

Abstract
Diabetes mellitus is a predisposing factor of melioidosis, contributing to higher mortality rates in diabetics infected with Burkholderia pseudomallei. To investigate how diabetes alters the inflammatory response, we established a streptozotocin (STZ) -induced diabetic murine acute-phase melioidosis model. Viable B. pseudomallei cells were consistently detected in the blood, liver and spleen during the 42-hr course of infection but the hyperglycaemic environment did not increase the bacterial burden. However, after 24 hr, granulocyte counts increased in response to infection, whereas blood glucose concentrations decreased over the course of infection. A genome-wide expression analysis of the STZ-diabetic murine acute melioidosis liver identified ∼ 1000 genes whose expression was altered in the STZ-diabetic mice. The STZ-diabetic host transcriptional response was compared with the normoglycaemic host transcriptional response recently reported by our group. The microarray data suggest that the presence of elevated glucose levels impairs the host innate immune system by delaying the identification and recognition of B. pseudomallei surface structures. Consequently, the host is unable to activate the appropriate innate immune response over time, which may explain the increased susceptibility to melioidosis in the STZ-diabetic host. Nevertheless, a general ‘alarm signal’ of infection as well as defence programmes are still triggered by the STZ-diabetic host, although only 24 hr after infection. In summary, this study demonstrates that in the face of a B. pseudomallei acute infection, poor glycaemic control impaired innate responses during the early stages of B. pseudomallei infection, contributing to the increased susceptibility of STZ-induced diabetics to this fatal disease.
Keywords: acute melioidosis, Burkholderia pseudomallei, diabetes, hyperglycaemia, innate immunity
Introduction
In 2010, diabetes mellitus (DM) affected 284 million people worldwide with a concomitant dramatic impact on health in terms of morbidity and mortality of affected individuals.1 The projection for 2030 indicates a prevalence of 439 million individuals comprising ∼ 7·7% of the world population.1 Diabetes has been identified as an important risk factor for infection, particularly Gram-negative infections,2–4 including melioidosis, an infection caused by the soil bacterium Burkholderia pseudomallei that is endemic in Southeast Asia and Northern Australia.5,6 The increased susceptibility of diabetics to infection has been suggested to be the result of defects in immunity such as impaired chemotaxis, phagocytosis, oxidative burst and killing activity, as well as of increased microbial adherence to diabetic cells.4,7 Little is known about the basis for the increased susceptibility of diabetic patients to B. pseudomallei infection but impaired neutrophil function is believed to be one of the possible causes of this increased prevalence of infection.6
Patients with DM (up to 60% are type 2 diabetes) present with a high incidence of melioidosis.6 Although insulin is thought to have a direct effect on the growth of B. pseudomallei,8 subsequent studies have attributed the inhibitory effect to a preservative used with insulin.9,10 Recently, Pongcharoen et al.11 reported that patients with DM have defective interleukin-17 (IL-17) production in response to B. pseudomallei infection, whereas Chanchamroen et al.12 reported that defective polymorphonuclear neutrophils (PMN) of diabetic subjects in the early phase of the inflammatory reaction against B. pseudomallei may contribute to increased susceptibility to melioidosis. Despite such clinical observations, little is known about how diabetes impairs protective immunity. Moreover, an intensive study on the immune response of diabetic hosts with respect to this bacterium is still not available. As prevalence of diabetes is expected to increase rapidly worldwide, there is potential for an increased number of individuals at risk of severe infection with B. pseudomallei. Hence, establishing a diabetes infection model is a logical first step to investigate the mechanism of impaired host defence in diabetes.
Diabetic mice established by multiple low-dose treatments with streptozotocin (STZ), a pancreatic islet β-cell toxin, have been widely used to study a number of infections.3,13 The multiple low-dose STZ treatments induce an autoimmune insulitis that leads to insulin insufficiency and diabetes that mimics several of the aetiological events that occur in the development of human type 1 diabetes.3 Although both type 1 and type 2 diabetes have different aetiologies, they share common clinical symptoms of hyperglycaemia, glucose intolerance, poor wound healing, nephropathy, vascular abnormalities and increased risk of infection.2,4,14 A study on Porphyromonas gingivalis infection demonstrated that the inflammatory response to this infection is not dependent upon the type of diabetes, but rather is the consequence of hyperglycaemia.4
The present study was conceived to investigate the exquisite interplay between the STZ-diabetic host response and B. pseudomallei. We established a systemic acute melioidosis infection of STZ-induced diabetic mice and performed transcriptional analysis of the liver and spleen isolated from diabetic mice infected over a 42-hr time period. Our study is the first to report on a global STZ-diabetic host–B. pseudomallei interaction by whole transcriptome analysis.
Materials and methods
Bacteria
The B. pseudomallei clinical isolate, referred to herein as BpD286, was obtained from the Pathogen Laboratory, School of Biosciences and Biotechnology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Malaysia, and was previously characterized based on biochemical tests as well as by 16S rRNA sequencing.15 Bacteria were grown in brain–heart infusion broth overnight at 37°. The cells were centrifuged at 10 000 g, suspended in brain–heart infusion broth containing 20% glycerol, frozen immediately in aliquots of 109 colony-forming units (CFU)/ml and stored at −80°.16
Mice
BALB/c male mice (5 to 7 weeks old) were purchased from the Institute for Medical Research, Malaysia. They were housed in High Temperature Polysufone (Tecniplast, Buguggiate, Italy) cages with a bedding of wood shavings, subjected to a 12-hr light/dark cycle and were fed on a diet of commercial pellets and distilled water ad libitum. All animal experiments were performed in accordance with the Universiti Kebangsaan Malaysia animal ethics guidelines and approved by the Universiti Kebangsaan Malaysia Animal Ethics Committee.
STZ-induced diabetes in BALB/c mice
Diabetic mice were established using the Low-Dose STZ Induction Protocol by Animal Models of Diabetic Complication Consortium as described elsewhere with minor modifications.3,13,17 BALB/c mice were rendered diabetic by treatment with STZ (50 mg/kg body weight) in 10 mm sodium citrate buffer (pH 4·5) by intra-peritoneal injection daily for 5 days. Mice were considered to be diabetic when blood glucose levels exceeded 14 mmol/l. Mice remained diabetic for 3–5 days before inoculation with bacteria. At the time the experiments were initiated, blood glucose levels ranged from 14·1 to 33·3 mmol/l. A second group of mice not treated with STZ (normoglycaemic mice) were used as non-diabetic controls. These mice had blood glucose levels that ranged from 5 to 8 mmol/l. Tail vein blood glucose levels were assessed with the glucometer Accu-Chek Active (Roche Diagnostics, Mannheim, Germany).
Development and characterization of acute melioidosis STZ-diabetic model
Infection experiments, determination of bacterial loads and leucocyte differential counts were performed as previously described.18 Mice were monitored for a period of 10 days based on the accepted protocol for observing mortality in B. pseudomallei acute-stage infection.19–21
Gene expression analyses and microarray data analysis
Microarray experiments were performed using the SentrixMouseRef-8 Expression BeadChips (Illumina, San Diego, CA), containing over 24 000 probes according to the instructions provided and as described previously.18 BeadStudio version 1.0 (Illumina) software was used to generate signal intensity values from the scans according to the standard procedure within the software. In brief, the sample intensities are scaled by a factor equal to the ratio of average intensity of virtual sample to the average intensity of the given sample. Background is subtracted before the scaling. The normalized data were analysed by GeneSpring GX7.3.1 Expression Analysis (Agilent Technologies, Santa Clara, CA) as previously described for acute normoglycaemic microarray data.18 In brief, normalization was applied in two steps: ‘per chip normalization’, by which each measurement was divided by the 50th percentile of all measurements in its array; and ‘per gene normalization’, by which all the samples were normalized against the median of the control samples (uninfected STZ-diabetic control tissues). The expression of each gene was reported as the ratio of the value obtained for each condition relative to the control condition after normalization of the data as previously described.18 The normalized data were grouped on the basis of the experimental conditions (organs and infection time-points). The Volcano Plot with parametric test was performed to determine differentially expressed genes. Differentially expressed genes were defined as those having a P-value ≤ 0·05 and an absolute change greater than twofold for B. pseudomallei-infected tissue at 16, 24 or 42 hr relative to the uninfected STZ-diabetic control tissue. The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through the GEO Series accession number GSE28683 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28683).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed in the Mastercycler® ep realplex (Eppendorf, Harburg, Germany) to quantify the expression of TLR2, TLR4, TLR5, IFNg, CXCL1 and CCL7 genes as previously described.18
Results
Melioidosis susceptibility of STZ-induced diabetic mice
Diabetes was successfully induced by multiple low-dose treatments with STZ. An average of 70% of the STZ-treated mice (21 of 30 mice) had blood glucose levels > 14 mmol/l and were considered diabetic.3 These STZ-diabetic mice exhibited signs of polydipsia and polyuria. Susceptibility to B. pseudomallei infection between STZ-diabetic and non-diabetic control mice was compared. Both groups of mice were infected with 1·2 × 103 CFU/ml B. pseudomallei D286 via the intravenous route and survival was monitored over 10 days post-infection. Burkholderia pseudomallei-infected STZ-diabetic mice and B. pseudomallei-infected normoglycaemic mice had similar survival percentages over the first 48-hr post-infection (p.i.) (Fig. 1). However, the B. pseudomallei-infected STZ-diabetic mice were more susceptible to melioidosis than the B. pseudomallei-infected normoglycaemic mice at the later stage of infection (Fig. 1). Mortality was first observed for the B. pseudomallei-infected STZ-diabetic mice on day 3 p.i. with a median survival of 7 days, whereas the B. pseudomallei-infected normoglycaemic mice had a longer median survival (9 days). By day 10 p.i., mortality of B. pseudomallei-infected STZ-diabetic mice was 75% (six of eight mice) compared with 50% for normoglycaemic mice (two of four mice) (Fig. 1). However, we postulate that the B. pseudomallei-infected STZ-diabetic group will continue to succumb to melioidosis based upon the day-10 post-mortem observation of abscesses on the spleens of surviving diabetic mice.
Figure 1.
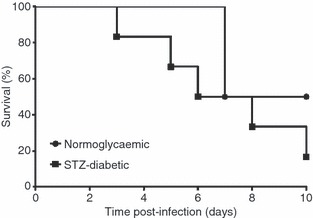
Melioidosis susceptibility of mice with streptozotocin (STZ)-induced diabetes. Mortality of STZ-diabetic mice (n = 8 mice) compared with normoglycaemic mice (n = 4 mice) following Burkholderia pseudomallei infection [Logrank (Mantel–Cox) test, P-value = 0·4483]. Animals were observed daily up to 10 days and the percentage survival was plotted against time. A representative of two independent experiments is shown. Mice were infected with 1·2 × 103 colony-forming units (CFU)/ml B. pseudomallei via the intravenous route.
Development and characterization of acute melioidosis in an STZ-diabetic mouse model
To characterize acute melioidosis in an STZ-diabetic mouse model, we monitored mouse blood glucose levels, bacterial loads in various organs and leucocyte differential counts during the course of infection in STZ-induced diabetic BALB/c mice infected with 9·36 × 103 CFU/ml B. pseudomallei D286. Non-fasting blood glucose levels were measured 2 weeks after STZ treatment (before infection). Mice with confirmed elevated blood glucose levels (14–33 mmol/l) were then injected with B. pseudomallei. Blood glucose levels were again measured 1 hr p.i. and at the conclusion of each time-point: 16, 24 and 42 hr p.i. (Fig. 2). Blood glucose levels of STZ-treated mice (mean = 24·51 mmol/l) remained > 14 mmol/l at 1 hr p.i., confirming that the infected mice from all the experimental groups were hyperglycaemic at the time of infection (Fig. 2). The mean blood glucose concentrations for B. pseudomallei-infected mice at 16, 24 and 42 hr p.i., were 23·36, 9·76 and 11·4 mmol/l, respectively. Blood glucose levels of B. pseudomallei-infected STZ-diabetic mice decreased gradually to hypoglycaemic levels before the mice succumbed to infection.
Figure 2.
