

Abstract
We report here the transmission of human prions to 18 new transgenic (Tg) mouse lines expressing 8 unique chimeric human/mouse prion proteins (PrP). Extracts from brains of two patients, who died of sporadic Creutzfeldt-Jakob disease (sCJD), contained either sCJD(MM1) or sCJD(VV2) prion strains and were used for inocula. Mice expressing chimeric PrP showed a direct correlation between expression level and incubation period for sCJD(MM1) prions irrespective of whether the transgene encoded methionine (M) or valine (V) at polymorphic residue 129. Tg mice expressing chimeric transgenes encoding V129 were unexpectedly resistant to infection with sCJD(VV2) prions, and when transmission did occur, it was accompanied by a change in strain type. The transmission of sCJD(MM1) prions was modulated by single amino acid reversions of each human PrP residue in the chimeric sequence. Reverting human residue 137 in the chimeric transgene from I to M prolonged the incubation time for sCJD(MM1) prions by more than 100 days; structural analyses suggest a profound change in the orientation of amino acid side chains with the I→M mutation. These findings argue that changing the surface charge in this region of PrP greatly altered the interaction between PrP isoforms during prion replication. Our studies contend that strain-specified replication of prions is modulated by PrP sequence-specific interactions between the prion precursor PrPC and the infectious product PrPSc.
INTRODUCTION
Human prion diseases have spontaneous, genetic, and infectious etiologies and are uniformly fatal. In all cases, the sole disease-causing agent is an aberrantly folded isoform of a normal cellular protein, termed the prion protein (PrP). Spontaneous PrP misfolding, assumed to be a stochastic event, results in sporadic Creutzfeldt-Jakob disease (sCJD); mutations in PrP make this conversion more likely to occur, giving rise to inherited prion diseases. Transmission from an exogenous source, such as ingestion of prion-infected human (kuru) or bovine (variant CJD) tissue, can initiate the misfolding cascade, resulting in an infectious etiology (31).
The transmissibility of prions enables an experimental paradigm for studying these devastating diseases. Initially, chimpanzees were used in laboratory studies (10, 11), but such experiments were extremely costly and time-consuming. Human prion transmissions to rodents led to alternate models; however, transmission of prions between species can lead to changes in the characteristics of the prion strain (12, 17). The introduction of transgenic (Tg) mouse models susceptible to human prions has enhanced the study of human prion diseases (3, 13, 16, 20, 43, 46).
Human PrP (HuPrP) is expressed as a 253-amino-acid polypeptide, with an N-terminal signal peptide for translocation, a C-terminal signal sequence for addition of a glycosylphosphatidyl inositol (GPI) lipid anchor, and two consensus sites for glycosylation. The resulting cellular glycoprotein (PrPC) has a predominantly α-helical structure and is localized to the outer leaflet of the cell membrane by the GPI moiety. In prion diseases, PrPC undergoes a major structural transformation, converting to β-sheet-rich, disease-causing PrPSc. This process is autocatalytic, with PrPSc driving the refolding of PrPC in a template-dependent manner.
A polymorphism at residue 129 in HuPrP encodes either a methionine (M) or valine (V) residue and has a major impact on susceptibility to prion disease (24). While homozygosity (MM or VV) at codon 129 occurs in approximately half the population, it accounts for almost 90% of the sCJD cases (26). This polymorphism also plays a role in the resultant strain type. Prion strains can be differentiated by biochemical and neuropathological analysis of PrPSc. Approximately 95% of sCJD(MM) cases exhibit an ∼21-kDa, unglycosylated protease-resistant PrP band on immunoblots, or “type 1” PrPSc; conversely, ∼95% of sCJD(VV) cases have an ∼19-kDa, unglycosylated PrP band, or “type 2” PrPSc. The different protease-resistant cores of type 1 and type 2 strains are believed to represent alternative conformations of PrPSc (41) and result from limited proteolysis at residues 82 and 97, respectively (27).
The first Tg mouse models expressing HuPrP were unexpectedly resistant to infection with CJD prions (42). This transmission barrier was abrogated by backcrossing Tg(HuPrP) mice to mice lacking expression of endogenous mouse PrP (MoPrP) (Prnp0/0) (43). Although the mature sequences of MoPrP and HuPrP are more than 90% identical, wild-type (wt) mice are largely resistant to infection with human prions, suggesting that a small number of residues must be responsible for prion susceptibility. In earlier studies, we found that mice expressing chimeric mouse/human PrP had abbreviated incubation periods for human prions (43), and reverting two or three additional human residues to mouse further shortened the incubation times (13, 20). Starting with this chimeric line expressing a construct that differs from MoPrP at seven residues, we investigated the role of expression level, the importance of the residue at polymorphic position 129, and the function of each remaining HuPrP residue, generating 18 novel Tg lines of mice. By inoculation with sCJD(MM1) and sCJD(VV2) prions into these mouse lines, we identified residues that are critical for the transmission of different human prion strains.
MATERIALS AND METHODS
Construct nomenclature.
The chimeric mouse/human construct, previously denoted MHu2M(M129), contains N- and C-terminal regions of MoPrP, with the central portion between residues 96 and 167 encoding the HuPrP sequence (with methionine at polymorphic position 129). This construct differs from MoPrP at nine residues: 96, 108, 111, 137, 142, 144, 154, 165, and 167 (43). For simplicity, this construct is here referred to as MHu(M129), and transgenic lines derived from this construct are referred to as Tg(MHu,M129). Reversion of human residues 165 and 167 from the MHu construct, previously denoted MHu2M(M165V,E167Q), led to transgenic mice with shorter incubation periods for sCJD(MM1) prions (20). Here, this construct is referred to as MHu#2, where #2 refers to the two reverted residues. This transgene was used as the base construct for all subsequent Tg mouse lines reported here. Subsequent reversion of HuPrP residues to the MoPrP sequence are denoted as additional mutations (Fig. 1).
Fig 1.

