

Abstract
G protein-coupled chemokine receptors and their peptidergic ligands are interesting therapeutic targets due to their involvement in various immune-related diseases, including rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, chronic obstructive pulmonary disease, HIV-1 infection and cancer. To tackle these diseases, a lot of effort has been focused on discovery and development of small-molecule chemokine receptor antagonists. This has been rewarded by the market approval of two novel chemokine receptor inhibitors, AMD3100 (CXCR4) and Maraviroc (CCR5) for stem cell mobilization and treatment of HIV-1 infection respectively. The recent GPCR crystal structures together with mutagenesis and pharmacological studies have aided in understanding how small-molecule ligands interact with chemokine receptors. Many of these ligands display behaviour deviating from simple competition and do not interact with the chemokine binding site, providing evidence for an allosteric mode of action. This review aims to give an overview of the evidence supporting modulation of this intriguing receptor family by a range of ligands, including small molecules, peptides and antibodies. Moreover, the computer-assisted modelling of chemokine receptor–ligand interactions is discussed in view of GPCR crystal structures. Finally, the implications of concepts such as functional selectivity and chemokine receptor dimerization are considered.
LINKED ARTICLES
This article is part of a themed section on the Molecular Pharmacology of G Protein-Coupled Receptors (GPCRs). To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.165.issue-6. To view the 2010 themed section on the same topic visit http://onlinelibrary.wiley.com/doi/10.1111/bph.2010.159.issue-5/issuetoc
Keywords: allosterism, G protein-coupled receptors, chemokine receptors, functional selectivity, dimerization, crystal structures, nanobody, ligand interactions, binding sites
Chemokines and their receptors
Chemokines and their receptors are key players in the immune defence by directing and controlling the migration, activation, differentiation and survival of the billion of leukocytes in our body (Viola and Luster, 2008). Some chemokine peptides are constitutively secreted in lymphoid tissues and involved in leukocyte homing during immune surveillance. The vast majority of chemokines, however, are secreted in response to inflammatory mediators or trauma, and function as paracrine chemoattractants to recruit leukocytes to sites of inflammation. To date, at least 45 chemokine subtypes have been identified in human, which are categorized into four classes (i.e. C, CC, CXC, CX3C) on the basis of the number and spacing of conserved cysteine residues in their N-termini (Figure 1) (Alexander et al., 2011). All chemokines share a similar tertiary protein fold that is stabilized by disulfide bonds between the four conserved cysteine residues (or two in the case of C-chemokines). The flexible N-terminus is followed by the C, CC, CXC or CX3C motif, and connected via an exposed N-loop to a highly structured core domain, which consists of a single-turn 310 helix, three antiparallel β-strands and a C-terminal α-helix (Figure 2). Soluble chemokines bind via their C-terminal α-helix to glycosaminoglycans on the surface of endothelial cells to form an immobilized chemotactic gradient, which guides passing immune cells towards the source of chemokine secretion (Proudfoot et al., 2003; Lau et al., 2004). On the other hand, CXCL16 and CX3CL1 are initially expressed as membrane-bound chemokines to serve as adhesion molecules for cells that express CXCR6 or CX3CR1 respectively, but can be cleaved by ADAM enzymes to become soluble chemokines (Schulte et al., 2007). Chemokines can form dimers as well as oligomers, which is essential for their in vivo but not their in vitro activity (Laurence et al., 2000; Proudfoot et al., 2003; Jin et al., 2007).
Figure 1.
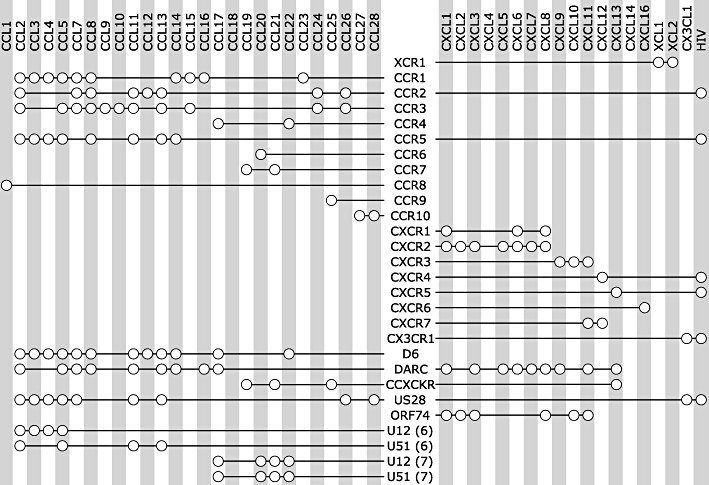
Overview of chemokine receptors (vertical) and their ligands (horizontal). An open circle indicates the specific ligand binds to the receptor connected to it by a black line. From top to bottom: human chemokine receptors (XCR1 to CX3CR1), chemokine decoy receptors (D6 to CCX CKR) and viral chemokine receptors (US28 to U51). For reasons of clarity, this figure does not show differences in affinity or activity between ligands for the same receptor. For more detailed information on ligand properties, visit the website of The International Union of Basic and Clinical Pharmacology (IUPHAR) (Murphy et al., 2011).
Figure 2.
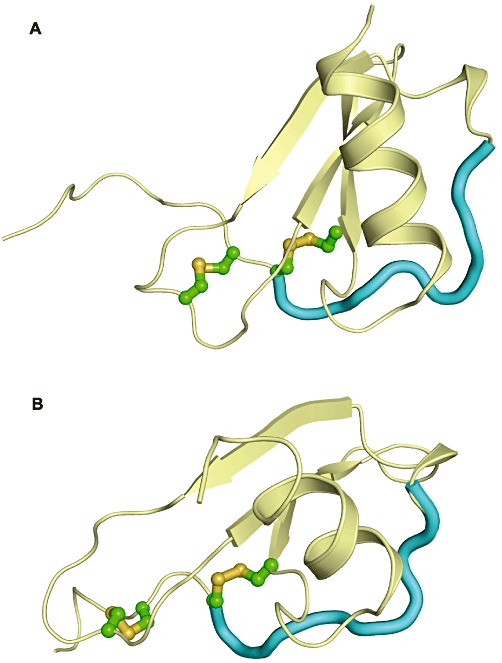
Overall tertiary structure of chemokines. (A) Structure of a CC-chemokine (CCL5). (B) Structure of a CXC-family chemokine (CXCL12). The disulfide bonds are indicated in green and yellow. The N-loop is shown in cyan. See text for more details.
Chemokine receptors are seven-transmembrane (7TM) receptors belonging to the superfamily of GPCRs (see below). The majority of chemokine receptors can bind a panel of chemokines, whereas some are highly specific (Figure 1). Chemokine receptors have been classified according to which chemokine subclass they bind, with one C, ten CC, seven CXC and one CX3C chemokine-binding receptors (Figure 1) (nomenclature follows Alexander et al., 2011). With the exception of CXCR7, which is exclusively biased towards β-arrestin-mediated signalling (Rajagopal et al., 2010), all C, CC, CXC and CX3C chemokine receptors signal at least through heterotrimeric G proteins (O'Hayre et al., 2008). Three non-G protein-signalling (so-called decoy) chemokine receptors (i.e. D6, DARC and CCX CKR) are thought to be primarily involved in scavenging a (wide) variety of inflammatory chemokines from the extracellular microenvironment, thereby limiting the recruitment of leukocytes (Mantovani et al., 2006). Differential expression of chemokine receptors on selective leukocyte (sub)populations allows these cells to sense and respond to local gradients of corresponding chemokines.
