

Abstract
Pharmacological activation of wild-type p53 has been found to protect normal cells in culture from cytotoxicity and nuclear aberrations caused by conventional cancer therapeutics. Hence, small-molecule p53 activators could have clinical benefits as chemoprotectants for cancer patients bearing p53-mutant tumors. We have evaluated 16 p53-based cyclotherapy regimes combining p53 activators tenovin-6, leptomycin B, nutlin-3 and low dose actinomycin D, with clinically utilized chemotherapeutic agents (S- and M-phase poisons), vinblastine, vinorelbine, cytosine arabinoside and gemcitabine. All the p53 activators induce reversible cell-cycle arrest in primary human fibroblasts and protect them from both S- and M-phase poisons. Furthermore, studies with p53-mutant cancer cell lines show that nutlin-3 and low dose actinomycin D do not affect the sensitivity of these cells to any of the chemotherapeutics tested. Thus, these two small molecules could be suitable choices for cyclotherapy regimes involving S- or M-phase poisons. In contrast, pre-incubation of p53-mutant cells with tenovin-6 or leptomycin B reduces the efficacy of vinca alkaloids, suggesting that these p53 activators could be effective as chemoprotectants if combined with S- but not M-phase poisons. Discrepancies were observed between the levels of protection detected immediately after treatment and following recovery in fresh medium. This highlights the need to assess both short- and long-term effects when evaluating compounds as potential chemoprotectants for cancer therapy.
Keywords: actinomycin D, chemoprotection, cyclotherapy, leptomycin B, Nutlin-3, p53, tenovin-6
Introduction
Chemotherapy approaches aim at developing treatments that eradicate cancer cells efficiently and selectively. However, many of the drugs currently used in the clinic cause DNA damage and indiscriminately target rapidly dividing cells in S- or M phase, thereby causing undesired mutagenic and cytotoxic events in proliferative normal tissues. As a consequence, patients not only suffer from immediate side effects such as nausea, hair loss, diarrhea and neutropenia, but are also subjected to an increased risk of developing resistance to treatment and/or second tumors later in life. Cyclotherapy strategies aim at improving the therapeutic window of conventional chemotherapy by protecting healthy tissues.1-3 For instance, if transient cell cycle arrest in G1 or G2 was induced in normal tissues only, their sensitivity to S- and M-phase poisons would be decreased while leaving the tumor vulnerable to the treatment.
Several small-molecule activators of the p53 tumor suppressor have been shown to induce a mild cytostatic response in normal cells in culture. It has therefore been proposed that such molecules could constitute ideal chemoprotectants for patients bearing p53-mutant tumors. In this context, p53 status provides a way to distinguish between normal cells, retaining wild-type p53 and cancer cells, lacking functional p53. Pre-treatment with specific p53 activators would selectively stop proliferation in healthy tissues, thereby shielding them from subsequent exposure to conventional chemotherapeutics without compromising the anticancer efficacy of the treatment.
Since cyclotherapy was first introduced as a promising concept in the year 2000, various publications have provided evidence supporting its potential (Table 1). The first studies used low doses (LD) of DNA damaging agents to induce p53-dependent cell cycle arrest. In this line, it has been shown that pre-incubation with LD doxorubicin (LDDOX) protects normal cells against tubulin poisons.4,5 Similarly, a LD of actinomycin D (LDactD), another clinically approved anticancer drug, has been used as a chemoprotectant in combination with the Aurora kinase inhibitor VX680.6 Unfortunately, both LDDOX and LDactD also protected p53-deficient cancer cell lines to a small extent. The remaining cyclotherapy studies summarized in Table 1 involve the specific p53 activator nutlin-3. This compound has been shown to selectively shield normal cells against S-phase poisons,8 tubulin poisons,9,10 a Polo-like kinase 1 inhibitor (PLK1I)12 and an Aurora kinase inhibitor.7 Most importantly, oral administration of nutlin-3 protects mice against PLK1I-induced neutropenia.12
Table 1. Existing p53-based cyclotherapy studies.
| Chemoprotectat | Chemotherapeutic | Protection | Reference | |
|---|---|---|---|---|
| |
|
p53 wild-type |
p53- deficient |
|
| Low dose doxorubicin |
Epitholones A and B |
Y |
N |
Blagosklonny et al.4 |
| Paclitaxel |
Y |
Y |
Blagosklonny5 |
|
| Vinblastine (VNB) |
Y |
N |
Blagosklonny et al.4 |
|
| Low dose actinomycin D |
Aurora kinase inhibitor VX680 |
Y |
Y |
Rao et al.6 |
| Nutlin-3 | Aurora kinase inhibitor VX680 |
Y |
N |
Cheok et al.7 |
| Cisplatin |
N |
? |
Kranz and Dobbelstein8 |
|
| Cytosine arabinoside (Ara-C) |
Y |
N |
Kranz and Dobbelstein8 |
|
| Doxorubicin (DOX) |
N |
? |
Kranz and Dobbelstein8 |
|
| Gemcitabine (GMTB) |
Y |
N |
Kranz and Dobbelstein8 |
|
| Nocodazole |
Y |
N |
Apontes et al.9 |
|
| Paclitaxel |
Y |
N |
Apontes et al.9 Carvajal et al.10 Tokalov and Abolmaali11 |
|
| Polo-like kinase inhibitor BI-2536 | Y | N | Sur et al.12 | |
(Y, protection against cytotoxicity caused by chemotherapeutic; N, no protection; and ?, not tested). Ideal chemoprotectants only shield p53 wild-type cells from cell-killing caused by chemotherapeutic agents.