(A) Structure of MHu#2(M129) based on that of mouse PrP (green; dashed line indicates unstructured region), with human PrP residues shown as orange spheres. (B) Schematic representation of mouse (green) and human (orange) PrP sequences, with human PrP residues (orange bars) highlighted in the chimeric sequences. Asterisk in human PrP sequence identifies the position of polymorphic residue 129 denoted methionine (M) or valine (V) in the chimeric sequences.
CJD inocula.
Both sCJD(MM1) and sCJD(VV2) brain tissues were obtained from neuropathologically confirmed cases. In both instances, the full PrP open reading frame was sequenced. Strain typing was performed by immunoblotting as described below.
Generation of transgenic mouse lines.
All animal procedures were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of California San Francisco.
Tg mouse lines were created as described previously, using the Cos.tet vector (34, 35). DNA constructs were generated by site-directed mutagenesis with the QuikChange II kit (Stratagene, La Jolla, CA). Constructs were then cloned into the Cos.tet vector and microinjected into Friend virus B/Prnp0/0 zygotes. Potential founders containing the transgene were identified and bred into Tg lines. The transgene sequence was confirmed for all new lines developed.
Homozygous Tg(HuPrP,V129+/+)152 mice on the FVB/Prnp0/0 background were produced by backcrossing Tg(HuPrP,V129)152/Prnp0/0 mice on a mixed C57BL/6;129/Sv background (43) to FVB/Prnp0/0 mice for 10 generations and then intercrossing the resulting mice. Potentially homozygous mice were selected by quantitative dot blot of genomic DNA and confirmed by backcrossing.
Expression levels were performed by sandwich enzyme-linked immunosorbent assay (ELISA) or immunoblotting. For the ELISA, 96-well Immulon 4HBX plates (Nunc, Rochester, NY) were coated with the anti-PrP Fab R1 antibody at a dilution of 1:10,000 in carbonate buffer (0.1 M NaCO3, pH 8.6) overnight at 4°C. The plates were washed 5 times with TBST (100 mM Tris, 65 mM NaCl, 0.5% Tween 20, pH 8.0) and then blocked with 1% nonfat dry milk in TBST. Two-fold serial dilution of brain homogenates from 2% to 0.0156% were incubated onto these plates and then washed with TBST. Horseradish peroxidase (HRP)-conjugated Fab HuM-P was used as the detection antibody, with tetramethylbenzidine as the substrate, and read at 450 nm using the Spectramax Plus plate reader (Molecular Devices). Imunoblot analyses were performed as described below but using the F20-108a monoclonal primary antibody (38) and an HRP-conjugated anti-mouse secondary antibody. Samples were measured in triplicate from two to four brains. Mean values are reported relative to wt FVB brains.
Bioassay of CJD prions.
Eight- to ten-week-old mice were intracerebrally inoculated with 30 μl of 1% brain homogenate in phosphate-buffered saline (PBS) containing 5% bovine serum albumin. Mice were monitored daily for health and twice a week for neurological signs characteristic of prion disease, as previously reported (5).
Statistical analysis.
For survival analysis, median incubation periods and 95% confidence intervals (CI) were determined as reported previously (12). Glycoform proportions were compared using seemingly unrelated regressions to perform joint regressions of monoglycosylated and diglycosylated values (the unglycosylated proportion being defined by the other two). Levels of significance (P values) were two-sided based on the Wald test combining effects in the two seemingly unrelated regressions. All calculations were performed with Stata 11 (Stata Corp., College Station, TX).
Immunoblotting.
Samples were prepared as described previously (13); briefly, frozen mouse brains were homogenized using a Precellys 24 beadbeater (MO BIO, Carlsbad, CA) to 10% (wt/vol) in PBS. Before being loaded onto 10% NUPAGE precast gels, samples were treated with 100 μg/ml of proteinase K (PK) for 1 h at 37°C and then resuspended in 2× lithium dodecyl sulfate sample buffer and boiled for 10 min. Immunoblotting was performed using the iBlot dry blotting system (Invitrogen) for 7 min and then blocked with 10% nonfat milk in Tris-buffered saline with Tween 20, pH 7.5. Results were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ) using the HRP-conjugated HuM-P Fab. Un-, mono-, and diglycosylated PK-resistant PrP bands were quantified with ImageJ (33).
Neuropathology.
Brains were immersion fixed in 10% (vol/vol) buffered formalin, paraffin-embedded, sectioned, and then stained with hematoxylin and eosin or processed by immunohistochemistry as described previously (23). Four sections were reviewed for each brain, corresponding to caudate nucleus, hippocampus/thalamus, hippocampus/midbrain, and cerebellum/pons, from at least three mice for each strain type, when available.
Analysis of protein structure.
The most complete and comparable data sets were those of the structured region (residues ∼120 to 231) of MoPrP (identifier [ID] 1XYX) (14) and HuPrP (ID 1QM3) (47), which were available from the Protein Data Bank as ensembles of 20 structures. The backbone root mean square deviation of residues 125 to 225 from all the ensemble structures was calculated using MOLMOL (19). Surface potential was calculated in a 10-Å box with 0 as the boundary condition, assuming simple charge, with dielectric constants of 80 and 2 for the solvent and molecule, respectively; the salt concentration was assumed to be 150 mM, and the salt radius was 2 Å.
RESULTS
New transgenic lines expressing chimeric mouse/human PrP.
The Tg22372 mouse line expresses the MHu#2(M129) transgene, which encodes a chimeric mouse/human PrP that differs from MoPrP at seven positions. These mice express the chimeric transgene at approximately the same level of PrP in wt FVB mice and had mean incubation periods of ∼110 days when inoculated with sCJD(MM1) prions (20). To determine whether we could further reduce the incubation period, we constructed additional lines of Tg mice. We began by producing Tg lines expressing higher levels of the MHu#2(M129) construct. Next, we investigated strain susceptibility in mice expressing the same construct but with the polymorphic residue 129 changed to valine: MHu#2(V129).
Finally, we examined the role of the other residues on the incubation period. Because there are 125 possible combinations of chimeric PrP with seven or fewer differences, we chose to study those that differ by a single residue from MHu#2. In studying the effects of single mutations, we cannot exclude the possibility that mutations may act synergistically. Reverting residue 96 from serine to asparagine was previously found to increase the incubation periods for sCJD(MM1) prions (20). We therefore reverted each of the six remaining HuPrP residues to the corresponding MoPrP residue and generated at least one Tg line for each construct (Fig. 1).