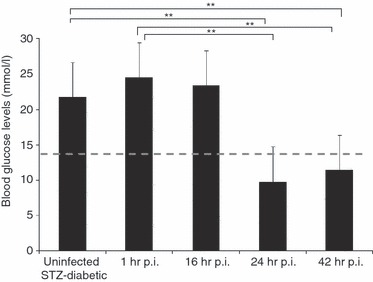
Mouse blood glucose concentrations. Non-fasting blood glucose levels of mice (n = 5 mice/group) were measured 2 weeks after streptozotocin (STZ) treatments, 1 hr post-infection (p.i.) and at the conclusion of each time-point: 16, 24 and 42 hr p.i., respectively. Diabetic mice had blood glucose levels > 14 mmol/l (above the dashed line). Data are mean ± standard deviation of five mice per group. Significance was determined using the Student's t-test (**P-value < 0·01).
The bacterial loads in liver, spleen and blood at 42 hr p.i. were significantly higher than bacterial loads at 16 hr p.i. (Fig. 3a–c), indicating propagation of intracellular bacteria in the infected host. The presence of high numbers of B. pseudomallei in the organs and blood confirms that systemic acute septicaemic melioidosis was successfully developed in STZ-diabetic BALB/c mice. No significant differences were observed in liver and spleen weights at all infection time-points (data not shown). To further characterize the STZ-diabetic host innate immune response to acute melioidosis, leucocyte counts and composition of blood samples taken at 16, 24 and 42 hr time-points during infection of STZ-diabetic mice were analysed. Analysis of the differential blood film after infection with 9·36 × 103 CFU/ml B. pseudomallei D286 revealed no changes in the neutrophil and leucocyte counts at 16 hr p.i. compared with the uninfected STZ-diabetic mice (Fig. 3d). Nonetheless, neutrophilia was observed at 24 hr p.i. onwards (Fig. 3d), indicating a delay in triggering the STZ-diabetic host innate immune response to acute B. pseudomallei infection compared with the infected normoglycaemic mice.18 Moreover, several blood cell abnormalities, including monocyte vacuolization, Pseudo Pelger, hypersegmentation of neutrophil nuclei and Rouleaux formation frequently seen in serious bacterial infections22,23 were also seen in the blood samples at 42 hr p.i. (Fig. 3e). This observation was not seen in normoglycaemic B. pseudomallei-infected mice, or in uninfected non-diabetic control mice, and has not been reported previously in any of the melioidosis cases. Peripheral blood cell morphology provides additional unique diagnostic information on B. pseudomallei infection.
Figure 3.
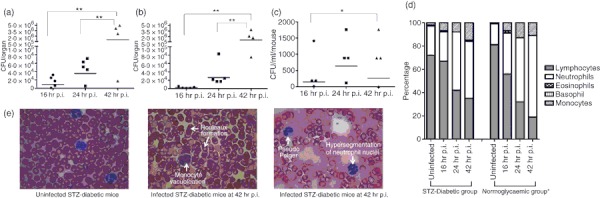
Characterization of streptozotocin (STZ) -induced diabetic acute melioidosis model. The bacterial loads in (a) liver; (b) spleen and (c) blood of STZ-induced diabetic BALB/c mice (n = 5 mice/group) at 16, 24 and 42 hr after intravenous infection with 9·36 × 103 colony-forming units (CFU)/ml Burkholderia pseudomallei. Each symbol represents one mouse; bar indicates geometric mean. Significance was determined using the Student's t-test (*P-value < 0·05; **P-value < 0·01). The control mice are not represented because no colonies were observed by plating. (d) Changes in differential leucocyte counts for both acute STZ-diabetic and acute normoglycaemic mice. Values are an average of pooled blood from three to five infected mice at a particular time-point. Data for the acute normoglycaemic infection model is adopted from Chin et al.18 (e) blood cell abnormalities.
Global transcriptional responses to acute-stage melioidosis in STZ-diabetic mice
STZ-diabetic BALB/c mice were infected with B. pseudomallei D286 intravenously. Gene expression profiles were obtained from a comparison of the transcriptome of infected STZ-diabetic liver and STZ-diabetic spleen with uninfected STZ-diabetic mice organs. We noted that an acute B. pseudomallei infection in STZ-diabetic mice results in more differentially expressed genes in the liver, particularly at 24 hr p.i. onwards (Fig. 4a), compared with the spleen. Surprisingly, very few genes were modulated in the STZ-diabetic spleen throughout the infection period (Fig. 4b). Analyses of the identified genes were further represented by Venn diagrams demonstrating the overlap between different experimental conditions in both liver (Fig. 4c) and spleen (Fig. 4d). There were only 34 and three common genes whose expression is consistently differentially modulated throughout the course of infection in STZ-diabetic liver and spleen, respectively. These expression profiles suggest that common responses, particularly the immune response to acute B. pseudomallei infection, are not well modulated when the diabetic host initially encounters the bacterium.
Figure 4.
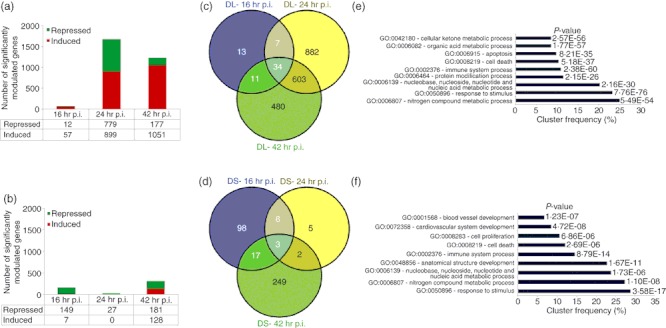
Differential gene expression of an acute melioidosis streptozotocin (STZ) -diabetic model over 42 hr relative to uninfected diabetic mice. Number of genes modulated during acute Burkholderia pseudomallei D286 infection in STZ-induced diabetic BALB/c mice at 16, 24 and 42 hr post-infection (p.i.) in both (a) liver and (b) spleen identified by Volcano plots with the cut-off of twofold change and P-value < 0·05; Venn diagrams demonstrating the overlap between different experimental conditions in both (c) STZ-diabetic liver (DL) and (d) STZ-diabetic spleen (DS) as determined by VENNY; Major biological processes modulated in acute diabetic model in (e) liver and (f) spleen as determined by GOTerm Finder analysis.
The microarray expression profile revealed differentially up-regulated genes (24 hr p.i. onwards) were clustered as those involved in immune response and cellular metabolism (Fig. 4e,f). The cytokine–cytokine receptor interaction, Jak-STAT signalling pathway, Toll-like receptor signalling pathway, chemokine signalling pathway, apoptosis, antigen processing and presentation were up-regulated as shown in Table 1. Concomitantly, the major down-regulated Kyoto Encyclopaedia of Genes and Genomes pathways include drug metabolism, cytochrome P450, glycine, serine and threonine metabolism, tryptophan metabolism, fatty acid metabolism and tricarboxylic acid cycle (Table 1). The identified genes were further categorized according to functional categories and the fold change relative to the uninfected diabetic control mice is presented as a heat map (Fig. 5 and Table 2). Kinetic profiles of the expression of host genes modulated by B. pseudomallei infection in normoglycaemic models18 is also included in Table 2 for comparison between the transcriptional expression responses in both models. As a result of the large number of significantly differentiated genes modulated during the infection, only data related to genes that have some functional information are shown and discussed below.
Table 1.
Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways regulated in Burkholderia pseudomallei-infected streptozotocin-induced diabetic liver analysed by GeneTrail
| Categories | KEGG pathways | P-value |
|---|---|---|
| Up-regulated | Cytokine–cytokine receptor interaction | 1·86698e−05 |
| Jak-signal transducer and activator of transcription signalling pathway | 1·92797e−05 | |
| Adipocytokine signalling pathway | 0·0131694 | |
| Toll-like receptor signalling pathway | 3·63952e−09 | |
| Apoptosis | 1·37271e−05 | |
| Chemokine signalling pathway | 1·37271e−05 | |
| Antigen processing and presentation | 3·38894e−05 | |
| Mitogen-activated protein kinase signalling pathway | 0·000283262 | |
| Systemic lupus erythematosus | 8·11437e−05 | |
| Cell adhesion molecules | 0·000293927 | |
| Down-regulated | Drug metabolism – cytochrome P450 | 1·81645e−12 |
| Glycine, serine and threonine metabolism | 2·68347e−06 | |
| Tryptophan metabolism | 2·76687e−06 | |
| Methane metabolism | 2·22093e−05 | |
| Alanine, aspartate and glutamate metabolism | 2·42568e−05 | |
| ABC transporters | 0·00317722 | |
| Nitrogen metabolism | 0·0039838 | |
| PPAR signalling pathway | 0·0171655 | |
| Fatty acid metabolism | 0·00368175 | |
| Tricarboxylic acid cycle | 0·0933579 |
Figure 5.
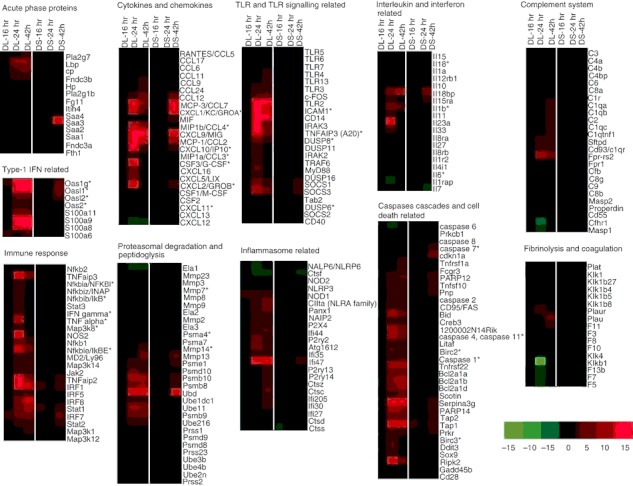
Transcriptional responses to acute Burkholderia pseudomallei infection in the mice with streptozotocin (STZ) -induced diabetes relative to uninfected diabetic mice. Hierarchical clustering of the expression profile of STZ-diabetic liver (DL) and STZ-diabetic spleen (DS) infected with B. pseudomallei at 16, 24 and 42 hr post-infection (p.i.) according to functional categories. The heat maps indicate the fold change in STZ-diabetic liver or STZ-diabetic spleen gene expression greater than (red) or less than (green) twofold at least once during the time course. Genes whose expression did not change are coloured in black. *Immune-related genes known to be associated with the general bacterial infection.
Table 2.