Although existing cyclotherapy studies using nutlin-3 are auspicious, this compound has a number of drawbacks: (1) its clinical use has not been approved yet; (2) its efficacy in vivo is low, with high doses needed for an effect in mice (i.e., 200 mg/kg orally administered nutlin-3);12,13 (3) its p53 selectivity is limited to a narrow window, with doses above 10 µM leading to DNA damage14,15 and doses below 2 µM having no detectable effect. This motivated us to explore other small molecules as possible alternatives. The advantages and drawbacks of every compound are reviewed in the discussion section. Like nutlin-3, they induce p53 protein and transcriptional activity, but they differ in their mechanisms of action and p53 activation kinetics,16 which might define their clinical suitability as chemoprotectants. In this paper, we systematically evaluate and compare the performance of tenovin-6,17 leptomycin B (LMB),18 nutlin-3 and LDactD in 16 different cyclotherapy regimes involving four clinically approved chemotherapeutics: the M-phase poisons vinblastine (VNB) and vinorelbine (VRL), and the S-phase poisons cytosine arabinoside (Ara-C) and gemcitabine (GMTB).
Results
Tenovin-6, leptomycin B, nutlin-3 and low-dose actinomycin D induce reversible cell cycle arrest in primary fibroblasts.
Tenovin-6, leptomycin B, nutlin-3 and LDactD are known to efficiently increase p53 protein levels and transcriptional activity in various normal and cancer-derived cell lines. In particular, they have been shown to induce the expression of p21 protein, which is indicative of G1 and G2 arrest. In this section, we investigate the impact of these compounds on the growth and viability of human normal dermal fibroblasts (HNDFs), paying special attention to the reversibility of the effects.
Tenovins were identified in a forward chemical genetic screen based on a p53-reporter assay. They activate p53 in a non-genotoxic manner and have been shown to inhibit the activity of sirtuins SirT1 and SirtT2.17 This compound has recently attracted a lot of attention, since it has been used successfully in pre-clinical models for chronic myelogenous leukemia (see discussion section). As can be seen from Figure 1A, treatment of HNDFs with 3 or 5 µM tenovin-6 caused no net changes in cell numbers. This could be due to tenovin-induced cell cycle arrest or a balance between cell proliferation and death. To discriminate between these possibilities, we performed cell cycle distribution analyses by FACS. Figure 2A confirms a dramatic reduction in the percentage of cells in S phase in response to 24-h tenovin-6 exposure as well as substantial increases in the percentages in both G1 and G2. The proportion of dead cells upon tenovin-6 treatment was negligible, further supporting that the effects are primarily cytostatic. Furthermore, as can be seen from Figure 2B, following removal of tenovin-6, HNDFs re-entered S phase, which proves that the cell cycle arrest is reversible. This is in agreement with the recovery experiments in Figures 1A and B, where cells resumed proliferation when tenovin-free medium was supplied.
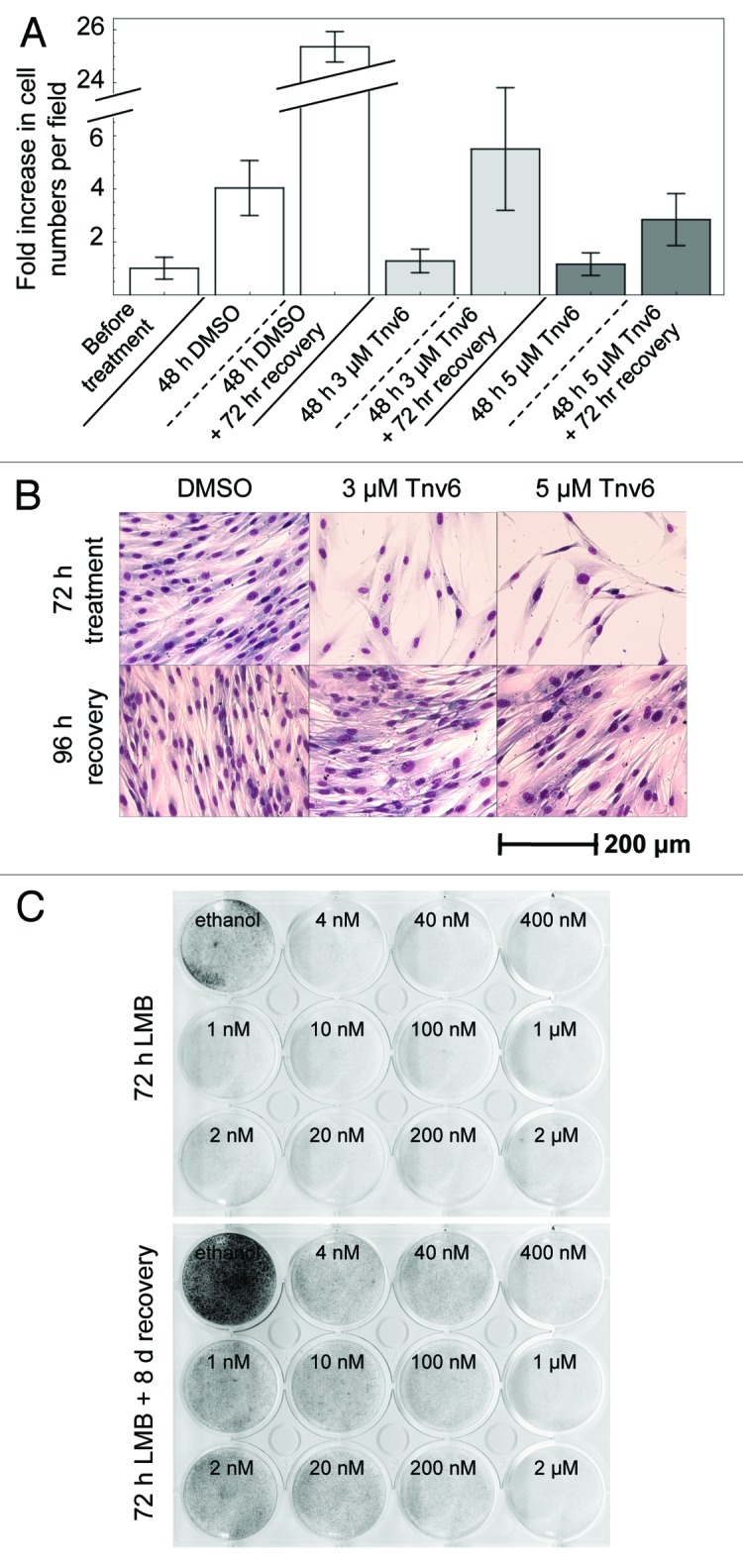
Figure 1. Reversible effects of tenovin-6 and leptomycin B on normal fibroblasts. (A) HNDFs were treated with tenovin-6 or vehicle (DMSO) for 48 h, and then left to recover for 72 h in fresh medium. Graph shows fold increase in cell numbers per field (n = 18) since treatment began. Error bars represent standard deviations. (B) HNDFs were treated with tenovin-6 or vehicle (DMSO) for 72 h, and then left to recover for 96 h in fresh medium before Giemsa staining. (C) HNDFs were treated with various concentrations of LMB or vehicle (absolute ethanol) for 72 h (upper plate) and then grown for 8 d in fresh medium (lower plate) before Giemsa staining.

