

Abstract
Stroke prevention efforts typically focus on either ischemic or hemorrhagic stroke. This approach is overly simplistic due to the frequent coexistence of ischemic and hemorrhagic cerebrovascular disease. This coexistence, termed “mixed cerebrovascular disease”, offers a conceptual framework that appears useful for stroke prevention strategies. Mixed cerebrovascular disease incorporates clinical and subclinical syndromes, including ischemic stroke, subclinical infarct, white matter disease of aging (leukoaraiosis), intracerebral hemorrhage, and cerebral microbleeds. Reliance on mixed cerebrovascular disease as a diagnostic entity may assist in stratifying risk of hemorrhagic stroke associated with platelet therapy and anticoagulants. Animal models of hemorrhagic cerebrovascular disease, particularly models of cerebral amyloid angiopathy and hypertension, offer novel means for identifying underlying mechanisms and developing focused therapy. Phosphodiesterase (PDE) inhibitors represent a class of agents that, by targeting both platelets and vessel wall, provide the kind of dual actions necessary for stroke prevention, given the spectrum of disorders that characterizes mixed cerebrovascular disease.
Keywords: Stroke, Cerebrovascular, Hemorrhage, Hemorrhagic transformation, Microbleeds, Leukoaraiosis, Amyloid, Hypertension, Phosphodiesterase inhibitor
“The way out is through the door. Why is it that no one will use this method?”
Confucius
Introduction
Stroke prevention efforts typically focus on either ischemic stroke or hemorrhagic stroke. It is understood that ischemic stroke prevention requires a comprehensive approach for the variety of stroke risk factors that a patient may encounter. Similarly, patients who have sustained hemorrhagic stroke will have targeted efforts directed against the vascular risk factor(s) thought to be most significant in the etiology of the hemorrhage, with hypertension being the typical culprit.
A principal focus for attention in stroke prevention involves medications that target various elements of coagulation pathways. For prevention of ischemic stroke, platelet anti-aggregant medications are a prime issue, as well as use of anticoagulants. Some degree of avoidance of these same medications is a frequent element of any clinical strategy for prevention of hemorrhagic stroke.
It is becoming increasingly clear that this approach is overly simplistic, because it tends to ignore the potential for coexistence of both entities. This coexistence is obvious in the presence of clinically evident intracerebral hemorrhage and ischemic stroke. But what happens when the coexistence is not obvious? What strategies are available when subclinical hemorrhagic or ischemic cerebrovascular disease may be present in the same individual patient? Or alternatively, when clinical and subclinical cerebrovascular disease coexist in a given patient?
The usual and customary conceptual framework for stroke, as used in stroke prevention efforts, begins to break down when the entity of “stroke” is examined critically. What kind of stroke are we trying to prevent, exactly? Is it possible to successfully navigate between the apparent extremes of hemorrhagic and ischemic cerebrovascular disease? It is the contention of this paper that these are the kinds of questions that will define stroke prevention for the coming decades of the 21st century. At the very least, a new conceptual framework for stroke seems worthy of consideration.
This paper will review a new framework for conceptualizing cerebrovascular disease. Mechanisms for hemorrhagic stroke will be discussed, with an emphasis on cerebral amyloid angiopathy. A pathway will be proposed for simultaneously addressing the “opposing” tendencies of thrombosis and hemorrhage as they may coexist in the same patient.
Mixed Cerebrovascular Disease: A New Conceptual Approach
The concept of “mixed cerebrovascular disease” has been proposed as a framework for better understanding stroke, and for improving stroke prevention efforts [1]. Mixed cerebrovascular disease incorporates clinical and subclinical stroke with hemorrhagic and ischemic stroke. The clinical stroke syndromes thus incorporate the typical variety of presentations encountered, with intracerebral hemorrhage and ischemic stroke subsets (small vessel disease, large vessel disease, cardiogenic, other). Subclinical stroke syndromes include silent ischemic stroke and cerebral microbleeds. Cerebral white matter disease (of aging) is an additional component of mixed cerebrovascular disease.
There are both strengths and weaknesses of this conceptual approach. A principal benefit of characterizing a given patient as having “mixed cerebrovascular disease” is that the clinician is immediately confronted with the fact that, going forward with stroke prevention efforts, this patient will require a complex strategy. Simply relying on “antiplatelet medications” for prevention of cerebral infarction will be insufficent.
The principal weakness of this approach is that it may simply be too inclusive. For example, cerebral white matter changes are ubiquitous in the population of age 65 and older, with more than 95 % of individuals showing at least some white matter change on magnetic resonance imaging (MRI) [2]. Only about one-third of these changes are probably sufficient to be characterized as “disease”, but the gradation between normal and pathological change remains unclear.
A similar problem may arise with the inclusion of cerebral microbleeds. Microbleeds are present in approximately 20 % of the population beginning at age 60, a proportion that increases to nearly 40 % by age 80 [3]. Pathological studies reveal a much higher prevalence over the age of 70 [4, 5], but it is unclear whether both MRI and neuropathological findings are demonstrating the same entity. However, this difficulty may be surmounted by simply focusing on the inclusion of MRI-demonstrable cerebral microbleeds, using gradient echo or the more sensitive susceptibility-weighted imaging sequences.
To summarize, the use of mixed cerebrovascular disease as a conceptual and clinical framework appears feasible. Inclusion of cerebral white matter disease may be problematic. Nevertheless, a characterization of stroke syndromes that incorporates clinical and subclinical ischemic and hemorrhagic disease may help quickly characterize the complexity of many stroke patients.
Intracerebral Hemorrhage and Ischemic Stroke Prevention: Platelet Therapy, Anticoagulants, and Statins
The risk of hemorrhagic stroke is known to increase with platelet therapy used for stroke prevention. For example, in stroke prevention trials the use of aspirin increased hemorrhagic stroke by relative risk of 84 %, or 12 hemorrhagic strokes per 10,000 persons [6]. This risk does not appear to significantly change with use of the platelet anti-aggregant clopidogrel [7]. However, combination treatment of aspirin with clopidogrel significantly increases hemorrhagic stroke risk beyond that encountered with clopidogrel alone [8]. Interestingly, addition of clopidogrel to aspirin does not appear to increase hemorrhagic stroke risk above aspirin alone [9].
Anticoagulation represents the other principal strategy for medical therapy in stroke prevention. Warfarin is the principal oral anticoagulant utilized, and carries a well-known risk for intracerebral hemorrhage. Prevention of cardiogenic stroke, particularly atrial fibrillation, is the primary indication for warfarin; in this population, the risk of intracerebral hemorrhage ranges from 0.3 % to 1 % annually [10].
Recent advances in oral anticoagulants have focused on substantially reducing this risk. Compared to warfarin, both the direct thrombin inhibitor dabigatran and the factor Xa inhibitor rivaroxaban have significantly less risk of intracerebral hemorrhage, while providing effective protection against cardiogenic stroke [11, 12]. For dabigatran, the annual risk of of intracerebral hemorrhage was 0.23–0.30 %, compared to 0.74 % for warfarin, a risk reduction of 70–77 % [11]. For rivaroxaban versus warfarin, risk of intracerebral hemorrhage was 0.5 versus 0.7 events/100 patient years, a risk reduction of approximately one-third [12]. It should be emphasized that these represent a reduction, but hardly an elimination, of risk of intracerebral hemorrhage with the new oral agents.
Statins represent another major component of therapy for prevention of ischemic stroke. The definitive clinical trial demonstrating benefits of atorvastatin showed divergent effects of the drug on ischemic versus hemorrhagic stroke [13]. On high dose atorvastatin, risk of ischemic stroke declined by 22 %, while risk of hemorrhagic stroke increased by 66 % [13]. Further analysis showed that some of this excess risk of hemorrhagic stroke originated from patients who had entered into the trial after a hemorrhagic stroke, and that risk of hemorrhagic stroke was not increased among patients who entered the trial following large vessel or cardiogenic stroke [14]. Nevertheless, risk of hemorrhagic stroke was nearly fivefold higher among patients with small vessel ischemic stroke, an increased risk that was even higher than that encountered in patients who had entered the trial with hemorrhagic stroke [14].
To summarize, routine preventive treatment for ischemic stroke generates a significant risk for hemorrhagic stroke. This increased risk includes use of platelet therapy, anticoagulants, and statins. Classification of ischemic stroke patients by risk for hemorrhagic stroke represents a potential benefit of moving toward the conceptual framework of mixed cerebrovascular disease.