Kinetic profiles of host genes expression modulated by Burkholderia pseudomallei infection in streptozotocin (STZ) -induced diabetes and normoglycaemic models for both liver and spleen
| Fold change in transcript at hour post-infection1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normoglycaemic2 | STZ-diabetic | ||||||||||||
| Liver | Spleen | Liver | Spleen | ||||||||||
| Gene symbol | GenBank | 16hr | 24hr | 42hr | 16hr | 24hr | 42hr | 16hr | 24hr | 42hr | 16hr | 24hr | 42hr |
| Toll-like receptor (TLR) and TLR-related | |||||||||||||
| TLR2 | NM_011905 | 107 | 72 | 25 | 7 | 8 | 3 | – | 93 | 23 | – | – | – |
| TLR3 | NM_126166 | 3 | 2 | 3 | – | – | – | – | 2 | 2 | – | – | – |
| TLR4 | NM_021297 | – | – | 2 | – | – | 3 | – | – | 2 | – | – | – |
| TLR5 | NM_016928 | −5 | −2 | −5 | – | – | – | – | – | – | – | – | – |
| TLR6 | NM_011604 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| TLR7 | NM_133211 | – | – | – | 3 | 5 | 3 | – | – | – | – | – | – |
| TLR13 | NM_205820 | – | – | 4 | – | 2 | 4 | – | – | 3 | – | – | – |
| c-FOS | NM_010234 | 5 | 4 | 3 | 7 | 8 | 3 | – | 4 | 6 | – | – | – |
| ICAM1 | NM_010493 | 28 | 45 | 18 | 5 | 3 | 3 | – | 51 | 12 | – | – | – |
| TRAF6 | NM_009424 | 4 | 3 | – | – | – | – | – | 2 | – | – | – | – |
| IRAK2 | NM_172161 | 3 | – | – | – | – | – | – | 4 | – | – | – | – |
| IRAK3 | NM_028679 | 30 | 39 | 28 | 6 | 8 | – | – | 30 | 6 | – | – | – |
| DUSP11 | NM_028099 | 2 | 2 | – | – | – | – | – | 2 | – | – | – | – |
| DUSP16 | NM_130447 | 2 | 2 | 2 | 5 | 5 | 2 | – | 4 | – | – | – | – |
| DUSP6 | NM_026268 | −3 | – | – | – | – | – | – | – | – | – | – | – |
| DUSP8 | NM_008748 | 8 | 4 | 2 | – | – | – | – | 8 | 3 | – | – | – |
| CD14 | NM_009841 | 106 | 259 | 151 | 10 | 20 | 5 | – | 171 | 48 | – | – | – |
| MyD88 | NM_010851 | 8 | 4 | 3 | 4 | 3 | 3 | – | 5 | – | – | – | – |
| CD40 | NM_170704 | 3 | – | – | – | 2 | – | – | – | – | – | – | – |
| Tab2 | NM_138667 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| SOCS1 | NM_009896 | 12 | 9 | 6 | 10 | 8 | 5 | – | 8 | 4 | – | – | 3 |
| SOCS2 | NM_007706 | – | 0 | 5 | – | 4 | – | – | – | – | – | – | – |
| SOCS3 | NM_007707 | 11 | 5 | – | 13 | 11 | – | – | 11 | 6 | – | – | 3 |
| TNFAIP3 (A20) | NM_009397 | 92 | 63 | 31 | – | – | – | – | 61 | 8 | – | – | – |
| Cyokines and chemokines | |||||||||||||
| MCP-1/CCL2 | NM_011333 | 28 | 69 | 39 | 39 | 51 | 11 | – | 90 | 13 | – | – | 10 |
| MIP1a/CCL3 | NM_011337 | 3 | 5 | – | 39 | 48 | 12 | – | 4 | – | – | – | – |
| MIP1b/CCL4 | NM_013652 | 20 | 30 | 27 | 39 | 35 | 10 | – | 12 | 8 | – | – | – |
| RANTES/CCL5 | NM_013653 | – | 11 | 14 | – | 3 | – | – | – | – | – | – | – |
| MCP-3/CCL7 | NM_013654 | 17 | 11 | 12 | 88 | 107 | 40 | – | 15 | 7 | – | – | 19 |
| CCL9 | NM_011338 | – | 2 | 4 | – | – | 4 | – | – | 4 | – | – | – |
| CCL12 | NM_011331 | 3 | 4 | 13 | 18 | 25 | 27 | – | 3 | 6 | – | – | 6 |
| CCL6 | NM_009139 | – | – | 5 | – | – | – | – | – | – | – | – | – |
| CCL24 | NM_019577 | – | – | 4 | – | – | – | – | – | 4 | – | – | – |
| CCL11 | NM_011330 | 2 | 4 | – | 13 | 16 | 2 | – | – | – | – | – | – |
| CCL17 | NM_011332 | – | – | – | 3 | 6 | – | – | – | – | – | – | – |
| CXCL1/KC/GROA | NM_008176 | 27 | 18 | 16 | 103 | 113 | 26 | – | 13 | 5 | – | – | 27 |
| CXCL10/IP10 | NM_021274 | 11 | 16 | – | 17 | 10 | – | – | 17 | 4 | – | – | 3 |
| CXCL11 | NM_019494 | – | – | – | 3 | 3 | – | – | – | – | – | – | – |
| CXCL12 | NM_013655 | −3 | −3 | – | – | – | – | – | −4 | −2 | – | – | – |
| CXCL13 | NM_018866 | – | 2 | – | – | – | 4 | – | – | – | – | – | – |
| CXCL16 | NM_023158 | – | 7 | 8 | – | 4 | 4 | – | 6 | – | – | – | – |
| CXCL2/GROB | NM_009140 | 7 | 11 | – | 36 | 41 | 6 | – | 24 | – | – | – | 4 |
| CXCL5/LIX | NM_009141 | 3 | 11 | – | 20 | 20 | 9 | – | 8 | – | – | – | – |
| CXCL9/MIG | NM_008599 | 67 | 67 | 57 | 20 | 22 | – | – | 41 | 38 | – | – | 16 |
| MIF | XM_147409 | – | – | – | – | 3 | 3 | – | – | – | – | – | 3 |
| CSF1/M-CSF | NM_007778 | 2 | 3 | 5 | 2 | 2 | – | – | – | – | – | – | – |
| CSF2 | NM_009969 | – | 2 | – | 7 | 5 | 2 | – | – | – | – | – | – |
| CSF3/G-CSF | NM_009971 | 9 | 15 | – | 29 | 62 | – | – | 24 | – | – | – | – |
| Immune response | |||||||||||||
| MD2/Ly96 | NM_016923 | – | 2 | – | – | – | – | – | 3 | 3 | – | – | – |
| Nfkb1 | NM_008689 | 4 | 4 | 3 | – | – | – | – | 7 | 4 | – | – | – |
| Nfkb2 | NM_019408 | 3 | 3 | 3 | – | – | – | – | – | – | – | – | – |
| Nfkbia/NFKBI | NM_010907 | 6 | 5 | – | – | 6 | – | – | 4 | – | – | – | – |
| Nfkbib/IkB | NM_010908 | 3 | 3 | – | – | – | – | – | 3 | – | – | – | – |
| Nfkbie/IkBE | NM_008690 | 3 | 13 | 13 | – | – | – | – | 13 | 6 | – | – | – |
| Nfkbiz/INAP | NM_030612 | 24 | 5 | 7 | – | 8 | – | – | 4 | – | – | – | – |
| IFNg | NM_008337 | 6 | 4 | – | 148 | 112 | 10 | – | 4 | – | – | – | – |
| TNFa | NM_013693 | 3 | 6 | 2 | 9 | 5 | 3 | – | 11 | – | – | – | – |
| TNFaip2 | NM_009396 | 8 | 17 | 6 | – | – | – | – | 31 | 7 | – | – | 2 |
| TNFaip3 | NM_009397 | 92 | 63 | 31 | – | – | – | – | 61 | 8 | – | – | – |
| NOS2 | NM_010927 | 3 | 11 | 3 | – | 3 | – | – | 27 | – | – | – | – |
| IRF1 | NM_008390 | 14 | 19 | 9 | 4 | 4 | 4 | – | 27 | 12 | 3 | – | 4 |
| IRF5 | NM_012057 | – | 3 | 2 | – | – | – | – | 4 | 4 | – | – | – |
| IRF7 | NM_016850 | 5 | 6 | 5 | 7 | 10 | 7 | 3 | 5 | 8 | – | – | 6 |
| IRF8 | NM_008320 | 8 | 6 | 6 | – | – | – | – | 8 | 13 | – | – | – |
| Jak2 | NM_008413 | 2 | 2 | 3 | – | 2 | – | – | 4 | 3 | – | – | – |
| Stat1 | NM_009283 | 11 | 6 | 9 | 4 | 5 | 5 | – | 6 | 10 | 2 | – | 3 |
| Stat2 | NM_019963 | – | – | 2 | 4 | 5 | 4 | – | 3 | 3 | – | – | 2 |
| Stat3 | NM_011486 | 6 | 3 | 3 | 3 | 3 | 3 | – | 2 | – | – | – | – |
| Map3k1 | NM_011945 | 3 | 2 | 5 | – | – | – | – | – | 3 | – | – | – |
| Map3k12 | NM_009582 | – | – | – | −2 | – | – | – | – | – | – | – | – |
| Map3k14 | NM_016896 | 3 | 2 | – | – | – | – | – | 3 | 2 | – | – | – |
| Map3k8 | NM_007746 | 4 | 3 | 2 | – | 6 | – | – | 3 | – | – | – | – |
| Interleukin and interleukin-related | |||||||||||||
| Il10 | NM_010548 | – | – | – | 2 | 7 | 4 | – | – | – | – | – | – |
| Il11 | NM_008350 | – | 3 | – | 2 | – | – | – | 3 | – | – | – | – |
| Il12rb1 | NM_008353 | – | – | – | 3 | 4 | – | – | – | – | – | – | – |
| Il15 | NM_008357 | – | – | – | 5 | 4 | 2 | – | – | – | – | – | – |
| Il15ra | NM_133836 | – | – | – | 4 | 7 | 4 | – | 3 | – | – | – | 2 |
| Il18 | NM_008360 | – | −3 | – | – | – | – | – | – | – | – | – | – |
| Il18bp | NM_010531 | – | 14 | 21 | 6 | 12 | 14 | – | 7 | 11 | – | – | 8 |
| Il1a | NM_010554 | – | – | – | 8 | 9 | – | – | – | – | – | – | – |
| Il1b | NM_008361 | 7 | 8 | 7 | 10 | 9 | – | – | 8 | 5 | – | – | – |
| Il1r2 | NM_010555 | 2 | 3 | 3 | 10 | 17 | 10 | – | – | – | – | – | – |
| Il1rap | NM_008364 | – | −6 | −5 | – | – | – | – | −4 | – | – | – | – |
| Il23a | NM_031252 | – | 11 | – | 2 | – | – | – | 20 | – | – | – | – |
| Il27 | NM_145636 | – | – | – | 4 | 4 | – | – | – | – | – | – | – |
| Il33 | NM_133775 | 3 | 5 | – | 6 | 7 | 5 | – | 6 | – | – | – | – |
| Il4i1 | NM_010215 | – | 15 | 15 | – | – | – | – | – | – | – | – | – |
| Il6 | NM_031168 | – | – | – | 83 | 75 | 6 | – | – | – | – | – | – |
| Il7 | NM_008371 | 4 | – | – | −3 | −3 | – | – | – | – | – | – | −2 |
| Il8ra | NM_178241 | – | – | – | 2 | 2 | 5 | – | – | – | – | – | – |
| Il8rb | NM_009909 | – | – | – | 3 | 5 | 5 | – | – | – | – | – | – |
| Caspases cascades and cell death | |||||||||||||
| caspase 1 | NM_009807 | 2 | 3 | 5 | – | – | – | – | 3 | 5 | – | – | – |
| caspase 2 | NM_007610 | 2 | – | 3 | – | – | – | – | 3 | 2 | – | – | – |
| caspase 4, caspase 11 | NM_007609 | 7 | 9 | 12 | 5 | 11 | 4 | – | 9 | 6 | – | – | – |
| caspase 6 | NM_009811 | – | −4 | – | −2 | – | – | – | −3 | – | – | – | – |
| caspase 7 | NM_007611 | – | 2 | – | – | 2 | 2 | – | – | – | – | – | 2 |
| caspase 8 | NM_009812 | – | – | – | – | 2 | – | – | – | – | – | – | – |
| PARP12 | NM_172893 | 6 | 5 | 4 | 8 | 8 | 4 | – | 5 | 5 | – | – | 4 |
| PARP14 | NM_145481 | 13 | 10 | 8 | 6 | 6 | 4 | – | 9 | 8 | – | – | 3 |
| Creb3 | NM_013497 | 3 | 3 | – | – | – | – | – | 4 | 2 | – | – | – |
| Prkcb1 | NM_008855 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| Bid | NM_007544 | 4 | 8 | 4 | – | – | – | – | 10 | 5 | – | – | – |
| Tnfrsf1a | NM_011609 | – | – | – | – | 2 | 3 | – | – | 3 | – | – | – |