Figure 2. Changes in HNDF cell-cycle distribution in response to small molecules. (A) Cells were treated with the indicated p53 activators for 24 h; and (B) Cells were treated for 24 h and then cultivated for 120 h in fresh medium. DNA synthesis and DNA content were evaluated by measuring BrdU incorporation and propidium iodide (PI) staining by FACS. Values correspond to percentage change compared with untreated controls prior to recovery [i.e., panel A(1)].
Leptomycin B is an antibiotic produced by Streptomyces sp. It inactivates the nuclear export receptor protein CRM1 (exportin-1), thereby causing nuclear accumulation of proteins containing a nuclear export signal.19,20 It is also an extremely potent activator of p53.18,21,22 In 1999, we reported indications that micromolar concentrations of LMB prevent proliferation of HNDFs but do not cause cell death.21Figures 2A and B show that LMB blocks HNDFs in the G1 and G2 phases of the cell cycle, and cells restart synthesizing DNA upon removal of the compound. Recovery from LMB treatment can also be seen in the clonogenic assay in Figure 1C. It is striking that these observations hold for concentrations ranging from 0.2–400 nM.
The synthetic compound nutlin-3 was identified in an in vitro biochemical screen for inhibitors of the interaction between p53 and its main negative regulator, Mdm2. The compound inserts into the p53-binding pocket on Mdm2,13,23-25 thereby promoting p53 stabilization. Nutlin-3 consists of a racemic mixture of two enantiomers, nutlin-3a and nutlin-3b (as only the former is active, 5 µM nutlin-3 corresponds to an effective dose of 2.5 µM nutlin-3a). This specific Mdm2 antagonist induces p53-dependent cell cycle arrest in normal cells in culture. The reversibility of these effects has been described as controversial because of studies reporting irreversible replicative senescence in primary murine embryonic fibroblasts (10 µM nutlin-3a for 24 h)26 and normal human fibroblasts (10 µM nutlin-3a for 7 d)27 in response to nutlin-3. However, it is generally believed that normal cells resume growth following withdrawal of the compound when doses below 10 µM of the racemic mixture are used. In this line, recovery from nutlin-3 treatment has been shown in 1043SK human skin fibroblasts (10 µM nutlin-3 for 7 d),10,13 WI-38-TERT human fibroblasts (20 µM nutlin-3a for 6 d),28 human prostate and mammary epithelial cells (10 µM nutlin-3a for 6 d)29 and human epidermal keratinocytes (4 µM nutlin-3 for 24 h).30 Providing further evidence supporting the reversibility of nutlin-3’s effects in normal cells, here we show that HNDFs treated with 5 µM nutlin-3 for 24 h stop proliferating (Fig. 2A), but re-enter S phase once the compound is removed (Fig. 2B). Hardly any nutlin-mediated cell death was observed in these experiments.
Actinomycin D is a classic clinically approved drug that is being widely used to treat various malignancies, including rhabdomyosarcomas31 and Wilms’ tumors.32 Like LMB, ActD is an antibiotic produced by Streptomyces sp, but its mechanism of action is very different: it binds to GC-rich regions in DNA, impeding RNA synthesis33-35 and, in particular, causing a reduction in the levels of rRNA. This is believed to lead to increased levels of free ribosomal proteins, such as L11 and L23, which can bind to Mdm2 and thereby interfere with p53 degradation.36-38 As a DNA-intercalating compound, ActD is highly unselective, cytotoxic and genotoxic at high doses. However, it does not induce DNA damage markers at low nanomolar concentrations,6,30 and LDactD can mimic nutlin-3 exposure in terms of p53-dependent mRNA expression profiles.30 It has been shown that human epidermal keratinocytes undergo reversible cell cycle arrest when exposed to 1 nM ActD for 24 h.30 Similarly, HNDFs incubated with 4 nM ActD for 72 h re-entered the cell cycle when cultivated further in fresh medium.6 In agreement with these observations, Figure 2A shows that HNDFs accumulate in G1 and G2 in response to LDactD, while Figure 2B confirms the reversibility of the cell cycle arrest.
Tenovin-6, leptomycin B, nutlin-3 and low-dose actinomycin D protect primary fibroblasts from adverse effects of S- and M-phase poisons.