Mixed Cerebrovascular Disease: Stratification of Ischemic Stroke Patients for Hemorrhagic Stroke Risk
Clinical management in stroke neurology relies on risk stratification, some of which is systematically formalized. The best known example of this is the scoring system based on presence of congestive heart failure, hypertension, age, diabetes, and prior stroke or transient ischemic attack (CHADS2) for estimating stroke risk in patients with atrial fibrillation [15]. Given the catastrophic nature of intracerebral hemorrhage, risk stratification focusing on avoidance of this entity is clearly an attractive option. Mixed cerebrovascular disease as a diagnostic entity would appear to provide that alternative.
Multiple studies have demonstrated that presence of cerebral microbleeds on MRI is clearly associated with intracerebral hemorrhage. Microbleeds are present in an estimated 68 % of patients with spontaneous intracerebral hemorrhage [16]. This has been shown in subjects with presumed cerebral amyloid angiopathy as well as chronic hypertension [16]. Moreover, association between cerebral microbleeds and intracerebral hemorrhage has been demonstrated in patients with prior ischemic stroke [17] and in subjects with recurrent intracerebral hemorrhage [18]. Current perspectives on cerebral microbleeds incorporate the understanding that cortical microbleeds reflect underlying amyloid angiopathy, while deep hemisphere microbleeds indicate the consequences of hypertensive vascular disease [3].
A relationship between use of platelet medications and microbleeds is to be expected, and in fact has been demonstrated. Wong and colleagues first demonstrated that intracerebral hemorrhage in aspirin users was more common in patients with presence of cerebral microbleeds, particularly when the microbleeds were more extensive [19]. In a larger, longitudinal study of more than 1000 subjects aged 60 and older, use of platelet medications was associated with approximately 70 % increased risk of cerebral microbleeds [20]. In a study of recurrent intracerebral hemorrhage, extent of microbleeds was associated with recurrent hemorrhage, as was aspirin use [18]. Among patients with chronic (>5 years) aspirin use, risk for cerebral microbleeds increased more than fivefold [21].
Similar to the microbleeds-platelet therapy relationship, warfarin use and microbleeds are substantially linked. Among anticoagulated patients developing intracerebral hemorrhage, presence of microbleeds was associated with more than sevenfold increased risk of intracerebral hemorrhage [22]. Lee et al. demonstrated an even more dramatic association, showing a more than 80-fold increased risk of intracerebral hemorrhage in warfarin-treated patients with cerebral microbleeds [23]. While a smaller study failed to show an association between warfarin use and microbleeds [24], meta-analysis confirmed a relationship between microbleeds and warfarin use in patients with intracerebral hemorrhage [25].
Another element that can be incorporated into risk stratification for drug-induced intracerebral hemorrhage is presence of cerebral white matter disease of aging, also known as leukoaraiosis. As noted previously, white matter changes are ubiquitous in an aging population [2] but, when excessive, show an important relationship to intracerebral hemorrhage in warfarin users. Smith et al carefully examined extent of cerebral white matter disease in ischemic stroke patients using warfarin [26]. The case control study demonstrated a nearly 13-fold increased risk for intracerebral hemorrhage conferred by leukoaraiosis among patients on warfarin [26].
Given the relationship between risk for intracerebral hemorrhage and both microbleeds and white matter disease, it is not surprising that an important relationship exists between leukoaraiosis and microbleeds. The association between leukoaraiosis and microbleeds has been reported by numerous investigators. Kato et al reported a strong correlation between number of microbleeds and extent of periventricular white matter changes [27]. Lee et al. [28], Naka et al. [29], Maia et al. [30], Gorner et al. [31], Gao et al. [32], and Pettersen et al. [33] all reported significant associations of varying strength between extent of microbleeds and severity of leukoaraiosis. Presence of microbleeds and leukoaraisosis both predict development of new microbleeds after ischemic stroke [34] and risk of intracerebral hemorrhage is particularly high in patients with hypertension, microbleeds, and leukoaraiosis [35].
The microbleed-white matter disease association has been further delineated by Yamada et al. [36], who studied the relationships between white matter disease, microbleeds, and age. This is a particularly important issue, given the age-dependence of both entities. However, in this study presence of microbleeds was more closely associated with white matter disease than with age [36]. Another intriguing finding was the single report linking low cholesterol levels and microbleeds [37], which may provide some insight into the increased risk of hemorrhagic stroke risk with high dose atorvastatin [14].
To summarize, cerebral microbleeds are a marker for increased risk of intracerebral hemorrhage. This includes risk of spontaneous intracerebral hemorrhage and risk of intracerebral hemorrhage associated with warfarin usage. Presence of microbleeds appears to be associated with use of platelet medications, and cerebral white matter disease of aging (leukoaraisosis) and microbleeds are closely associated. This latter relationship, between leukoaraiosis and microbleeds, has been shown so consistently that a common underlying mechanism appears likely. This mechanism appears most likely to be a microvascular origin [38].
Mechanisms of Intracerebral Hemorrhage: Animal Models
In order to investigate the molecular and cellular mechanisms involved in intracerebral hemorrhage, as well as develop effective therapies, it is important to have animal models that replicate the critical features of the human cerebrovascular pathogenesis. Moreover, to develop effective therapies that do not exacerbate one type of cerebrovascular disorder at the expense of the other (mixed cerebrovascular disease), it is imperative that experiments are performed in animal models that replicate this conundrum. Because animal models of ischemic stroke are so well-developed, we have chosen to focus on mouse models that develop spontaneous pathological lesions that may complicate therapeutic intervention for ischemic stroke, e.g., cerebral amyloid angiopathy and cerebral microhemorrhages, or by experimental manipulation to induce hypertension, which can contribute to ischemic stroke or intracerebral hemorrhage.
Robust amyloid-beta (Aβ) depositions in the walls of arterial blood vessels can result to noncellular thickening of the vessel wall that can be visualized by hematoxylin and eosin staining. However, histochemical or anti-Aβ antibody immunohistochemical staining are required to detect cerebral amyloid angiopathy (Fig. 1) [39]. Modest vascular Aβ deposits, as well as capillary involvement, can only be visualized by immunohistochemistry. Most of the human mutant amyloid precursor protein (APP) transgenic mouse models deposit parenchymal plaques but also develop varying degrees of cerebral amyloid angiopathy (Fig. 2) [39, 40]. Characterization of the vascular pathology shows prominent accumulation of Aβ40 peptide, smooth muscle degeneration and loss, and evidence of microhemorrhage, all of which are found associated with human cerebral amyloid angiopathy. Generally, the cerebral amyloid angiopathy has been found in the larger cortical and meningeal vessels reminiscent of cerebral amyloid angiopathy-type 2 observed in humans. In 1999, Jucker and colleagues reported on their APP23 transgenic mice, which were generated with human APP751 with human double Swedish (Sw) mutation, which enhances β-secretase cleavage [43, 44]. They utilized the murine thymocyte differentiation antigen 1 (Thy-1 or CD90) Thy-1.2 promoter to drive expression of the transgene in a C57B/6 background. The mice overexpress APP sevenfold and first show Aβ deposits at 6 months of age. They also reported significant deposition of Aβ in the cerebral vasculature that had striking similarities to that observed in human aging and Alzheimer’s disease. Amyloid deposition occurred preferentially in arterioles and capillaries and within individual vessels showed a wide heterogeneity [41]. The APP23 transgenic mice were the first to show that anti-amyloid immunotherapy in older mice exacerbated both cerebral amyloid angiopathy and microhemorrhages [42].
Fig. 1.
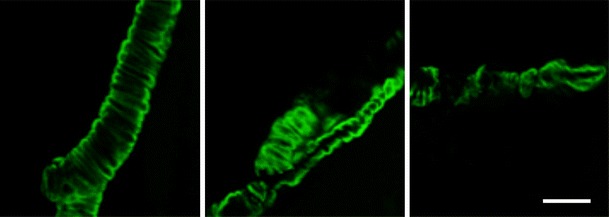
Examples of cerebral amyloid angiopathy in cortical arteries in 24-month-old Tg2576 mice detected by staining with Thioflavine S, which binds to fibrillar forms of amyloid-beta (Aβ). (Scale bar: 50 μm)
Fig. 2.
