

Abstract
Short tandem repeats (STRs) have a wide range of applications, including medical genetics, forensics, and genetic genealogy. High-throughput sequencing (HTS) has the potential to profile hundreds of thousands of STR loci. However, mainstream bioinformatics pipelines are inadequate for the task. These pipelines treat STR mapping as gapped alignment, which results in cumbersome processing times and a biased sampling of STR alleles. Here, we present lobSTR, a novel method for profiling STRs in personal genomes. lobSTR harnesses concepts from signal processing and statistical learning to avoid gapped alignment and to address the specific noise patterns in STR calling. The speed and reliability of lobSTR exceed the performance of current mainstream algorithms for STR profiling. We validated lobSTR's accuracy by measuring its consistency in calling STRs from whole-genome sequencing of two biological replicates from the same individual, by tracing Mendelian inheritance patterns in STR alleles in whole-genome sequencing of a HapMap trio, and by comparing lobSTR results to traditional molecular techniques. Encouraged by the speed and accuracy of lobSTR, we used the algorithm to conduct a comprehensive survey of STR variations in a deeply sequenced personal genome. We traced the mutation dynamics of close to 100,000 STR loci and observed more than 50,000 STR variations in a single genome. lobSTR's implementation is an end-to-end solution. The package accepts raw sequencing reads and provides the user with the genotyping results. It is written in C/C++, includes multi-threading capabilities, and is compatible with the BAM format.
Short tandem repeats (STRs), also known as microsatellites, are a class of genetic variations with repetitive elements of 2–6 nucleotides (nt) that consist of approximately a quarter million loci in the human genome (Benson 1999). The repetitive structure of those loci creates unusual secondary DNA conformations that are prone to replication slippage events and result in high variability in the number of repeat elements (Mirkin 2007). The spontaneous mutation rate of STRs exceeds that of any other type of known genetic variation and can reach 1/500 mutations per locus per generation (Walsh 2001; Ballantyne et al. 2010), 200-fold higher than the rate of spontaneous copy number variations (CNV) (Lupski 2007) and 200,000-fold higher than the rate of de novo SNPs (Conrad et al. 2011).
STR variations have been instrumental in wide-ranging areas of human genetics. STR expansions are implicated in the etiology of a variety of genetic disorders, such as Huntingon's Disease and Fragile-X Syndrome (Pearson et al. 2005; Mirkin 2007). Forensics DNA fingerprinting relies on profiling autosomal STR markers and Y-chromosome STR (Y-STR) loci (Kayser and de Knijff 2011). STRs have been extensively used in genetic anthropology, where their high mutation rates create a unique capability to link recent historical events to DNA variations, including the well-known Cohen Modal Haplotype that segregates in patrilineal lines of Jewish priests (Skorecki et al. 1997; Zhivotovsky et al. 2004). Another relatively recent application of STR analysis is tracing cell lineages in cancer samples (Frumkin et al. 2008).
Despite the plurality of applications, STR variations are not routinely analyzed in whole-genome sequencing studies, mainly due to a lack of adequate tools (Treangen and Salzberg 2011). STRs pose a remarkable challenge to mainstream HTS analysis pipelines. First, not all reads that align to an STR locus are informative (Supplemental Fig. 1A). If a single or paired-end read partially encompasses an STR locus, it provides only a lower bound on the number of repeats. Only reads that fully encompass an STR can be used for exact STR allelotyping. Second, mainstream aligners, such as BWA, generally exhibit a trade-off between run time and tolerance to insertions/deletions (indels) (Li and Homer 2010). Thus, profiling STR variations—even for an expansion of three repeats in a trinucleotide STR—would require a cumbersome gapped alignment step and lengthy processing times (Supplemental Fig. 1B). Third, PCR amplification of an STR locus can create stutter noise, in which the DNA amplicons show false repeat lengths due to successive slippage events of DNA polymerase during amplification (Supplemental Fig. 1C; Hauge and Litt 1993; Ellegren 2004). Since PCR amplification is a standard step in library preparation for whole-genome sequencing, an STR profiler should explicitly model and attempt to remove this noise to enhance accuracy.
Here, we present lobSTR, a rapid and accurate algorithm for STR profiling in whole-genome sequencing data sets (Fig. 1). Briefly, the algorithm has three steps. The first step is sensing: lobSTR swiftly scans genomic libraries, flags informative reads that fully encompass STR loci, and characterizes their STR sequence. This ab initio procedure relies on a signal processing approach that uses rapid entropy measurements to find informative STR reads followed by a Fast Fourier Transform to characterize the repeat sequence. The second step is alignment: lobSTR uses a divide-and-conquer strategy that anchors the nonrepetitive flanking regions of STR reads to the genome to reveal the STR position and length. We use a modified reference that takes advantage of the information extracted from the sensing step to increase the alignment specificity. This step avoids a cumbersome gapped alignment and, importantly, is virtually indifferent to the magnitude of STR variations. Finally, in the third step, the pipeline allelotypes the STRs using a statistical learning approach that models the stutter noise in order to enhance the signal of the true allelic configuration. For full details about the lobSTR algorithm, see the Supplemental Material, Supplemental Figures 2–5, and Supplemental Table 1.
Figure 1.

lobSTR algorithm overview. lobSTR consists of three steps. The sensing step detects informative STR reads and determines their repeat motif. The alignment step maps the STRs' flanking regions to the reference. The allelotyping step determines the STR alleles present at each locus.
lobSTR implementation offers a complete solution that takes raw sequencing data and reports the alleles present at each profiled STR locus. The program's input is one or more sequencing libraries in FASTA/FASTQ or BAM format. The output is the alignment of STR reads in BAM format and the most likely alleles for each STR locus in a custom tab-delimited text format. lobSTR supports multi-threaded processing. lobSTR is available at http://jura.wi.mit.edu/erlich/lobSTR/.