Mice from each line were monitored for onset of spontaneous disease. The Tg17062 line expressing MHu#2(M129) at 4.9× developed an ataxic phenotype at ∼300 days, and two Tg lines expressing the MHu#2(M129,Y144W) transgene showed circling behavior, tremor, and ataxia at ∼550 days. However, none of these mice had any PK-resistant PrP in their brains, and neuropathological examination showed no abnormalities. All other lines remained healthy for at least 600 days (Tables 1 and 2).
Table 1.
Influence of the codon 129 polymorphism on the susceptibility to sCJD(MM1) and sCJD(VV2) prions in transgenic micea
| Mouse line | Expression level | Uninoculated |
sCJD(MM1) prions |
sCJD(VV2) prions |
|||
|---|---|---|---|---|---|---|---|
| No. of days (95% CI) until spontaneous disease | n/n0 | Incubation period, days (95% CI) | n/n0 | Incubation period, days (95% CI) | n/n0 | ||
| Tg(HuPrP,M129)440b | 2 | >600 | 0/9 | 162 (159, 170) | 33/33 | >600 | 6/21e |
| Tg(MHu#2,M129)22372b | 0.8 | >600 | 0/12 | 111 (107, 112) | 49/49 | >600 | 1/19f |
| Tg(MHu#2,M129)17103 | 1.2 | >600 | 0/6 | 103 (99, 104) | 7/7 | ND | |
| Tg(MHu#2,M129+/+)22372c | 1.6 | >600 | 0/17 | 124 (123, 126) | 34/34 | ND | |
| Tg(MHu#2,M129)17051 | 2.8 | >600 | 0/7 | 167 (165, 194) | 8/8 | ND | |
| Tg(MHu#2,M129)17062 | 4.9 | 289 (226, 312) | 6/6 | 244 (235, 255)d | 18/18 | ND | |
| Tg(HuPrP,V129+/+)152 | 4 | >600 | 0/7 | 211 (193, 214) | 7/7 | 193 (167, 208) | 8/8 |
| Tg(MHu#2,V129)10355 | 1.1 | >600 | 0/7 | 125 (117, 148) | 8/8 | >600 | 0/6 |
| Tg(MHu#2,V129)7104 | 1.3 | >600 | 0/7 | 133 (130, 134) | 8/8 | >600 | 0/7 |
| Tg(MHu#2,V129)6550 | 1.8 | >600 | 0/7 | 209 (193, 227) | 8/8 | >600 | 1/5g |
| Tg(MHu#2,V129)7110 | 2.4 | >600 | 0/7 | 207 (196, 214) | 8/8 | >600 | 4/16h |
PrP expression level in brain relative to FVB mice, reported as mean fold expression from two to four brains. Spontaneous disease from time of birth and incubation periods from time of inoculation are reported as median time to onset of clinical signs in days, with 95% confidence intervals, calculated using Kaplan-Meier statistics. n, number of mice with clinical signs of disease; n0, number of mice monitored; ND, not determined.
Includes data previously reported in reference 20.
Includes data previously reported in reference 28.
When recalculated from date of birth, durations not significantly different from spontaneous disease in uninoculated mice.
Individual mice showed clinical disease at 329, 336, 339, 339, 482, and 535 dpi.
One mouse showed clinical disease at 449 dpi.
One mouse showed clinical disease at 476 dpi.
Individual mice showed clinical disease at 473, 494, 509, and 540 dpi.
Table 2.
Susceptibility of Tg mice expressing chimeric mouse/human PrP to sCJD(MM1) prionsa
| Line | Expression level | No. of days (95% CI) until spontaneous disease | n/n0 | Incubation period, days (95% CI) | n/n0 |
|---|---|---|---|---|---|
| Tg(MHu#2,M129,M108L)1208 | 0.3 | >600 | 0/7 | 193 (189,194) | 7/7 |
| Tg(MHu#2,M129,M108L)1284 | 0.3 | >600 | 0/5 | 187 (179, 201) | 8/8 |
| Tg(MHu#2,M129,M111V)1014b | 2.8 | >600 | 0/6 | 77 (74, 81) | 20/20 |
| Tg(MHu#2,M129,I137 M)10027 | 1.8 | >600 | 0/5 | 237 (224, 292) | 6/6 |
| Tg(MHu#2,M129,I137 M)10025 | 2.0 | >600 | 0/8 | 239 (214, 253) | 4/4 |
| Tg(MHu#2,M129,S142N)3018 | 1.1 | >600 | 0/7 | 118 (106, 123) | 22/22 |
| Tg(MHu#2,M129,S142N)3061 | 1.5 | >600 | 0/6 | 152 (134, 152) | 7/7 |
| Tg(MHu#2,M129,Y144W)16914 | <0.05 | 550 (508, 585) | 6/7 | 558 (419, >600)c | 4/4 |
| Tg(MHu#2,M129,Y144W)17421 | <0.05 | 555 (548, >600) | 3/6 | 553 (438, >600)c | 6/6 |
| Tg(MHu#2,M129,H154Y)4561 | 0.3 | >600 | 0/4 | 231 (200, 249) | 5/5 |
| Tg(MHu#2,M129,H154Y)5099 | 0.1 | >600 | 0/4 | 391 (201, 439) | 7/7 |
PrP expression level in brain relative to FVB mice, reported as mean fold expression from two to four brains. Spontaneous disease from time of birth and incubation periods from time of inoculation are reported as median time to onset of clinical signs in days, with 95% confidence intervals, calculated using Kaplan-Meier statistics. n, number of mice with clinical signs of disease; n0, number of mice monitored; ND, not determined.
Includes data previously reported in reference 13.
When recalculated from date of birth, durations not significantly different from spontaneous disease in uninoculated mice.
Direct correlation of incubation time and expression level.