| CD95/FAS | NM_007987 | – | 4 | 4 | 4 | 5 | – | – | 3 | 3 | – | – | – |
| Tnfrsf22 | NM_023680 | 3 | 6 | 12 | – | – | – | – | 10 | 10 | – | – | – |
| Tnfsf10 | NM_009425 | 2 | 4 | 3 | 4 | 4 | 2 | – | 3 | 4 | – | – | 3 |
| 1200002N14Rik | NM_027878 | 10 | 22 | 23 | 4 | 5 | 5 | – | 9 | 6 | – | – | – |
| Bcl2a1a | NM_009742 | 8 | 6 | 8 | – | – | – | – | 5 | 7 | – | – | – |
| Bcl2a1b | NM_007534 | 11 | 9 | 12 | – | – | – | – | 8 | 10 | – | – | – |
| Bcl2a1d | NM_007536 | 12 | 10 | 13 | – | – | – | – | 7 | 9 | – | – | – |
| Birc2 | NM_007465 | 5 | 5 | 6 | – | – | – | – | 5 | 2 | – | – | – |
| Birc3 | NM_007464 | 5 | 4 | – | – | – | – | – | 5 | – | – | – | – |
| Cd28 | NM_007642 | – | – | 6 | – | – | – | – | – | – | – | – | – |
| Cdkn1a | NM_007669 | 29 | 12 | 12 | 11 | 13 | 5 | – | – | – | – | – | 7 |
| Ddit3 | NM_007837 | – | 5 | 6 | – | – | – | – | 5 | – | – | – | – |
| Fcgr3 | NM_010188 | – | 2 | 5 | – | – | – | – | – | 3 | – | – | – |
| Gadd45b | NM_008655 | 4 | 4 | 2 | 9 | 9 | 4 | – | – | – | – | – | – |
| Litaf | NM_019980 | 6 | 3 | 3 | 3 | 4 | 3 | – | 5 | 3 | – | – | – |
| Pnp | NM_013632 | 4 | 3 | 3 | 5 | 5 | 3 | – | 3 | 2 | – | – | 3 |
| Prkr | NM_011163 | 8 | 4 | 3 | 7 | 6 | 4 | – | 5 | 3 | – | – | 2 |
| Ripk2 | NM_138952 | 31 | 17 | 6 | 3 | – | – | – | 38 | 5 | – | – | – |
| Scotin | NM_025858 | 3 | 5 | 5 | – | – | – | – | 4 | 5 | – | – | – |
| Serpina3g | XM_354694 | 39 | 30 | 28 | 7 | 8 | 8 | – | 24 | 30 | – | – | 3 |
| Sox9 | NM_011448 | 6 | 4 | 19 | – | – | – | – | 3 | – | – | – | – |
| Tap1 | NM_013683 | 7 | 7 | 6 | 3 | 3 | – | – | 14 | 14 | 2 | – | 3 |
| Tap2 | NM_011530 | 6 | 7 | 8 | – | – | – | – | 6 | 6 | – | – | 2 |
| Inflammasome-related | |||||||||||||
| NAIP2 | NM_010872 | – | 2 | 4 | – | – | – | – | 2 | 2 | – | – | – |
| NLRP3 | NM_145827 | – | – | – | 2 | – | – | – | – | – | – | – | – |
| CIIta (NLRA family) | NM_007575 | – | 4 | 4 | – | – | – | – | 4 | 7 | – | – | – |
| NALP6/NLRP6 | NM_133946 | – | −4 | −4 | – | – | – | – | −2 | – | – | – | – |
| NOD1 | NM_172729 | 3 | 2 | 3 | – | 3 | 4 | 2 | 3 | 3 | – | – | – |
| NOD2 | NM_145857 | – | – | – | 3 | 3 | 2 | – | – | – | – | – | – |
| P2X4 | NM_011026 | – | 2 | 2 | – | – | – | – | 3 | 3 | – | – | – |
| P2ry13 | NM_028808 | 2 | 2 | 3 | – | – | – | – | – | 3 | – | – | – |
| P2ry14 | NM_133200 | 2 | 3 | 4 | – | – | – | – | – | 4 | – | – | – |
| P2ry2 | NM_008773 | 3 | 3 | – | 4 | 5 | 3 | – | 6 | 2 | – | – | – |
| Panx1 | NM_019482 | – | 6 | 6 | – | – | – | – | 4 | 6 | – | – | – |
| Ctsz | NM_022325 | – | – | – | – | – | 3 | – | – | 3 | – | – | – |
| Ctss | NM_021281 | – | – | 4 | – | – | – | – | – | – | −2 | – | – |
| Ctsc | NM_009982 | – | – | 5 | – | – | 3 | – | – | 5 | – | – | – |
| Ctsf | NM_019861 | – | −3 | −3 | – | – | – | – | −2 | −2 | – | – | −2 |
| Ctsd | NM_009983 | – | – | – | – | – | 3 | – | – | – | – | – | – |
| Atg16l2 | XM_133655 | 6 | 6 | 5 | – | – | – | – | 7 | 3 | – | – | – |
| Ifi205 | NM_172648 | 4 | 2 | 3 | 3 | 3 | – | – | – | 3 | – | – | – |
| Ifi27 | NM_029803 | – | 2 | 5 | – | 2 | 3 | – | – | – | – | – | – |
| Ifi30 | NM_023065 | – | – | 4 | – | – | – | – | – | 4 | – | – | – |
| Ifi35 | NM_027320 | 3 | 3 | – | – | 4 | 2 | – | 3 | 3 | – | – | – |
| Ifi44 | NM_133871 | 5 | 5 | 4 | 3 | – | 2 | – | 4 | 5 | – | – | – |
| Ifi47 | NM_008330 | 28 | 18 | 15 | – | – | – | – | 31 | 20 | – | – | 3 |
| Type 1 interferon (IFN) related | |||||||||||||
| Oas1g | NM_011852 | 8 | 22 | 10 | – | 9 | 6 | 2 | 16 | 17 | – | – | 6 |
| Oas2 | NM_145227 | 2 | 3 | 2 | 9 | 10 | 6 | – | 2 | 2 | – | – | – |
| Oasl1 | NM_145209 | 13 | 22 | 12 | 19 | 18 | 6 | – | 15 | 15 | – | – | 7 |
| Oasl2 | NM_011854 | 3 | 4 | 4 | 6 | 7 | 5 | – | 5 | 6 | – | – | 5 |
| S100a9 | NM_009114 | 27 | 31 | 55 | – | – | – | – | 31 | 24 | – | – | – |
| S100a8 | NM_013650 | 26 | 37 | 60 | – | – | – | – | 33 | 27 | – | – | – |
| S100a6 | NM_011313 | – | – | 6 | – | – | – | 2 | – | 4 | – | – | – |
| S100a11 | NM_016740 | – | 5 | 5 | – | – | 2 | – | 5 | 5 | – | – | – |
| Acute-phase proteins | |||||||||||||
| Fgl1 | NM_145594 | 3 | 3 | 3 | – | – | – | – | – | – | – | – | – |
| Itih4 | NM_018746 | – | 2 | – | – | – | – | – | – | – | – | – | – |
| Cp | NM_007752 | 5 | 4 | 6 | 2 | 3 | 4 | – | 5 | 6 | – | – | – |
| Hp | NM_017370 | – | 5 | 5 | – | 7 | 5 | – | – | – | – | – | – |
| Pla2g7 | NM_013737 | – | 2 | 3 | – | – | – | – | – | 2 | – | – | – |
| Pla2g1b | NM_011107 | – | – | – | 25 | 29 | – | – | – | – | – | – | – |
| Lbp | NM_008489 | 7 | 6 | 8 | – | – | – | – | 8 | 8 | – | – | – |
| Saa1 | NM_009117 | 13 | 13 | 13 | – | – | – | – | – | – | – | – | – |
| Saa2 | NM_011314 | 92 | 86 | 74 | – | – | – | – | – | – | – | – | – |
| Saa3 | NM_011315 | 124 | 121 | 112 | 36 | 91 | 94 | – | – | – | – | – | 56 |
| Saa4 | NM_011316 | 3 | 2 | – | – | – | – | – | – | – | – | – | – |
| Fndc3b | NM_173182 | 6 | 3 | 5 | – | 3 | – | – | 2 | 3 | – | – | – |
| Fndc3a | NM_207636 | – | – | – | 3 | 3 | – | – | – | – | – | – | – |
| Fth1 | NM_010239 | – | – | – | – | 2 | 2 | – | – | – | – | – | – |
| Proteasomal degradation and peptidoglysis | |||||||||||||
| Mmp13 | NM_008607 | 3 | – | 6 | 49 | 26 | 10 | – | – | 6 | – | – | 6 |
| Mmp14 | NM_008608 | – | – | 6 | 2 | 4 | 3 | – | – | 3 | – | – | – |
| Mmp2 | NM_008610 | −2 | – | – | −4 | −3 | – | – | – | – | – | – | – |
| Mmp23 | NM_011985 | – | – | – | −3 | −3 | – | – | – | – | – | – | – |
| Mmp3 | NM_010809 | – | – | – | 8 | 41 | 35 | – | – | – | – | – | – |
| Mmp7 | NM_010810 | – | 2 | 3 | – | – | – | – | – | – | – | – | – |
| Mmp8 | NM_008611 | – | – | 3 | 3 | 10 | 13 | – | – | – | – | – | – |
| Mmp9 | NM_013599 | – | 2 | 4 | – | 3 | 5 | – | – | – | – | – | – |
| Ela2 | NM_007919 | – | – | – | 20 | 21 | – | – | – | – | – | – | – |
| Ela1 | NM_033612 | −7 | −12 | – | 14 | 14 | – | – | −2 | −2 | – | – | – |
| Ela3 | NM_026419 | – | – | – | 34 | 57 | – | – | – | – | – | – | – |
| Ubd | NM_023137 | 137 | 192 | 203 | 28 | 33 | 30 | – | 262 | 218 | – | – | 14 |
| Psma4 | NM_011966 | – | – | – | – | 2 | 2 | – | – | – | – | – | – |
| Psma7 | NM_011969 | – | – | – | – | 3 | 2 | – | – | – | – | – | – |
| Psmb10 | NM_013640 | 7 | 10 | 7 | 3 | 4 | 2 | – | 11 | 9 | – | – | 5 |
| Psmb8 | NM_010724 | 4 | 9 | 8 | – | – | – | – | 7 | 8 | – | – | 2 |
| Psmb9 | NM_013585 | 7 | 7 | 8 | – | – | – | – | 6 | 7 | – | – | – |
| Psmd10 | NM_016883 | 4 | 5 | 5 | – | – | – | – | 5 | 3 | – | – | – |
| Psmd8 | NM_026545 | – | 2 | 2 | – | – | – | – | – | – | – | – | – |
| Psmd9 | NM_026000 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Psme1 | NM_011189 | – | 4 | 3 | – | 3 | 3 | – | 3 | 4 | – | – | 3 |
| Prss1 | NM_053243 | – | – | – | 45 | 67 | – | – | – | – | – | – | – |
| Prss2 | NM_009430 | – | – | – | 21 | 28 | – | – | – | – | – | – | – |
| Prss23 | NM_029614 | – | – | – | – | 3 | 2 | – | – | – | – | – | – |
| Ube1dc1 | NM_025692 | 2 | 2 | – | – | – | – | – | 3 | 3 | – | – | – |
| Ube1l | NM_023738 | 6 | 11 | 8 | 3 | 3 | 2 | – | 9 | 8 | – | – | – |
| Ube2l6 | NM_019949 | – | 3 | 3 | – | – | – | – | 3 | 3 | – | – | – |
| Ube2n | NM_080560 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Ube3b | NM_054093 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Ube4b | NM_022022 | 2 | – | – | – | – | – | – | – | – | – | – | – |
| Complement system | |||||||||||||
| C1r | NM_023143 | – | – | – | – | 2 | – | – | – | – | – | – | – |
| C1qa | NM_007572 | – | – | – | – | – | 3 | – | – | 3 | – | – | – |
| C1qb | NM_009777 | – | – | – | – | – | 3 | – | – | 4 | – | – | – |
| C1qc | NM_007574 | – | – | – | – | – | 3 | – | – | 4 | – | – | – |
| C1qtnf1 | NM_019959 | – | – | – | – | – | 2 | – | – | 2 | – | – | – |
| C2 | NM_013484 | – | 2 | 2 | – | 4 | 5 | – | – | 2 | – | – | – |
| C3 | NM_009778 | – | – | – | – | 2 | 3 | – | – | – | – | – | – |
| C4a | NM_011413 | – | – | – | – | – | 3 | – | – | – | – | – | – |
| C4b | NM_009780 | – | – | – | – | – | 3 | – | – | – | – | – | – |
| C4bp | NM_007576 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| C6 | NM_016704 | – | – | −4 | – | – | – | – | – | – | – | – | – |
| C8a | NM_146148 | – | −3 | – | – | – | – | – | – | – | – | – | – |
| C8b | NM_133882 | – | −3 | −6 | – | – | – | – | – | – | – | – | – |
| C9 | NM_013485 | – | −3 | −4 | – | – | – | – | – | – | – | – | – |
| C8g | XM_130127 | – | −2 | −4 | – | – | – | – | – | – | – | – | – |