As a next step, we investigated whether the transient halt in proliferation induced by p53 activators in normal cells is able to prevent the cytotoxicity and nuclear aberrations caused by classic S- and M-phase poisons. As S-phase poisons we chose two clinically approved nucleotide analogs, GMTB and Ara-C. Both compounds are transported into the nucleus by nucleoside transporters and are enzymatically modified by deoxycytidine kinase into active metabolites, which incorporate into DNA in a similar manner.39-41 However, it has been shown that GMTB also inhibits ribonucleotide reductase,42,43 whereas Ara-C does not. The working doses (i.e., 50–100 nM for GMTB and 5–10 µM for Ara-C) are comparable to those used in Kranz and Dobbelstein.8 As M-phase poisons we chose two vinca alkaloids, VNB and VRL, that are in clinical use for various malignancies.44 The working doses of these compounds (i.e., 4–25 nM for VNB and 20–40 nM for VRL) were decided based upon careful titrations in normal and cancer cells (not shown).
Our cyclotherapy protocol, which we performed for each of the 16 drug combinations, is described in Materials and Methods. In short, cells were seeded on day 0 and treated with p53 activators and chemotherapeutic drugs on days 1 and 2, respectively. On day 4, the cells were rinsed and given the chance to recover from the treatment in drug-free medium for several days. In the case of HNDFs, the difference between chemotherapy and cyclotherapy is striking. Representative images are shown in Figure 3. Following chemotherapy, i.e., treatment with S- or M-phase poisons alone, cells showed gross nuclear aberrations, abnormal morphologies and impaired growth. In strong contrast, if pre-incubated with p53 activators, cells resembled untreated “healthy” fibroblasts with normal nuclei, morphology, size and viability.

Figure 3. p53 activators protect HNDFs from the adverse effects of S- and M-poisons. Cells were pre-incubated with the indicated small-molecule p53 activators (or vehicle) for 24 h prior to treatment with S- or M-phase poisons for 48 h. After recovery in drug-free medium (4−9 d, depending on drug combination), cells were fixed with methanol−acetone and stained with Giemsa.
To further highlight the quality of the protection conferred to normal cells, next we describe two illustrative examples. First, Figure S1 depicts results for HNDFs exposed to a tenovin-6 and vinca alkaloid combination. As can be seen in Figure S1A, treatment with 25 nM VNB led to high levels of cell death (left part) and re-growth was poor following withdrawal of the drug (right part). However, when pre-incubated with tenovin-6 for 24 h prior to VNB addition, the level of cell death was negligible, and cells successfully resumed proliferation upon removal of the compounds. Moreover, Figure S1B and C support that the tenovin-based cyclotherapy regime not only protects HNDFs from the cell death, but also from the nuclear damage caused by vinca alkaloids. Second, Figure 4A shows results for HNDFs subjected to an LDactD and GMTB combination. Samples for FACS analysis were harvested 48 h post-seeding. Exposure to GMTB alone resulted in cell death and accumulation of cells in S-phase, whereas LDactD alone efficiently induced G1 and G2 arrest, in agreement with the data presented in Figure 2A. There was no difference in the cell cycle distribution of HNDFs treated with LDactD alone or with both LDactD and GMTB (Fig. 4A), suggesting that the impact of GMTB on cells pre-incubated with LDactD is negligible. The response of cells lacking functional p53 to the same regime was remarkably different (Fig. 4B), as will be discussed in the next section.

Figure 4. Changes in cell-cycle distribution in response to a cyclotherapy regime combining low dose actinomycin D with gemcitabine. (A) HNDFs and (B) MDA-MB-231 cells were treated as indicated. DNA synthesis and DNA content were evaluated by measuring BrdU incorporation and PI staining by FACS.
Effects of p53 activators and cyclotherapy regimes on p53-mutant and p53-null cancer cells.
Ideally, a compound to be used as a chemoprotectant has to be able to reduce side effects without interfering with the efficacy of the anticancer treatment. Hence, we tested whether the four p53 activators confer p53-mutant cells with resistance against S- or M-phase poisons. For this purpose, we used the MDA-MB-231 (p53R280K) and MDA-MB-468 (p53R273H) breast adenocarcinoma-derived cell lines.
Images of the clonogenic assays performed for each of the 16 drug combinations in MDA-MB-231 and MDA-MB-468 cells are provided in Figure 5A and Figure S2, respectively, while the table in Figure 6A gives an overview of all results. By comparing the columns in this table, the corresponding p53 activators can be classified into two groups, namely (1) tenovin-6 and LMB and (2) nutlin-3 and LDactD. FACS analyses showed that MDA-MB-231 cells are irresponsive to nutlin-3 and LDactD (Fig. 5B) and, as expected, these small molecules did not protect p53-mutant cell lines against any of the S- and M-phase poisons tested (Fig. 6A). In contrast, tenovin-6 and LMB induced G1 arrest in MDA-MB-231 cells (Fig. 5B) and protected both p53-mutant cell lines against vinca alkaloids (Fig. 6A). Regarding S-phase poisons, only weak protection from GMTB with tenovin-6 in MDA-MB-231 cells was observed (Fig. 5A), suggesting that tenovin-6 and LMB might perform better in cyclotherapy regimes when combined with S- than with M-phase poisons.

Figure 5. Studies with MDA-MB-231 cells. (A) Cells were subjected to the cyclotherapy protocol described in Materials and Methods, and then stained with Giemsa. (B) Changes in cell-cycle distribution in response to exposure to small-molecule p53 activators for 24 h. DNA synthesis and DNA content were evaluated by measuring BrdU incorporation and PI staining by FACS. Values correspond to percentage change compared with untreated controls [i.e., panel B(1)].

Figure 6. Summary tables for cancer cell lines. (A) MDA-MB-231 and MDA-MB-468 cells and (B) HCT116-p53wt and HCT116-p53ko cells. Cells were subjected to the cyclotherapy protocol described in Materials and Methods. Protection level was decided based upon data recorded after recovery. See also Figures 5A; Figs. S2 and S5.