We inoculated sCJD(MM1) prions into four Tg lines expressing MHu#2(M129) at different levels and paradoxically found longer incubation times for mice expressing higher levels of chimeric PrP (Table 1). Hemizygous Tg22372 mice, expressing MHu#2(M129) at 0.8× that of MoPrP in wt FVB mice, demonstrated median incubation times of 111 days. Tg17103 mice expressing the same transgene product at 1.2× exhibited a median incubation time of 103 days. Homozygous Tg22372 mice expressing chimeric MHu#2(M129) at 1.6× showed incubation times of 124 days. Finally, Tg17051 mice expressing the same transgene product at 2.8× had a median incubation time of 167 days. Incubation times plotted against PrP expression levels from these four different lines of Tg(MHu#2, M129) mice showed a strong correlation (Fig. 2; R2 = 0.91). This direct relationship was observed previously with Tg mice expressing the original chimeric MHu(M129) construct (20).
Fig 2.

Incubation periods for sCJD(MM1) prions plotted against expression levels in Tg mice expressing chimeric MHu#2. Tg(MHu#2,M129) lines (filled symbols) show a strong, direct correlation between expression level and incubation period (solid line; R2 = 0.91). Similarly, mouse lines expressing the MHu#2(V129) construct (open symbols) also show a direct correlation (dashed line; R2 = 0.80). Points indicate median incubation period, and error bars indicate 95% confidence intervals.
A similar direct correlation was also observed when sCJD(MM1) prions were inoculated into 4 lines of Tg mice expressing MHu#2(V129) at different levels (Table 1, Fig. 2), between 1.1× and 2.4× that of wt FVB mice. Tg10355 and Tg7104 mice, expressing MHu#2(V129) at 1.1× and 1.3×, respectively, exhibited median incubation times of 125 and 133 days, respectively. Tg6550 and Tg7110 mice expressing the same transgene product at 1.8× and 2.4× had similar incubation times of 209 and 207 days, respectively. As with lines expressing the MHu#2(M129) construct, there was a direct correlation between incubation period and expression level of MHu#2(V129) (Fig. 2; R2 = 0.80).
Susceptibility of Tg mice to sCJD(MM1) prions.
Immunoblotting of the brain homogenates from ill mice expressing HuPrP(M129), HuPrP(V129), MHu#2(M129), or MHu#2(V129) all resulted in a type 1 PrPSc banding pattern (Fig. 3A). Each construct led to a similar proportion of monoglycosylated PrP (∼35%), but proportions of unglycosylated and diglycosylated PrP varied with each construct (Fig. 3B). However, compared to the inoculum, only glycoform proportions for the MHu#2(M129) construct were significantly different (P < 0.01). Neuropathological analysis of Tg(MHu#2,V129) lines inoculated with sCJD(MM1) prions showed moderate vacuolation, finely granular PrPSc deposition, and astrocytic gliosis (Fig. 3C to E), analogous to that previously reported for Tg(MHu#2,M129) mice (20). Together, these observations suggest that there was minimal change to the sCJD(MM1) strain type after transmission to Tg mice.
Fig 3.

Characterization after transmission of sCJD(MM1) prions to Tg mice expressing human (Hu) or chimeric (MHu#2) PrP with methionine (M) or valine (V) at polymorphic codon 129. (A) Immunoblot of brain homogenates: lane 1, inoculum; lane 2, Tg(HuPrP,M129)440; lane 3, Tg(MHu#2,M129)22372; lane 4, Tg(HuPrP,V129+/+)152; and lane 5, Tg(MHu#2,V129)6550 mice. Samples were treated with 100 μg/ml of PK for 1 h at 37°C prior to being loaded on gels and probed with the anti-PrP HuM-P antibody. Apparent molecular masses of migrated protein standards are shown in kilodaltons. (B) Proportions of unglycosylated (black), monoglycosylated (gray), and diglycosylated (white) PK-resistant PrP for each construct from multiple samples: 1, sCJD(MM1) (1 sample; 18 replicates); 2, HuPrP(M129) (5 samples; 8 total replicates); 3, MHu#2(M129) (6 samples; 11 total replicates); 4, HuPrP(V129) (1 sample; 4 replicates); 5, MHu#2(V129) (8 samples; 10 total replicates). Bars represent means, and error bars represent standard deviations; statistical difference from inoculum (lane 1) indicated above each bar: n.s., not significant; **, P < 0.01. (C to E) Micrographs of the hypothalamus of ill Tg Tg(MHu#2,V129)7110 mice stained with hematoxylin and eosin (H&E) (C) and stained immunohistochemically for PrPSc (D) or glial fibrillary acidic protein (E). Bar in panel E represents 100 μm and applies to all micrographs.
Susceptibility of Tg mice to sCJD(VV2) prions.
In previous studies, Tg mice expressing HuPrP(V129) were highly susceptible to sCJD(VV2) prion strains (3, 8, 43), whereas mice expressing HuPrP(M129), MHu(M129), or MHu#2(M129) were not (1, 3, 20). To test whether our novel Tg lines expressing chimeric MHu#2,V129 were susceptible, we inoculated them with sCJD(VV2) prions (Table 1). Among four Tg lines expressing MHu#2(V129) and injected with sCJD(VV2) prions, only 5 of 34 mice developed signs of neurologic dysfunction, after extended incubation periods (>470 days; Table 1). Biochemically, only passage to Tg(HuPrP,V129+/+)152 retained the type 2 strain, characterized by the same-sized unglycosylated PrP band (Fig. 4A). The glycoform ratio of this brain homogenate also closely paralleled that of the inoculum (Fig. 4B). Neuropathologically, this strain was characterized by severe vacuolation, coarsely granular PrPSc deposition, and intense astrocytic gliosis (Fig. 4C to E).
Fig 4.