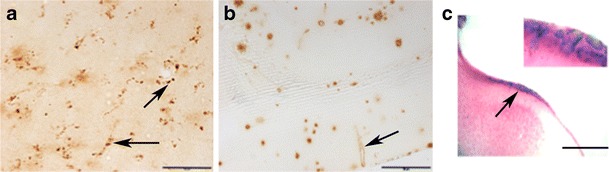
Types of cerebral amyloid angiopathy pathology in two amyloid precursor protein (APP) transgenic (Tg) mouse models. a Punctate amyloid-β40 (Aβ40) deposits (arrows) on microvessels of 12-month-old APP transgenic mice with three human APP mutations, the Swedish, Dutch and Iowa (Tg-SwDI; scale bar: 20 μm). b Cerebral amyloid angiopathy Aβ40 deposit (arrows) in large penetrating artery in 20-month-old Tg2576 mouse (bar: 100 μm). c Spontaneous microhemorrhages (arrow) in 24 = month = old Tg2576 mouse, visualized using Prussian blue staining. (Scale bar: 200 μm). Inset: A higher magnification image (200×) of spontaneous microhemorrhages
Another useful APP transgenic (Tg) model is the Tg2576 mouse (C57B/6-SJL) that express human APP695, which also contains the Sw APP mutation. The transgene is driven by the hamster prion protein gene promoter and Tg2576 mice express APP at 5.6 times more than wild-type mouse APP. Old Tg2576 mice (18+ months) develop cerebral amyloid angiopathy in the larger vessels such as leptomeningeal and pial, and develop spontaneous microhemorrhages. The Tg2576 mouse model has been used extensively to investigate age-related cerebral amyloid angiopathy deposition and microhemorrhages, as well as anti-Aβ immunotherapy induced microhemorrhages (Fig. 3) [45–48].
Fig. 3.
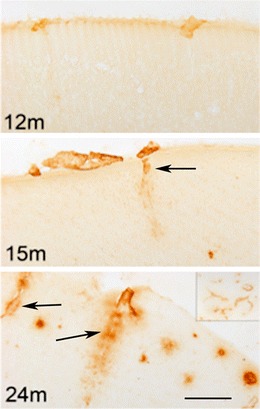
The progression of cerebral amyloid angiopathy pathology in amyloid precursor protein Tg2576 mice at 12, 15 and 24 months of age (12, 15, and 24 m). Immunostaining is performed using an antibody specific for Aβ40, which is the predominant amyloid peptide deposited in cerebral amyloid angiopathy. Parenchyma- penetrating blood vessels with amyloid angiopathy are noted by arrows. (Scale bar: 100 μm). A lower magnification image (4×) of frontal cortex with extensive cerebral amyloid angiopathy is shown in the top right corner of 24-month-old mouse
There have been efforts to generate cerebrovascular amyloid models in the absence of significant parenchymal amyloid deposition. Transgenic mouse lines were developed utilizing mutations within human Aβ that are found in familial forms of cerebral amyloid angiopathy. For example, transgenic mice were generated that produce the familial cerebral amyloid angiopathy Dutch E22Q variant of human Aβ in brain resulting in a model of significant larger meningeal and cortical vessel cerebral amyloid angiopathy in absence of parenchymal amyloid plaques. There was also smooth muscle cell degeneration, hemorrhages, and neuroinflammation [49].
Another very useful transgenic model that deposits Aβ in cerebral vessels is the Tg-SwDI (C57B/6; B line, Thy-1.2 promoter), which contains the human APP-Sw mutation, but in addition contains two human vasculotropic mutations (the Dutch and the Iowa mutations) in the Aβ sequence [50, 51]. This mouse (hemizygous) begins to develop microvessel Aβ deposits, reminiscent of cerebral amyloid angiopathy-type 1 in humans, at 4–5 months of age in several cortical areas. As the mice age, the microvessel deposition becomes more widespread, and copious diffuse deposits develop throughout the cortex. The only glial activation in the central nervous system in the Tg-SwDI mice is associated with the vascular deposition of Aβ.
Interestingly, two recent reports have established the feasibility of actually imaging cerebral microhemorrhages in APP transgenic mouse models [52, 53]. Luo et al. [52] reported on magnetic resonance imaging detection and time course of cerebral microhemorrhages during passive immunotherapy in living amyloid precursor protein transgenic mice. Beckmann et al. [53] used superparamagnetic iron oxide particles to enhance the magnetic resonance imaging detection of cerebral amyloid angiopathy-related microvascular alterations in APP transgenic mouse models of Alzheimer’s disease.
As mentioned above, hypertension has long been understood to cause ischemic strokes [54, 55] as well as intracerebral hemorrhage [56, 57] and white matter disease [58] that have been linked small vessel disease [59, 60]. More recently, however, vascular risk factors such as hypertension have been proposed to play multiple roles in shaping the trajectory to dementia in the elderly [61]. Several prospective cohort studies have provided compelling data suggesting that higher blood pressure levels are associated with an increased risk for dementia in the elderly [62–65], and high midlife blood pressure levels have been correlated with late-life cognitive deficits [66]. Finally, with regard to risk for dementia of the Alzheimer’s disease-type, data from the Rotterdam Scan Study indicate that apolipoprotein E4 carriers are at increased risk for white matter lesions if they have hypertension [67]. In summary, midlife hypertension increases the risk for cognitive impairment [63, 68, 69], and atrophy of the hippocampus [70, 71], white matter disease [72], amyloid plaques, and vascular lesions [73].
Growing evidence indicates that hypertension-induced vascular injury contributes to a chronic low-grade inflammatory process and that inflammation may play a significant role in the pathogenesis of hypertension [74]. In vitro, angiotensin II has been shown to modulate the function of various adhesion molecules, chemokines, cytokines and growth factors, and ultimately contributes to cell proliferation, hypertrophy and inflammation. Angiotensin II influences the inflammatory response by increasing vascular permeability via prostaglandins and vascular endothelial growth factor [75], among other factors. Importantly, angiotensin II-induced vascular inflammation is mediated through different and countervailing vascular wall effectors via two angiotensin II receptor (AT) subtypes (proinflammatory AT1 and anti-inflammatory AT2) [74].
Chronic hypertension models resemble most key features of small vessel disease, and share the major risk factors of hypertension and age with human small vessel disease. The most widely used model has been the stroke-prone spontaneously hypertensive rat (SHRSP) [76]. Interestingly, the SHRSP rat can develop both hemorrhagic and ischemic strokes. However, genetic factors appear to contribute to stroke susceptibility in SHRSP independent of blood pressure [76]. None of the animal models fully mimics all features of the human cerebrovascular disease. The optimal choice of model depends on the aspect of pathophysiology being studied [77]. For example, the SHRSP rat model does not develop cerebral amyloid angiopathy, and is not conducive to breeding with other cerebrovascular models, which are rather limited in rats. Hypertensive mouse models do not appear to develop stroke spontaneously, although there is a report of spontaneous unilateral brainstem infarction in non-inbred Swiss mice [78].
While APP transgenic mice have not been shown to develop spontaneous ischemic stroke, there are several publications demonstrating increased susceptibility of these animals to ischemic insult. In 1997, Zhang et al. [79] demonstrated that middle cerebral artery occlusion produced enlarged infarct volume and reduced cerebral blood flow in Tg2576 mice compared with wild-type littermates; this was attributed to diminished endothelium-dependent vascular reactivity. Koistinaho and colleagues [80] showed increased susceptibility to brain ischemia in APP751 mice overexpressing the 751-amino acid isoform of human APP, and suggested that this was mediated by increased microglial activation and inflammation. Recently, experimental models based on angiotensin II infusion have been reported to better replicate human diseases, such as postmenopausal hypertension, preeclampsia, vascular remodeling, vascular aging, and neovascularization [81]. Mechanisms that lead to intracerebral hemorrhage during hypertension, however, remain poorly understood, in part because of a paucity of experimental models of spontaneous intracerebral hemorrhage in mice [82].
The first experimental model of spontaneous intracerebral hemorrhage in hypertensive mice utilized double transgenic mice with overexpression of human rennin and human angiotensinogen, treated with No-nitro-l-arginine methyl ester (l-NAME), an inhibitor of nitric oxide synthase, and high-salt diet [83, 84]. But because this model requires crossing the two transgenic mouse lines, it is not easily applied to other transgenic mouse models, such as many APP transgenic mouse models. More recently, Heistad and colleagues [82] developed an experimental model of hypertension-induced intracerebral hemorrhage (acute and chronic insult) that facilitates studies in genetically altered mice. They suggested that acute hypertension, induced by angiotensin II or norepinephrine, may be critical for spontaneous intracerebral hemorrhage during chronic hypertension, possibly via oxidative stress and matrix metalloproteinase 9 (MMP-9) [82]. They induced chronic hypertension with angiotensin II using subcutaneously implanted mini-osmotic pumps, and then delivered acute doses via subcutaneous injections. The incidence of intracerebral hemorrhage and levels of oxidative stress and MMP-9 were greater in mice with acute hypertension produced by angiotensin II than by norepinephrine. Another less aggressive hypertension protocol is the slow-pressor angiotensin II model of hypertension, which has been validated by several groups [75, 85, 86], and is pertinent because this model of hypertension mimics essential hypertension found in humans [87]. One constraint of this type of experimental approach is the limited duration, typically 4 weeks, that hypertension can be maintained with an Alzet pump (www.alzet.com).