Figures 4B and Figure S3 provide additional data for drug combinations in p53-mutant cells. First, Figure 4B shows the results of FACS analyses performed with MDA-MB-231 cells exposed to a cyclotherapy regime involving LDactD and GMTB. By comparing the samples, it can be seen that GMTB-treated MDA-MB-231 cells have the same cell cycle distribution independent of the presence or absence of LDactD, indicating that LDactD failed to protect these cells against GMTB. This observation is in agreement with the corresponding clonogenic assay in Figure 5A. However, the levels of protection observed immediately after treatment and following recovery do not always match. An illustrative example is given in Figure S3, where MDA-MB-231 and MDA-MB-468 cells were treated with tenovin-6 and vinca alkaloids. Cell counts after treatment (Fig. S3A) suggest that tenovin-6 offered no protection, but recovery assays (Fig. S3B) reveal that cells pre-incubated with tenovin-6 possess increased viability compared with cells treated with the M-phase poisons alone. In particular, Figure S3B shows that tenovin-6 provides MDA-MB-468 cells with strong protection against both VNB and VRL.
Next, we investigated the impact of p53 activators and drug combinations on the isogenic colorectal cancer-derived cell lines HCT116-p53wt and HCT116-p53ko. Nutlin-3 interacts directly with p53’s main negative regulator and, consequently, the biological effects of this compound are strongly p53-dependent.7,45 As expected, we observed no effects in HCT116-p53ko cells in response to nutlin-3 treatment (Fig. S4). In contrast, tenovin-6, LMB and LDactD, which activate p53 through indirect mechanisms, have both p53-dependent and p53-independent effects. In particular, they can affect p53-null cells (Fig. S4) and can even confer better protection against anticancer drugs in the absence than in the presence of p53 (Figs. 6B; Fig. S5). LDactD, for instance, protected HCT116-p53ko but not HCT116-p53wt cells against the cytotoxicity of vinca alkaloids. It is worth noting here that the impact of the same drug combinations on p53-mutant and p53-null cells can be remarkably different.
Discussion
In this paper, we describe cyclotherapy studies involving four small-molecule p53 activators (i.e., tenovin-6, LMB, nutlin-3 and LDactD) and four widely used chemotherapeutics (i.e., VNB, VRL, Ara-C and GMTB). All the p53 activators induced reversible cell cycle arrest in primary human fibroblasts (Figs. 1 and 2) and effectively protected them against S- and M-phase poisons (Fig. 3). This held true even when fibroblasts were treated with doses of S- and M-phase poisons above those required to kill tumor cells. Despite these comparable results with normal cells, we identified differences in the effects of the various drug combinations on p53-mutant and p53-null cancer cell lines (Fig. 6). As discussed above, this can partly be understood in terms of the each compound’s selectivity for the p53 pathway.
With regard to our results in primary fibroblasts, it is worth noticing that there is a substantial accumulation of HNDFs with 8N DNA content in response to all four small-molecule p53 activators (Fig. 2A). This phenomenon, which became even more noticeable following recovery from treatment (Fig. 2B), has been previously reported in nutlin-treated cancer cell lines expressing wild-type p53.45 The possibility that tenovin-6, LMB, nutlin-3 and LDactD could promote polyploidy and, subsequently, aneuploidy and genomic instability46,47 constitutes a concern from a therapeutic point of view. We can envisage at least two possible explanations for these experimental observations. According to a first scenario, the p53-dependent cell cycle arrest results in a “memory loss” in G2 phase of the cell cycle and, as a consequence, a number of tetraploid cells enter S phase instead of undergoing cell division. In this situation, the compounds would actively cause endoreduplication. Alternatively, the changes in the proportion of 8N cells could simply indicate cell cycle arrest in a pre-existing polyploid population. This would imply that polyploid cells are continuously being produced under culture conditions. To gain further insight, we subjected HNDFs to serum deprivation, one of the mildest procedures available to halt cell proliferation. As can be seen in Figure S4, this also led to a substantial increase in the proportion of cells with 8N DNA content following recovery in the presence of serum. These data support the hypothesis that polyploid cells were present in the HNDF population prior to treatment. Hence, it is unlikely that the p53 activators induced an increase in the endoreduplication frequency.
Tenovin-6 is a novel p53 activator that was first described in 2008.17 This small molecule is comparable to nutlin-3 according to its dose range and performance as a chemoprotectant in normal cells, and, importantly, no associated genotoxicity has been detected. Moreover, several independent studies have validated tenovin-6’s modes of action, and, very recently, it has been shown to eradicate chronic myelogenous leukemia (CML) stem cells resistant to Imatinib.48-50 Elimination of leukemic stem cells is essential to achieve cure. Here we have shown that within the low micromolar dose range, this compound causes mild cytostatic effects in HNDFs (Figs. 1 and 2), thereby successfully safeguarding these cells from the toxicity of VNB, VRL, Ara-C and GMTB (Fig. 3; Fig. S1). Supporting its potential in cyclotherapy, tenovin-6 does not protect p53-mutant cancer cells against the S-phase poison Ara-C, and only weak protection from GMTB was observed in MDA-MB-231 cells. However, tenovin-6 shielded both p53-mutant cell lines against vinca alkaloids, and, in particular, strong protection was observed in MDA-MB-468 cells (Fig. S2). This suggests that tenovin-6 should not be used in combination with tubulin poisons.