Characterization after transmission of sCJD(VV2) prions to Tg mice expressing human (Hu) or chimeric (MHu#2) PrP with methionine (M) or valine (V) at polymorphic codon 129. (A) Immunoblot of brain homogenates: lane 1, inoculum; lane 2, Tg(HuPrP,M129)440; lane 3, Tg(MHu#2,M129)22372; lane 4, Tg(HuPrP,V129+/+)152; and lane 5, Tg(MHu#2,V129)6550 mice. Samples were treated with 100 μg/ml of PK for 1 h at 37°C prior to being loaded on gels and probed with the anti-PrP HuM-P antibody. Apparent molecular masses of migrated protein standards are shown in kilodaltons. (B) Proportions of unglycosylated (black), monoglycosylated (gray), and diglycosylated (white) PK-resistant PrP for each construct from multiple samples: 1, sCJD(VV2) (1 sample; 13 replicates); 2, HuPrP(M129) (3 samples; 7 total replicates); 3, MHu#2(M129) (1 sample; 6 replicates); 4, HuPrP(V129) (1 sample; 4 replicates); 5, MHu#2(V129) (5 samples; 14 total replicates). Bars represent means, and error bars represent standard deviations; statistical difference from inoculum (lane 1) indicated above each bar: n.s., not significant; **, P < 0.01. (C to H) Micrographs of brain tissue from ill Tg mice, showing the most dramatic neuropathological changes: the thalamus of Tg(HuPrP,V129)152 mice (C to E) and piriform cortex of Tg(MHu#2,V129)7110 mice (F to H). Samples were stained with H&E (C, F) and stained immunohistochemically for PrPSc (D, G) or glial fibrillary acidic protein (E, H). Bar in panel H represents 100 μm and applies to all micrographs. (I, J) Vacuolation score histogram, an estimate of the brain area occupied by vacuoles, for Tg(HuPrP,V129)152 mice (I) and Tg(MHu#2,V129)7110 mice (J). NC, neocortex; Pir, piriform cortex; Hp, hippocampus; Th, thalamus; Hab, habenula; Hy, hypothalamus; Cd, caudate nucleus; Cb-G, cerebellar cortex, granular cell layer; Bs, brainstem. (K, L) Kaplan-Meier survival graphs for first (black) and second (gray) transmissions of sCJD(VV2) prions in Tg(MHu#2,V129)7110 (second passages of two independent brains shown) (K) and Tg(MHu#2,V129)6550 (L) mice.
For other constructs, when transmission did occur, brains of the resulting mice had a larger unglycosylated fragment. Immunoblot analysis of the brain homogenates from ill Tg(MHu#2,V129) mice showed that an intermediate band appeared between type 1 and type 2 (Fig. 4A), which was consistent for all 5 mice analyzed. Neuropathological analysis showed vacuolation predominantly in the neocortex and piriform cortex, an intense degree of PrPSc staining, and moderate astrocytic gliosis (Fig. 4F to H), differing from that of Tg(HuPrP,V129) mice. This difference in strain type between sCJD(VV2) passaged in mice expressing HuPrP(V129) versus MHu#2(V129) was also supported by the vacuolation profiles throughout the resulting brains (Fig. 4I and J).
Brain homogenates from sick Tg(MHu#2,V129) mice injected with sCJD(VV2) prions were prepared and inoculated into the same Tg lines for serial passage. Two mice from the Tg7110 line, sick at 494 and 509 days postinfection (dpi), resulted in efficient transmission to other Tg7110 mice, with median incubation times of 320 and 386 days, respectively (Fig. 4K). Brain extract from the Tg6550 mouse, which developed clinical disease at 476 dpi, transmitted to all Tg6550 mice with a median incubation period of 389 days (Fig. 4L). For all second passages, incubation periods were shorter than for the initial transmissions. This phenomenon is one indication that a strain barrier was posed (36).
Role of individual residues in transmission of MM1 prions.
Because increasing the expression level did not reduce the incubation period for Tg(MHu#2,M129) mice, we investigated the role of the remaining HuPrP residues in MHu. Since previous studies showed that the S→N reversion at residue 96 lengthened the incubation times (20), we examined the remaining six human residues and reverted each one to the mouse sequence in the MHu#2 transgene.
Tg mice with MHu#2 constructs containing reverted residues 108 or 154 had low expression levels (<0.3×) of chimeric PrP. Tg mice with MHu#2 constructs containing reverted residues 111, 137, or 142 expressed chimeric PrP at ∼1 to 3×. In Tg mice with reverted residue 144, protein expression levels could not be detected despite a high copy number of transgene integration; most of these Tg(MHu#2,M129,Y144W) mice developed spontaneous disease at ∼550 days of age. All 11 Tg lines were inoculated with sCJD(MM1) prions (Table 2). In the 2 Tg lines with the residue 144 reversion, all infected mice died in ∼550 days. Given that this duration is similar to onset of spontaneous disease, we conclude that these Tg(MHu#2,M129,Y144W) mice developed spontaneous illness rather than prion disease. Reverting residue 142 had little effect on the incubation periods in the Tg(MHu#2,M129,S142N) lines compared to in Tg mice expressing MHu#2(M129): incubation times were ∼120 to 150 days. Tg lines with reverted residue 108 and 0.3× expression levels showed slightly longer incubation periods for sCJD(MM1) prions (∼190 days). Reversion of residue 137 in Tg10027 and Tg10025 mice expressing MHu#2(M129,I137M) resulted in an ∼70% increase in the incubation period compared to that for Tg(MHu#2,M129) mice with a similar expression level. Tg lines expressing reverted residue 154 also demonstrated extended incubation periods, from 230 to 390 days; however, these lines had low expression levels, making direct comparison difficult. As reported earlier, restoring residue 111 in the Tg(MHu#2,M129,M111V)1014 line reduced the incubation time to 77 days, a >50% decrease compared to in Tg(MHu#2,M129) mice with a similar expression level (Table 2) (13).
Immunoblotting of the brain homogenates from ill Tg mice showed type 1 PrPSc (Fig. 5A). The ratio of the glycosylated forms was slightly different for each construct, with the proportion of diglycosylated PrP increased and that of unglycosylated PrP reduced compared to the inoculum (Fig. 5B). These differences were significant for the MHu#2(M129,M108L) (P < 0.01), MH#2(M129,I137M) (P < 0.05), and MHu#2(M129,H154Y) (P < 0.01) constructs but not significant for MHu#2(M129,M111V) and MHu#2(M129,S142N) constructs, suggesting that residues differing between HuPrP and MHu#2 can impact glycosylation.