Recently we have begun to focus on mouse models of mixed cerebrovascular pathology to address some of the clinically relevant questions regarding therapeutic intervention in these complicated cases. Our initial approach has been to examine how cerebral amyloid angiopathy affects hypertension-induced adverse changes in cerebral blood vessels, together with how the associated increases in neuroinflammation may exacerbate microhemorrhages and larger intracerebral hemorrhages. Furthermore, we speculate that Aβ and tau pathology in Alzheimer’s disease transgenic mouse models may also be amplified due to chronic hypertension. In preliminary studies we have used both Heistad and colleagues’ aggressive protocol, in which spontaneous intracerebral hemorrhage occurs in wild-type mice when exposed to acute and chronic hypertension [82], as well as the somewhat milder slow-pressor angiotensin II model of hypertension [74, 85, 86]. In preliminary studies using the acute and chronic hypertension model we found that hypertensive Tg2576 mice were more prone to develop intracerebral hemorrhages in response to hypertension than non-Tg littermates. Regardless of whether the mice developed intracerebral hemorrhages, there was robust inflammation and increased vascular amyloid (Fig. 4] in the gray matter of Alzheimer’s disease mouse models, showing that hypertension may affect gray as well as white matter in the brain.
Fig. 4.
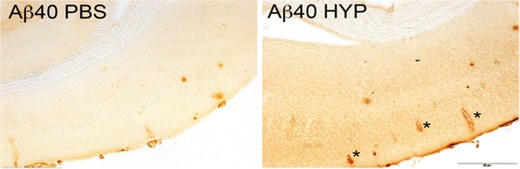
Hypertension induced by chronic angiotensin II + No-nitro-l-arginine methyl ester (L-NAME) treatment and acute angiotensin II injections (HYP) increases cerebral amyloid angiopathy relative to phosphate buffered saline (PBS) only-treated control 15-month-old Tg2576 mice. Detection of cerebral amyloid angiopathy by immunohistochemical staining with an antibody specific for Aβ40. Note cerebral amyloid angiopathy involvement in the cortex of the hypertensive Tg2576 mice (asterisks). (Scale bar: 200 μm)
Perhaps surprisingly, plasma Aβ40 levels have recently been independently associated with small vessel disease [88], and these results are consistent with a role for Aβ40 in producing disruption of vascular endothelial function [89, 90]. Age-related alterations in transport across the blood–brain barrier, as well as a reduction in the efficacy of the perivascular drainage pathway, have been proposed to explain enhanced accumulation of cerebral parenchymal and vascular amyloid deposits in the elderly [91–95]. Modeling suggest that vessel pulsations provide the force to drive perivascular drainage, and age-related stiffening of arteries has been hypothesized to reduce flow and thus enhancing Aβ deposition in the central nervous system [94].
Another crucial area of investigation where animal models have been particularly useful is hemorrhagic transformation following ischemic strokes [96]. Cerebral hemorrhagic transformation, which occurs in 30–40 % of all ischemic strokes, is a highly complex phenomenon in which secondary hemorrhagic lesion(s) are produced with increased permeability of the blood–brain barrier, extravasation into surrounding tissue, and exacerbation of the brain injury [96]. Because hemorrhagic transformation has been linked to reperfusion injury, the widespread use of thrombolytic medications may further increase the incidence of secondary hemorrhagic lesions in ischemic stroke patients [97, 98]. Animal models have been quite useful in identifying underlying mechanisms, triggers, and molecular pathways which contribute to hemorrhagic transformation including inflammation, oxidative stress, and matrix metalloproteinases as potential facilitators of hemorrhagic transformation due to their ability to disrupt the blood–brain barrier [96, 99]. Moreover, animal models have been instrumental in testing potential therapies, such as statins, angiotensin receptor antagonists, and minocycline, designed to reduce the incidence of hemorrhagic transformation [100–102].
The important roles of hypertension [103–106] and hyperglycemia [107–109] in development of hemorrhagic transformation have been extensively investigated. Acute hypertension can induce hemorrhagic transformation in embolic models [103, 104], while chronic hypertension can provoke hemorrhagic transformation with thrombolysis [105]. In spontaneously hypertensive rats, hemorrhagic transformation has been attributed to larger ischemic lesions and more blood–brain barrier disruption and vasogenic edema [106]. Mechanisms of hyperglycemia-induced hemorrhagic transformation include increased oxidative stress, upregulation of MMP-9, and enhanced blood–brain barrier disruption, as observed in models of diabetes and/or acute hyperglycemia [107–109]. Moreover, the possible neuroprotective agent isofluorane has been shown to actually worsen hyperglycemia-induced hemorrhagic transformation [110].
An example of how a mouse model can be used to investigate clinically relevant issues of therapeutic intervention in older stroke patients involved the use of tissue plasminogen activator in APP23 transgenic mice. Winkler et al. [111] reported that the intravenous administration of tissue plasminogen activator in APP23 mice produces increased cerebral amyloid angiopathy-associated microhemorrhages and can also generate subarachnoid and parenchymal hematomas. They concluded that tissue plasminogen activator use, in the presence of cerebral amyloid angiopathy, carries an increased risk for cerebral hemorrhage in mice. Moreover, they emphasized that more work is needed on tissue plasminogen activator-induced hemorrhage and cerebral amyloid angiopathy in elderly patients and in subjects with Alzheimer’s disease, in whom extensive cerebral amyloid angiopathy is quite common [111].
While management of hypertension and atherosclerosis can reduce the incidence of intracerebral hemorrhage, there are currently no approved therapies for attenuating cerebral amyloid angiopathy. Thus there is a critical need for new strategies that improve blood–brain barrier function and limit the development of cerebral amyloid angiopathy.
PDE Inhibitors, Stroke Prevention, and Mixed Cerebrovascular Disease
Mixed cerebrovascular disease represents the Scylla and Charybdis of modern stroke neurology. The stroke neurologist must navigate between the apparent extremes of ischemic and hemorrhagic processes. For the most part, the patient will come to the attention of the neurologist with symptoms suggesting ischemic stroke, and cerebral microbleeds and white matter disease will be identified incidentally. Given its relationship to hemorrhagic stroke, the presence of cerebral microbleeds is particularly challenging.
An attractive therapeutic strategy for mixed cerebrovascular disease is one that targets both the coagulation system and the vessel wall. Platelet agents may be focused on receptor antagonism, but inhibition of signal transduction pathways is an important alternative strategy for inhibition of platelet activation pathways. Inhibition of platelet signal transduction can be achieved by manipulation of platelet second messenger pathways and/or amplification of effects of endothelial-derived molecules (e.g., prostacyclin and nitric oxide) that activate cyclases producing elevated levels of intracellular cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) [112]. Importantly, cAMP pathways have well-described major roles in development of the blood–brain barrier [113].
Platelet levels of cyclic nucleotides have critical regulatory function, so that elevated levels of cAMP and cGMP interfere with all known platelet activation pathways [112]. Signaling of cyclic nucleotides is modulated by their hydrolysis by phosphodiesterases (PDEs), with the latter regulated by any of the more than 60 isoforms of the eleven families of PDE inhibitors [112]. Notably, the PDE inhibitors dipyridamole and cilostazol (Fig. 5) have been shown to have beneficial actions for ischemic stroke prevention [114–117]. However, neither drug has been considered a first-line agent for stroke prevention efforts.
Fig. 5.
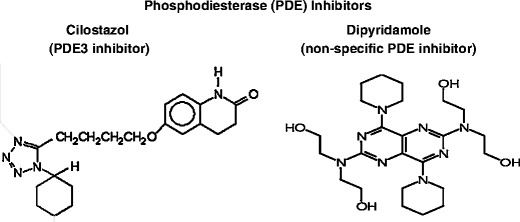
Two phosphodiesterase (PDE) inhibitors used for stroke prevention efforts, cilostazol, and dipyridamole
Dipyridamole, a relatively nonspecific PDE inhibitor with effects on both PDE3 and PDE5 [112], has been shown beneficial for the prevention of ischemic stroke, with stroke risk reduction comparable to that seen with aspirin [114]. Dipyridamole is known to have dual actions, combining platelet anti-aggregant effects and vessel wall protection [118]. Platelet actions are generated by adenosine-mediated effects, along with potentiation of the platelet anti-aggregatory effects of prostacyclin and nitric oxide [118]. Dipyridamole inhibits red blood cell uptake of adenosine, resulting in elevated plasma adenosine leading to stimulation of platelet adenylyl cyclase and increased platelet cAMP [112, 118]. Vessel wall protection effects of dipyridamole are produced via anti-oxidative effects and by inhibition of platelet–monocyte interactions [118].