Inappropriate or prolonged expression of chemokines and/or chemokine receptors results in an excessive infiltration of (certain) leukocytes into (inflamed) source tissue or confers chemokine sensitivity to cells that are normally not responsive to chemokines, respectively, resulting in chronic inflammation, autoimmune diseases, tumour growth, survival and metastasis (Figure 3) (Balkwill, 2004; O'Hayre et al., 2008; Kraneveld et al., 2011). In addition, the CCR5 and CXCR4 function as co-receptors for HIV entry into CD4+ cells (Deng et al., 1996; Dragic et al., 1996; Feng et al., 1996).
Figure 3.

Chemokine receptors, their cellular expression profile and association with disease. The cellular expression profiles of the chemokine receptors are shown by open circles on the left-hand side. The cells are divided in haematopoietic cells and miscellaneous cells. Meaning of abbreviations: VSMC, vascular smooth muscle cells; VE, vascular endothelial cells; LE, lymphatic endothelial cells; BSMC, bronchial smooth muscle cells. The association of a particular receptor with disease is shown on the right-hand side of the figure. The diseases are categorized into inflammatory diseases and cancer. Abbreviations: IBD, inflammatory bowel disease; COPD, chronic obstructive pulmonary disease.
Many humans are latently infected by one or more herpesvirus species. Many of these herpesviruses encode GPCRs that are constitutively active but also responsive to human chemokines. These virally encoded chemokine receptors are thought to contribute to immune evasion and viral dissemination, but some of them are also involved in the development and/or progression of herpesvirus-associated inflammatory diseases and cancer (Figures 1 and 3) (Maussang et al., 2009; Bongers et al., 2010; Slinger et al., 2010).
Considering the key role of (viral) chemokine receptors in the pathogenesis of autoimmune and inflammatory diseases, cancer and HIV infection, it is not surprising that these receptors gained increasing attention the last decade by both academia and pharmaceutical industry in their quest to develop drugs to treat such diseases.
Redundancy or (functional) selectivity?
The chemokine system is often ‘accused’ of showing significant redundancy, due to the fact that a single receptor binds multiple ligands, and conversely, a single ligand can bind several chemokine receptors (Figure 1) (Allen et al., 2007). However, differential spatio-temporal expression patterns for different chemokines and receptors in our body indicate that they probably have distinct roles in vivo (Mantovani, 1999) (Figure 3). Furthermore, heteromerization of chemokine receptors may enable selective fine-tuning of chemokine receptor signalling (see section on ‘cross-modulation within chemokine receptor oligomers’). Moreover, activation of a single receptor by different agonists might lead to differential signalling or functional selectivity, as observed now for different chemokine receptors, including CCR1, CCR2, CCR5, CCR7 and CXCR3 (Oppermann et al., 1999; Kohout et al., 2004; Tian et al., 2004; Kouroumalis et al., 2005; Leach et al., 2007; Berchiche et al., 2010). For example, both CCR7 chemokines CCL19 and CCL21 promote [35S]-GTPγS binding and calcium signalling with similar potencies. However, only CCL19 induced efficient phosphorylation, β-arrestin recruitment and receptor internalization (Kohout et al., 2004). A more recent study showed that CCR7 engages different GPCR kinases (GRKs) in response to CCL19 or CCL21, explaining the observed differences in receptor regulation (Zidar et al., 2009). As a consequence, the signalling outcome does not depend on the receptor and ligand alone, but also on the expression levels of different signalling proteins in a specific cell, giving texture to the responses mediated by ligands. Along these lines, growing evidence supports differential binding of chemokines to a single receptor, and a single chemokine to different receptors, suggesting that these ligands have distinct roles (Ahuja et al., 1996; Pakianathan et al., 1997; Cox et al., 2001; Blanpain et al., 2003; Casarosa et al., 2005; Jensen et al., 2008). Overall, the evidence seems to refute the notion of redundancy in the chemokine receptor system. It rather indicates that the interplay between the timing and location of expression of ligands/receptors in the body, in combination with functional selectivity, is a mechanism for selectivity within the chemokine receptor family.
The two-step model of chemokine-receptor activation
The binding interactions of endogenous ligands in some class A GPCRs, such as the aminergic receptors, are relatively well known, especially with the recent successful crystallization of the β1- and β2-adrenoceptor (ADRB1/2), adenosine A2A, and the dopamine D3 receptor (DRD3) (Cherezov et al., 2007; Jaakola et al., 2008; Warne et al., 2008; Chien et al., 2010). In contrast, binding modes of peptide ligands, such as chemokines, are less well characterized, due to their relatively large size and associated challenges in obtaining structural information. However, several studies have highlighted important regions in both chemokines and receptors that are involved in binding and function. The interaction of chemokines with their receptors is generally considered to be a two-step process (Allen et al., 2007). First, the chemokine binds with its core region, including the N-loop (binding domain), to the N-terminus and extracellular loops (ELs) of the receptor (Figure 4B). We propose to use the term chemokine recognition site 1 (CRS1), instead of site I often used in the literature, to avoid confusion with binding sites in the transmembrane (TM) pockets for small molecules. The binding to CRS1 is dominated by ionic interactions between positively charged residues in the chemokine and negatively charged amino acids at the N-terminus and extracellular surface of the receptor, including sulfonated tyrosines (Fernandez and Lolis, 2002; Colvin et al., 2006; Allen et al., 2007; Veldkamp et al., 2008). In the second step, the flexible N-terminus (triggering domain) of the chemokine is positioned in such a way that it interacts with a second site (CRS2), formed by parts of the ELs and/or TM domains, resulting in receptor activation (Figure 4C) (Monteclaro and Charo, 1996; Wu et al., 1996; Pease et al., 1998; Gupta et al., 2001; Blanpain et al., 2003; Xanthou et al., 2003; Ott et al., 2004; Rajagopalan and Rajarathnam, 2004). This is supported by truncations or mutations in the N-termini of chemokines, generally leading to a loss in agonist activity, while often retaining high receptor binding affinity (Gong and Clark-Lewis, 1995; Proudfoot et al., 1996; Clark-Lewis et al., 2003; Richter et al., 2009). In the case of CCR5, several reports indicate that a TXP motif in TM2 and surrounding aromatic residues in TM2 and 3 are involved in chemokine-mediated activation of CCR5, but not in high-affinity binding, suggesting that in step two the N-terminus interacts with residues in this TM region (Govaerts et al., 2001; 2003; Blanpain et al., 2003). As this motif is conserved among chemokine receptors, it is hypothesized that the TM2–TM3 interface in these receptors takes part in a common mechanism of ligand-induced conformational rearrangements leading to movements of helices, notably TM2 and TM3, and thereby chemokine receptor activation. For CXCR4, different studies have demonstrated that the core region of its ligand CXCL12 binds to the extracellular regions of CXCR4, while the N-terminus has additional interactions with TM residues, including D972.63 and E2887.39 in TM2 and TM7 respectively (Kofuku et al., 2009). Ballesteros–Weinstein numbering is used in superscript throughout the text to enable the comparison of residue positions between receptors (Ballesteros and Weinstein, 1995) (Table 1). Another recently described numbering scheme is used for residues in EL2 (de Graaf et al., 2008).
Figure 4.