Fig 5.

Passage of sCJD(MM1) prions to Tg mice expressing MHu#2(M129) and an additional reversion to the HuPrP sequence. (A) Immunoblot of brain homogenates: lane 1, inoculum; lane 2a, Tg1208 mice expressing MHu#2(M129,M108L); lane 2b, Tg1284 mice expressing MHu#2(M129,M108L); lane 3, Tg1014 expressing MHu#2(M129,M111V); lane 4a, Tg10027 expressing MHu#2(M129,I137M); lane 4b, Tg10025 expressing MHu#2(M129,I137M); lane 5a, Tg3018 expressing MHu#2(M129,S142N); lane 5b, Tg3061 expressing MHu#2(M129,S142N); lane 6a, Tg4561 expressing MHu#2(M129,H154Y); and lane 6b, Tg5099 expressing MHu#2(M129,H154Y). Samples were treated with 100 μg/ml of PK for 1 h at 37°C prior to loading on gels and probed with the anti-PrP HuM-P antibody. Apparent molecular masses of migrated protein standards are shown in kilodaltons. (B) Proportions of unglycosylated (black), monoglycosylated (gray), and diglycosylated PK-resistant PrP for each construct from multiple repetitions: 1, sCJD(MM1) (1 sample; 18 replicates); 2, MHu#2(M129,M108L) (4 samples; 10 replicates); 3, MHu#2(M129,M111V) (4 samples; 8 total replicates); 4, MHu#2(M129,I137M) (8 samples; 17 total replicates); 5, MHu#2(M129,S142N) (8 samples; 17 total replicates); and 6, MHu#2(M129,H154Y) (8 samples; 15 total replicates). Bars represent means, and error bars represent standard deviations; statistical difference from inoculum (lane 1) indicated above each bar: n.s., not significant; *, P < 0.05; **, P < 0.01. (C to E) Kaplan-Meier survival curves for first (black) and second (gray) passages of sCJD(MM1) prions in Tg(MHu#2,M129)22372 (C), Tg(MHu#2,M129,M111V)1014 (D), and Tg(MHu#2,M129,S142N)3018 (E) mice.
Brains of ill Tg22372 (Fig. 5C), Tg1014 (Fig. 5D), and Tg3018 (Fig. 5E) mice were inoculated into the same lines for second passages. Second passage of sCJD(MM1) prions reduced the incubation periods in Tg22372 and Tg3018 mice. For Tg22372 mice, the median incubation time diminished from 111 days to 96 days (95% CI of 89, 99); for Tg3018 mice, the incubation time decreased from 118 days to 91 days (95% CI of 85, 98). In contrast, serial passage of sCJD(MM1) prions in Tg1014 mice showed no shortening of the incubation time: on initial passage, the median incubation time was 77 days, and on second passage, it was 76 days (95% CI of 74, 82).
Structural analysis.
To determine why Tg mice expressing chimeric PrP that differs from MoPrP at only six or seven residues are susceptible to sCJD(MM1) prions, while wt mice are largely resistant to human prions, we compared the structures of HuPrP and MoPrP (14, 47). In all PrP structures determined to date, the N terminus is largely flexible and the region from residue ∼120 to the C terminus has a well-defined structure. As expected from the high degree of sequence similarity, the three-dimensional (3D) structures of HuPrP and MoPrP are globally similar, with a backbone root mean square deviation of 1.02 Å for all ensemble members from the two structures. Of the seven residues that differ between MHu#2 and MoPrP, four are in the structured region. These residues delineate an area on one face of the PrPC molecule (Fig. 1A).
For residues 137, 142, 144, and 154 (MoPrP numbering), we compared the orientations of the side chains for all members of the nuclear magnetic resonance (NMR) structural ensembles. MoPrP N142 and W144, as well as the corresponding HuPrP residues S143 and Y145, occupy similar regions of space, and all ensemble members have similar orientations. In structural determinations at pH 4, HuPrP H155 has a slightly different orientation from MoPrP Y154: H155 is charged and repelled by R156. At physiologic pH, H155 would be neutral. In a structure determined at pH 7 (4), H155 occupies a similar position to that occupied by Y154 in MoPrP at pH 4. The orientation of MoPrP Y154 is unlikely to be influenced by pH. The most marked difference between the structures of MoPrP and HuPrP is the orientation of MoPrP M137 compared to that of HuPrP I138; they occupy different regions of space (Fig. 6A and B). Further analysis showed a large difference of side chain orientation between the two sequences for the conserved arginine residue at position 135/136, due to steric hindrance from I138 in HuPrP (Fig. 6A and B). Changes in the location of the charged residues on the surface of PrP impact the electrostatic potential of the protein (Fig. 6C and D). This analysis correlates closely with the incubation periods of the various transgenes in which the reversion I137M had the biggest extension of incubation period at the same PrP expression levels.
Fig 6.
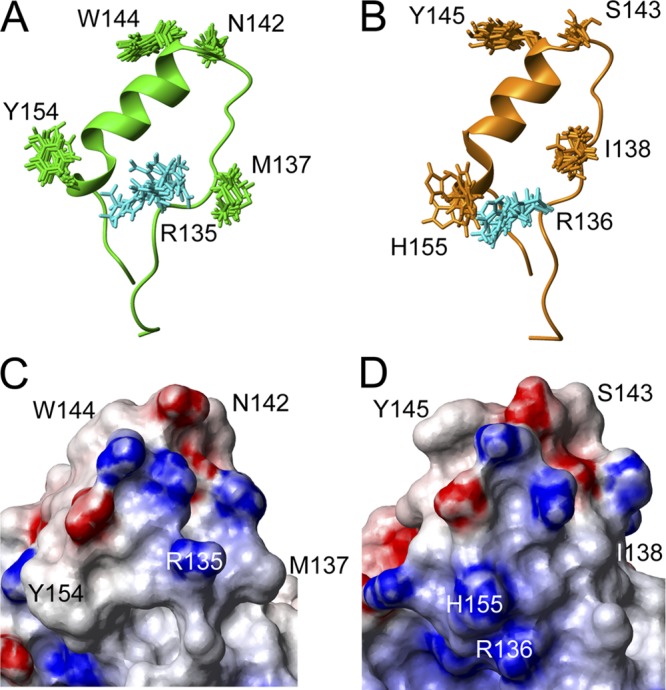
Structural analysis of MoPrP and HuPrP, in same orientation as that in Fig. 1A. (A, B) Diagrams of the regions around helix 1 in MoPrP(131–158) (A) and HuPrP(132–159) (B), showing all side-chain conformations from the structure ensembles of the residues differing between the two species, plus the conserved arginine residue 135/136 shown in cyan. (C, D) Molecular surface diagrams of the most stable conformer for MoPrP (C) and HuPrP (D), for the same regions shown in panels A and B, colored by electrostatic potential. Red indicates negative charges; blue shows positive charges.