Recent animal work has emphasized the potential for dipyridamole in stroke prevention as well as treatment. Acute administration of intravenous dipyridamole post-infarct produced reduction of infarct volume in a rat model of experimental stroke [119]. In a study particularly relevant for the treatment of mixed cerebrovascular disease, oral supplementation of dipyridamole, at clinically relevant doses, did not worsen cerebral microscopic hemorrhage in a mouse model of cerebral amyloid angiopathy [120]. This was observed even after accelerated development of microscopic hemorrhages using immunotherapy in aged transgenic animals [120].
PDE3 has high affinity for both cAMP and cGMP, but behaves as a relatively specific cAMP PDE due to its low hydrolysis efficacy for cGMP [112]. Cilostazol, a specific PDE3 inhibitor with both platelet and vessel wall effects, has been shown effective for prevention of ischemic stroke versus aspirin [112, 116, 117]. Cilostazol inhibits shear stress-induced platelet activation as well as collagen-, adenosine diphosphate (ADP)-, arachidonic acid-, and epinephrine-induced platelet aggregation [112]. Nevertheless, in a stroke prevention trial, hemorrhagic events (including intracerebral hemorrhage) were reduced by more than one half in cilostazol-treated patients compared to aspirin [117]. In a placebo-controlled stroke prevention trial, cilostazol-treated patients tended to have fewer intracranial hemorrhages than placebo (four versus seven) [115]. However, more work is needed in the evaluation of hemorrhagic events in clinical trials using cilostazol as well as dipyridamole.
Vessel wall effects of cilostazol have been shown in clinical and preclinical studies. These include reports of reduction of hemorrhagic conversion of ischemic events in murine models of experimental stroke, an effect observed with and without treatment with tissue plasminogen activator [121, 122]. In a clinical study of patients with intracranial atherosclerosis, cilostazol appeared to have beneficial effects against progression of the disorder [123]. Potential mechanisms of anti-atherogenic effects of cilostazol include modulation of expression of MMP-9 and tissue inhibitor of metalloproteinase-1 (TIMP-1) by monocytes-macrophages [124]. In vitro studies have shown enhancement of endothelial barrier function and protective effects against injury with cilostazol in preparations using human brain endothelial cells [125].
In summary, an attractive therapeutic strategy for mixed cerebrovascular disease is one that utilizes agents acting on both platelets and vessel wall. Several PDE inhibitors have those dual actions. Dipyridamole and cilostazol have already been shown effective in ischemic stroke prevention clinical trials and may be particularly useful for patients with mixed cerebrovascular disease.
Conclusion
Mixed cerebrovascular disease consists of ischemic and hemorrhagic phenomena, both clinically evident disease as well as subclinical processes. Ischemic stroke, subclinical infarct, and white matter disease of aging (or leukoaraiosis) combined with intracerebral hemorrhage and cerebral microbleeds constitute this entity. The incorporation of these processes into a single entity creates a novel concept in stroke diagnostics.
The treatment of mixed cerebrovascular disease presents the stroke clinician with a profound dilemma. What is the way out of this conundrum? Use of PDE inhibitors, combining platelet and vessel wall effects, provides one possible strategy. There are, no doubt, other approaches that will become increasingly obvious in the coming decades.
Acknowledgment
This study was supported by the National Institutes of Health (NIH) NS20989 (M.F.), NIH AG020241 (D.H.C.), NIH AG00538 (D.H.C.), NIH NS050895 (D.H.C.), and Alzheimer’s Association grant IIRG11-204835 (D.H.C.).
M.F. has received support from Boehringer-Ingelheim (research grants, speakers’ bureau, honoraria) and Otsuka Pharmaceutical (research grant and honoraria). D.H.C. has received support from Boehringer-Ingelheim (research grant).
Contributor Information
Mark Fisher, Phone: +1-714-4566856, FAX: +1-714-4566894, Email: mfisher@uci.edu.
Vitaly Vasilevko, Email: vvasilev@uci.edu.
David H. Cribbs, Email: cribbs@uci.edu
References
- 1.Fisher M. The challenge of mixed cerebrovascular disease. Ann NY Acad Sci. 2010;1207:18–22. doi: 10.1111/j.1749-6632.2010.05758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Longstreth WT, Manolio TA, Arnold A, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. Stroke. 1996;27:1274–1282. doi: 10.1161/01.STR.27.8.1274. [DOI] [PubMed] [Google Scholar]
- 3.Vernooij MW, Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: The Rotterdam scan study. Neurology. 2008;70:1208–1214. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 4.Cullen KM, Kocsi Z, Stone J. Pericapillary haem-rich deposits: Evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab. 2005;25:1656–1667. doi: 10.1038/sj.jcbfm.9600155. [DOI] [PubMed] [Google Scholar]
- 5.Fisher M, French S, Ji P, Kim RC. Cerebral microbleeds in the elderly: A pathological analysis. Stroke. 2010;41:2782–2785. doi: 10.1161/STROKEAHA.110.593657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He J, Whelton PK, Vu B, Klag MJ. Aspirin and risk of hemorrhagic stroke: A meta-analysis of randomized controlled trials. JAMA. 1998;280:1930–1935. doi: 10.1001/jama.280.22.1930. [DOI] [PubMed] [Google Scholar]
- 7.CAPRIE Steering Committee A randomized, blinded trial of clopidogrel versus aspirin in patients at risk of ischemic events (CAPRIE) Lancet. 1996;348:1329–1339. doi: 10.1016/S0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 8.Diener HC, Bogousslavsky J, Brass LM, et al. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): Randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:331–337. doi: 10.1016/S0140-6736(04)16721-4. [DOI] [PubMed] [Google Scholar]
- 9.Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706–1717. doi: 10.1056/NEJMoa060989. [DOI] [PubMed] [Google Scholar]
- 10.Flaherty ML. Anticoagulant-associated intracerebral hemorrhage. Semin Neurol. 2010;30:565–572. doi: 10.1055/s-0030-1268866. [DOI] [PubMed] [Google Scholar]
- 11.Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 12.Patel MR, Mahaffey KW, Garg J. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883–891. doi: 10.1056/NEJMoa1009638. [DOI] [PubMed] [Google Scholar]
- 13.The stroke prevention by aggressive reduction in cholesterol levels (SPARCL) investigators High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–559. doi: 10.1056/NEJMoa061894. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein LB, Amarenco P, Szarek M, Callahan A., 3rd Hennerici M, Sillesen H, Zivin JA, Welch KM, SPARCL investigators. Hemorrhagic stroke in the stroke prevention by aggressive reduction in cholesterol levels study. Neurology. 2008;70:2364–2370. doi: 10.1212/01.wnl.0000296277.63350.77. [DOI] [PubMed] [Google Scholar]
- 15.Oldgren J, Alings M, Darius H, Diener HC, Eikelboom J, Ezekowitz MD, Kamensky G, Reilly PA, Yang S, Yusuf S, Wallentin L, Connolly SJ. rely investigators. Risks for stroke, bleeding, and death in patients with atrial fibrillation receiving dabigatran or warfarin in relation to the CHADS2 score: A subgroup analysis of the RE-LY trial. Ann Intern Med. 2011;155:660–667. doi: 10.7326/0003-4819-155-10-201111150-00004. [DOI] [PubMed] [Google Scholar]
- 16.Koennecke HC. Cerebral microbleeds on MRI: Prevalence, associations, and potential clinical implications. Neurology. 2006;66:165–171. doi: 10.1212/01.wnl.0000194266.55694.1e. [DOI] [PubMed] [Google Scholar]
- 17.Fan YH, Zhang L, Lam WW, Mok VC, Wong KS. Cerebral microbleeds as a risk factor for subsequent intracerebral hemorrhages among patients with acute ischemic stroke. Stroke. 2003;34:2459–2462. doi: 10.1161/01.STR.0000090841.90286.81. [DOI] [PubMed] [Google Scholar]