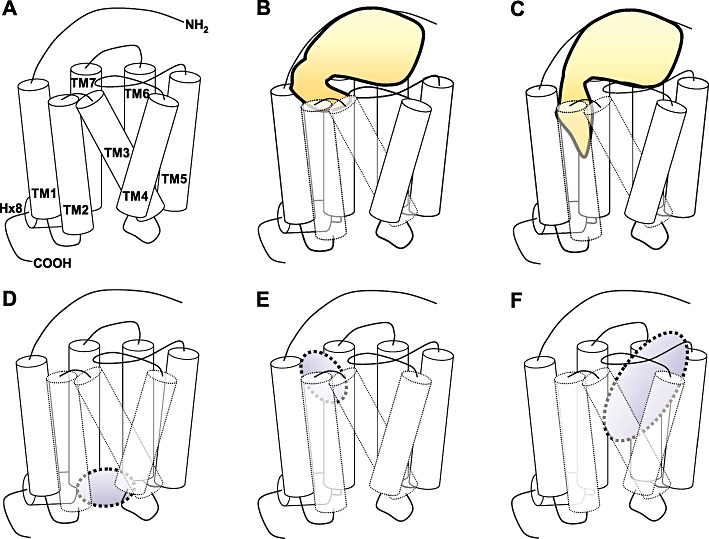
Schematic overview of GPCRs, their activation and ligand-binding sites. (A) The overall topology of a GPCR structure, with annotated TM helices, and N- and C-terminus. (B,C) Schematic representation of the two-step model of chemokine receptor activation. (B) The first step: chemokine binding to the extracellular surface of the chemokine receptor, including the N-terminus (CRS1). (C) The second step: the flexible N-terminus of the chemokine is positioned to interact with EL and TM residues (CRS2), mediating receptor activation. (D) Intracellular allosteric binding pocket used by CXCR2 and CCR4 ligands. Also the G protein binds in this region. (E) Binding pocket for small-molecule ligands in the chemokine receptor between TM1, 2, 3 and 7 (TMS1). (F) Binding pocket for small-molecule ligands in chemokine receptors between TM3, 4, 5, 6 and 7 (TMS2). Table 1 and Figure 5 give an overview of the interaction residues of small ligands binding to chemokine receptors.
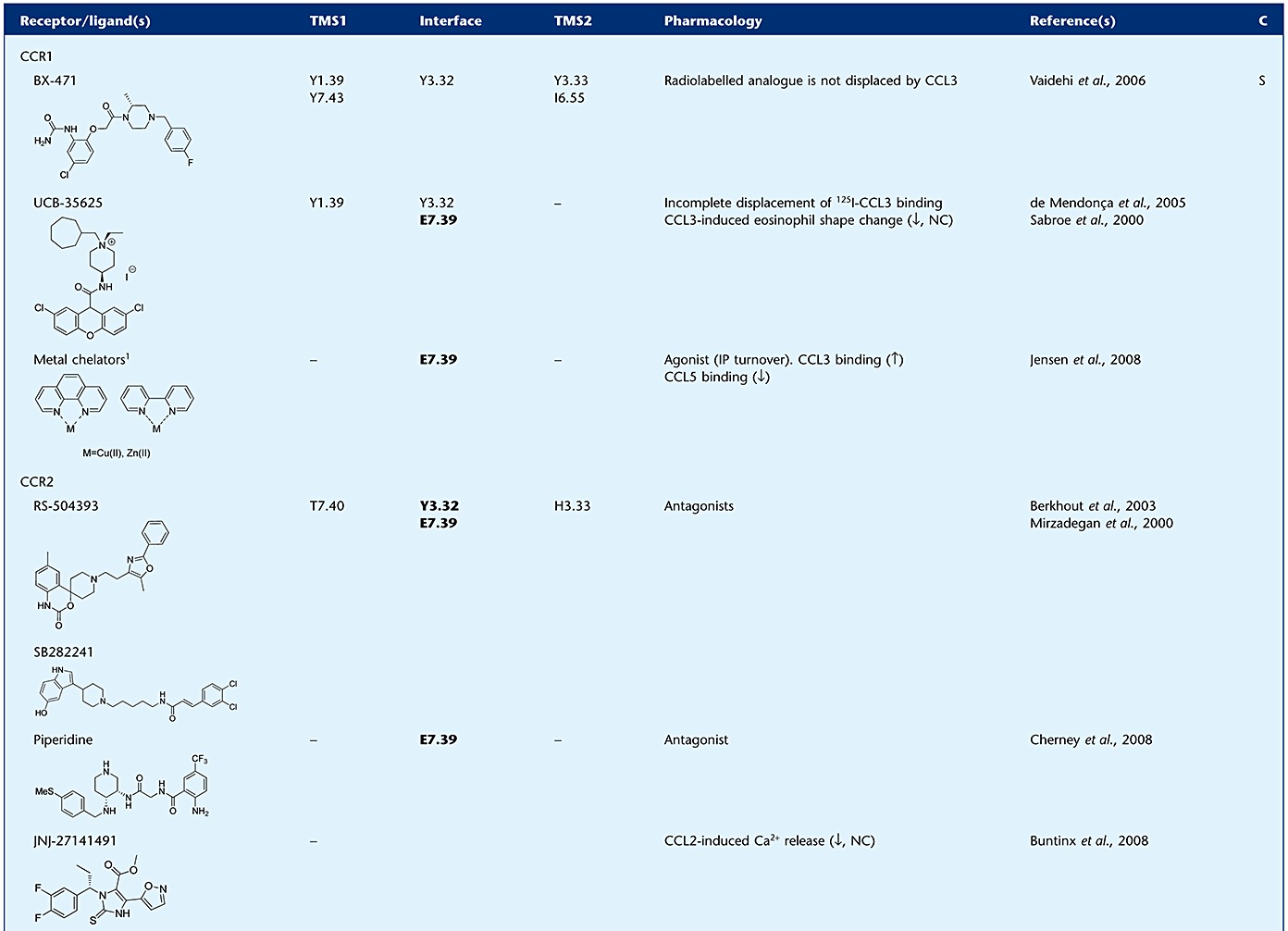
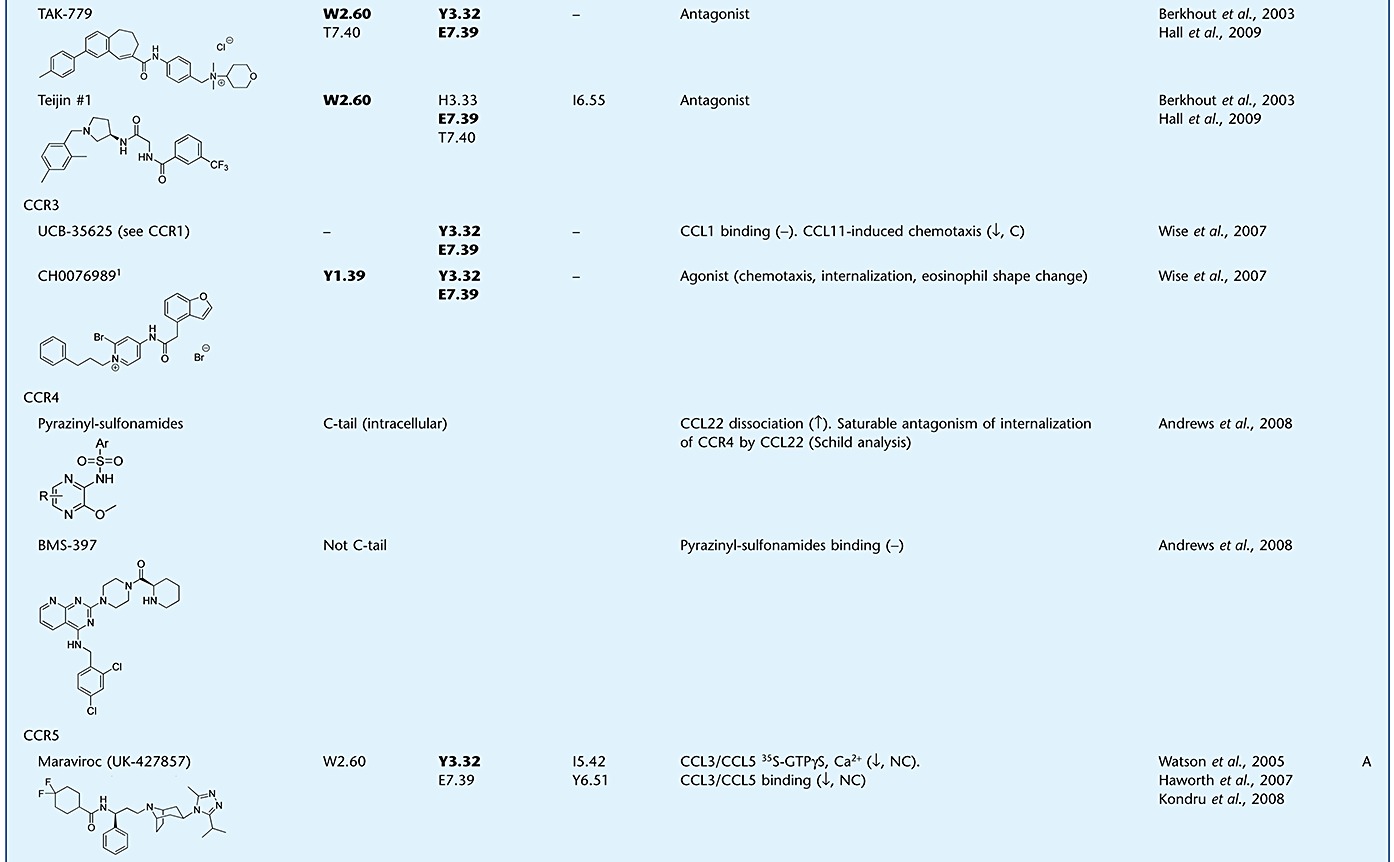
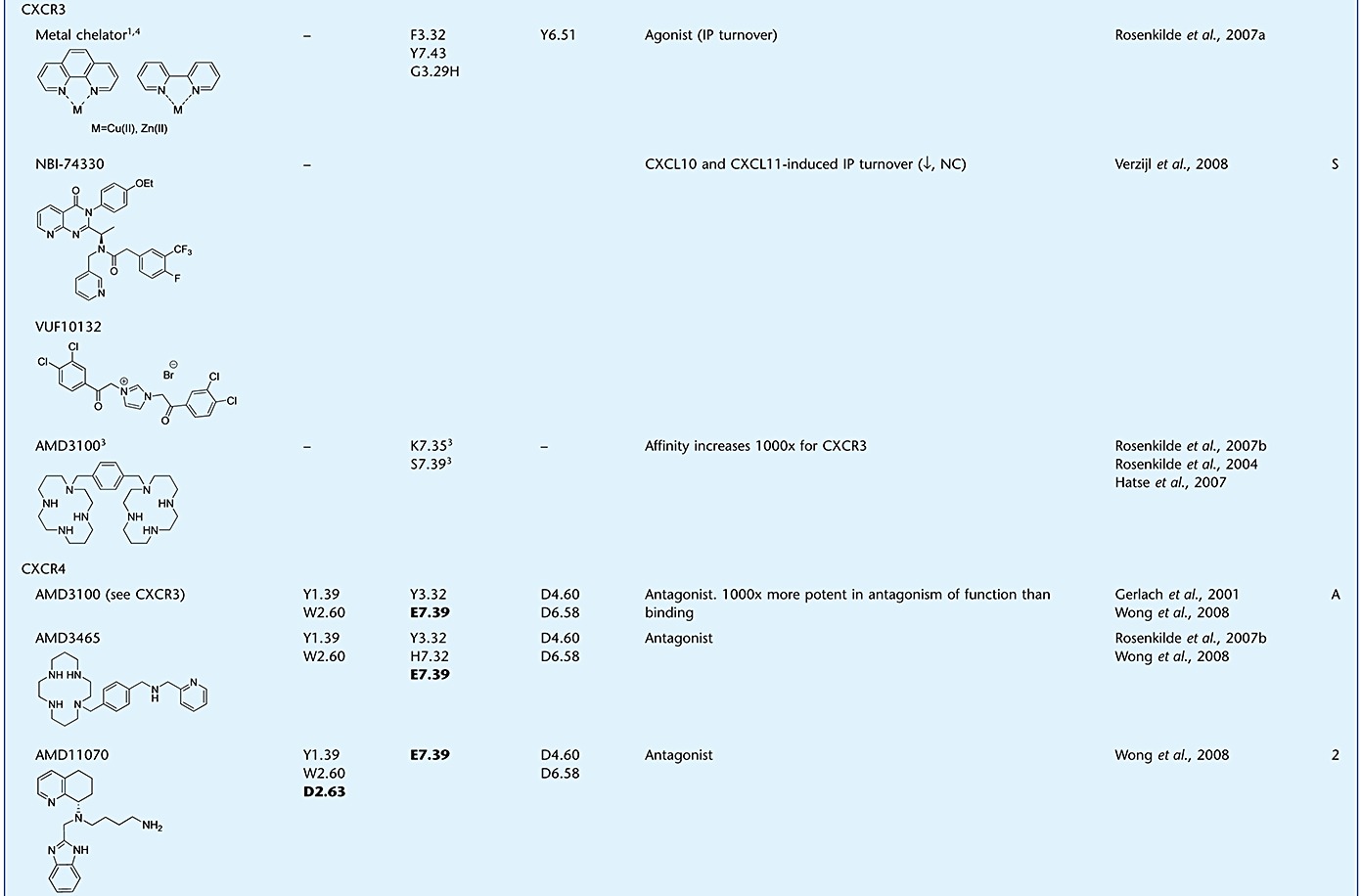
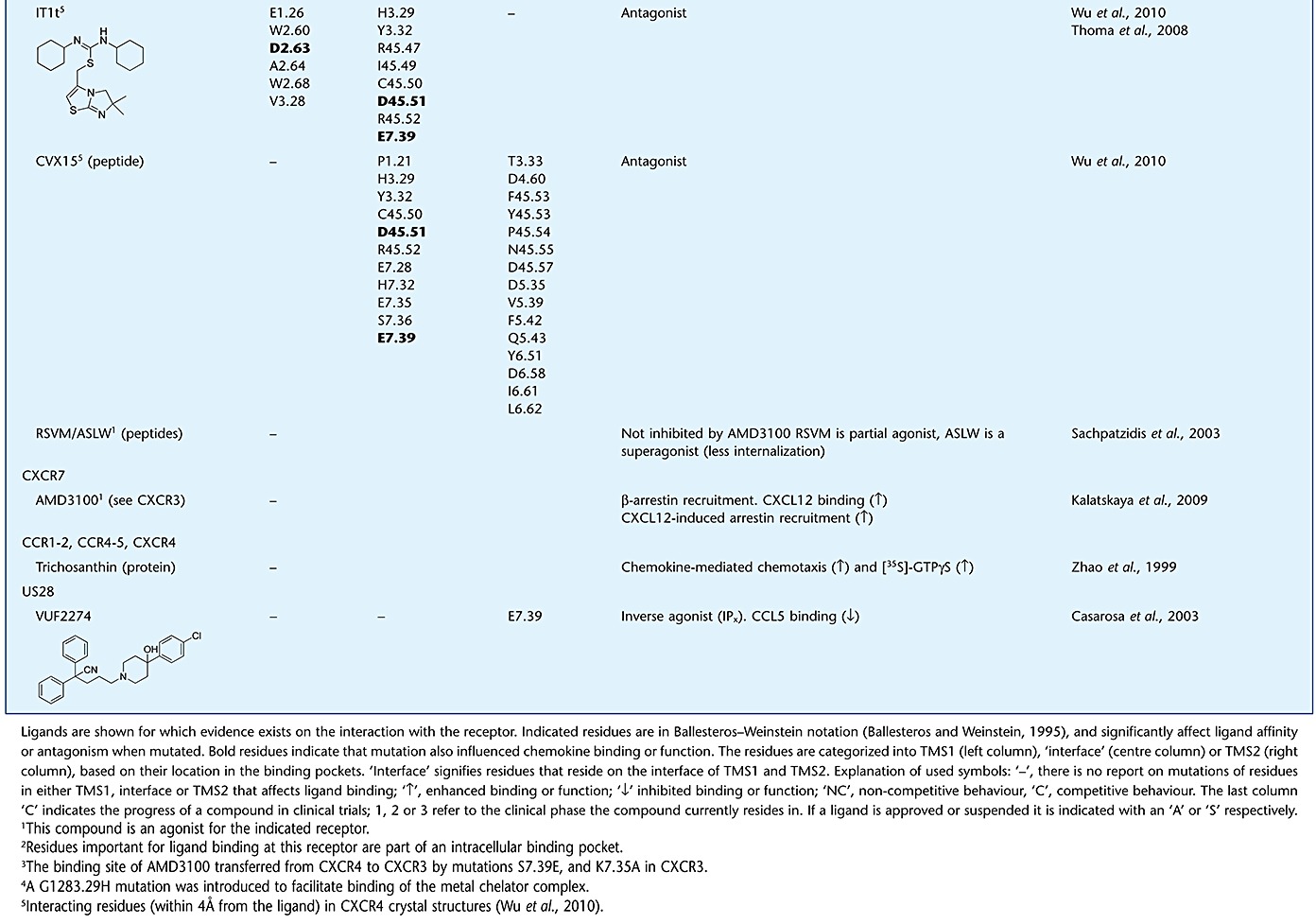
Table 1.
Evidence supporting the mechanism of action for small-molecule chemokine ligands
 |
 |
 |
 |
 |
 |
TM2 has also been suggested to regulate functional selectivity through an extended allosteric interface by an hydrogen bonding network (Nygaard et al., 2010; Rosenkilde et al., 2010), as the mutation of a conserved proline in TM2 in the angiotensin II receptor (P2.60 in chemokine receptors) led to a loss in Gq coupling for the agonist angiotensin II, while functional selectivity for the biased agonist [Sar1-Ile4-Ile8]angiotensin II was lost at this same mutation (Reis et al., 2007; Rosenkilde et al., 2010). The triggering domain of chemokines is thought to interact with residues in this region as well (CRS2) (Table 1). Together with the finding that (modified) chemokines show functional selectivity at a single receptor, including CCR5 (Mack et al., 1998), it can be hypothesized that this region is involved in functional selectivity of chemokines.