As tenovin-6 reduces the activity of sirtuins SirT1 and SirT2, its use as a chemoprotectant could be questioned on the basis of recent evidence indicating that SirT1 and SirT2 inhibition has genome destabilizing effects. SirT1 is involved in DNA damage repair response and acts as a tumor suppressor in mice.51 SirT2 regulates the anaphase-promoting complex (APC), and SIRT2-deficient mice show increased tumor incidence compared with wild-type counterparts.52 However, whether these effects occur when inactivation of SirT1 and/or SirT2 is transient still needs to be investigated.
The performance of the nuclear export inhibitor LMB as a chemoprotectant was comparable to that of tenovin-6 in all cyclotherapy combinations tested. In normal cells LMB’s effects were primarily cytostatic and reversible (Figs. 1 and 2), and the compound provided good protection against VNB, VRL, Ara-C and GMTB in these cells (Fig. 3). In p53-mutant cell lines, like tenovin-6, this small molecule provided protection against the vinca alkaloids but not the nucleotide analogs (Fig. 6A). This suggests that LMB could be used in combination with S- but not M-phase poisons. LMB is generally regarded as a compound with strong anticancer potency53 but poor in vivo efficacy due to toxicity. A phase I trial with LMB revealed dose-limiting toxicity in the form of nausea, vomiting, anorexia and malaise.54 However, these observations might not be relevant when using LMB at very low doses in a cyclotherapy setting, and, importantly, there are now semi-synthetic LMB derivatives available that may not have such in vivo tolerance limitations.55 The best analog has a maximum tolerated dose (MTD) in mice that is 15-fold higher than that of LMB. Furthermore, as the toxicity of LMB greatly depends on the presence of wild-type p53, topical application has been proposed as a treatment strategy for human papillomavirus (HPV)-positive or p53-mutant lesions.56,57 Finally, LMB is a Michael acceptor that reacts with cysteine residues in proteins.19 This fact, per se, is not enough reason to discard LMB as a potential drug, as Michael acceptors can show selectivity for cancer cells and outstanding efficacy in preclinical models. This is the case, for example, with piperlongumine.58 Perhaps the major concern with LMB is that it may have genome destabilizing effects, as inactivation of CRM1 has been shown to result in centrosome duplication and multipolar spindles.59,60
The highly specific p53 activator nutlin-3 induces reversible G1 and G2 arrest in HNDFs (Fig. 2), which is in agreement with previous observations in other normal cell lines.9,10 Moreover, nutlin-3 protects HNDFs from the adverse effects of VNB and VRL, and, in particular, it clearly reduces the number of cells with gross nuclear aberrations caused by the tubulin poisons (Fig. 3). Similarly, nutlin-3 also protects primary fibroblasts against two S-phase poisons, Ara-C and GMTB, as previously described in reference 8. In contrast, nutlin-3 does not induce cell cycle arrest in p53-mutant or p53-null cancer cells, thereby leaving them vulnerable to subsequent treatment with M- or S-phase poisons (Fig. 6). In line with the literature summarized in Table 1, these results support that nutlin-3 could constitute an ideal chemoprotectant for patients bearing p53-mutant tumors. Like with LMB, concerns have been raised about possible undesired side effects, as nutlin-3 has been described to cause DNA damage at doses above 10 µM.14,15 At present, however, the main drawback with nutlin-based cyclotherapy may be that this small molecule has not been clinically approved yet. It is therefore of great interest that the nutlin-like compound RG7112 is currently undergoing a phase I clinical trial (NCT00623870) to treat hematological neoplasms.61
When looking for other p53 activators as alternatives to nutlin-3, LDactD constitutes an obvious choice. Actinomycin D is a clinically approved drug, and, at low doses, it has been shown to mimic the effects of nutlin-3.30 LDactD (1–4 nM) has a reversible cytostatic effect on HNDFs (Fig. 2 and Rao et al.6), and, like the other p53 activators tested, it protects HNDFs from cytotoxicity and appearance of aberrant nuclei caused by VNB, VRL, Ara-C and GMTB (Fig. 3). At a dose of 2 nM, ActD not only failed to protect two p53-mutant cell lines from any of the above S- and M-phase poisons, but also caused additional cell death in co-treated cultures (Fig. 5A; Fig. S2). These observations, which extend our previous work using LDactD in combination with the Aurora kinase inhibitor VX680,6 suggest that the performance of nutlin-3 and LDactD in cyclotherapy is comparable. However, Figure 6 shows that, unlike nutlin-3, LDactD shields HCT116-p53ko cells from the cytotoxicity caused by Ara-C and vinca alkaloids. This indicates that p53-null and p53-mutant cells respond differently to cyclotherapy regimes involving LDactD.
Finally, we would like to highlight again the importance of assessing both short- and long-term effects when selecting possible chemoprotectants for cancer therapy. Protection of cancer cells lacking functional p53 may not become apparent until cultures are given the chance to re-grow after treatment. In Figure S3, this is exemplified for p53-mutant cells exposed to tenovin-6 and vinca alkaloids. Immediately after treatment, there is no significant difference between cells pre-incubated with tenovin-6 or treated with the tubulin poisons alone, suggesting that no protection occurs. Yet, if the drugs are removed, and cancer cells are left to recover in fresh medium, cell viability is higher under cyclotherapy than chemotherapy conditions.
Materials and Methods
Cells and reagents.
HCT116 p53ko and p53wt cells (kind gift from B. Vogelstein) were cultivated in McCoy’s 5A medium (#M8408, Sigma) supplemented with 1% penicillin-streptomycin (P/S), 10% Fetal Bovine Serum (FBS) and 15% L-Glutamine (#SH30034.01, Thermo Scientific). HNDFs (#C-12300, PromoCell) and MDA-MB-231 and MDA-MB-468 cells (CRUK) were grown in DMEM (#SH30243.01, Thermo Scientific) supplemented with 1% P/S and 10% FBS. Nutlin-3 (#N6287, Sigma) consisting of two enantiomers 3a and 3b (1:1), only one being active, was stored at -20°C as 40 mM solution in DMSO (#D8418, Sigma). Actinomycin D (#A9415), cytosine arabinoside (#C1768), gemcitabine (#G6423), vinblastine (#V1377) and vinorelbine (#V2264) were also purchased from Sigma and stored under the same conditions. Leptomycin B was obtained from Cancer Research UK and stored as 10 mM solution in absolute ethanol. Tenovin-6 was synthesized in house and stored at as 40 mM solution in DMSO.