- 18.Biffi A, Halpin A, Towfighi A, Gilson A, Busl K, Rost N, Smith EE, Greenberg SM, Rosand J, Viswanathan A. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology. 2010;75:693–698. doi: 10.1212/WNL.0b013e3181eee40f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong KS, Chang YL, Liu JY, Gao S, Lam WW. Asymptomatic microbleeds as a risk factor for aspirin-associated intracerebral hemorrhages. Neurology. 2003;60:511–513. doi: 10.1212/01.WNL.0000046583.40125.20. [DOI] [PubMed] [Google Scholar]
- 20.Vernooij MW, Haag MD, Lugt A, Hofman A, Krestin GP, Stricker BH, Breteler MM. Use of antithrombotic drugs and the presence of cerebral microbleeds: The Rotterdam Scan Study. Arch Neurol. 2009;66:714–720. doi: 10.1001/archneurol.2009.42. [DOI] [PubMed] [Google Scholar]
- 21.Ge L, Niu G, Han X, Gao Y, Wu Q, Wu H, Zhang Y, Guo D. Aspirin treatment increases the risk of cerebral microbleeds. Can J Neurol Sci. 2011;38:863–868. doi: 10.1017/s0317167100012440. [DOI] [PubMed] [Google Scholar]
- 22.Ueno H, Naka H, Ohshita T, Kondo K, Nomura E, Ohtsuki T, Kohriyama T, Wakabayashi S, Matsumoto M. Association between cerebral microbleeds on T2*-weighted MR images and recurrent hemorrhagic stroke in patients treated with warfarin following ischemic stroke. AJNR Am J Neuroradiol. 2008;29:1483–1486. doi: 10.3174/ajnr.A1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology. 2009;72:171–176. doi: 10.1212/01.wnl.0000339060.11702.dd. [DOI] [PubMed] [Google Scholar]
- 24.Orken DN, Kenangil G, Uysal E, Forta H. Cerebral microbleeds in ischemic stroke patients on warfarin treatment. Stroke. 2009;40:3638–3640. doi: 10.1161/STROKEAHA.109.559450. [DOI] [PubMed] [Google Scholar]
- 25.Lovelock CE, Cordonnier C, Naka H, Al-Shahi Salman R, Sudlow CL, Edinburgh Stroke Study Group. Sorimachi T, Werring DJ, Gregoire SM, Imaizumi T, Lee SH, Briley D, Rothwell PM. Antithrombotic drug use, cerebral microbleeds, and intracerebral hemorrhage: A systematic review of published and unpublished studies. Stroke. 2010;41:1222–1228. doi: 10.1161/STROKEAHA.109.572594. [DOI] [PubMed] [Google Scholar]
- 26.Smith EE, Rosand J, Knudsen KA, Hylek EM, Greenberg SM. Leukoaraiosis is associated with warfarin-related hemorrhage following ischemic stroke. Neurology. 2002;59:193–197. doi: 10.1212/WNL.59.2.193. [DOI] [PubMed] [Google Scholar]
- 27.Kato H, Izumiyama M, Izumiyama K, Takahashi A, Itoyama Y. Silent cerebral microbleeds on T2*-weighted MRI: Correlation with stroke subtype, stroke recurrence, and leukoaraiosis. Stroke. 2002;33:1536–1540. doi: 10.1161/01.STR.0000018012.65108.86. [DOI] [PubMed] [Google Scholar]
- 28.Lee SH, Bae HJ, Ko SB, Kim H, Yoon BW, Roh JK. Comparative analysis of the spatial distribution and severity of cerebral microbleeds and old lacunes. J Neurol Neurosurg Psychiatry. 2004;75:423–427. doi: 10.1136/jnnp.2003.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naka H, Nomura E, Wakabayashi S, Kajikawa H, Kohriyama T, Mimori Y, Nakamura S, Matsumoto M. Frequency of asymptomatic microbleeds on T2*-weighted MR images of patients with recurrent stroke: Association with combination of stroke subtypes and leukoaraiosis. AJNR Am J Neuroradiol. 2004;25:714–719. [PMC free article] [PubMed] [Google Scholar]
- 30.Maia LF, Vasconcelos C, Seixas S, Magalhães R, Correia M. Lobar brain hemorrhages and white matter changes: Clinical, radiological and laboratorial profiles. Cerebrovasc Dis. 2006;22:155–161. doi: 10.1159/000093245. [DOI] [PubMed] [Google Scholar]
- 31.Görner A, Lemmens R, Schrooten M, Thijs V. Is leukoaraiosis on CT an accurate surrogate marker for the presence of microbleeds in acute stroke patients? J Neurol. 2007;254:284–289. doi: 10.1007/s00415-006-0311-z. [DOI] [PubMed] [Google Scholar]
- 32.Gao T, Wang Y, Zhang Z. Silent cerebral microbleeds on susceptibility-weighted imaging of patients with ischemic stroke and leukoaraiosis. Neurol Res. 2008;30:272–276. doi: 10.1179/016164107X251556. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK. St George-Hyslop P, Rogaeva E, Black SE. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 34.Jeon SB, Kwon SU, Cho AH, Yun SC, Kim JS, Kang DW. Rapid appearance of new cerebral microbleeds after acute ischemic stroke. Neurology. 2009;73:1638–1644. doi: 10.1212/WNL.0b013e3181bd110f. [DOI] [PubMed] [Google Scholar]
- 35.Lee SH, Heo JH, Yoon BW. Effects of microbleeds on hemorrhage development In leukoaraiosis patients. Hypertens Res. 2005;28:895–899. doi: 10.1291/hypres.28.895. [DOI] [PubMed] [Google Scholar]
- 36.Yamada S, Saiki M, Satow T, Fukuda A, Ito M, Minami S, Miyamoto S. Periventricular and deep white matter leukoaraiosis have a closer association with cerebral microbleeds than age. Eur J Neurol. 2012;19:98–104. doi: 10.1111/j.1468-1331.2011.03451.x. [DOI] [PubMed] [Google Scholar]
- 37.Lee SH, Bae HJ, Yoon BW, Kim H, Kim DE, Roh JK. Low concentration of serum total cholesterol is associated with multifocal signal loss lesions on gradient-echo magnetic resonance imaging: Analysis of risk factors for multifocal signal loss lesions. Stroke. 2002;33:2845–2849. doi: 10.1161/01.STR.0000036092.23649.2E. [DOI] [PubMed] [Google Scholar]
- 38.Fisher M. Cerebral microbleeds and white matter disease: Separated at birth? Eur J Neurol. 2012;19:2–3. doi: 10.1111/j.1468-1331.2011.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attems J, Jellinger K, Thal DR, Nostrand W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- 40.Herzig MC, Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloidangiopathy: Murine and cellular models. Brain Pathol. 2006;16:40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 43.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 44.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. Neuroscience. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflamm. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrushina I, Ghochikyan A, Mkrtichyan M, Mamikonyan G, Movsesyan N, Ajdari R, Vasilevko V, Karapetyan A, Lees A, Agadjanyan MG, Cribbs DH. Mannan-Abeta28 conjugate prevents Abeta-plaque deposition, but increases microhemorrhages in the brains of vaccinated Tg2576 (APPsw) mice. J Neuroinflammation. 2008;5:42. doi: 10.1186/1742-2094-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neuroll Disord Drug Targets. 2009;8:50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nature Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 50.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 51.Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/S0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo F, Rustay NR, Seifert T, Roesner B, Hradil V, Hillen H, Ebert U, Severin JM, Cox BF, Llano DA, Day M, Fox GB. Magnetic resonance imaging detection and time course of cerebral microhemorrhages during passive immunotherapy in living amyloid precursor protein transgenic mice. J Pharmacol Exp Ther. 2010;335:580–588. doi: 10.1124/jpet.110.172932. [DOI] [PubMed] [Google Scholar]
- 53.Beckman N, Gerard C, Abramowski D, Cannet C, Staufenbiel M. Noninvasive magnetic resonance imaging detection of cerebral amyloid angiopathy-related microvascular alterations using superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer’s disease: application to passive Abeta immunotherapy. J Neurosci. 2011;31:1023–1031. doi: 10.1523/JNEUROSCI.4936-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, Abbot R, Godwin J, Dyer A, Stamler J. Blood pressure, stroke, and coronary heart disease. Part 1. Prolonged differences in blood pressure: Prospective observational studies corrected for the regression dilution bias. Lancet. 1990;335:765–774. doi: 10.1016/0140-6736(90)90878-9. [DOI] [PubMed] [Google Scholar]