Results
Comparing lobSTR to mainstream aligners
We benchmarked lobSTR's alignment performance with reads from an Illumina whole-genome sequencing library with 101-bp reads (Methods). To demonstrate its added value for STR profiling over mainstream aligners, we also ran BWA, Novoalign, and Bowtie on the same input data with and without the GATK local indel realignment tool. In addition, we ran BLAT (Kent 2002) to characterize STR alignment by a tool that is centered on sensitivity rather than speed. BWA and Novoalign were tested with the default parameters that can detect up to 5-bp and 7-bp indels, respectively. Bowtie has no indel tolerance and was evaluated as a control condition with tolerance of up to two mismatches. BLAT was tested with the default parameters that can tolerate up to 10% divergence from the reference, which corresponds to ∼10-bp indels. To focus on the pure algorithm speed-up, all tests were executed on a single CPU.
lobSTR excelled in all of the parameters required for efficient STR alignment (Table 1). First, lobSTR processed the reads 2.2 times faster than Bowtie, 22 times faster than BWA, 70 times faster than Novoalign, and almost 1000 times faster than BLAT (Fig. 2A). These results indicate that there is a minimal computational payment in running lobSTR in parallel to mainstream aligners in order to augment variation calling to include STR polymorphisms. Second, as required, lobSTR reported only informative reads that fully encompass STR loci. On the other hand, the mainstream aligners reported between 2000 and 5000 noninformative STR reads per million input sequences, which may confound downstream calling algorithms if not removed. Third, lobSTR detected the largest number of informative reads with STR variations compared with mainstream aligners (Fig. 2B). The other aligners showed a strong tendency to report STR reads with the reference allele vis-à-vis with their indel tolerance. Bowtie did not report any STR variation. After GATK local realignment, BWA and Novoalign, respectively, reported that 20% and 25% of the informative reads have STR variations. BLAT reported that 37% of the informative reads have STR variations, compared with 50% in lobSTR. Analyzing data collected from a large number of randomly ascertained STR loci (Utah Marker Development Group 1995; Payseur et al. 2011) demonstrates that 33%–66% of STR sequence reads should exhibit a nonreference allele (see Methods). This suggests that lobSTR's results are more representative of the true rate of STR variations than mainstream alignment tools.
Table 1.
STR Alignment performance across different algorithms
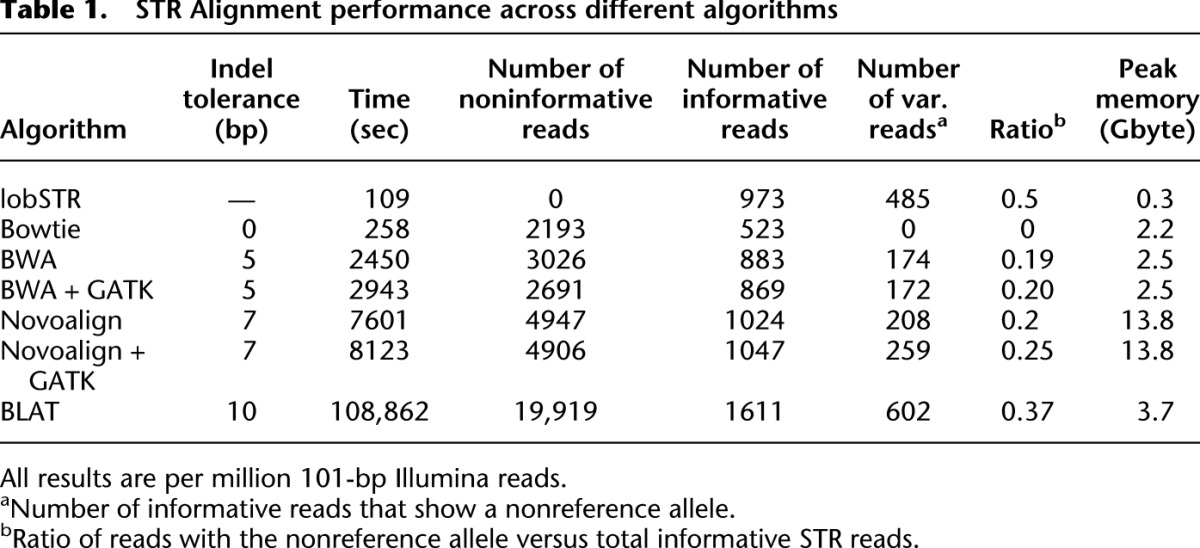
Figure 2.

lobSTR shows an added value for STR profiling over mainstream techniques. (A) Alignment speed (reads per second) of lobSTR, mainstream aligners, and BLAT. lobSTR processes reads between 2.5 and 1000 times faster than alternative methods. (B) The sensitivity of detecting STR variations of different alignment strategies. Only BLAT detected more STR variations than lobSTR. (C) lobSTR accurately detects pathogenic trinucleotide expansions that are discarded by mainstream aligners. The figure shows simulation results of the HOXD13 heterozygous locus with a normal and a pathogenic allele that contains seven additional alanine insertions. BWA reports only the normal allele. Reads exhibiting a pathogenic STR expansion are not detected. lobSTR identifies both alleles present at the simulated locus. All positions are according to hg18.