Despite the increasing evidence supporting the two-step model for chemokine receptor binding and activation, the exact regions of CRS1 and CRS2 used for these interactions seem to be different not only between receptors, but also for different ligands binding to the same receptor (Cox et al., 2001; Blanpain et al., 2003; Casarosa et al., 2005; Jensen et al., 2008).
Allosteric interactions in chemokine receptors and their consequences
Chemokines bind with high affinity to their receptors involving numerous interactions with the receptor extracellular surface (Allen et al., 2007). Interestingly, low-molecular weight ligands (500–600 Da) are often able to disrupt binding or function of the roughly 100-fold larger chemokine ligands (or HIV gp120 envelope glycoprotein) with nanomolar potencies. From the size differences, it seems evident that these small ligands probably do not act via simple steric hindrance or competition, but rather operate in an allosteric manner. In general, allosteric ligands bind to sites that are topographically distinct from the orthosteric endogenous ligand-binding site. Only since the past decade we have begun to appreciate the different mode-of-action between allosteric and orthosteric ligands. The ability of allosteric ligands to change receptor conformations via distant, yet conformationally linked sites can positively or negatively impact the affinity as well as the efficacy of endogenous (orthosteric) ligands (Christopoulos and Kenakin, 2002).
Modulation by allosteric ligands is saturable, meaning that its maximum is attained with full occupancy of the allosteric sites on the receptor. In addition, this maximum effect further depends on the level of cooperativity between the two ligands. Furthermore, allosteric ligands exert effects that are generally probe-dependent, meaning that these effects are not the same towards all orthosteric agonists. This may be exemplified by allosteric CCR1 agonists, which enhance the binding of CCL3, while they inhibit the binding of CCL5 at the same receptor (Jensen et al., 2008). In addition, an allosteric modulator can differentially affect receptor signalling mediated by orthosteric agonists, by selective potentiation of one signalling pathway while inhibiting a second, and leaving a third unaltered, reflecting the permissive nature of allosterism. Next to orthosteric ligand modulation, allosteric ligands can also exhibit