electrophilic prostanoids with different structure are site-selective. Biochemistry. 46: 6607–6616 [DOI] [PubMed] [Google Scholar]
- 15.Oeste C. L., Diez-Dacal B., Bray F., Garcia de Lacoba M., de la Torre B. G., Andreu D., Ruiz-Sanchez A. J., Perez-Inestrosa E., Garcia-Dominguez C. A., Rojas J. M., et al. 2011. The C-terminus of H-Ras as a target for the covalent binding of reactive compounds modulating Ras-dependent pathways. PLoS ONE. 6: e15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson A. D., Subbanagounder G., Welsbie D. S., Faull K. F., Navab M., Jung M. E., Fogelman A. M., Berliner J. A. 1999. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. J. Biol. Chem. 274: 24787–24798 [DOI] [PubMed] [Google Scholar]
- 17.Berliner J. A., Territo M. C., Sevanian A., Ramin S., Kim J. A., Bamshad B., Esterson M., Fogelman A. M. 1990. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J. Clin. Invest. 85: 1260–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navab M., Imes S. S., Hama S. Y., Hough G. P., Ross L. A., Bork R. W., Valente A. J., Berliner J. A., Drinkwater D. C., Laks H., et al. 1991. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J. Clin. Invest. 88: 2039–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawasaki K., Watabe T., Sase H., Hirashima M., Koide H., Morishita Y., Yuki K., Sasaoka T., Suda T., Katsuki M., et al. 2008. Ras signaling directs endothelial specification of VEGFR2+ vascular progenitor cells. J. Cell Biol. 181: 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamauchi J., Miyamoto Y., Tanoue A., Shooter E. M., Chan J. R. 2005. Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc. Natl. Acad. Sci. USA. 102: 14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waku T., Shiraki T., Oyama T., Fujimoto Y., Maebara K., Kamiya N., Jingami H., Morikawa K. 2009. Structural insight into PPARgamma activation through covalent modification with endogenous fatty acids. J. Mol. Biol. 385: 188–199 [DOI] [PubMed] [Google Scholar]
- 22.Roy M., Li Z., Sacks D. B. 2005. IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell. Biol. 25: 7940–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell-Parikh L. C., Ide T., Lawson J. A., McNamara P., Reilly M., FitzGerald G. A. 2003. Biosynthesis of 15-deoxy-delta12,14–PGJ2 and the ligation of PPARgamma. J. Clin. Invest. 112: 945–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H., Shi W., Tontonoz P., Wang S., Subbanagounder G., Hedrick C. C., Hama S., Borromeo C., Evans R. M., Berliner J. A., et al. 2000. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ. Res. 87: 516–521 [DOI] [PubMed] [Google Scholar]
- 25.Boyault S., Bianchi A., Moulin D., Morin S., Francois M., Netter P., Terlain B., Bordji K. 2004. 15-Deoxy-delta(12,14)-prostaglandin J(2) inhibits IL-1beta-induced IKK enzymatic activity and IkappaBalpha degradation in rat chondrocytes through a PPARgamma-independent pathway. FEBS Lett. 572: 33–40 [DOI] [PubMed] [Google Scholar]
- 26.Kadl A., Meher A. K., Sharma P. R., Lee M. Y., Doran A. C., Johnstone S. R., Elliott M. R., Gruber F., Han J., Chen W., et al. 2010. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 107: 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R., Chen W., Yanes R., Lee S., Berliner J. A. 2007. OKL38 is an oxidative stress response gene stimulated by oxidized phospholipids. J. Lipid Res. 48: 709–715 [DOI] [PubMed] [Google Scholar]
- 28.Gargalovic P. S., Gharavi N. M., Clark M. J., Pagnon J., Yang W. P., He A., Truong A., Baruch-Oren T., Berliner J. A., Kirchgessner T. G., et al. 2006. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26: 2490–2496 [DOI] [PubMed] [Google Scholar]
- 29.Harrison D., Griendling K. K., Landmesser U., Hornig B., Drexler H. 2003. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 91: 7A–11A [DOI] [PubMed] [Google Scholar]