Reporting STR reads with nonreference alleles is crucial for profiling pathogenic mutations. We further explored whether lobSTR can correctly detect disease alleles of dominant trinucleotide repeat expansion disorders. As test cases, we focused on two conditions that can be theoretically profiled using standard Illumina runs. The first condition was a GCN expansion in PABPN1 that causes oculopharyngeal muscular dystrophy (OPMD) (Brais et al. 1998), where the normal allele exhibits 10 repeats and the pathogenic allele spectrum for the dominant form is between 12 and 17 repeats (Pearson et al. 2005). The second condition was a GCG expansion in HOXD13 that is implicated in synpolydactyly (Muragaki et al. 1996), a severe limb malformation, where the normal allele is 15 repeats and the documented pathogenic allele spectrum is between 22 and 29 repeats (Pearson et al. 2005). To simulate each condition, we generated 100 reads of length 101 bp that were equally sampled from the disease locus consisting of a normal and pathogenic allele with 100 bp flanking upstream and downstream regions with a 1% sequencing error rate. For both simulated disease conditions, lobSTR accurately aligned the normal and pathogenic reads to the correct location in the genome. All aligned reads were informative, and the allelotyping step correctly assigned a heterozygous state to the disease loci with the correct repeat lengths: (10, 15) for PABPN1 and (15, 22) for HOXD13. In stark contrast, BWA failed to correctly align reads from the pathogenic alleles of both loci. Only reference reads were reported (Fig. 2C).
Measuring lobSTR concordance using biological replicates
To explore the precision of lobSTR, we conducted genome-wide STR profiling of blood and saliva samples from the same individual (Lam et al. 2012). These samples were sequenced using Illumina HiSeq 2000 with 101-bp PE to a mean autosomal coverage of 50× and 102×, respectively. lobSTR ran with default parameters on 20 CPUs and analyzed the two data sets within 12 and 22 h, respectively. After filtering loci with low-quality calls, 143,793 shared STRs were covered in the two data sets with at least one read, and 79,771 STRs were covered with 10 reads or more (Fig. 3A).
Figure 3.

(A–C) Measuring lobSTR consistency from two samples of the same individual; (green) period 2; (orange) period 3; (red) period 4; (blue) period 5; (purple) period 6; (black) all. (A) Loci covered in both samples at increasing coverage thresholds. (B) The genotype discordance rate as a function of coverage threshold. (C) The allelic discordance rate as a function of coverage threshold. (D) Number of repeat differences at heterozygous loci. (Blue) No difference; (red) integer numbers of repeat differences; (green) noninteger numbers of repeat differences. Most discordance calls consist of a single repeat unit difference between calls in the two samples. Distance was measured as the second minimum distance between alleles of the two samples. The y-axis is given in a square root scale.
We quantified the rate of discordant autosomal calls between the two samples. We focused on two measurements: the genotype discordance rate and the allelic discordance rate (Pompanon et al. 2005). The former reports as an error any mismatch between corresponding calls, whereas the latter reports only the fraction of discordant alleles in corresponding calls. For example, consider a locus that is called (A, B) in the saliva sample and (A, C) in the blood sample. This locus shows a single genotype discordance, but only 0.5 allelic discordance, since the A allele was correct.
Both types of error greatly diminished with sufficient coverage (Fig. 3B,C). At 5× coverage, the genotype discordance rate was 11%, and the allelotype discordance was 5%. At 21× coverage, the genotype discordance rate was 3%, and the allelotype discordance rate was 2%. Similar to STR studies with capillary platforms (Weber and Broman 2001), most of the errors were generated in dinucleotide STR loci, whereas other types of STRs showed moderate and similar error rates. The dinucleotide error rates presumably stem from two factors: First, these loci usually show the highest heterozygosity rates (Chakraborty et al. 1997; Brinkmann et al. 1998; Pemberton et al. 2009). Therefore, they require on average more sequence reads to be correctly called. Second, dinucleotide STRs are more prone to stutter noise (Ellegren 2004), and their higher error rates might be due to residual noise after lobSTR stutter deconvolution.
We further analyzed the STR length differences in discordant calls. To avoid analyzing errors that are simply due to allele drop-outs, we focused on discordant calls that were both heterozygous in blood and saliva. At a coverage of ≥5×, >90% of the errors showed a single repeat unit difference, and 99% of the errors were within two repeat units (Fig. 3D). This indicates that incorrect alignment of STRs has a minimal effect on allelotyping results and that stutter is likely the main source of noise. We also found that only 0.8% of calls at heterozygous loci showed a difference due to an incomplete repeat unit. This highlights that lobSTR can determine STR alleles at a single-base-pair resolution.
Tracing Mendelian inheritance using lobSTR
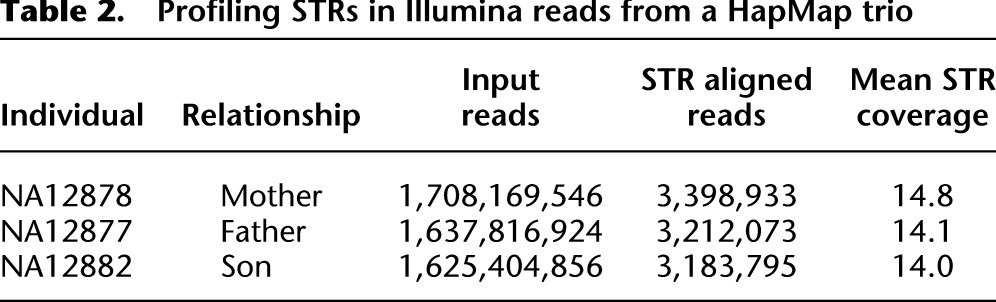
To further explore lobSTR performance, we conducted a genome-wide STR profiling of a HapMap trio—a father (NA12877), mother (NA12878), and son (NA12882)—from the CEU population that were sequenced using 100PE reads on a HiSeq 2000 (Table 2). The average autosomal coverage was 50×, and the average STR coverage was 14×. At the ≥10× coverage threshold, 57% of the STRs in the CEU trio had a nonreference allele.