agonistic activity in the absence of an orthosteric agonist, which is also referred to as allosteric agonism (Saita et al., 2006; May et al., 2007). This may include selective or biased activation of signalling pathways, also referred to as functional selectivity, or biased agonism (Galandrin et al., 2007; Kenakin, 2010a). Altogether, allosterism adds another layer of complexity to GPCR pharmacology, which has forced us to reconsider the approaches to identify and optimize ligands in drug discovery and development programmes.
Allosteric ligands have been identified for different chemokine receptors, including CC and CXC chemokine receptors (Sachpatzidis et al., 2003; Watson et al., 2005; Vaidehi et al., 2006; Rosenkilde et al., 2007a; Jensen et al., 2008; Salchow et al., 2010). These ligands include not only small molecules, but also metal chelators and peptides. In the next sections we will discuss some of the evidence supporting allosteric modulation and functional selectivity of chemokine receptors and the implications of receptor dimerization. In addition, development of biologicals for the treatment of chemokine-associated disease is discussed.
Allosteric small-molecule chemokine receptor antagonists
The growing evidence implicating chemokine receptors and their ligands in disease has boosted the discovery and development of related therapeutics in the past decade. About 10 years ago, Berlex Biosciences entered clinical trials with the first antagonist for a chemokine receptor, BX-471, a selective, potent and orally available CCR1 antagonist. However, the compound failed in a phase II trial with multiple sclerosis (MS) patients due to lack of efficacy. Nevertheless, it had set the stage for the clinical development of other chemokine receptor antagonists. Since then, several chemokine receptor antagonists have been developed but failed in clinical trials due to failure of reaching clinical endpoints (Proudfoot et al., 2010). However, the recent approval of maraviroc (CCR5 antagonist) and AMD3100 (CXCR4 antagonist), together with numerous clinical trials currently being conducted with small molecules and biologicals, reflects the faith in the chemokine system as a tractable therapeutic target (Proudfoot et al., 2010) (Tables 1 and 2).
Table 2.