DISCUSSION
A wealth of evidence argues that the sole component of mammalian prions is the conformational isoform PrPSc. To account for the different properties of prion strains, one of us (SBP) suggested that variations in the conformation of PrPSc might be the site where biological information for the prion is enciphered (30). Over the past 2 decades, numerous findings support the contention that the characteristics of both mammalian and fungal prion strains are enciphered in the conformation of the prion protein (2, 6, 7, 9, 21, 22, 29, 40, 41, 44).
Relationship between incubation period and expression level.
In Tg mice expressing full-length PrP from various species, an inverse relationship exists between expression level and incubation period (32, 37, 39). However, this relationship is not linear over the full range of expression levels but is asymptotic: increased expression level shortens the incubation period to a minimum value characterized by the particular strain. Very high expression levels frequently lead to spontaneous disease, which has rarely been shown to correspond to infectivity and likely reflects cellular malfunction due to protein overexpression.
In the chimeric transgenes reported here, we observed a direct relationship between expression level and incubation period. However, we do not expect this relationship to be linear: very low expression levels are unlikely to yield very rapid incubation periods. Rather, the relationship may be parabolic, with a particular transgene expression level related to a minimum incubation period, and lower and higher expression levels leading to longer incubation times. Such a phenomenon could be explained biologically by a stoichiometric relationship with a cofactor, such as the postulated “protein X” (43). However, why chimeric transgenes are more susceptible to such a mechanism remains to be determined, and studies to address this are under way.
Transmission characteristics of sCJD(MM1) and sCJD(VV2) prions.
Prion strains can be characterized in a number of ways, including incubation period in certain hosts, intensities of PK-resistant PrP glycoform bands, and vacuolation profile and PrP distribution throughout the brain. A growing body of data supports the concept that the six potential sCJD subtypes (MM1, MM2, MV1, MV2, VV1, VV2) represent four distinct strains, termed M1, M2, V1, and V2 (3, 15, 25, 45). Here, we studied the two most common forms of sCJD, MM1 and VV2, representing strain types M1 and V2.
Transmission of the M1 strain was not hindered by the polymorphism at residue 129 in mice expressing HuPrP constructs. Conversely, transmission of the V2 strain to Tg(HuPrP,V129) mice was efficient, but transmission to Tg(HuPrP, M129) mice was inefficient and accompanied by change in strain properties, identifying the expression of V129 as essential for the transmission of the V2 strain. Other recent studies on the transmission of human prion strains have similarly observed a change in strain phenotype following transmission of V2 prions to mice expressing HuPrP(M129) (3, 18).
The chimeric constructs reported here offer additional insight into the transmission of M1 and V2 strains. Because the M1 strain transmitted efficiently to mice expressing HuPrP(M129) and MHu#2(M129) transgenes, and the V2 strain transmitted efficiently to mice expressing the HuPrP(V129) transgene, we generated mice expressing the MHu#2(V129) transgene and inoculated them with V2 prions. We found that only 5 of 34 mice among four Tg lines expressing chimeric MHu#2(V129) developed clinical signs of prion disease. Moreover, when transmission did occur, it was accompanied by a change in the strain type, as determined both biochemically and neuropathologically (Fig. 4). This novel strain type was retained upon serial passage.
Our findings show that the differences in transmission between V2 and M1 prions into Tg mice expressing MHu#2 are not a function of the polymorphic residue in the host but rather the conformation of the two prion strains and how they interact with PrPC. While V129 is mandatory for propagation of the V2 strain, it is not sufficient, and other human PrP residues in the N- and/or C-terminal regions are required. Our data argue that the conformations of sCJD(VV2) and sCJD(MM1) prions differ in their abilities to bind to PrPC during the propagation of nascent prions.
Our studies contend that strain-specified replication of prions is modulated by PrP sequence-specific interactions between the prion precursor PrPC and the infectious isoform PrPSc.
Impact of chimeric transgenes on incubation periods.
Reverting human-encoded amino acid residues to mouse in the chimeric MHu transgene had profound effects on the length of the incubation time and the strain-specified properties of PrPSc. For example, reverting residues 165 and 167 from human to mouse decreased the incubation time from ∼200 days to ∼110 days for sCJD(MM1) prions (20). Reverting a third residue at 111 reduced the incubation time still more, to ∼75 days (13).
The conservative substitution of isoleucine for methionine at residue 137 in the chimeric transgene resulted in an ∼70% increase in incubation period. Structural analysis suggested that this may result in an alteration to the electrostatic charge distribution on the protein surface (Fig. 6). A similar argument was used to explain the impact of the E200K mutation associated with familial CJD, since the overall structure of PrP remained largely unchanged (48).
In all cases, transmission of sCJD(MM1) prions resulted in a type 1 strain that produced similar neuropathological changes, suggesting that these mutations affected the kinetics of prion replication.
Conclusions.
The Tg mice described here that express chimeric mouse/human PrP genes provide a novel set of tools with which to study strains of prions. These same mice provide a means of evaluating human prion infectivity relatively rapidly; such bioassays may play a pivotal role in the development of therapeutics for treating patients dying of CJD.