Table 2.
Profiling STRs in Illumina reads from a HapMap trio
In general, deviations of offspring's STR alleles from Mendelian inheritance (MI) indicate a potential calling error (Ewen et al. 2000). With 5× coverage across all trio members, the MI rate was 95%; with 10× coverage, the MI rate was 97%; and with a coverage threshold of 15 or more, the MI rate was 99% (Fig. 4A). We also repeated the analysis only with discordant parental sites (for example, A/B call in one parent and A/C call in another parent). We noticed a drop to 93% in the MI patterns with a low coverage threshold of 5×, which is mainly because of partial coverage of heterozygous sites in the parents. The MI rate was recovered to the same level with a higher coverage threshold. At 17× coverage, 99% of sites showed a perfect Mendelian segregation pattern (Fig. 4B).
Figure 4.

Validating lobSTR by Mendelian inheritance in a HapMap trio. Mendelian inheritance (blue and cyan) rose to 99% above 17× coverage. (Dark and light red) The number of covered loci at each coverage threshold. (A) Mendelian inheritance of all covered loci. (B) Mendelian inheritance of loci with discordant parental allelotypes.
Validating lobSTR accuracy with DNA electrophoresis
We sought to compare lobSTR calls with the results of DNA electrophoresis, which is considered the gold standard for STR allelotyping. First, we focused on a set of STR markers that are used for forensic DNA fingerprinting. As an input for lobSTR, we sequenced a male genome from our laboratory collection with three runs of Illumina GAIIx for 101PE cycles that yielded ∼740 million reads. The autosomal sequencing coverage was 36× according to alignment with mainstream algorithms. lobSTR identified 1.6 million informative reads that mapped to ∼140,000 STR loci, with an average of 4.91× coverage of diploid STR loci. In parallel, we used a commercial forensic kit to genotype 14 autosomal STR markers on a capillary electrophoresis platform. Thirteen out of 14 markers were covered by at least a single sequence read, and eight markers were covered by at least three sequence reads. The marker that was not covered spanned >129 bp, exceeding the limit for detecting informative reads with the 101-bp sequence reads.
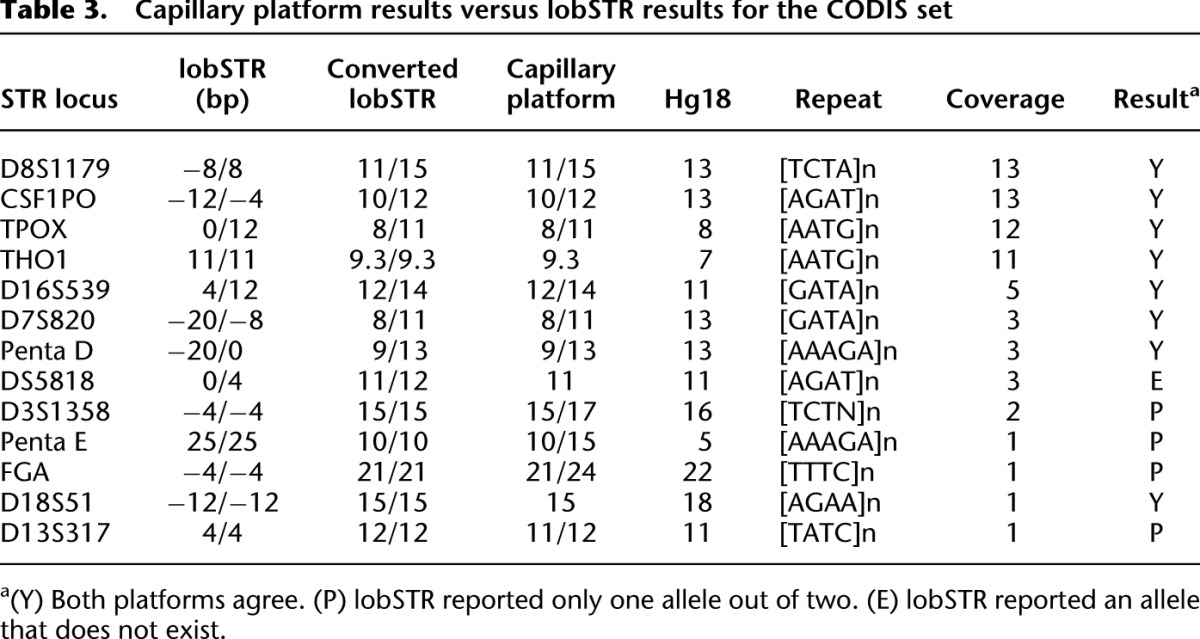
We observed good concordance between lobSTR and the capillary results (Table 3). lobSTR correctly called all but one of the eight markers that were covered by at least three reads and most of the alleles in loci that were covered with two or less reads. Remarkably, some of these markers, such as D8S1179, displayed two heterozygous alleles that did not match the reference. Other alleles, such as in Penta D and Penta E, correctly returned 20-bp and 25-bp length differences from the reference allele, respectively. The capillary results of one tetranucleotide marker, THO1, exhibited a noninteger number of copies (9 repeats + 3 bp). lobSTR reported exactly the same results, further demonstrating that STRs can be called within a single-base-pair resolution. lobSTR also correctly called a homozygous STR that was covered by a single read. In another four markers with a coverage of ≤2×, lobSTR correctly called one allele and missed the other allele due to sequencing coverage. We observed only a single erroneous call due to stutter noise in the D5S818 locus. This homozygous locus was covered by three sequence reads: two correct and one with a single repeat expansion. With such a low sequencing coverage, the allelotyping algorithm was not able to identify the noisy read and assigned a heterozygous state to the locus.