Clinical trials with mAbs targeting chemokine receptors
| Receptor/mAbs | Epitope | Isotype | Pathological condition(s) | Phase | Trial (PMID) | Status |
|---|---|---|---|---|---|---|
| CCR2 | ||||||
| MLN1202 | N.a. | IgG1 | Metastatic cancer | II | NCT01015560 | S |
| Atherosclerotic cardiovascular disease | II | NCT00715169 | C | |||
| Multiple sclerosis | II | NCT01199640 | C | |||
| CCR4 | ||||||
| KW-0761 | Nt | IgG1 | Adult T-cell leukaemia-lymphoma and peripheral T-cell lymphoma | I + II | NCT00920790 | O |
| NCT00355472 | O | |||||
| NCT00888927 | O | |||||
| KW-0761 + multidrug chemotherapy | Peripheral and cutaneous T-Cell lymphoma | II | NCT01226472 | R | ||
| Peripheral T/NK-cell lymphoma | II | NCT01192984 | R | |||
| Adult T-cell leukaemia-lymphoma | II | NCT01173887 | R | |||
| CCR5 | ||||||
| HGS004/CCR5mAb004 | EL2 | IgG4 | HIV infection | I | NCT00114699 | C |
| PRO140 | Nt & EL2 | IgG1 | HIV infection | I + II | NCT00110591 | C |
| NCT00613379 | C | |||||
| NCT00642707 | C | |||||
| PRO140 + oral antiretroviral therapy | HIV infection | II | NCT01272258 | R | ||
| CXCR4 | ||||||
| MDX-1338 | N.a. | N.a. | Acute myeloid leukaemia | I | NCT01120457 | R |
Info obtained in January 2011 on http://www.clinicaltrials.gov. Epitope indicates the receptor region involved in mAb recognition: Nt, N-terminus; N.a., information not available. Status of clinical trials: C, completed; O, ongoing; R, recruiting; S, suspended.
Small molecules generally interact with residues in the TM helices of the receptor. Two binding pockets in these helices can be distinguished: the minor and the major binding pocket, formed by residues from TM1, 2, 3, 7, or TM3, 4, 5, 6 respectively (Figure 4E,F) (Surgand et al., 2006). To avoid confusion with the chemokine recognition sites, we refer to these sites as TM site 1 (TMS1) and TMS2 respectively. Many ligands bind in both TMS1 and TMS2, but some seem to bind exclusively to TMS1 or TMS2 (Table 1). The mechanisms by which small-molecule ligands modulate chemokine affinity and/or efficacy are largely unknown. Although some ligands have been demonstrated to act in an allosteric manner, there are also studies that show overlap in binding sites of chemokines and small molecules, supporting a competitive component in the mechanism of action. However, ligands with partially overlapping binding sites, such as the CXCR3 chemokines CXCL10 and CXCL11, also show non-competitive ‘allosteric’ behaviour (Cox et al., 2001; Xanthou et al., 2003). Blanpain and co-workers showed that mutations in TM2 and 3 of CCR5 affect the function but not the binding of CCL3, while CCL5 binding and function remains unchanged (Blanpain et al., 2003). As many small-molecule ligands for CCR5 have also shown to bind in this region (e.g. Y371.39, W862.60, Y1083.32 and E2837.39) including maraviroc, aplaviroc, vicriviroc, SCH-C and TAK-779 (Maeda et al., 2006; Kondru et al., 2008; Hall et al., 2009), it can be hypothesized that these compounds compete with the CCL3 N-terminus to bind in the TM region and, thereby block its ability to activate CCR5 (Figure 4B,E). However, a simple competition hypothesis is not sufficient to explain the fact that these CCR5 antagonists are able to inhibit the binding of both CCL3 and CCL5 in an insurmountable allosteric manner (Watson et al., 2005). In addition, aplaviroc showed behaviour deviating from the other CCR5 antagonists, in that it only displaced 20% of CCL5 even at 10 µM (Ki± 3 nM), while the calcium response mediated by the chemokine was completely blocked by only 10 nM of the ligand. The latter suggests an allosteric mode of action for aplaviroc, illustrated by the saturability and probe dependence of the effects observed for this CCR5 antagonist (Watson et al., 2005).
A study of the CCR1-specific compound BX-471 showed that mutation of residues in both TMS1 and TMS2 affect ligand binding, including residues Y411.39, Y1133.32, Y1143.33, I2596.55 and Y2917.43, which are not important for CCL3 binding and function (Vaidehi et al., 2006). Nevertheless, the compound was still able to displace [125I]-CCL3 from the receptor, indicating an allosteric mechanism of inhibition. In contrast, the chemokine did not affect the binding of a radiolabelled analogue of BX-471. UCB-35625 is another small-molecule CCR1 antagonist, for which residues Y411.39, Y1133.32 and E2877.39 (TMS1) were shown to be involved in ligand binding, sharing residues Y411.39 and Y1133.32 with BX-471, indicating that these two small molecules bind to different yet overlapping sites (de Mendonça et al., 2005). UCB-35625 has a potency in the picomolar range to block CCL3-induced eosinophil shape change, while it has a ∼1000-fold lower potency in displacing the chemokine from the receptor (Sabroe et al., 2000). Moreover, the displacement of CCL3 is incomplete, even at saturating concentrations of the compound. The latter suggests an allosteric inhibition of efficacy but not affinity. In contrast to CCL3, CCL5-induced activation of CCR1 is dependent on E2877.39 (Jensen et al., 2008), and although not investigated, it might be speculated that the observed effects of UCB-35625 on CCL3 binding and activity would have been different when CCL5 was used as a probe. Also CCR2 and CCR3 ligands, like RS-504393, TAK-779 and UCB-35625, interact with TM residues shown to be involved in chemokine-induced receptor activation (Mirzadegan et al., 2000; Berkhout et al., 2003; de Mendonça et al., 2005; Wise et al., 2007).
Evidence for the mode of action of antagonists is not only found for CC, but also for CXC chemokine receptors, including CXCR4. As for many other chemokine receptors, CXCL12 binding to CXCR4 follows the two-step activation model (Kofuku et al., 2009). CXCR4 CRS2 contains residues from EL2 and TM domains, such as D18745.51 (EL2), D972.63 and E2887.39 that are involved in both chemokine binding and activation (Brelot et al., 2000). In the recent crystal structures of CXCR4, the small-molecule antagonist IT1t is shown to bind to these same residues, and it was suggested that it competitively blocks the interactions of the CXCL12 N-terminus with CXCR4 (Wu et al., 2010). Similarly, the binding site of the well-studied bicyclam antagonist AMD3100 is mainly lined by three acidic TM residues, D1714.60, D2626.58 and E2887.39 (Gerlach et al., 2001; Hatse et al., 2003; Rosenkilde et al., 2004; Wong et al., 2008). Although the binding site of AMD3100 and IT1t do not seem to overlap completely, AMD3100 might bind partly to CRS2 where the N-terminus of CXCL12 interacts with the receptor (Kofuku et al., 2009). Altogether these findings might, at least in part, suggest a competitive mechanism of action for these compounds, by preventing the binding of the CXCL12 to CRS2 on the receptor, leading to the observed antagonism of functional responses. Along with the data presented above for CC chemokine receptors, competition of small molecules with this triggering domain of the chemokine might pose a general mechanism of action for chemokine receptor antagonists that have effects on efficacy rather than affinity. Table 1 shows an overview of evidence for binding mechanisms of small ligands targeting chemokine receptors.