ACKNOWLEDGMENTS
We thank the staff of the Hunter's Point animal facility and Hang Nguyen for expert editorial assistance.
This work was supported by grants from the National Institutes of Health (AG02132, AG10770, AG031220, and AG021601) as well as by gifts from the Sherman Fairchild Lincy Foundation, Schott Foundation for Public Education, Rainwater Charitable Foundation, and Robert Galvin.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Asante EA, et al. 2002. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 21:6358–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bessen RA, Marsh RF. 1992. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66:2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bishop MT, Will RG, Manson JC. 2010. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. U. S. A. 107:12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calzolai L, Zahn R. 2003. Influence of pH on NMR structure and stability of the human prion protein globular domain. J. Biol. Chem. 278:35592–35596 [DOI] [PubMed] [Google Scholar]
- 5. Carlson GA, et al. 1986. Linkage of prion protein and scrapie incubation time genes. Cell 46:503–511 [DOI] [PubMed] [Google Scholar]
- 6. Colby DW, et al. 2009. Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. U. S. A. 106:20417–20422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936 [DOI] [PubMed] [Google Scholar]
- 8. Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 383:685–690 [DOI] [PubMed] [Google Scholar]
- 9. Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144:1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gajdusek DC, Gibbs CJ, Jr, Alpers M. 1966. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 209:794–796 [DOI] [PubMed] [Google Scholar]
- 11. Gibbs CJ, Jr, et al. 1968. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161:388–389 [DOI] [PubMed] [Google Scholar]
- 12. Giles K, et al. 2008. Resistance of bovine spongiform encephalopathy (BSE) prions to inactivation. PLoS Pathog. 4:e1000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giles K, et al. 2010. Human prion strain selection in transgenic mice. Ann. Neurol. 68:151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. 2005. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc. Natl. Acad. Sci. U. S. A. 102:646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haik S, Brandel JP. 2011. Biochemical and strain properties of CJD prions: complexity versus simplicity. J. Neurochem. 119:251–261 [DOI] [PubMed] [Google Scholar]
- 16. Hill AF, et al. 1997. The same prion strain causes vCJD and BSE. Nature 389:448–450 [DOI] [PubMed] [Google Scholar]
- 17. Kimberlin RH, Cole S, Walker CA. 1987. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 68:1875–1881 [DOI] [PubMed] [Google Scholar]
- 18. Kobayashi A, Asano M, Mohri S, Kitamoto T. 2007. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J. Biol. Chem. 282:30022–30028 [DOI] [PubMed] [Google Scholar]
- 19. Koradi R, Billeter M, Wuthrich K. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 20. Korth C, et al. 2003. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc. Natl. Acad. Sci. U. S. A. 100:4784–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Legname G, et al. 2006. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U. S. A. 103:19105–19110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mastrianni JA, et al. 1999. Prion protein conformation in a patient with sporadic fatal insomnia. N. Engl. J. Med. 340:1630–1638 [DOI] [PubMed] [Google Scholar]
- 23. Muramoto T, et al. 1997. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an α-helix. Nat. Med. 3:750–755 [DOI] [PubMed] [Google Scholar]
- 24. Palmer MS, Dryden AJ, Hughes JT, Collinge J. 1991. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 352:340–342 [DOI] [PubMed] [Google Scholar]
- 25. Parchi P, et al. 2010. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 133:3030–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parchi P, et al. 1999. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46:224–233 [PubMed] [Google Scholar]
- 27. Parchi P, et al. 2000. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. U. S. A. 97:10168–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peretz D, et al. 2006. Inactivation of prions by acidic sodium dodecyl sulfate. J. Virol. 80:322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peretz D, et al. 2002. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34:921–932 [DOI] [PubMed] [Google Scholar]
- 30. Prusiner SB. 1991. Molecular biology of prion diseases. Science 252:1515–1522 [DOI] [PubMed] [Google Scholar]
- 31. Prusiner SB. 2001. Shattuck lecture—neurodegenerative diseases and prions. N. Engl. J. Med. 344:1516–1526 [DOI] [PubMed] [Google Scholar]
- 32. Prusiner SB, et al. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673–686 [DOI] [PubMed] [Google Scholar]
- 33. Rasband WS. 1997–2011. ImageJ. U.S. National Institutes of Health, Bethesda, MD: http://imagej.nih.gov/ij/ [Google Scholar]
- 34. Scott M, et al. 1989. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 59:847–857 [DOI] [PubMed] [Google Scholar]
- 35. Scott MR, Köhler R, Foster D, Prusiner SB. 1992. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1:986–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scott MR, Peretz D, Nguyen H-OB, DeArmond SJ, Prusiner SB. 2005. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J. Virol. 79:5259–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott MR, et al. 1997. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 94:14279–14284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stanker LH, et al. 2010. Conformation-dependent high-affinity monoclonal antibodies to prion proteins. J. Immunol. 185:729–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tamgüney G, et al. 2006. Transmission of elk and deer prions to transgenic mice. J. Virol. 80:9104–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka M, Collins SR, Toyama BH, Weissman JS. 2006. The physical basis of how prion conformations determine strain phenotypes. Nature 442:585–589 [DOI] [PubMed] [Google Scholar]
- 41. Telling GC, et al. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082 [DOI] [PubMed] [Google Scholar]
- 42. Telling GC, et al. 1994. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc. Natl. Acad. Sci. U. S. A. 91:9936–9940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Telling GC, et al. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79–90 [DOI] [PubMed] [Google Scholar]
- 44. Toyama BH, Kelly MJ, Gross JD, Weissman JS. 2007. The structural basis of yeast prion strain variants. Nature 449:233–237 [DOI] [PubMed] [Google Scholar]
- 45. Uro-Coste E, et al. 2008. Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 4:e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wadsworth JD, Asante EA, Collinge J. 2010. Review: contribution of transgenic models to understanding human prion disease. Neuropathol. Appl. Neurobiol. 36:576–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zahn R, et al. 2000. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U. S. A. 97:145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD. 2000. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J. Biol. Chem. 275:33650–33654 [DOI] [PubMed] [Google Scholar]