Cyclotherapy cell culture assays.
Cells were seeded on 6-well plates on day 0 and treated with small-molecule p53 activators or vehicle on day 1. After 24 h pre-incubation (day 2), cells were treated with S- or M-phase poisons for 48 h or left untreated. Thereafter (day 4), the medium was removed, the cells were rinsed with PBS and left to recover in fresh medium for several days. In the case of tenovin-6, cells were also washed with PBS prior to exposure to S- or M-phase poisons to mimic the relative short half-life of tenovin-6 in vivo.17 Data were recorded at three time points: before and after treatment, and after recovery. Each of the 16 cyclotherapy combinations was tested in at least two independent experiments in every cell line.
Giemsa staining and image capture.
Cells were seeded on 6-well plates and treated with compounds as indicated. At the end of the experiment, cells were fixed with ice-cold methanol-acetone (1:1) for 8 min at -20°C and then left to dry at RT. Cells were stained with 5–10% Giemsa (#48900, Sigma) in PBS for 5–20 min washed with lukewarm tap water to remove excess stain and then air-dried. Photographs were taken using a Zeiss AxioVert 40C microscope equipped with a high-resolution AxioCam MRc5 camera.
Cell cycle analysis.
Cells were seeded on 6-well plates and treated with compounds as indicated. Thereafter, cells were incubated with 30 µM bromodexoyuridine (BrdU, #B9285, Sigma) for 30 min at 37°C, and then subsequently trypsinised, washed with PBS, and fixed in 3 ml of ice-cold ethanol for at least one hour at -20°C. Cells were digested in pre-warmed 1 mg/ml pepsin (#P6887, Sigma) in 30 mM HCl (pH 1.5) for 30 min at 37°C with gentle shaking, and then incubated in 2 M HCl for 20 min at RT. Next, cells were washed with PBS and antibody buffer (0.5% w/v BSA, 0.5% v/v Tween-20 in PBS) and incubated with mouse primary antibody against BrdU (#347580, Becton Dickinson) in antibody buffer for one hour at RT. After washing with PBS, samples were incubated with secondary sheep anti-mouse IgG FITC-conjugated antibody (#F3008, Sigma) for 30 min at RT in the dark. Finally, cells were washed one more time with PBS and then stained with 25 µg/ml propidium iodide (#P3566, Invitrogen). Fluorescence-activated cell sorting (FACS) was performed using a Becton Dickinson FACScan operated by the CELLQuest software.
Supplementary Material
Acknowledgements
S.L. and I.v.L. gratefully acknowledge the support provided by the AICR (Research Grant #10-0043), Cancerfonden, Karolinska Institutet and the Centre for Advanced Cancer Therapies (ACT). B.R. and M.S. were funded by Cancer Research UK and Karolinska Institutet, respectively.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20254
References
- 1.Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res. 2001;61:4301–5. [PubMed] [Google Scholar]
- 2.Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell Cycle. 2002;1:375–82. doi: 10.4161/cc.1.6.259. [DOI] [PubMed] [Google Scholar]
- 3.van Leeuwen IM, Laín S. Pharmacological manipulation of the cell cycle and metabolism to protect normal tissues against conventional anticancer drugs. Oncotarget. 2011;2:274–6. doi: 10.18632/oncotarget.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000;105:533–9. doi: 10.1172/JCI8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blagosklonny MV. Sequential activation and inactivation of G2 checkpoints for selective killing of p53-deficient cells by microtubule-active drugs. Oncogene. 2002;21:6249–54. doi: 10.1038/sj.onc.1205793. [DOI] [PubMed] [Google Scholar]
- 6.Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, et al. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget. 2010;1:639–50. doi: 10.18632/oncotarget.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ. 2010;17:1486–500. doi: 10.1038/cdd.2010.18. [DOI] [PubMed] [Google Scholar]
- 8.Kranz D, Dobbelstein M. Nongenotoxic p53 activation protects cells against S-phase-specific chemotherapy. Cancer Res. 2006;66:10274–80. doi: 10.1158/0008-5472.CAN-06-1527. [DOI] [PubMed] [Google Scholar]
- 9.Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011;2:222–33. doi: 10.18632/oncotarget.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005;65:1918–24. doi: 10.1158/0008-5472.CAN-04-3576. [DOI] [PubMed] [Google Scholar]
- 11.Tokalov SV, Abolmaali ND. Protection of p53 wild type cells from taxol by nutlin-3 in the combined lung cancer treatment. BMC Cancer. 2010;10:57. doi: 10.1186/1471-2407-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr., Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–9. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 14.Verma R, Rigatti MJ, Belinsky GS, Godman CA, Giardina C. DNA damage response to the Mdm2 inhibitor nutlin-3. Biochem Pharmacol. 2010;79:565–74. doi: 10.1016/j.bcp.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentine JM, Kumar S, Moumen A. A p53-independent role for the MDM2 antagonist Nutlin-3 in DNA damage response initiation. BMC Cancer. 2011;11:79. doi: 10.1186/1471-2407-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Leeuwen IM, Higgins M, Campbell J, Brown CJ, McCarthy AR, Pirrie L, et al. Mechanism-specific signatures for small-molecule p53 activators. Cell Cycle. 2011;10:1590–8. doi: 10.4161/cc.10.10.15519. [DOI] [PubMed] [Google Scholar]
- 17.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–63. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laín S, Midgley C, Sparks A, Lane EB, Lane DP. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res. 1999;248:457–72. doi: 10.1006/excr.1999.4433. [DOI] [PubMed] [Google Scholar]
- 19.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–7. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meissner T, Krause E, Vinkemeier U. Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett. 2004;576:27–30. doi: 10.1016/j.febslet.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 21.Smart P, Lane EB, Lane DP, Midgley C, Vojtesek B, Laín S. Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. Oncogene. 1999;18:7378–86. doi: 10.1038/sj.onc.1203260. [DOI] [PubMed] [Google Scholar]