Table 3.
Capillary platform results versus lobSTR results for the CODIS set
Next, we evaluated lobSTR calls made in 12 low-pass sequenced genomes from the Human Genome Diversity Project (HGDP) (Green et al. 2010; Reich et al. 2010). Five genomes had a coverage of 1.4×–1.9× with 109-bp reads, and the other seven had a coverage of 4.8×–7.7× with 77-bp reads (Supplemental Table 3). One hundred and ninety-five STRs with equivalent entries in the lobSTR reference have been genotyped in these genomes using DNA electrophoresis as part of the CEPH–HGDP panel (Ramachandran et al. 2005; Pemberton et al. 2009). Combining lobSTR results from all data sets gave 59 comparable markers with a coverage of three to five reads with a median coverage of 3× (Supplemental Table 4). Despite the low coverage, lobSTR correctly returned 75% of the genotypes and 85% of the allele calls. Most of the alleles showed at least 5-bp difference from the reference, and some alleles showed a difference of 24 bp and were correctly called. We did not observe a significant correlation between errors and the size of the variation.
Genome-wide STR profiling confirms previously locus-centric observations
Encouraged by the accuracy and speed of lobSTR, we harnessed our pipeline to establish a reliable reference for future studies. Our input data set was a male individual that, as of today, has been sequenced to the highest coverage of 126-fold from a blood sample (Ajay et al. 2011). Fourteen billion sequencing reads were obtained from 100-bp PE runs on Illumina GAIIx and HiSeq 2000. lobSTR ran for 26 h using 25 CPUs. It aligned ∼6 million reads to ∼180,000 STR loci out of the 249,000 in the Tandem Repeat Table reference with an average coverage of 20.82 for autosomal loci. The average reference allele length of undetected loci was 150 bp, whereas the mean reference length of detected loci was 41 bp. Therefore, in most cases, the undetected loci could not physically be spanned by a single read of the current sequencing length.
We assigned each autosomal STR to one of four allelotype categories: Both alleles match the reference (homozygous reference), one allele matches the reference (heterozygous reference), both alleles do not match the reference but are the same (homozygous nonreference), and both alleles are different and do not match the reference (heterozygous nonreference). In all previous experiments, a coverage threshold of 20× resulted with near-perfect STR calling even for dinucleotide loci. To increase the reliability of our results, we focused the analysis on the 97,844 loci that were called with at least this sequencing coverage. The length distribution of these alleles in the reference was mainly between 25 and 50 bp with a low number of very long STRs (Fig. 5A).
Figure 5.

Genome-wide STR profile of an individual. (A) Distribution of STRs with 20× coverage or more as a function of the allele size in hg18. (B) Distribution of allele size differences from reference in lobSTR calls. The average difference was 6.3 bp away from the reference. (C) STR polymorphism as a function of period. The number of STR alleles matching the reference sequence increases with increasing repeat unit length. (Red) Homozygous reference; (blue) heterozygous nonreference/reference; (green) homozygous nonreference/nonreference; (orange) heterozygous nonreference/nonreference. (D) Longer STR regions are more polymorphic. The median STR length (thick black line) increases with the number of variant alleles. (*) A significant (p < 0.05) difference according to a one-sided Mann–Whitney test. Boxes denote the interquartile range, and whiskers denote three times the interquartile range. (E) lobSTR shows mutational trends at single-base-pair resolution. The number of base pairs different from the reference modulo period size versus the number of alleles detected (in logarithmic scale) is shown for each period; (green) period 2; (orange) period 3; (red) period 4; (blue) period 5; (purple) period 6. Incomplete STR unit differences tend to differ by a full unit ±1 bp from the reference. (F) Fraction of trinucleotide STRs with nonreference alleles in introns versus exons. The 95% confidence intervals are given by the error bars.
Similar to the other genomes in this study, 55% (52,338) of the STR loci differed from the reference: 22,271 (23%) loci were heterozygous reference, 15,515 (16%) loci were homozygous nonreference, and 14,552 (15%) loci were heterozygous nonreference. The other 43,335 (45%) loci were homozygous reference. Some of the variations reached to a 49-bp difference from the reference allele. On average, STR variations showed a 6.3-bp difference from the reference allele, and 41% of the variations were >5 bp away from the reference (Fig. 5B). Thus, mainstream-dependent analysis pipelines that can tolerate only a few nucleotide indels, such as BWA, are likely to miss most STR variations.
The genome-wide STR dynamics reported by lobSTR confirm previous findings of locus-centric studies. The rate of STR polymorphism showed a striking correlation with the repeat unit length (Fig. 5C). Dinucleotide STRs are nearly equally likely to fall into any of the above four categories, whereas hexanucleotide STRs are most likely to match the reference. This trend matches results of a previous study that measured the mutation rate of a few hundreds of Y-STR loci as a function of repeat unit length (Jarve et al. 2009). Similar to our results, the investigators showed that penta- and hexanucleotide repeats mutate at half the rate of tri- and tetranucleotide repeats. We also found that the rate of STR polymorphism is significantly correlated to the length of the STR allele in the reference (Fig. 5D). The nonreference loci (n = 52,338) had significantly greater lengths than loci that are homozygous reference (n = 43,335; p < 0.05, one-sided Mann–Whitney test for each allelotype category vs. reference) as previously reported in studies that analyzed a few dozen STRs (Brinkmann et al. 1998; Ellegren 2000).
We also used lobSTR to determine genome-wide trends of STRs at single-base-pair resolution (Fig. 5E). Overall, 99% of alleles varying from the reference allele showed differences that were complete multiples of the STR unit. This trend varied by period, with dinucleotide STRs least likely (0.3%) to differ by an incomplete motif unit and hexanucleotide STRs most likely (4.7%).