| Cfb | NM_008198 | – | – | – | 6 | 9 | 9 | – | – | – | – | – | – |
| Cfhr1 | NM_015780 | – | −3 | −2 | – | – | – | – | −6 | – | – | – | – |
| Masp1 | NM_008555 | – | −4 | −3 | – | – | – | – | −3 | – | – | – | – |
| Masp2 | XM_358353 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Properdin | XM_135820 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| Cd55 | NM_010016 | – | – | 3 | – | – | – | – | – | – | – | – | – |
| Cd93/c1qr | NM_010740 | – | 2 | 4 | – | 4 | 3 | – | 3 | 3 | – | – | – |
| Sftpd | NM_009160 | 2 | 3 | 9 | – | – | 2 | – | 3 | 4 | – | – | – |
| Fpr-rs2 | NM_008039 | 13 | 12 | 17 | 7 | 10 | 10 | – | 10 | 7 | – | – | – |
| Fpr1 | NM_013521 | – | – | 2 | – | 6 | 5 | – | – | – | – | – | – |
| Fibrinolysis and coagulation | |||||||||||||
| Plau | NM_008873 | – | – | – | – | – | 5 | – | – | 6 | – | – | – |
| Plaur | NM_011113 | 6 | 3 | – | 12 | 9 | 4 | – | 4 | – | – | – | – |
| Plat | NM_008872 | – | – | – | 3 | 9 | 3 | – | – | – | – | – | – |
| Klkb1 | NM_008455 | −3 | −28 | −5 | – | – | – | – | −15 | – | – | – | – |
| Klk1 | NM_010639 | – | – | – | – | 27 | – | – | – | – | – | – | – |
| Klk1b27 | NM_020268 | – | – | – | 9 | 22 | – | – | – | – | – | – | – |
| Klk1b4 | NM_010915 | – | – | −3 | 10 | 21 | – | – | – | – | – | – | – |
| Klk1b5 | NM_008456 | – | – | – | 14 | 26 | – | – | – | – | – | – | – |
| Klk1b8 | NM_008457 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| Klk4 | NM_019928 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| F10 | NM_007972 | – | −2 | – | 3 | 6 | 4 | – | – | – | – | – | – |
| F11 | NM_028066 | – | – | 2 | – | – | – | – | – | 2 | – | – | – |
| F13b | NM_031164 | – | −7 | −4 | – | – | – | – | −3 | – | – | – | – |
| F3 | NM_010171 | – | −2 | – | 6 | 5 | – | – | – | – | – | – | – |
| F5 | NM_007976 | – | −3 | – | – | – | – | – | −3 | – | – | – | – |
| F7 | NM_010172 | – | −2 | – | – | – | – | – | −3 | – | – | – | – |
| F8 | NM_007977 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Oxidative and anti-oxidative | |||||||||||||
| Gpx2 | NM_030677 | – | – | 6 | – | – | – | – | – | 3 | – | – | – |
| Gpx3 | NM_008161 | – | 2 | 4 | – | – | 5 | – | 3 | – | – | – | – |
| Gpx7 | NM_024198 | – | – | 2 | – | – | – | – | – | 2 | – | – | – |
| Gsta4 | NM_010357 | – | −3 | −2 | – | – | – | – | – | – | – | – | – |
| Gstm1 | NM_010358 | – | −3 | – | – | – | – | – | −2 | – | – | – | – |
| Gstm2 | NM_008183 | – | – | – | – | – | 2 | – | – | – | – | – | – |
| Gstm3 | NM_010359 | – | −2 | – | – | – | – | – | −2 | – | – | – | – |
| Gstm4 | NM_026764 | −3 | −5 | −3 | – | – | – | – | −4 | – | – | – | – |
| Gstm6 | NM_008184 | – | −4 | −2 | – | – | – | – | −3 | – | – | – | – |
| Gstm7 | XM_359308 | – | −5 | – | – | – | – | – | −3 | – | – | – | – |
| Gsto1 | NM_010362 | – | −3 | – | – | – | – | – | −2 | – | – | – | – |
| Gstt1 | NM_008185 | – | −5 | −6 | – | – | – | – | – | −3 | – | – | – |
| Gstt2 | NM_010361 | −2 | −3 | – | – | – | – | – | – | – | – | – | – |
| Gstt3 | NM_133994 | – | −6 | −2 | – | – | – | – | – | – | – | – | – |
| Gstz1 | NM_010363 | – | −3 | – | – | – | – | – | −2 | – | – | – | – |
| Mgst3 | NM_025569 | – | −3 | −3 | – | −11 | – | – | – | – | – | – | – |
| Gsr | NM_010344 | – | – | – | – | – | 2 | – | – | – | – | – | – |
| Sod2 | NM_013671 | – | 2 | – | 5 | 6 | 4 | – | 2 | – | – | – | – |
| Tgm2 | NM_009373 | 6 | 7 | 7 | 3 | 5 | 5 | – | 9 | 9 | – | – | 5 |
| Noxo1 | NM_027988 | – | 13 | 6 | – | – | – | – | 6 | #N/A | – | – | – |
| Vnn3 | NM_011979 | 13 | 25 | 10 | 2 | – | – | – | 27 | 11 | – | – | – |
| Glycolysis/gluconeogenesis | |||||||||||||
| Hk2 | NM_013820 | 7 | 4 | 6 | 5 | 6 | 4 | – | 6 | 4 | – | – | – |
| Hk3 | NM_001033245 | 3 | 3 | 4 | – | 4 | 6 | – | 4 | 7 | – | – | – |
| Khk | NM_008439 | −2 | – | −9 | – | −2 | – | – | – | – | – | – | – |
| Pfkl | NM_008826 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| Pfkp | NM_019703 | – | – | – | – | 3 | 3 | – | – | 3 | – | – | – |
| Pgk1 | NM_008828 | – | – | – | – | 4 | 3 | – | – | – | – | – | – |
| Pfkm | NM_021514 | −4 | −4 | −3 | – | – | – | – | −3 | – | – | – | – |
| Pgam1 | NM_023418 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Eno1 | NM_023119 | – | −2 | – | – | – | – | 2 | – | – | – | – | – |
| Eno2 | NM_013509 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| Aldoa | NM_007438 | −4 | −3 | −3 | – | – | 3 | – | – | – | – | – | – |
| Aldoc | NM_009657 | −2 | −4 | −5 | – | – | – | – | – | – | – | – | – |
| Gapdh | NM_008084 | – | – | – | – | 2 | – | – | – | – | – | – | 2 |
| Pkm2 | NM_011099 | – | 5 | 7 | – | 2 | 3 | – | 6 | 11 | – | – | – |
| Ldha | NM_010699 | – | – | – | – | 2 | – | – | – | – | – | – | 2 |
| Pdk4 | NM_013743 | – | – | – | – | 3 | – | – | – | – | – | – | – |
| Pdhb | NM_024221 | – | −2 | −3 | – | – | – | – | – | – | – | – | – |
| Pdk1 | NM_172665 | −3 | −4 | −3 | – | – | – | – | −4 | – | – | – | – |
| Pdk3 | NM_145630 | – | – | 2 | – | – | – | – | 2 | 2 | – | – | – |
| Pdk2 | NM_133667 | −2 | −3 | – | – | – | – | – | −3 | – | – | – | – |
| Glycogen breakdown | |||||||||||||
| Pygl | NM_133198 | – | −10 | −4 | – | – | 3 | – | – | – | – | – | – |
| Pgm2 | NM_028132 | – | −2 | – | – | 4 | – | – | – | – | – | – | – |
| Glycogen synthesis | |||||||||||||
| Ugp2 | NM_139297 | −4 | −7 | −3 | – | – | – | – | −6 | – | – | – | – |
| Ugt2b1 | NM_152811 | – | −9 | −41 | – | – | – | – | −5 | – | – | – | – |
| Ugt2b34 | NM_153598 | – | −6 | −2 | – | – | – | – | −6 | – | – | – | – |
| Ugt3a2 | NM_144845 | – | −6 | −10 | – | – | – | – | −3 | – | – | – | – |
| Ugt1a7c | NM_201410 | – | – | – | −2 | – | – | – | −4 | – | – | – | – |
| Tricarboxylic acid cycle | |||||||||||||
| Aco1 | NM_007386 | – | −3 | – | – | – | – | – | −3 | – | – | – | – |
| Idh1 | NM_010497 | – | – | −3 | – | – | – | – | −3 | – | – | – | – |
| Idh3b | NM_130884 | – | −2 | −2 | – | – | – | – | – | – | – | – | – |
| Idh3a | NM_029573 | −2 | – | – | – | – | – | – | – | – | – | – | – |
| Sdhb | NM_023374 | – | −3 | −4 | – | – | – | – | – | −2 | – | – | – |
| Sdhc | NM_025321 | – | −2 | – | – | – | – | – | −2 | – | – | – | – |
| Sdhd | NM_025848 | – | −3 | – | – | – | – | – | −3 | – | – | – | – |
| Mdh1 | NM_008618 | – | −4 | −3 | – | – | – | – | −3 | – | – | – | – |
| Fatty acid metabolism | |||||||||||||
| Ehhadh | NM_023737 | – | −6 | – | – | – | – | – | – | – | – | – | – |
| Hadh | NM_008212 | – | −3 | −2 | – | – | – | – | −2 | – | – | – | – |
| Acat1 | NM_144784 | – | −6 | −7 | – | – | – | – | −3 | −4 | – | – | – |
| Acat2 | NM_009338 | – | −2 | −3 | – | – | – | – | – | – | – | – | – |
| Acat3 | NM_153151 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| Valine, leucine and Isoleucine degradation | |||||||||||||
| Hmgcs1 | NM_145942 | – | – | −2 | – | – | – | – | – | – | – | – | – |
| Hmgcs2 | NM_008256 | – | −7 | – | −3 | – | – | – | – | – | – | – | – |
| Aldh1a1 | NM_013467 | – | −7 | −2 | – | – | – | – | −4 | – | – | – | – |
| Aldh1a7 | NM_011921 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Aldh1b1 | NM_028270 | −3 | −9 | – | – | – | – | – | −5 | – | – | – | – |
| Aldh1l1 | NM_027406 | – | −3 | −4 | – | – | – | – | – | – | – | – | – |
| Aldh3b1 | NM_026316 | – | 3 | 5 | – | – | – | – | – | 3 | – | – | – |
| Aldh4a1 | NM_175438 | −2 | −14 | −6 | – | – | – | – | −5 | – | – | – | – |
| Aldh5a1 | NM_172532 | – | −2 | – | – | – | – | – | – | – | – | – | – |
| Aldh6a1 | NM_134042 | – | −3 | – | – | – | – | – | −2 | – | – | – | – |
| Aldh8a1 | NM_178713 | −5 | −8 | – | – | – | – | – | −3 | – | – | – | – |
| Cytochrome b | |||||||||||||
| Cyp17a1 | NM_007809 | – | −3 | – | – | – | – | – | – | – | – | – | – |
| Cyp1b1 | NM_009994 | – | – | – | – | 5 | 5 | – | 3 | – | – | – | – |
| Cyp1a2 | NM_009993 | −3 | −11 | −88 | – | – | – | – | −7 | −14 | – | – | – |
| Cyp27a1 | NM_024264 | – | −2 | −5 | −2 | −2 | – | – | – | – | – | – | – |
| Cyp2a12 | NM_133657 | – | −3 | – | – | – | – | – | – | – | – | – | – |
| Cyp2a4 | NM_009997 | −4 | −5 | −3 | – | – | – | – | −6 | – | – | – | – |
| Cyp2a5 | NM_007812 | −8 | – | −10 | – | – | – | – | −8 | −4 | – | – | – |
| Cyp2b10 | NM_009999 | – | – | 2 | – | – | – | – | – | – | – | – | – |
| Cyp2c29 | NM_007815 | – | −6 | −49 | – | – | – | – | −3 | – | – | – | – |
| Cyp2c37 | NM_010001 | – | −8 | −112 | – | – | – | – | −4 | – | – | – | – |