The success story of chemokine receptor antagonists: CCR5 and CXCR4
The quest for therapeutics targeting chemokine receptors has been catalysed by their significant involvement in various human diseases. In the 1990s, it was shown that chemokine receptors were promising targets for treatment in HIV-1 infection (Deng et al., 1996; Feng et al., 1996). Genetic evidence was provided by the impact of the naturally occurring mutation CCR5-Δ32 that encodes a truncated, non-functional form of CCR5 with no apparent deleterious consequences. It was found that CCR5-Δ32 is significantly underrepresented in the HIV-1 infected groups, and individuals homozygous for the mutation are only rarely infected with HIV-1 (Gorry et al., 2002) supporting the role of CCR5 in HIV-1 entry. In addition, CXCR4 was found as a second co-receptor for HIV-1. Namely, CCR5 and CXCR4 facilitate HIV-1 entry to macrophages and T-cells respectively (Berger et al., 1999). In the first stages of infection the virus mainly uses CCR5 as a co-receptor (CCR5-tropic). These findings paved the way for discovery and development of small-molecule antagonists for CCR5. TAK-779 was the first to be discovered, showing inhibition of HIV-1 infection in vitro and in vivo (Baba et al., 1999). Since then, several CCR5 antagonists have entered clinical trials, including aplaviroc, maraviroc and vicriviroc. Maraviroc (Celsentri™, developed by Pfizer) is the first CCR5 antagonist approved by the European Medicines Agency (EMA) for use in treatment-experienced patients harbouring only CCR5-tropic viruses. It represents a novel class of anti-retroviral drugs, as it is the first therapeutic targeting a cellular rather than a viral protein. Maraviroc is a potent inhibitor of CCL4 binding to the CCR5 receptor (IC50= 2 nM) and a potent antiviral agent (EC90= 1 nM) (Wood and Armour, 2005). In addition, Maraviroc has been demonstrated to behave as an allosteric antagonist. It binds to a site in the receptor that is topographically distinct to the site where the viral gp120 envelope protein binds (Watson et al., 2005; Muniz-Medina et al., 2009) and that involves key interactions with the TM domains of CCR5 (Kondru et al., 2008) (Table 1). More recently, a second-generation maraviroc analogue has been described, PF-232798, which retains the attractive antiviral effect combined with improved absorption profiles in rat and dog (Stupple et al., 2010) and is currently in phase II clinical trials. In addition, vicriviroc also showed long-term potent antiviral activity and is currently in phase III clinical trials (Klibanov, 2009).
During the course of disease, HIV-1 shifts its tropism from CCR5 to CXCR4, a hallmark of the symptomatic stage when the disease progresses to AIDS (Jekle et al., 2003). Consequently, there has been an increased interest in the discovery and development of CXCR4 antagonists able to block the interaction of HIV-1 with CXCR4, preventing subsequent infection of cells. One of the early compounds showing anti-HIV activity was AMD3100 (De Clercq, 2003). However, despite its efficacy in clinical trials, AMD3100 treatments in HIV-1 patients were discontinued due to several events of cardiac toxicity. A serendipitous finding during these trials was that AMD3100 promoted mobilization of hematopoietic stem cells from the bone marrow to the periphery. Subsequently, AMD3100 (plerixafor, Mozobil™) has been successfully developed by Genzyme as an effective therapeutic for autologous bone marrow transplantations in patients suffering from non-Hodgkin's lymphoma and multiple myeloma (De Clercq, 2010). As can be seen from the blocking of CXCR4 with AMD3100, the CXCL12/CXCR4 axis is involved in multiple homeostatic processes. These include T cell trafficking and homing, stem cell localization and organ development (Tsibris and Kuritzkes, 2007). Since CXCR4 or CXCL12/SDF-1α knockout mice are not viable because of significant defects in B-cell lymphopoiesis and bone-marrow myelopoiesis (Nagasawa et al., 1996), long-term CXCR4 antagonism might lead to severe adverse effects. Future in vivo studies are required to answer the question whether CXCR4 can actually be targeted safely for the (long-term) treatment of CXCR4-tropic HIV-1 infection.
Allosteric agonists for chemokine receptors and functional selectivity
Despite the therapeutic focus on chemokine antagonists, the process of screening for and optimization of chemokine receptor antagonists has led to the discovery of several small-molecule agonists for different chemokine receptors, such as CCR1, CCR3, CCR5, CCR8, CXCR3 and CXCR4 (Sachpatzidis et al., 2003; Saita et al., 2006; Stroke et al., 2006; Jensen et al., 2007; Rosenkilde et al., 2007a; Wise et al., 2007). Despite their relatively small size, these ligands are generally able to fully activate receptor signalling. Similarly to small-molecule antagonists, residues involved in receptor binding have been shown to reside in TMS1 and TMS2 of the receptors (Table 1). For example, CH0076989, a small-molecule agonist for CCR3, activates multiple signalling pathways including chemotaxis and receptor internalization by interacting with residues in TMS1 (i.e. Y411.39, Y1133.32 and E2877.39) (Wise et al., 2007). Since these residues are also important for CCL11-induced receptor activation, this suggests that CH0076989 activates the receptor in a similar manner as the chemokine, probably by interacting with the TM2–TM3 interface (Blanpain et al., 2003; Govaerts et al., 2003). As pointed out earlier, this receptor region might be involved in ligand-biased signalling (Rosenkilde et al., 2010). Indeed, CCR3 agonist CH0076989 seems to bind in that region (TMS1) (Table 1). Interestingly, while equal receptor internalization was observed when stimulating CCR3 with either CH0076989 or CCL11, the efficacy of the small agonist to induce chemotaxis was significantly lower than for the chemokine, suggesting functional selectivity. YM-370749, a small-molecule agonist for the CCR5 receptor, also exhibited functional selectivity, while it binds to TMS2 and not TMS1. This compound promoted calcium mobilization and receptor internalization, but was unable to induce chemotaxis (Saita et al., 2006). Importantly, YM-370749 inhibited HIV-1 replication. The use of functionally selective agonists that down-regulate the receptor without concomitant undesired side effects, such as chemotaxis, might pose a novel therapeutic avenue for the treatment of diseases like HIV-1 infection. As chemokine receptors can initiate more signalling pathways than described here, including Janus kinase/signal transducers and activators of transcription (JAK/STAT) (Kehrl et al., 2009), it would be interesting to see whether chemokine receptor agonists, for example CCR8 agonist LMD-009 (binds in TMS1), show selectivity in activation of these other signalling pathways. Moreover, there is accumulating evidence for GPCRs suggesting that selective activation of specific signalling routes, that is, G proteins versus β-arrestins, might be beneficial over non-biased agonists (Kenakin and Miller, 2010), again highlighting the therapeutic potential of such functionally selective ligands.
Intracellular binding sites in chemokine receptors
GPCR signalling is allosteric by nature, in which extracellular endogenous agonists act as positive allosteric modulators on the coupling of intracellular G proteins, and vice versa (Kenakin, 2010b). Indeed, high-affinity chemokine binding to a number of tested receptors is G protein dependent as revealed by experiments in which Gi/o proteins are uncoupled using, e.g. GTPγS, Gpp(NH)p, or Pertussis toxin (Cox et al., 2001; Staudinger et al., 2001; Springael et al., 2006; Bradley et al., 2009; Nijmeijer et al., 2010). Agonist-induced or constitutive coupling of a GPCR to G proteins can limit the availability of a shared G protein pool to interact with other receptors, which may subsequently hamper high-affinity agonist binding to the latter receptors (Nijmeijer et al., 2010).