- 55.Napoli M, Papa F. Systemic inflammation, blood pressure, and stroke outcome. J Clin Hypertens (Greenwich) 2006;8:187–194. doi: 10.1111/j.1524-6175.2005.04590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert JJ, Vinters HV. Cerebral amyloid angiopathy: incidence and complications in the aging brain: I. Cerebral hemorrhage. Stroke. 1983;14:915–923. doi: 10.1161/01.STR.14.6.915. [DOI] [PubMed] [Google Scholar]
- 57.Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology. 1998;51:690–694. doi: 10.1212/WNL.51.3.690. [DOI] [PubMed] [Google Scholar]
- 58.Havlik RJ, Foley DJ, Sayer B, Masaki K, White L, Launer LJ. Variability in midlife systolic blood pressure is related to late-life brain white matter lesions: the Honolulu–Asia Aging study. Stroke. 2002;33:26–30. doi: 10.1161/hs0102.101890. [DOI] [PubMed] [Google Scholar]
- 59.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 60.Ogata J, Yamanishi H, Ishibashi-Ueda H. Review: Role of cerebral vessels in ischaemic injury of the brain. Neuropathol Appl Neurobiol. 2011;37:40–55. doi: 10.1111/j.1365-2990.2010.01141.x. [DOI] [PubMed] [Google Scholar]
- 61.Dickstein DL, Walsh J, Brautigam H, Stockton SD, Jr, Gandy S, Hof PR. Role of vascular risk factors and vascular dysfunction in Alzheimer’s disease. Mt Sinai J Med. 2010;77:82–102. doi: 10.1002/msj.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ. Midlife blood pressure and dementia: the Honolulu–Asia aging study. Neurobiol Aging. 2000;21:49–55. doi: 10.1016/S0197-4580(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 63.Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular risk factors and late-life mild cognitive impairment: a population-based study. Neurology. 2001;56:683–1689. doi: 10.1212/WNL.56.12.1683. [DOI] [PubMed] [Google Scholar]
- 64.Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64:277–281. doi: 10.1212/01.WNL.0000149519.47454.F2. [DOI] [PubMed] [Google Scholar]
- 65.Qiu C, Xu W, Winblad B, Fratiglioni L. Vascular risk profiles for dementia and Alzheimer’s disease in very old people: a population-based longitudinal study. J Alzheimers Dis. 2010;20:293–300. doi: 10.3233/JAD-2010-1361. [DOI] [PubMed] [Google Scholar]
- 66.Qiu C, Winblad B, Fratiglioni L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005;4:487–499. doi: 10.1016/S1474-4422(05)70141-1. [DOI] [PubMed] [Google Scholar]
- 67.Leeuw FE, Richard F, Groot JC, Duijn M, Hofman A, Gijn J, Breteler MM. Interaction between hypertension, apoE, and cerebral white matter lesions. Stroke. 2004;35:1057–1060. doi: 10.1161/01.STR.0000125859.71051.83. [DOI] [PubMed] [Google Scholar]
- 68.Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension is related to cognitive impairment: a 20-year follow-up of 999 men. Hypertension. 1998;31:780–786. doi: 10.1161/01.hyp.31.3.780. [DOI] [PubMed] [Google Scholar]
- 69.Launer LJ, Masaki K, Petrovitch H, Foley D, Havlik RJ. The association between midlife blood pressure levels and late-life cognitive function. The Honolulu–Asia aging study. JAMA. 1995;274:1846–1851. doi: 10.1001/jama.1995.03530230032026. [DOI] [PubMed] [Google Scholar]
- 70.Korf ES, White LR, Scheltens P, Launer LJ. Midlife blood pressure and the risk of hippocampal atrophy: The Honolulu Asia aging study. Hypertension. 2004;44:29–34. doi: 10.1161/01.HYP.0000132475.32317.bb. [DOI] [PubMed] [Google Scholar]
- 71.Heijer T, Launer LJ, Prins ND, Dijk EJ, Vermeer SE, Hofman A, Koudstaal PJ, Breteler MM. Association between blood pressure, white matter lesions, and atrophy of the medial temporal lobe. Neurology. 2005;64:263–267. doi: 10.1212/01.WNL.0000149641.55751.2E. [DOI] [PubMed] [Google Scholar]
- 72.DeCarli C, Miller BL, Swan GE, Reed T, Wolf PA, Garner J, Jack L, Carmelli D. Predictors of brain morphology for the men of the NHLBI twin study. Stroke. 1999;30:529–536. doi: 10.1161/01.STR.30.3.529. [DOI] [PubMed] [Google Scholar]
- 73.Petrovitch H, White LR, Izmirilian G, Ross GW, Havlik RJ, Markesbery W, Nelson J, Davis DG, Hardman J, Foley DJ, Launer LJ. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu–Asia aging study. Neurobiol Aging. 2000;21:57–62. doi: 10.1016/s0197-4580(00)00106-8. [DOI] [PubMed] [Google Scholar]
- 74.Marchesi C, Paradis P, Schiffrin EL. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 75.Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–858. doi: 10.1016/j.neuroscience.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 76.Nabika T, Cui Z, Masuda J. The stroke-prone spontaneously hypertensive rat: how good is it as a model for cerebrovascular diseases? Cell Mol Neurobiol. 2004;24:639–646. doi: 10.1023/B:CEMN.0000036402.79129.2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hainsworth AH, Markus HS. Do in vivo experimental models reflect human cerebral small vessel disease? A systematic review. J Cereb Blood Flow Metab. 2008;28:1877–1891. doi: 10.1038/jcbfm.2008.91. [DOI] [PubMed] [Google Scholar]
- 78.Southard T, Brayton CF. Spontaneous unilateral brainstem infarction in Swiss mice. Vet Pathol. 2011;48:726–729. doi: 10.1177/0300985810370155. [DOI] [PubMed] [Google Scholar]
- 79.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koistinaho M, Kettunen MI, Goldsteins G, Keinänen R, Salminen A, Ort M, Bures J, Liu D, Kauppinen RA, Higgins LS, Koistinaho J. Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc Natl Acad Sci U S A. 2002;99:1610–1615. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qin Z. Newly developed angiotensin II-infused experimental models in vascular biology. Regul Pept. 2008;150:1–6. doi: 10.1016/j.regpep.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 82.Wakisaka Y, Chu Y, Miller JD, Rosenberg GA, Heistad DD. Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab. 2010;30:56–69. doi: 10.1038/jcbfm.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iida S, Baumbach GL, Lavoie JL, Faraci FM, Sigmund CD, Heistad DD. Spontaneous stroke in a genetic model of hypertension in mice. Stroke. 2005;36:1253–1258. doi: 10.1161/01.str.0000167694.58419.a2. [DOI] [PubMed] [Google Scholar]
- 84.Wakisaka Y, Miller JD, Chu Y, Baumbach GL, Wilson S, Faraci FM, Sigmund CD, Heistad DD. Oxidative stress through activation of NAD(P)H oxidase in hypertensive mice with spontaneous intracranial hemorrhage. J Cereb Blood Flow Metab. 2008;28:1175–1185. doi: 10.1038/jcbfm.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol. 2002;13:2860–2868. doi: 10.1097/01.ASN.0000035087.11758.ED. [DOI] [PubMed] [Google Scholar]
- 86.Welch WJ, Chabrashvili T, Solis G, Chen Y, Gill PS, Aslam S, Wang X, Ji H, Sandberg K, Jose P, Wilcox CS. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension. 2006;48:934–941. doi: 10.1161/01.HYP.0000242928.57344.92. [DOI] [PubMed] [Google Scholar]
- 87.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2003;284:R893–R912. doi: 10.1152/ajpregu.00491.2002. [DOI] [PubMed] [Google Scholar]
- 88.Gomis M, Sobrino T, Ois A, Millan M, Rodriguez-Campello A. Perez de la Ossa N, Rodriguez-Gonzalez R, Jimenez-Conde J, Cuadrado-Godia E, Roquer J, Davalos A. Plasma beta-amyloid 1-40 is associated with the diffuse small vessel disease subtype. Stroke. 2009;40:3197–3201. doi: 10.1161/STROKEAHA.109.559641. [DOI] [PubMed] [Google Scholar]
- 89.Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: role of inflammation. Neurol Res. 2003;25:642–651. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- 90.Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M. Proinflammatory and vasoactive effects of Abeta in the cerebrovasculature. Ann N Y Acad Sci. 2002;977:65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x. [DOI] [PubMed] [Google Scholar]