| Cyp2c39 | NM_010003 | – | −9 | – | – | – | – | – | – | – | – | – | – |
| Cyp2c50 | NM_134144 | – | −11 | −22 | – | – | – | – | −3 | −3 | – | – | – |
| Cyp2c54 | NM_206537 | – | −4 | −5 | – | – | – | – | −5 | −10 | – | – | – |
| Cyp2c55 | NM_028089 | – | – | −3 | – | – | – | – | – | – | – | – | – |
| Cyp2d10 | NM_010005 | – | −3 | −3 | – | – | – | – | – | – | – | – | – |
| Cyp2d13 | NM_133695 | −3 | −5 | −6 | – | – | – | – | −3 | – | – | – | – |
| Cyp2d22 | NM_019823 | – | −4 | −3 | – | – | 2 | – | −2 | −3 | – | – | – |
| Cyp2d26 | NM_029562 | – | −4 | −2 | – | – | – | – | −3 | – | – | – | – |
| Cyp2d9 | NM_010006 | – | −3 | −3 | – | – | – | – | – | – | – | – | – |
| Cyp2e1 | NM_021282 | – | −3 | −12 | – | – | – | – | −2 | – | – | – | – |
| Cyp2f2 | NM_007817 | – | −6 | −11 | – | – | – | – | – | – | – | – | – |
| Cyp2g1 | NM_013809 | −3 | −3 | −3 | – | – | – | – | −3 | – | – | – | – |
| Cyp2j9 | NM_028979 | – | −3 | – | – | – | – | – | −3 | – | – | – | – |
| Cyp3a11 | NM_007818 | – | −6 | −5 | – | – | – | – | −3 | – | – | – | – |
| Cyp3a25 | NM_019792 | – | −7 | −3 | – | – | – | – | −3 | – | – | – | – |
| Cyp4a10 | NM_010011 | – | −7 | – | – | – | – | – | – | – | – | – | – |
| Cyp4a12 | NM_177406 | – | −2 | −2 | – | – | – | – | – | – | – | – | – |
| Cyp4a12b | NM_172306 | – | −4 | −4 | – | – | – | – | – | – | – | – | – |
| Cyp4b1 | NM_007823 | −2 | – | – | – | −3 | – | – | – | – | – | – | – |
| Cyp4f14 | NM_022434 | −2 | −12 | −50 | – | – | – | – | −4 | – | – | – | – |
| Cyp4f15 | NM_134127 | −5 | −14 | −4 | – | – | – | – | −12 | −4 | – | – | – |
| Cyp4v3 | NM_133969 | −3 | −6 | −3 | – | – | 2 | – | −3 | −3 | – | – | – |
| Cyp51 | NM_020010 | – | – | −3 | – | – | – | – | – | – | – | – | – |
| Cyp7a1 | NM_007824 | −176 | −228 | −211 | – | – | – | – | −98 | −39 | – | – | – |
| Cyp7b1 | NM_007825 | – | – | – | – | 3 | 4 | – | – | – | – | – | – |
| Cyp8b1 | NM_010012 | – | −154 | −137 | – | – | – | – | −68 | – | – | – | – |
| Miscellaneous | |||||||||||||
| Faah | NM_010173 | −3 | −36 | −5 | – | – | – | – | −10 | – | – | – | – |
| Car1 | NM_009799 | −2 | −4 | −5 | – | – | −6 | −2 | −6 | −7 | – | – | – |
| Car13 | NM_024495 | 4 | 7 | 8 | 5 | 8 | 4 | – | 4 | 3 | – | – | – |
| Car14 | NM_011797 | −4 | −13 | −10 | – | – | – | – | −3 | −4 | – | – | – |
| Car2 | NM_009801 | – | – | – | – | – | −11 | – | – | – | – | – | – |
| Car3 | NM_007606 | – | −6 | −263 | – | – | – | – | −4 | −34 | – | – | – |
| Car4 | NM_007607 | 2 | 12 | – | – | 5 | 2 | – | – | – | – | – | – |
| Car5a | NM_007608 | −7 | −26 | −11 | – | – | – | – | −19 | – | – | – | – |
| Akr1b3 | NM_009658 | – | – | 3 | – | – | – | – | – | – | – | – | – |
| Akr1b7 | NM_009731 | – | – | 4 | – | – | – | – | – | – | – | – | – |
| Akr1c14 | NM_134072 | −8 | −23 | −12 | – | – | – | – | −26 | – | – | – | – |
| Akr1c19 | NM_001013785 | – | −5 | −8 | – | – | – | – | −7 | −6 | – | – | – |
| Akr1c6 | NM_030611 | – | −17 | −24 | – | – | – | – | −5 | – | – | – | – |
| Akr1e1 | NM_018859 | – | −3 | – | – | – | – | – | −2 | – | – | – | – |
| Akr7a5 | NM_025337 | – | −4 | −2 | – | – | – | – | – | – | – | – | – |
| Ddc | NM_016672 | −14 | −31 | −4 | – | – | – | – | −13 | – | – | – | – |
| Apoa2 | NM_013474 | −3 | −6 | – | – | – | – | – | – | – | – | – | – |
| Apoa5 | NM_080434 | – | −24 | – | – | – | – | – | −11 | – | – | – | – |
| Aqp1 | NM_007472 | −3 | – | – | – | −4 | −12 | – | – | – | – | – | – |
| Aqp11 | NM_175105 | −5 | −5 | −2 | – | – | – | – | −3 | −2 | – | – | – |
| Aqp4 | NM_009700 | – | −3 | −2 | – | – | – | – | −2 | – | – | – | – |
| Aqp8 | NM_007474 | −6 | – | −6 | – | – | – | – | – | – | – | – | – |
| Aqp9 | NM_022026 | −5 | −24 | −4 | – | – | −4 | – | −8 | – | – | – | – |
| Arg1 | NM_007482 | – | −5 | – | – | 3 | 5 | – | −4 | – | – | – | – |
| Arg2 | NM_009705 | – | 19 | 11 | 4 | 6 | 3 | – | 12 | – | – | – | – |
Modulated transcripts are classified according to functional groups. Only genes with a more than twofold change in B. pseudomallei-infected liver or spleen versus uninfected control tissue are shown; ‘–’, no significant change in gene expression.
Data adopted from our recent work on genome-wide expression profiling of a murine acute melioidosis model.18
Delayed activation of host defence responses to B. pseudomallei infection in STZ-diabetic mice correlates with the delayed Toll-like receptor2 signature
Our recent genome-wide expression study of B. pseudomallei infected normoglycaemic mice revealed that the Toll-like receptor (TLR2) -mediated signalling pathway is responsible for recognition and initiation of the inflammatory response, leading to the elevation of a broad range of innate immune mechanisms, including the ‘core host immune response’ genes commonly seen in general inflammatory infections.18 To unravel the susceptibility of DM patients to melioidosis, we compared the STZ-diabetic host transcriptional response, particularly the innate immune response, to the normoglycaemic host transcriptional response.18Figure 6 shows the fold change levels of TLR2 and several major transcription factors [interferon-γ (IFN-γ), tumour necrosis factor (TNF), nuclear factor-κB1 (NF-κB1), interferon regulatory factor 1 (IRF-1), IRF-7, signal transducer and activator of transcription 1 (STAT1) and STAT2] that are responsible for regulating various defence mechanisms in both STZ-diabetic and normoglycaemic infected mice relative to the uninfected mice, respectively. In response to acute B. pseudomallei infection, these genes were induced in normoglycaemic mice as early as 16 hr p.i. but were only induced 24 hr p.i. in the STZ-diabetic host. Concomitantly, induction of ‘common host immune response’ genes representing a general ‘alarm signal’ for inflammatory infections by several different human pathogens was delayed (after 24 hr) in the STZ-diabetic liver (Fig. 7) compared with normoglycaemic mice. This cluster of genes includes the pro-inflammatory mediators [TNF, IL1b, colony stimulating factor 1 (CSF1) and CSF3], the chemokines (CCL3, CCL4, CXCL1, CXCL2, CXCL3), IFN-stimulated genes (ISGs) (OAS) and the IFN-inducible chemokine genes (CCL9, CXCL10, CXCL11) (Fig. 5 and Table 2). However, many of these defence genes [intercellular adhesion molecule 1 (ICAM1), TNF-α-induced protein 3 (TNFAIP3), macrophage inflammatory protein 1a (MIP1a), MIP1b, dual specificity phosphatase (DUSP) 8, CXCL1, CXCL2, CXCL10, CSF3, NF-κB family members (Nfkbia, Nfkbib, Nfkbie), IFNg, TNF, IL1b and mitogen-activated protein kinase kinase kinase 8 (MAP3K8)] were suppressed at 42 hr p.i. in the infected STZ-diabetic liver (Fig. 5 and Table 2). On the other hand, in the STZ-diabetic spleen, a small number of immune response genes (CXCL1, CXCL2, CXCL10, CASPASE7, OAS1g, OASL1 and OASL2) were mildly elevated at 42 hr p.i. (Fig. 5 and Table 2), including the ‘common host immune response’ genes (Fig. 8). The relative expression of selected differentially regulated host-cell genes was analysed by qRT-PCR on the same samples as those analysed by microarray analysis. The samples from both STZ-diabetic and normoglycaemic mice were verified by qRT-PCR as up-regulated or down-regulated, albeit with magnitudes different from those recorded by the microarray analysis (see Supplementary material, Data S2).
Figure 6.
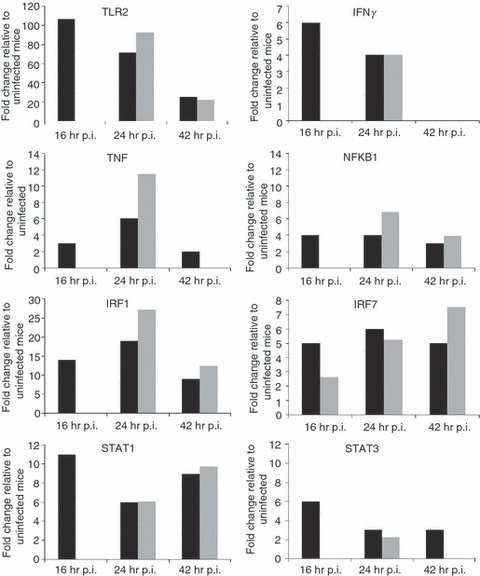
Expression profiles of Toll-like receptors and transcription factors. Toll-like receptor 2 and various transcription factors expression profiles (fold change relative to uninfected mice) during acute melioidosis infection in liver for both the streptozotocin (STZ) -induced diabetes (grey bars) and normoglycaemic infection models (black bars), respectively. Data for the acute normoglycaemic infection model is adopted from Chin et al.18
Figure 7.
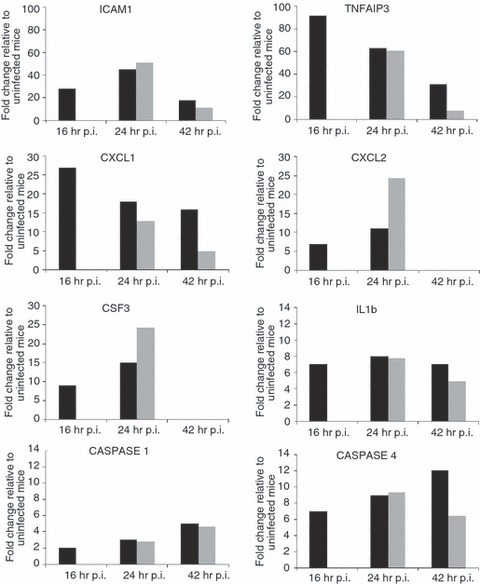
Expression profiles of ‘core immune response’ genes in the Burkholderia pseudomallei-infected liver. Several ‘common core immune response’ gene expression profiles (fold change relative to uninfected mice) during acute melioidosis infection in liver for the streptozotocin (STZ) -induced diabetes (grey bars) and normoglycaemic infection models (black bars), respectively. Data for the acute normoglycaemic infection model is adopted from Chin et al.18
Figure 8.