Finally, lobSTR reported significant differences between repeat variations in intronic and exonic regions (Fig. 5F). Intronic trinucleotide STRs were twice as likely to exhibit at least one nonreference allele than exonic regions (0.480–0.502, 95% CI; and 0.179–0.336, 95% CI for introns and exons, respectively), and nearly five times as likely to exhibit two nonreference alleles (0.107–0.119, 95% CI; and 0–0.047, 95% CI for introns and exons, respectively). Significantly, lobSTR reported that 1.9% (62 out of 3276) of the intronic trinucleotide STRs showed length differences that were not a multiple of three nucleotides. On the other hand, all reported exonic trinucleotide variants retained the reading frame. In addition, lobSTR allelotyped 34,667 intronic and seven exonic non-trinucleotide STRs. Of the intronic non-trinucleotide STRs, 18,277 (53%) showed at least one allele with a frameshift deviation, and 8686 (25%) showed two frameshifted alleles. Surprisingly, three of the seven exonic loci, all tetranucleotides, showed expansions by units of 4 bp, which would result in a frameshift mutation. In one case, in exon 8 of DCHS2, the frameshift variation was homozygous. This call was supported by 33 independent reads, showing a potential loss of function in this gene.
Taken together, the overall findings of lobSTR in this genome serve as a biological validation for the accuracy and utility of genome-wide STR profiling using our technique.
Discussion
STR profiling techniques have changed very little in the past two decades, relying on the faithful yet cumbersome capillary electrophoresis technique to scan a few dozen loci at a time. The advent of HTS has ushered in the opportunity to conduct genome-wide STR variation analyses. Here, we present an end-to-end solution for this task. Our solution bypasses the gapped alignment problem, has no inherent indel limitation, and can reliably profile highly polymorphic STRs at a single-base-pair resolution. We provide a detailed comparison between lobSTR and popular mainstream aligners and show that even with long reads, these aligners are significantly biased toward the detection of the reference allele. We have established the feasibility of lobSTR to profile STR loci from a total of 20 genomic data sets and demonstrated the strategy's accuracy by analyzing its consistency and ability to trace Mendelian inheritance, and by comparing its results to orthogonal molecular techniques. Moreover, our genome-wide STR analysis confirms previous biological observations, which further highlights the algorithmic validity.
lobSTR results from the trio genomes and the Ajay et al. (2011) genome consistently showed genome-wide polymorphism rates of 55%–57% for STRs with lengths 25 bp and over. A recent study by McIver et al. (2011) evaluated the performance of STR calling using post-BWA alignment files with a set of quality rules. Using a mixture of Illumina 45- to 100-bp reads and 454 Life Sciences (Roche) reads from two trios in the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010), they reported that 1.1% of the STRs with lengths of 20 bp and over were polymorphic. We wondered if the polymorphism discrepancy between the studies could be explained by the shorter reading lengths in the McIver study that biased their calls to very short, less polymorphic STRs. However, when we ran lobSTR on the 1000 Genomes CEU trio data sets (Methods), we found again that 57% of the STRs were polymorphic (25,885 out of 45,461 STRs that were called with ≥5× coverage at the three genomes). These results suggest that STR profiling that is restricted by the default BWA indel tolerance—5 bp for the Illumina data sets in the McIver et al. (2011) study—can significantly reduce the sensitivity for observing STR variations.
We envision that lobSTR will be used in parallel to conventional analysis pipelines in order to augment variation calling to include STR loci. The fast running time of our algorithm should not impose a significant computational burden on users. A low-coverage genome of 5× takes about an hour on a standard server with 25 CPUs, a high-coverage genome of 30× takes 8 h using the same settings, and an ultra-high-covered genome of 126× takes 26 h (Supplemental Table 2).
Currently, the major barrier for STR profiling is the sequencing read length, because the number of detectable STRs is limited to those that are entirely spanned by a single read. To test the effect of genomic coverage on STR profiling, we sampled reads from the 126× genome and calculated the amount of reported STRs (Supplemental Fig. 6). With genome-wide coverage of 40×, there are more than 100,000 STRs that will pass an STR-coverage threshold of 10×. However, higher genomic coverage does not linearly improve the number of STRs that pass this threshold, marking a potential upper bound of sequencing read lengths of 100 bp. We also explored the utility of the longer reads by Sanger, 454, and IonTorrent for STR profiling of personal genomes using lobSTR (Supplemental Table 5; Supplemental Material). Longer reads, indeed, increased the number of reported STR loci compared with the same autosomal coverage by Illumina. However, out of these, Sanger seemed to be the only method to produce reliable STR reads. We expect that as sequencing reads continue to increase in both length and quality, lobSTR's performance will further improve and allow inclusion of a larger number of STR variations. Ultimately, these will include large pathogenic expansion, such as those in Huntington's Disease, which can span >100 bp.
As of today, sequence analysis algorithms can detect almost any type of genetic variations, from SNPs (Goya et al. 2010) and indels (Koboldt et al. 2009; Goya et al. 2010) to CNVs and chromosomal translocations (Chen et al. 2009). lobSTR adds a new layer of information with tens of thousands of highly polymorphic genetic variations that have a multitude of applications, from personal genomics, to population studies, forensics analysis, and cancer genome profiling.
Methods
Comparing lobSTR to mainstream aligners
All alignment strategies were tested in a Linux environment, on a server with four 12-core AMD Opteron 6100 and 128 Gbyte of RAM. The following software versions were tested: BWA version 0.5.7, Bowtie version 0.12.7, and Novoalign freeware version 2.7.13, BLAT version 34, and GATK version 1.3-21.