- 91.Deane R, Wu Z, Sagare A, Davis J, Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 92.Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 93.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH. Weller RO Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 94.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clapham R, O’Sullivan E, Weller RO, Carare RO. Cervical lymph nodes are found in direct relationship with the internal carotid artery: significance for the lymphatic drainage of the brain. Clin Anat. 23:43–7. [DOI] [PubMed]
- 96.Wang X, Lo EH. Triggers and mediators of hemorrhagic transformation in cerebral ischemia. Mol Neurobiol. 2003;28:229–244. doi: 10.1385/MN:28:3:229. [DOI] [PubMed] [Google Scholar]
- 97.Pfeilschifter W, Spitzer D, Pfeilschifter J, Steinmetz H, Foerch C. Warfarin anticoagulation exacerbates the risk of hemorrhagic transformation after rt-PA treatment in experimental stroke: therapeutic potential of PCC. PLoS One. 2011;6:e26087. Epub 2011 Oct 19. [DOI] [PMC free article] [PubMed]
- 98.Pfeilschifter W, Bohmann F, Baumgarten P, Mittelbronn M, Pfeilschifter J, Lindhoff-Last E, Steinmetz H, Foerch C. Thrombolysis with recombinant tissue plasminogen activator under dabigatran anticoagulation in experimental stroke. Ann Neurol. 2012 Feb 10. doi:10.1002/ana.23558. [Epub ahead of print] [DOI] [PubMed]
- 99.Del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, Wang X, Hosomi N, Mabuchi T, Koziol JA. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab. 2012 Feb 22. doi:10.1038/jcbfm.2012.11. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 100.Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- 101.Guan W, Kozak A, Fagan SC. Drug repurposing for vascular protection after acute ischemic stroke. Acta Neurochir Suppl. 2011;111:295–298. doi: 10.1007/978-3-7091-0693-8_49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke. 2008;39:3372–3377. doi: 10.1161/STROKEAHA.108.514026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bowes MP, Zivin JA, Thomas GR, Thibodeaux H, Fagan SC. Acute hypertension, but not thrombolysis, increases the incidence and severity of hemorrhagic transformation following experimental stroke in rabbits. Exp Neurol. 1996;141:40–46. doi: 10.1006/exnr.1996.0137. [DOI] [PubMed] [Google Scholar]
- 104.Fagan SC, Bowes MP, Lyden PD, Zivin JA. Acute hypertension promotes hemorrhagic transformation in a rabbit embolic stroke model: effect of labetalol. Exp Neurol. 1998;150:153–158. doi: 10.1006/exnr.1997.6756. [DOI] [PubMed] [Google Scholar]
- 105.Tejima E, Katayama Y, Suzuki Y, Kano T, Lo EH. Hemorrhagic transformation after fibrinolysis with tissue plasminogen activator: evaluation of role of hypertension with rat thromboembolic stroke model. Stroke. 2001;32:1336–1340. doi: 10.1161/01.STR.32.6.1336. [DOI] [PubMed] [Google Scholar]
- 106.Henning EC, Latour LL, Hallenbeck JM, Warach S. Reperfusion-associated hemorrhagic transformation in SHR rats: Evidence of symptomatic parenchymal hematoma. Stroke. 2008;39:3405–3410. doi: 10.1161/STROKEAHA.108.520304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: Relation to blood–brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Elgebaly MM, Ogbi S, Li W, Mezzetti EM, Prakash R, Johnson MH, Bruno A, Fagan SC, Ergul A. Neurovascular injury in acute hyperglycemia and diabetes: A comparative analysis in experimental stroke. Transl Stroke Res. 2011;2:391–398. doi: 10.1007/s12975-011-0083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xing Y, Jiang X, Yang Y, Xi G. Hemorrhagic transformation induced by acute hyperglycemia in a rat model of transient focal ischemia. Acta Neurochir Suppl. 2011;111:49–54. doi: 10.1007/978-3-7091-0693-8_9. [DOI] [PubMed] [Google Scholar]
- 110.Hu Q, Ma Q, Zhan Y, He Z, Tang J, Zhou C, Zhang J. Isoflurane enhanced hemorrhagic transformation by impairing antioxidant enzymes in hyperglycemic rats with middle cerebral artery occlusion. Stroke. 2011;42:1750–1756. doi: 10.1161/STROKEAHA.110.603142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Winkler DT, Biedermann L, Tolnay M, Allegrini PR, Staufenbiel M, Wiessner C, Jucker M. Thrombolysis induces cerebral hemorrhage in a mouse model of cerebral amyloid angiopathy. Ann Neurol. 2002;51:790–793. doi: 10.1002/ana.10210. [DOI] [PubMed] [Google Scholar]
- 112.Gresele P, Momi S, Falcinelli E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634–646. doi: 10.1111/j.1365-2125.2011.04034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, et al. A cell culture model of the blood–brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European stroke prevention study: 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci. 1996;143:1–13. doi: 10.1016/S0022-510X(96)00308-5. [DOI] [PubMed] [Google Scholar]
- 115.Gotoh F, Tohgi H, Hirai S, Terashi A, Fukuuchi Y, Otomo E, Shinohara Y, Itoh E, Matsuda T, Sawada T, Yamaguchi T, Nishimaru K, Ohashi Y. Cilostazol stroke prevention study: A placebo-controlled double-blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis. 2000;9:147–157. doi: 10.1053/jscd.2000.7216. [DOI] [PubMed] [Google Scholar]
- 116.Huang Y, Cheng Y, Wu J, Li Y, Xu E, Hong Z, Li Z, Zhang W, Ding M, Gao X, Fan D, Zeng J, Wong K, Lu C, Xiao J, Yao C. Cilostazol versus aspirin for secondary ischaemic stroke prevention cooperation investigators. Cilostazol as an alternative to aspirin after ischaemic stroke: a randomized, double-blind, pilot study. Lancet Neurol. 2008;7:494–499. doi: 10.1016/S1474-4422(08)70094-2. [DOI] [PubMed] [Google Scholar]
- 117.Shinohara Y, Katayama Y, Uchiyama S, Yamaguchi T, Handa S, Matsuoka K, Ohashi Y, Tanahashi N, Yamamoto H, Genka C, Kitagawa Y, Kusuoka H, Nishimaru K, Tsushima M, Koretsune Y, Sawada T, Hamada C. CSPS 2 group. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol. 2010;9:959–968. doi: 10.1016/S1474-4422(10)70198-8. [DOI] [PubMed] [Google Scholar]
- 118.Kim HH, Liao JK. Translational therapeutics of dipyridamole. Arterioscler Thromb Vasc Biol. 2008;28:s39–s42. doi: 10.1161/ATVBAHA.107.160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garcia-Bonilla L, Sosti V, Campos M, Penalba A, Boada C, Sumalla M, et al. Effects of acute post-treatment with dipyridamole in a rat model of focal cerebral ischemia. Brain Res. 2011;1373:211–220. doi: 10.1016/j.brainres.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 120.Fisher M, Vasilevko V, Passos GF, Ventura C, Quiring D, Cribbs DH. Therapeutic modulation of cerebral microhemorrhage in a mouse model of cerebral amyloid angiopathy. Stroke. 2011;42:3300–3303. doi: 10.1161/STROKEAHA.111.626655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hase Y, Okamoto Y, Fujita Y, Kitamura A, Nakabayashi H, Ito H, Maki T, Washida K, Takahashi R, Ihara M. Cilostazol, a phosphodiesterase inhibitor, prevents no-reflow and hemorrhage in mice with focal cerebral ischemia. Exp Neurol. 2012;233:523–533. doi: 10.1016/j.expneurol.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 122.Kasahara Y, Nakagomi T, Matsuyama T, Stern D, Taguchi A. Cilostazol reduces the risk of hemorrhagic infarction after administration of tissue-type plasminogen activator in a murine stroke model. Stroke. 2012;43:499–506. doi: 10.1161/STROKEAHA.111.635417. [DOI] [PubMed] [Google Scholar]
- 123.Kato T, Sakai H, Takagi T. Nishimura Y. AJNR Am J Neuroradiol: Cilostazol prevents progression of asymptomatic carotid artery stenosis in patients with contralateral carotid artery stenting; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chuang SY, Yang SH, Chen TY, Pang JH. Cilostazol inhibits matrix invasion and modulates the gene expressions of MMP-9 and TIMP-1 in PMA-differentiated THP-1 cells. Eur J Pharmacol. 2011;670:419–426. doi: 10.1016/j.ejphar.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 125.Liu S, Yang F, Yu C, Paganini-Hill A, Fisher M. Phosphodiesterase inhibitors modulate human brain microvascular endothelial cell barrier properties and response to injury. Ann Neurol. 2011;70(Suppl 15):S6. [Google Scholar]