- 22.Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–93. doi: 10.1128/mcb.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol. 2010;17:982–9. doi: 10.1038/nsmb.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassilev LT. p53 Activation by small molecules: application in oncology. J Med Chem. 2005;48:4491–9. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 25.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007;67:7350–7. doi: 10.1158/0008-5472.CAN-07-0200. [DOI] [PubMed] [Google Scholar]
- 27.Kumamoto K, Spillare EA, Fujita K, Horikawa I, Yamashita T, Appella E, et al. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 2008;68:3193–203. doi: 10.1158/0008-5472.CAN-07-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle. 2009;8:3777–81. doi: 10.4161/cc.8.22.10121. [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res. 2009;7:1497–509. doi: 10.1158/1541-7786.MCR-09-0144. [DOI] [PubMed] [Google Scholar]
- 30.Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–8. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 31.Walterhouse D, Watson A. Optimal management strategies for rhabdomyosarcoma in children. Paediatr Drugs. 2007;9:391–400. doi: 10.2165/00148581-200709060-00006. [DOI] [PubMed] [Google Scholar]
- 32.Wilms’ tumor: status report, 1990. By the National Wilms’ Tumor Study Committee. J Clin Oncol. 1991;9:877–87. doi: 10.1200/JCO.1991.9.5.877. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg IH. The interaction of actinomycin with DNA. Antibiot Chemother. 1971;17:67–86. doi: 10.1159/000392364. [DOI] [PubMed] [Google Scholar]
- 34.Goldberg IH, Rabinowitz M, Reich E. Basis of actinomycin action. II. Effect of actinomycin on the nucleoside triphosphate-inorganic pyrophosphate exchange. Proc Natl Acad Sci U S A. 1963;49:226–9. doi: 10.1073/pnas.49.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg IH, Reich E. Actinomycin inhibition of RNA synthesis directed by DNA. Fed Proc. 1964;23:958–64. [PubMed] [Google Scholar]
- 36.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–87. doi: 10.1016/S1535-6108(03)00134-X. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–80. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plunkett W, Huang P, Gandhi V. Preclinical characteristics of gemcitabine. Anticancer Drugs. 1995;6(Suppl 6):7–13. doi: 10.1097/00001813-199512006-00002. [DOI] [PubMed] [Google Scholar]
- 40.Sigmond J, Kamphuis JA, Laan AC, Hoebe EK, Bergman AM, Peters GJ. The synergistic interaction of gemcitabine and cytosine arabinoside with the ribonucleotide reductase inhibitor triapine is schedule dependent. Biochem Pharmacol. 2007;73:1548–57. doi: 10.1016/j.bcp.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 41.Grant S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res. 1998;72:197–233. doi: 10.1016/S0065-230X(08)60703-4. [DOI] [PubMed] [Google Scholar]
- 42.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–72. [PubMed] [Google Scholar]
- 43.Bergman AM, Eijk PP, Ruiz van Haperen VW, Smid K, Veerman G, Hubeek I, et al. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005;65:9510–6. doi: 10.1158/0008-5472.CAN-05-0989. [DOI] [PubMed] [Google Scholar]
- 44.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 45.Shen H, Moran DM, Maki CG. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008;68:8260–8. doi: 10.1158/0008-5472.CAN-08-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King RW. When 2+2=5: the origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Wang L, Li L, Wang Z, Ho Y, McDonald T, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–81. doi: 10.1016/j.ccr.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011;7:e1002135. doi: 10.1371/journal.pgen.1002135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Lüscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012;109:E187–96. doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–23. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20:487–99. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laín S, Xirodimas D, Lane DP. Accumulating active p53 in the nucleus by inhibition of nuclear export: a novel strategy to promote the p53 tumor suppressor function. Exp Cell Res. 1999;253:315–24. doi: 10.1006/excr.1999.4672. [DOI] [PubMed] [Google Scholar]
- 54.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74:648–9. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69:510–7. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gray LJ, Bjelogrlic P, Appleyard VC, Thompson AM, Jolly CE, Lain S, et al. Selective induction of apoptosis by leptomycin B in keratinocytes expressing HPV oncogenes. Int J Cancer. 2007;120:2317–24. doi: 10.1002/ijc.22591. [DOI] [PubMed] [Google Scholar]
- 57.Hietanen S, Lain S, Krausz E, Blattner C, Lane DP. Activation of p53 in cervical carcinoma cells by small molecules. Proc Natl Acad Sci U S A. 2000;97:8501–6. doi: 10.1073/pnas.97.15.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–4. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, et al. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282–92. doi: 10.1128/MCB.23.15.5282-5292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Budhu AS, Wang XW. Loading and unloading: orchestrating centrosome duplication and spindle assembly by Ran/Crm1. Cell Cycle. 2005;4:1510–4. doi: 10.4161/cc.4.11.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andreeff M, Kojima K, Ruvolo V, Younes A, Wei W, Konopleva M, et al. Pharmacodynamic biomarkers in the Phase 1 Trial of RG7112, a small-molecule MDM2 antagonist in leukemia. 52nd Annual Meeting of the American Society of Hematology, Orlando, FL, USA 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.