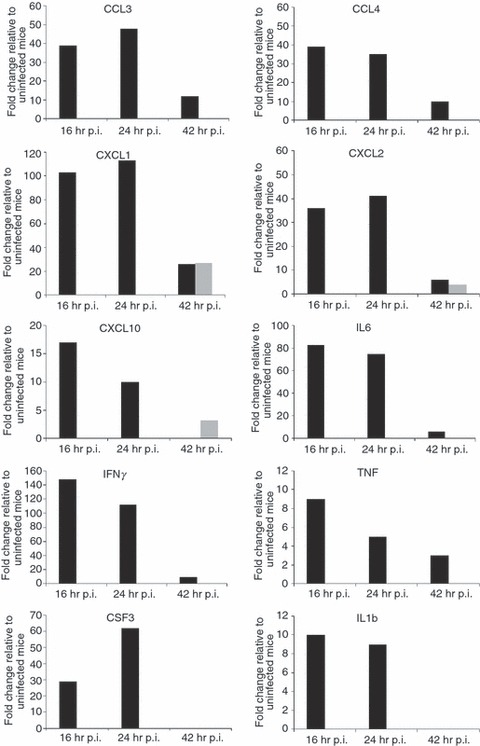
Expression profiles of ‘core immune response’ genes in the Burkholderia pseudomallei-infected spleen. Several ‘common core immune response’ gene expression profiles (fold change relative to uninfected mice) during acute melioidosis infection in spleen for the streptozotocin (STZ) -diabetic (grey bars) and normoglycaemic infection models (black bars), respectively. Data for the acute normoglycaemic infection model is adopted from Chin et al.18
Discussion
Diabetes mellitus has a dramatic impact on health; its complications cause a high degree of morbidity and mortality3 and it is an important predisposing factor for melioidosis.6 The mechanism for the increased susceptibility of patients with DM to melioidosis still remains to be fully understood. In the present study, we addressed this using an animal model of DM, the first mouse model of DM for acute melioidosis in understanding the STZ-diabetic host response to infection, globally.
Our study first described how blood glucose levels of the STZ-diabetic hosts were influenced by B. pseudomallei infection (Fig. 2). The STZ-diabetic mice continued to be hypoglycaemic (< 5 mmol/l) before succumbing to B. pseudomallei infection, suggesting that hypoglycaemia is associated with a prognosis of severe acute illness. In addition, high blood glucose levels (> 30 mmol/l) were associated with high mortality in B. pseudomallei infection (unpublished data). A recent cohort study by Peralta et al.24 reported that alteration of the blood glucose concentration is associated with risk of death among patients with community-acquired gram-negative rod bacteraemia. Patients with blood glucose concentrations between 7·77 and 9·43 mmol/l appeared to have low mortality rates of 3·3%, whereas patients with blood glucose concentrations < 5·16 or > 12·06 mmol/l had the highest mortality (12·05%), suggesting a direct relationship between sepsis-related mortality with high or low blood glucose concentrations.24 Hence, alteration of blood glucose levels is potentially detrimental, although the precise relationship with the eventual outcome in melioidosis patients has yet to be determined.
Some microorganisms become more virulent in a hyperglycaemic environment4 and this could explain the increased susceptibility to infections in diabetic patients. Geerlings et al.25 reported that glucosuria enhances the growth of different Escherichia coli strains, leading to increased incidence of urinary tract infections in diabetic patients. However, bacterial loads were not increased over the first 24 hr following B. pseudomallei infection in our STZ-diabetic model (Fig. 3a–c) when compared with the acute normoglycaemic model (see Supplementary material, Data S1). As there was no correlation between blood glucose levels and bacterial replication, the hyperglycaemic environment most likely does not promote B. pseudomallei growth at this early phase of infection. Martens et al.17 reported that hyperglycaemia per se does not directly promote Mycobacterium tuberculosis growth in the STZ-diabetic mouse with acute tuberculosis. Nevertheless, the effect of hyperglycaemia on cellular and metabolic functions of intracellular B. pseudomallei remains to be elucidated.
During bacterial infections, these pathogens intimately engage the defence response by inducing various inflammatory responses; this observation is also true for an acute melioidosis infection. Surprisingly, this genome-wide study clearly demonstrated that the STZ-diabetic host does not initiate the innate immune system at the early onset of infection as a means to eliminate B. pseudomallei. Nevertheless, this transcriptional study on the infected STZ-diabetic host is consistent with previous B. pseudomallei-infected in vivo and in vitro studies.26,27 This transcriptional analysis strongly suggests that TLR2 is also responsible for the initiation of the STZ-diabetic host defence response to B. pseudomallei infection as previously seen in the acute normoglycaemic model.18
Very few immune response genes were modulated in the STZ-diabetic spleen (Figs 5 and 8), indicating a possible malfunction of the STZ-diabetic spleen with a limited ability to respond to B. pseudomallei infection over a 42-hr period of infection. Dysfunction of the STZ-diabetic spleen correlates with uncontrolled spread of intracellular bacteria in multiple organs (Fig. 3a–c) and ultimately, increased the presentation of susceptibility to infection in diabetics. In addition, several ‘common host immune response’ genes [IL6, IL18, CXCL11, matrix metallopeptidase 7 (MMP7), proteosome (prosome, macropain) subunit alpha (PSMA4) and DUSP6] were not modulated in this study. Among these, IL6 is the chief stimulator of most acute-phase proteins (APPs)28 and we noted that production of APPs was strongly induced in the acute normoglycaemic mice infected with B. pseudomallei.18 These proteins are believed to be the cause of the severe tissue damage commonly seen in acute melioidosis as a result of an overwhelmed inflammatory response.18 The expression profiles demonstrate delayed activation of appropriate immune response-related genes at the early stage of infection, as well as suppression of potent inflammation-related genes (Fig. 4), contributing to intracellular bacterial propagation and dissemination (Fig. 3).
Acute forms of melioidosis that lead to sepsis, multiple organ failure and death are thought to result from an uncontrolled inflammatory reaction that ultimately may lead to excessive inflammation29 and eventually tissue injury in the B. pseudomallei-infected host. Recently, Koh et al.30 reported reduced mortality in diabetic patients with melioidosis who were treated with glyburide, a drug prescribed for diabetes that acts via an anti-inflammatory route. These findings support our data demonstrating an overwhelmed inflammatory response that is contributing to increased mortality in acute melioidosis. Acute inflammation studies have focused on the liver, the centre of the acute-phase response and the major target site for pro-inflammatory cytokines.31 The APPs are commonly used as an early indicator to diagnose occurrence of inflammation and disease.31 They are important in providing protective functions at sites of tissue injury,28 neutralizing the pathogens, preventing further pathogen entry while minimizing tissue damage and promoting repair processes. This then permits host homeostatic mechanisms to rapidly restore normal physiological functions.32 We previously reported that prolonged expression of APP may lead to tissue injury, as numerous APPs [ceruloplasmin (CP), lipopolysaccharide binding protein (LBP), haptoglobin, platelet-activating factor acetylhydrolase, serum amyloid A (SAA) and fibronectin type III domain containing 3B (FNDC3B)] were induced in response to the B. pseudomallei acute infection in normoglycaemic mice. Both SAA2 and SAA3 were highly induced throughout the infection period.18 However, in this study, only CP, LBP and FNDC3B were elevated 24 hr p.i. with expression levels similar to that in normoglycaemic infected mice (Fig. 5 and Table 2). This suggests that CP, LBP and FNDC3B are specific signatures of an acute B. pseudomallei infection regardless of host metabolism. Expression profiles of these APP genes, the acute-phase responses factor genes (STAT3 and IL6) as well as proteasomal degradation and proteolysis-related genes suggest that the B. pseudomallei-infected STZ-diabetic host does not encounter consequences attributed by an overwhelming inflammatory response as noted in the normoglycaemic infection model.
Neutrophils are the host frontline defence system, essential in initiating the responses to pathogens and orchestrating later immune system by releasing cytokines and chemokines, which attract other cells to the site of infection.33 The reduction in bactericidal activity, impaired phagocytosis, reduced production of reactive oxygen species and decreased release of lysosomal enzymes contribute to the high susceptibility to and severity of infections in DM.34 Previously, diabetic PMNs exhibited impaired B. pseudomallei phagocytosis and reduced migration in response to IL8, a major chemokine responsible for this function.12 In this study, CXCL1, CXCL2 and CSF3, which play an important role in neutrophil migration and mobilization were elevated 24 hr p.i. in the diabetic host (Figs 7 and 8). These expression profiles were consistent with increased production of neutrophils 24 hr p.i. (Fig. 3d) in the B. pseudomallei-infected STZ-diabetic mice. Zinc deficiency disturbs the lymphocyte response and impaired chemotaxis of diabetic PMN and patients with type 1 and type 2 DM are known to have low plasma zinc levels.4,35 This may explain the static leucocyte counts in the diabetic mice following B. pseudomallei acute infection at the early stage (16 hr p.i.) (Fig. 3d). These results revealed that the STZ-diabetic host fails to activate potent chemokines in time, leading to delayed PMN infiltration, which is pivotal for removing intracellular pathogens by phagocytosis, and eventually favours bacterial survival at the later stage of infection (Fig. 3). In addition, neutrophil counts for B. pseudomallei-infected STZ-diabetic mice were lower when compared with infected normoglycaemic mice, particularly at 42 hr p.i. (Fig. 3d), suggesting that inadequate neutrophil production increases the severity of infection. Taken together, the expression profiles suggest that elevated glucose levels impair the STZ-diabetic host innate immune system by delaying the identification and recognition of the B. pseudomallei surface structure. Consequently, the host is unable to activate the appropriate innate immune response over time, hence, increasing susceptibility to melioidosis in an STZ-diabetic host.
Cytokine expression as a result of an infection in diabetics has been the subject of considerable controversy; some reports indicated diminished inflammatory cytokine expression, while others reported enhanced expression upon bacterial infections.3,4,13 Our study on diabetes and B. pseudomallei infection suggests an impaired immune response at an early stage, similar to a report on reduced interleukin-17 expression in PMN of diabetics infected with B. pseudomallei and B. thailandensis.11 Furthermore, Williams et al.36 recently reported that diabetic mice with an extended period of uncontrolled hyperglycaemia (chronic diabetic) have impaired innate immune responses to B. pseudomallei. The decreased expression of IL12 and IL18 by bone marrow-derived dendritic cells isolated from chronic diabetic compared with non-diabetic-derived bone marrow-derived dendritic cells suggested inadequate stimulation of T helper type 1 protective responses during a B. pseudomallei infection.36 Yamashiro et al.13 reported that production of inducible nitric oxide synthase (iNOS), IL-12 and IFN-γ were lower in diabetic mice in response to Mycobacterium tuberculosis infection when compared with normal mice. However, in this study, expression of iNOS was highly induced (27-fold change) at 24 hr p.i. in the STZ-diabetic liver when compared with the normoglycaemic infected mice (11-fold change) at the same time-point (Table 2).18 In contrast, IFN-γ production was completely attenuated in the B. pseudomallei-infected STZ-diabetic spleen (Fig. 8) when compared with the infected-normoglycaemic mice, whereas the expression of IL12 was not elevated in either acute diabetic or acute normoglycaemic studies.