- 30.Steinberg D., Parthasarathy S., Carew T. E., Khoo J. C., Witztum J. L. 1989. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 320: 915–924 [DOI] [PubMed] [Google Scholar]
- 31.Witztum J. L., Steinberg D. 1991. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Invest. 88: 1785–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witztum J. L. 1994. The oxidation hypothesis of atherosclerosis. Lancet. 344: 793–795 [DOI] [PubMed] [Google Scholar]
- 33.Parthasarathy S., Santanam N. 1994. Mechanisms of oxidation, antioxidants, and atherosclerosis. Curr. Opin. Lipidol. 5: 371–375 [DOI] [PubMed] [Google Scholar]
- 34.Vogiatzi G., Tousoulis D., Stefanadis C. 2009. The role of oxidative stress in atherosclerosis. Hellenic J. Cardiol. 50: 402–409 [PubMed] [Google Scholar]
- 35.Stocker R., Keaney J. F., Jr 2005. New insights on oxidative stress in the artery wall. J. Thromb. Haemost. 3: 1825–1834 [DOI] [PubMed] [Google Scholar]
- 36.Ross A. C., Zolfaghari R. 2011. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 31: 65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herdeg C., Oberhoff M., Baumbach A., Schroeder S., Leitritz M., Blattner A., Siegel-Axel D. I., Meisner C., Karsch K. R. 2003. Effects of local all-trans-retinoic acid delivery on experimental atherosclerosis in the rabbit carotid artery. Cardiovasc. Res. 57: 544–553 [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Ramirez M., Martinez-Gonzalez J., Juan-Babot J. O., Rodriguez C., Badimon L. 2005. Transcription factor SOX18 is expressed in human coronary atherosclerotic lesions and regulates DNA synthesis and vascular cell growth. Arterioscler. Thromb. Vasc. Biol. 25: 2398–2403 [DOI] [PubMed] [Google Scholar]
- 39.Dulin E., Garcia-Barreno P., Guisasola M. C. 2010. Extracellular heat shock protein 70 (HSPA1A) and classical vascular risk factors in a general population. Cell Stress Chaperones. 15: 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scher J. U., Pillinger M. H. 2005. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin. Immunol. 114: 100–109 [DOI] [PubMed] [Google Scholar]
- 41.Bishop-Bailey D., Hla T. 1999. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J. Biol. Chem. 274: 17042–17048 [DOI] [PubMed] [Google Scholar]
- 42.Powell W. S. 2003. 15-Deoxy-delta12,14–PGJ2: endogenous PPAR-gamma ligand or minor eicosanoid degradation product? J. Clin. Invest. 112: 828–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garzón B., Oeste C. L., Diez-Dacal B., Perez-Sala D. 2011. Proteomic studies on protein modification by cyclopentenone prostaglandins: expanding our view on electrophile actions. J. Proteomics. 74: 2243–2263 [DOI] [PubMed] [Google Scholar]
- 44.Subbanagounder G., Leitinger N., Schwenke D. C., Wong J. W., Lee H., Rizza C., Watson A. D., Faull K. F., Fogelman A. M., Berliner J. A. 2000. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler. Thromb. Vasc. Biol. 20: 2248–2254 [DOI] [PubMed] [Google Scholar]
- 45.Podrez E. A., Byzova T. V., Febbraio M., Salomon R. G., Ma Y., Valiyaveettil M., Poliakov E., Sun M., Finton P. J., Curtis B. R., et al. 2007. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 13: 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oskolkova O. V., Afonyushkin T., Preinerstorfer B., Bicker W., von Schlieffen E., Hainzl E., Demyanets S., Schabbauer G., Lindner W., Tselepis A. D., et al. 2010. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J. Immunol. 185: 7706–7712 [DOI] [PubMed] [Google Scholar]
- 47.Weerapana E., Wang C., Simon G. M., Richter F., Khare S., Dillon M. B., Bachovchin D. A., Mowen K., Baker D., Cravatt B. F. 2010. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 468: 790–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.