The input was 5 million Illumina reads of the male sample from our laboratory collection. BLAT results were filtered to include only the top hit for each read. We suppressed multi-mappers in all other tools. Informative STR reads were identified by the intersectBed tool of the Bedtools packages (Quinlan and Hall 2010). We converted CIGAR scores to the number of base pairs difference from the reference allele by counting any insertions or deletions falling within and directly adjacent to the STR region. Simulating reads from pathogenic STR loci was conducted using a simple Python script (available by request from the authors).
Determining the expected number of nonreference reads
A previous study by the Utah Marker Development Group 1995 has shown that 70% of thousands of randomly chosen tetranucleotide STR loci are polymorphic. We also re-analyzed Payseur et al. (2011) data to infer the polymorphism rate in STRs with length ≥25 bp in the assembled genome of Craig Venter using results reported in their Supplemental Tables 1–5. Concordant with the Utah study, this rate was 66%.
The rate of nonreference STR reads is bounded between two extreme cases. The lower bound is that all polymorphic STRs are heterozygous with a reference allele. Thus, only half of the reads from variable loci will show a nonreference allele, which gives 33% as a lower bound. The upper bound is that all polymorphic STRs are different from the reference in their two alleles. In this case, every read from a variable locus will show a nonreference allele, which gives 66% as an upper bound.
Biological replicates analysis
Raw reads for blood-derived and saliva-derived genomic DNA from the same individual were downloaded from the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) with accessions SRX097307 and SRX097312, respectively. Loci in which (1) <75% of reads agreed with the allelotype call in both samples or (2) the locus was covered in either sample by more than three times the mean coverage level were removed from the analysis.
CEU trio data for Mendelian inheritance
The HapMap CEU trio was NA12877 (father), NA12878 (mother), and NA12882 (son). Raw reads were downloaded from the European Sequence Read Archive (http://www.ebi.ac.uk/ena/) with accession numbers ERP001228, ERP001229, and ERP001230, respectively. To determine if an STR followed Mendelian inheritance, we required that the alleles detected in the son could be explained by inheriting one allele from each parent. Low-quality loci were filtered as described in “Biological Replicates Analysis” above.
Validating lobSTR accuracy using capillary electrophoresis
Four Catch-All buccal swabs (Epicenter, QEC89100) were used to collect the DNA sample according to the manufacturer's protocol. gDNA was extracted by QuickExtract (Epicentre), followed by phenol:chloroform purification and ethanol precipitation. Library preparation was performed according to the standard Illumina protocol. Three runs of 101-bp paired-end reads were generated on the Illumina GAIIx platform. The study was approved by MIT's Committee on the Use of Humans as Experimental Subjects (COUHES). The general sequencing coverage was analyzed as previously reported (Erlich et al. 2011).
Capillary electrophoresis results were obtained from the Sorenson Genomics laboratory using the commercial Promega PowerPlex 16 system. To find the genomic positions of these loci, we downloaded corresponding primers that target these loci from the Short Tandem Repeats Internet Database (STRBASE) website (http://www.cstl.nist.gov/strbase/) of the U.S. National Institute of Standards and Technology (NIST) and used the In Silico PCR tool on the UCSC Genome Browser to reveal their location. Two loci had proprietary primers, and their genomic locations could not be identified. The STR repeats in the sequencing file were converted to the PowerPlex allele nomenclature using the NIST definitions.
Obtaining CEPH–HGDP STR allelotypes
STR allelotypes along with a table of RefSeq reference alleles were downloaded from the Rosenberg laboratory site (http://www.stanford.edu/group/rosenberglab/repeatsDownload.html). The allelotypes were given as the number of repeats converted from PCR product size as described in Pemberton et al. (2009). The repeat number is given as the reference repeat number plus the difference in product size from the reference divided by the motif size. Sequence data were downloaded from the NCBI Short Read Archive with accession numbers ERX004003, ERX004002, ERX004001, ERX004000, ERX0039999, ERX004007, ERX007978, ERX007977, ERX007976, ERX007975, ERX007974, ERX007973, and ERX007972.
Using the STS marker table available from the UCSC Genome Browser, we converted the Pemberton et al. (2009) markers to hg18 genomic coordinates and annotated them using the TRF table. lobSTR calls that are supported by three or more reads were converted to the Pemberton results. Noninteger repeats reported by lobSTR were rounded to the smallest integer for compatibility with Pemberton data. Markers that could not be faithfully annotated were removed from the analysis.
Genome-wide STR profiling of a deeply sequenced personal genome
Raw sequencing reads for accession number ERP000765 were downloaded from the European Nucleotide Archive's Sequence Read Archive (http://www.ebi.ac.uk/ena/). The Mann–Whitney test was performed using the wilcox.test function in R. Confidence intervals were calculated using a normal approximation to the Poisson distribution, with a 95% confidence interval of λ ± 1.96√λ, where λ is the estimated mean of the distribution. Only loci with greater than 20-fold coverage were included in the analysis. Exon and intron coordinates were obtained from the UCSC Table Browser for human genome build hg18.
1000 Genomes data analysis for the McIver study
The HapMap CEU trio was NA12878 (daughter), NA12891 (father), and NA12892 (mother). Raw sequencing reads for the CEU HapMap trios with length of at least 47 bp were downloaded from the 1000 Genomes NCBI ftp site (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/). Two hundred twenty-eight, 274, and 214 files were included for individuals NA12878, NA12891, and NA12892. To accommodate the shorter read lengths, lobSTR was run with nondefault parameters -fft-window-size 20, -fft-window-step 10, -maxflank 100, and -extend-flank 5.
Software access
lobSTR is available at http://jura.wi.mit.edu/erlich/lobSTR/.