The complement system of the vertebrate host forms a powerful immune barrier for invading microbes, and many pathogens have used multiple evasion strategies to interfere with and to inactivate the complement attack.37 Our previous work also described for the first time that suboptimal activation and function of the downstream complement system promotes uncontrolled spread of B. pseudomallei.18 In this diabetes study, the complement-related genes were not modulated over the course of infection (Fig. 5 and Table 2), suggesting that the membrane-attack complex formation may fail to remove the intracellular pathogen. It has been shown that the capsular polysaccharide renders B. pseudomallei resistant to in vitro phagocytosis by reducing C3b deposition on the bacterial surfaces upon infection.12 Hence, this study suggests that poor glycaemic control impairs the complement system of an STZ-diabetic host rendering it unable to eliminate intracellular bacteria, hence increasing the susceptibility of diabetics to infection. Nonetheless, the B. pseudomallei-infected STZ-diabetic host over-expressed many cell death-related, inflammasone-related and proteasomal degradation genes 24 hr p.i. in the STZ-diabetic liver (Fig. 5 and Table 2). These expression profiles are similar to our previous work on the acute normoglycaemic model18 although activation is delayed. Hence, the B. pseudomallei-infected host most likely triggers cell death programmes and proteolysis to limit a favourable niche for the intracellular pathogen regardless of host metabolic conditions.
In conclusion, we have provided the first genome-wide expression profile on an STZ-diabetic mouse model in response to acute B. pseudomallei infection. The STZ-diabetic and normoglycaemic host immune response to acute B. pseudomallei infection is summarized in Table 3. Our transcriptional analysis suggests that pattern recognition receptors of the STZ-diabetic host are defective in sensing pathogens during early infection (16 hr) leading to delayed activation of an appropriate innate immune response. Nonetheless, various inflammatory and immune responses as well as the general ‘alarm signal’ of infection were still elevated 24 hr p.i. and were mainly triggered via the TLR2 pathway, as seen in the acute normoglycaemic host. Malfunction of the immune response of the STZ-diabetic spleen also correlates with uncontrolled spread of intracellular bacteria in multiple organs. We believe that the impaired innate immunity in diabetics during early B. pseudomallei infection contributes to their increased susceptibility to this fatal disease.
Table 3.
Immune responses towards acute Burkholderia pseudomallei infection: acute streptozotocin (STZ) -induced diabetes model versus acute normoglycaemic model
| Acute melioidosis infection (within 42-hr infection period) | ||
|---|---|---|
| Immune responses towards B. pseudomallei infection | STZ-diabetic model (this study) | Normoglycaemic model18 |
| The TLR2 is responsible for recognition and initiation of defence response | TLR2 and several transcription factors were elevated 24hr p.i. in the diabetic liver | Rapid induction of TLR2. Several transcription factors were elevated as early as 16hr p.i. in both liver and spleen |
| Induction of various immune response genes, including the ‘core immune response’ genes to general inflammation infections | Most of the inflammatory genes were elevated only 24hr p.i. in the diabetic liver, but were mildly elevated after 42hr p.i. in the diabetic spleen | Rapid and overwhelmed inflammatory response throughout the infection period. Several potent chemokines were suppressed at 42hr p.i. |
| Activation of APPs lead to occurrence of tissue damage | Mild elevation of some APPs and proteasomal degradation-related genes after 24hr p.i. the liver | High induction of many APPs in the liver. Peptidoglysis and proteasomal degradation-related genes were elevated throughout the infection period in both liver and spleen |
| Activation of complement pathway | Not activated in response to infection | Complement pathway-related genes were mildly elevated after 24hr p.i. but some key genes of membrane attack complex formation were suppressed |
| Activation of various cell death mechanisms | Caspase and cell death-related genes were elevated in the liver 24hr p.i. | Caspase and inflammasome-related genes were elevated in both liver and spleen as early as 16hr p.i. |
APP, acute-phase protein; p.i., post-infection; TLR, Toll-like receptor.
Acknowledgments
We would like to thank Mohd. Nor Hasan for his technical assistance in animal handling. We are grateful to the Animal House of Universiti Kebangsaan Malaysia for the animal facilities. This study was supported by the Malaysia Genome Institute–Stanford University International Research Grant awarded to S.N. by the Ministry of Science, Technology and Innovation, Malaysia. C.Y.C. was supported by the National Science Fellowship from the Ministry of Science, Technology and Innovation, Malaysia.
Disclosures
There is no conflict of interest.
Supporting Information
Data S1. Bacterial loads inB. pseudomallei- infected STZ-diabetic andnormoglycemic mice.
Data S2. qRT-PCR analysis of STZ-diabetic and normoglycemic host genes found to be differentially expressed by microarray analysis.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Farag YM, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrol Dial Transplant. 2011;26:35. doi: 10.1093/ndt/gfq576. [DOI] [PubMed] [Google Scholar]
- 2.Naguib G, Al-Mashat H, Desta T, Graves DT. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 2004;123:87–92. doi: 10.1111/j.0022-202X.2004.22711.x. [DOI] [PubMed] [Google Scholar]
- 3.Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. Inflammation is more persistent in type 1 diabetic mice. J Dent Res. 2005;84:324–8. doi: 10.1177/154405910508400406. [DOI] [PubMed] [Google Scholar]
- 4.Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM) FEMS Immunol Med Microbiol. 1999;26:259–65. doi: 10.1111/j.1574-695X.1999.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 5.Peacock SJ. Melioidosis. Curr Opin Infect Dis. 2006;19:421–8. doi: 10.1097/01.qco.0000244046.31135.b3. [DOI] [PubMed] [Google Scholar]
- 6.Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18:383–416. doi: 10.1128/CMR.18.2.383-416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 8.Woods DE, Jones AL, Hill PJ. Interaction of insulin with Pseudomonas pseudomallei. Infect Immun. 1993;61:4045–50. doi: 10.1128/iai.61.10.4045-4050.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simpson AJ, Wuthiekanun V. Interaction of insulin with Burkholderia pseudomallei may be caused by a preservative. J Clin Pathol. 2000;53:159–60. doi: 10.1136/jcp.53.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson AJ, Newton PN, Chierakul W, Chaowagul W, White NJ. Diabetes mellitus, insulin, and melioidosis in Thailand. Clin Infect Dis. 2003;36:e71–2. doi: 10.1086/367861. [DOI] [PubMed] [Google Scholar]
- 11.Pongcharoen S, Ritvirool PN, Sanguansermsri D, et al. Reduced interleukin-17 expression of Burkholderia pseudomallei-infected peripheral blood mononuclear cells of diabetic patients. Asian Pac J Allergy Immunol. 2008;26:63–9. [PubMed] [Google Scholar]
- 12.Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, Lertmemongkolchai G. Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun. 2009;77:456–63. doi: 10.1128/IAI.00503-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamashiro S, Kawakami K, Uezu K, Kinjo T, Miyagi K, Nakamura K, Saito A. Lower expression of Th1-related cytokines and inducible nitric oxide synthase in mice with streptozotocin-induced diabetes mellitus infected with Mycobacterium tuberculosis. Clin Exp Immunol. 2005;139:57–64. doi: 10.1111/j.1365-2249.2005.02677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227–39. doi: 10.2741/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SH, Chong CE, Lim BS, Chai SJ, Sam KK, Mohamed R, Nathan S. Burkholderia pseudomallei animal and human isolates from Malaysia exhibit different phenotypic characteristics. Diagn Microbiol Infect Dis. 2007;58:263–70. doi: 10.1016/j.diagmicrobio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Hoppe I, Brenneke B, Rohde M, Kreft A, Haussler S, Reganzerowski A, Steinmetz I. Characterization of a murine model of melioidosis: comparison of different strains of mice. Infect Immun. 1999;67:2891–900. doi: 10.1128/iai.67.6.2891-2900.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martens GW, Arikan MC, Lee J, Ren F, Greiner D, Kornfeld H. Tuberculosis susceptibility of diabetic mice. Am J Respir Cell Mol Biol. 2007;37:518–24. doi: 10.1165/rcmb.2006-0478OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chin CY, Monack DM, Nathan S. Genome wide transcriptome profiling of a murine acute melioidosis model reveals new insights into how Burkholderia pseudomallei overcomes host innate immunity. BMC Genomics. 2010;11:672. doi: 10.1186/1471-2164-11-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu B, Koo GC, Yap EH, Chua KL, Gan YH. Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect Immun. 2002;70:504–11. doi: 10.1128/IAI.70.2.504-511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulett GC, Currie BJ, Clair TW, et al. Burkholderia pseudomallei virulence: definition, stability and association with clonality. Microbes Infect. 2001;3:621–31. doi: 10.1016/s1286-4579(01)01417-4. [DOI] [PubMed] [Google Scholar]
- 21.Barnes JL, Ketheesan N. Route of infection in melioidosis. Emerg Infect Dis. 2005;11:638–9. doi: 10.3201/eid1104.041051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potasman I, Prokocimer M. The added value of peripheral blood cell morphology in the diagnosis and management of infectious diseases – part 2: illustrative cases. Postgrad Med J. 2008;84:586–9. doi: 10.1136/pgmj.2008.069617. [DOI] [PubMed] [Google Scholar]
- 23.Prokocimer M, Potasman I. The added value of peripheral blood cell morphology in the diagnosis and management of infectious diseases – part 1: basic concepts. Postgrad Med J. 2008;84:579–85. doi: 10.1136/pgmj.2008.069609. [DOI] [PubMed] [Google Scholar]
- 24.Peralta G, Sanchez MB, Garrido JC, Ceballos B, Mateos F, De Benito I, Roiz MP. Altered blood glucose concentration is associated with risk of death among patients with community-acquired Gram-negative rod bacteremia. BMC Infect Dis. 2010;10:181. doi: 10.1186/1471-2334-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geerlings SE, Brouwer EC, Gaastra W, Verhoef J, Hoepelman AI. Effect of glucose and pH on uropathogenic and non-uropathogenic Escherichia coli: studies with urine from diabetic and non-diabetic individuals. J Med Microbiol. 1999;48:535–9. doi: 10.1099/00222615-48-6-535. [DOI] [PubMed] [Google Scholar]
- 26.Wiersinga WJ, Wieland CW, Dessing MC, et al. Toll-like receptor 2 impairs host defense in gram-negative sepsis caused by Burkholderia pseudomallei (Melioidosis) PLoS Med. 2007;4:e248. doi: 10.1371/journal.pmed.0040248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feterl M, Govan BL, Ketheesan N. The effect of different Burkholderia pseudomallei isolates of varying levels of virulence on toll-like-receptor expression. Trans R Soc Trop Med Hyg. 2008;102(Suppl. 1):S82–8. doi: 10.1016/S0035-9203(08)70021-X. [DOI] [PubMed] [Google Scholar]
- 28.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 29.Gan YH. Interaction between Burkholderia pseudomallei and the host immune response: sleeping with the enemy? J Infect Dis. 2005;192:1845–50. doi: 10.1086/497382. [DOI] [PubMed] [Google Scholar]
- 30.Koh GC, Maude RR, Schreiber MF, et al. Glyburide is anti-inflammatory and associated with reduced mortality in melioidosis. Clin Infect Dis. 2011;52:717–25. doi: 10.1093/cid/ciq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peatman E, Terhune J, Baoprasertkul P, et al. Microarray analysis of gene expression in the blue catfish liver reveals early activation of the MHC class I pathway after infection with Edwardsiella ictaluri. Mol Immunol. 2008;45:553–66. doi: 10.1016/j.molimm.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 32.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–23. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 33.Ekman AK, Cardell LO. The expression and function of Nod-like receptors in neutrophils. Immunology. 2010;130:55–63. doi: 10.1111/j.1365-2567.2009.03212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, Curi R, Sannomiya P. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res. 2007;40:1037–44. doi: 10.1590/s0100-879x2006005000143. [DOI] [PubMed] [Google Scholar]
- 35.Moutschen MP, Scheen AJ, Lefebvre PJ. Impaired immune responses in diabetes mellitus: analysis of the factors and mechanisms involved. Relevance to the increased susceptibility of diabetic patients to specific infections. Diabetes Metab. 1992;18:187–201. [PubMed] [Google Scholar]
- 36.Williams NL, Morris JL, Rush C, Govan BL, Ketheesan N. Impact of streptozotocin-induced diabetes on functional responses of dendritic cells and macrophages towards Burkholderia pseudomallei. FEMS Immunol Med Microbiol. 2011;61:218–27. doi: 10.1111/j.1574-695X.2010.00767.x. [DOI] [PubMed] [Google Scholar]
- 37.Zipfel PF, Wurzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol. 2007;44:3850–7. doi: 10.1016/j.molimm.2007.06.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.