Acknowledgments
Y.E. is an Andria and Paul Heafy Family Fellow. This publication was supported by the National Defense Science & Engineering Graduate Fellowship (M.G.) and by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University (D.G.). D.G. and S.R. acknowledge support from Israeli Science Foundation grant ISF 1227/09 and an IBM Open Collaborative Research grant. We thank Mona Sheikh, Dina Esposito, and Alon Goren for useful comments on the manuscript; Assaf Gordon for his assistance with multi-threading programming; Cole Trapnell for his assistance with preparing lobSTR executables; Mona Sheikh and Sam Sinai for testing lobSTR code; and Dina Esposito for preparing samples for genotyping.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.135780.111.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH 2011. Accurate and comprehensive sequencing of personal genomes. Genome Res 21: 1498–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne KN, Goedbloed M, Fang R, Schaap O, Lao O, Wollstein A, Choi Y, van Duijn K, Vermeulen M, Brauer S, et al. 2010. Mutability of Y-chromosomal microsatellites: Rates, characteristics, molecular bases, and forensic implications. Am J Hum Genet 87: 341–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res 27: 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, et al. 1998. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18: 164–167 [DOI] [PubMed] [Google Scholar]
- Brinkmann B, Klintschar M, Neuhuber F, Huhne J, Rolf B 1998. Mutation rate in human microsatellites: Influence of the structure and length of the tandem repeat. Am J Hum Genet 62: 1408–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R 1997. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci 94: 1041–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H 2000. Heterogeneous mutation processes in human microsatellite DNA sequences. Nat Genet 24: 400–402 [DOI] [PubMed] [Google Scholar]
- Ellegren H 2004. Microsatellites: Simple sequences with complex evolution. Nat Rev Genet 5: 435–445 [DOI] [PubMed] [Google Scholar]
- Erlich Y, Edvardson S, Hodges E, Zenvirt S, Thekkat P, Shaag A, Dor T, Hannon GJ, Elpeleg O 2011. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res 21: 658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen KR, Bahlo M, Treloar SA, Levinson DF, Mowry B, Barlow JW, Foote SJ 2000. Identification and analysis of error types in high-throughput genotyping. Am J Hum Genet 67: 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumkin D, Wasserstrom A, Itzkovitz S, Stern T, Harmelin A, Eilam R, Rechavi G, Shapiro E 2008. Cell lineage analysis of a mouse tumor. Cancer Res 68: 5924–5931 [DOI] [PubMed] [Google Scholar]
- Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, Senz J, Crisan A, Marra MA, Hirst M, et al. 2010. SNVMix: Predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics 26: 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328: 710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge XY, Litt M 1993. A study of the origin of ‘shadow bands’ seen when typing dinucleotide repeat polymorphisms by the PCR. Hum Mol Genet 2: 411–415 [DOI] [PubMed] [Google Scholar]
- Jarve M, Zhivotovsky LA, Rootsi S, Help H, Rogaev EI, Khusnutdinova EK, Kivisild T, Sanchez JJ 2009. Decreased rate of evolution in Y chromosome STR loci of increased size of the repeat unit. PLoS ONE 4: e7276 doi: 10.1371/journal.pone.0007276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M, de Knijff P 2011. Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 12: 179–192 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L 2009. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25: 2283–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, Clark MJ, Chen R, Natsoulis G, O'Huallachain M, Dewey FE, Habegger L, Ashley EA, Gerstein MB, Butte AJ, et al. 2012. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol 30: 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Homer N 2010. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform 11: 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR 2007. Genomic rearrangements and sporadic disease. Nat Genet 39: S43–S47 [DOI] [PubMed] [Google Scholar]
- McIver LJ, Fondon JW III, Skinner MA, Garner HR 2011. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics 97: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM 2007. Expandable DNA repeats and human disease. Nature 447: 932–940 [DOI] [PubMed] [Google Scholar]
- Muragaki Y, Mundlos S, Upton J, Olsen BR 1996. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science 272: 548–551 [DOI] [PubMed] [Google Scholar]
- Payseur BA, Jing P, Haasl RJ 2011. A genomic portrait of human microsatellite variation. Mol Biol Evol 28: 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD 2005. Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet 6: 729–742 [DOI] [PubMed] [Google Scholar]
- Pemberton TJ, Sandefur CI, Jakobsson M, Rosenberg NA 2009. Sequence determinants of human microsatellite variability. BMC Genomics 10: 612 doi: 10.1186/1471-2164-10-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompanon F, Bonin A, Bellemain E, Taberlet P 2005. Genotyping errors: Causes, consequences and solutions. Nat Rev Genet 6: 847–859 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci 102: 15942–15947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468: 1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorecki K, Selig S, Blazer S, Bradman R, Bradman N, Waburton PJ, Ismajlowicz M, Hammer MF 1997. Y chromosomes of Jewish priests. Nature 385: 32. [DOI] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL 2011. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat Rev Genet 13: 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Utah Marker Development Group 1995. A collection of ordered tetranucleotide-repeat markers from the human genome. Am J Hum Genet 57: 619–628 [PMC free article] [PubMed] [Google Scholar]
- Walsh B 2001. Estimating the time to the most recent common ancestor for the Y chromosome or mitochondrial DNA for a pair of individuals. Genetics 158: 897–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JL, Broman KW 2001. Genotyping for human whole-genome scans: Past, present, and future. Adv Genet 42: 77–96 [DOI] [PubMed] [Google Scholar]
- Zhivotovsky LA, Underhill PA, Cinnioglu C, Kayser M, Morar B, Kivisild T, Scozzari R, Cruciani F, Destro-Bisol G, Spedini G, et al. 2004. The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am J Hum Genet 74: 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]