In addition, GPCRs can interact with multiple other interacting partners, such as receptor activity-modifying proteins (RAMPs), β-arrestins, GRKs and other GPCRs (Kenakin and Miller, 2010), via regions that do not overlap with the binding site of endogenous ligands. Experimental evidence for such binding sites was presented for CCR4 and CXCR2 where some small-molecule antagonists appeared to bind along the intracellular surface of the GPCRs instead of the TM domains (Andrews et al., 2008; Nicholls et al., 2008; Salchow et al., 2010). Nicholls et al. (2008) studied two classes of CXCR2 antagonists that had a 1000-fold higher affinity for CXCR2 compared to CXCR1. By constructing different chimeras between CXCR1 and CXCR2, they found a reversal of antagonism when switching the intracellular C-terminal tails. Using a similar approach, evidence was presented for an intracellular binding site in CCR4 (Andrews et al., 2008). In the case of CXCR2, the point mutant K320N7.59 in Hx8 of CXCR2 led to a 10-fold decrease in affinity for the compounds, while mutation of N311K7.59 at the same position in CXCR1 led to a 100-fold increase in affinity, providing additional evidence for an intracellular binding mode (Nicholls et al., 2008). Moreover, other groups, including our own, have presented pharmacological evidence for an allosteric binding mode for these and other classes of CXCR2 ligands (Gonsiorek et al., 2007; Bradley et al., 2009; de Kruijf et al., 2009). These include the inability of chemokines to displace a small-molecule antagonist with similar chemical structure, and insurmountable inhibition of CXCL8-promoted β-arrestin recruitment and [125I]-CXCL8 binding. Site-directed mutagenesis of different intracellular residues was performed to further delineate the binding pocket for these CXCR2 ligands (Salchow et al., 2010). Salchow and co-workers identified several key CXCR2 residues involved in interaction of CXCR2 antagonist SCH-527123, a ligand already suggested to bind in an allosteric manner (Gonsiorek et al., 2007), and compounds similar to those used in the previous study (Nicholls et al., 2008). The binding pocket seems to be lined by T832.39, D842.40, D1433.49 (E/DRY motif), Y3147.53 and K3207.59 along the intracellular surface of the TM helices (see Table 1 and Figure 4D). Since studied mutations are in close proximity to the site of G protein coupling, or to a region that is involved in receptor signal transduction, this might in fact govern a mechanism of allosteric inhibition (Figure 4D).
Recently, pharmacological modulation through interactions with intracellular parts of CXCR4 has also been described by Tchernychev and colleagues who identified the pepducin ATI-2341 as a potent agonist of this receptor (Tchernychev et al., 2010). Pepducins are synthetic molecules that are composed of a peptide derived from the amino acid sequence of an intracellular loop of the target GPCR coupled to a lipid tether. The peptide component of the pepducin confers receptor-modulating activity whilst the lipid counterpart facilitates cell penetration and access to the intracellular face of the target GPCR. The ATI-2341 is derived from IL1 of CXCR4 and activates CXCR4-mediated signalling pathways, induces receptor internalization, and promotes both in vitro and in vivo chemotaxis. Interestingly, ATI-2341 acts as functional antagonist in vivo, leading to a similar mobilization of hematopoietic stem cells from the bone marrow as is observed for the CXCR4 antagonist AMD3100 (Tchernychev et al., 2010). The mechanism responsible for these seemingly contradictory effects requires further investigation. Although more evidence is needed regarding the molecular determinants of these ligand–receptor interactions, these studies indicate that targeting of allosteric regions other than the classical major and minor TM-binding pockets is feasible within the class of chemokine receptors.
Biological therapeutics targeting chemokine receptors
Monoclonal antibody (mAb)-derived therapeutics have been proven to be an excellent paradigm as high-affinity biopharmaceuticals in the diagnosis and treatment of cancer and inflammatory diseases, as exemplified by the acquisition of mAb technology companies by major drug companies (Nelson et al., 2010). During the last 2–3 decades, 42 engineered mAbs that target growth factors and receptor tyrosine kinases have gained US Food and Drug Administration (FDA) approval (Walsh, 2010). Hitherto, no mAb-derived therapeutic against GPCRs has been approved for clinical use. The difficulty to develop such therapeutics may have been due to the intrinsic nature of GPCRs. Their limited availability as purified proteins as well as their low immunogenicity as membrane-embedded proteins render GPCRs difficult antigens for the generation of antibodies that recognize their targets with high specificity and affinity (Olson and Jacobson, 2009; Hutchings et al., 2010). However, several attempts have been successful and clinical trials are currently evaluating the therapeutical potential of mAbs targeting chemokine receptors (Table 2).
Therapeutic antibodies can act via two different mechanisms. First, mAbs can bind and block the target protein, directly interfering with its function (i.e. direct targeted therapy). Alternatively, the mAb triggers an indirect biological activity upon recognition of its antigen by recruiting cytotoxic monocytes/macrophages (i.e. antibody-dependent cell-mediated cytotoxicity) or by binding complement factors (i.e. complement-dependent toxicity). In addition, other proteins or drugs that are conjugated to such targeting mAbs can induce cellular responses (Satoh et al., 2006; Schrama et al., 2006).
MLN1202 is a genetically engineered human IgG1 mAb targeting CCR2 that has been developed by Millenium Pharmaceuticals, and optimized to reduce antibody- and complement-dependent cytotoxicity. MLN1202 inhibits chemokine-induced CCR2 signalling in transfected cells (Vergunst et al., 2008). This mAb has been in clinical trials for the treatment of various inflammatory diseases involving CCR2-expressing monocytes/macrophages (Figure 2 and Table 2). Treatment of patients at risk for atherosclerotic diseases with MLN1202 significantly reduced median serum levels of C-reactive protein (Gilbert et al., 2011), which is considered to be a predictive biomarker of inflammation associated with cardiovascular diseases (Ridker, 2007). In contrast, MLN1202 failed to block CCR2-mediated infiltration of macrophages into the inflamed synovium of rheumatoid arthritis patients or reduce the expression of synovial pro-inflammatory cytokines (Vergunst et al., 2008). This failure may have been due to the incomplete receptor occupancy (86–94%) by MLN1202 or the fact that CCR2 is not the correct/only therapeutical target for this pathological condition (Proudfoot, 2008; Vergunst et al., 2008). Finally, clinical trials in multiple sclerosis patients have also been conducted with MLN1202 but no results are publicly available. Also, a phase 2 clinical trial for the treatment of bone metastasis by MLN1202 had been initiated but was recently suspended.
Two mAbs targeting CCR5 have been developed by Human Genome Science (HGS004) and Progenics Pharmaceuticals (PRO 140) and have been investigated in the context of CCR5-mediated HIV-1 infection (Weiss and Clapham, 1996). HGS004 binds to EL2 of CCR5 and inhibits chemokine-induced signalling as well as HIV co-receptor activity (Lalezari et al., 2008). Phase 1 clinical studies demonstrated that HGS004 reduces plasma HIV-1 RNA levels ≥10-fold in 54% of treated patients (Lalezari et al., 2008). However, the lack of a clear dose–response relationship indicated that the anti-HIV potency of HGS004 as a single agent might be suboptimal. Also, some patients treated with high doses of HGS004 showed a shift from CCR5- to CXCR4-tropic viruses or dual strains.
PRO 140 binds to the N-terminal region (residue D2) and EL2 (R16845.40, Y17645.48) of CCR5. Interestingly, PRO 140 is more potent in inhibiting HIV-1 co-receptor activity than antagonizing chemokine-induced signalling, giving the possibility to inhibit HIV-1 infection without affecting CCR5-mediated signalling, an example of permissive antagonism (Olson et al., 1999). Phase 1 and 2 clinical studies demonstrated that a single intravenous injection of PRO 140 could reduce HIV-1 viral loads in 100% of treated patients (Jacobson et al., 2008; 2010a). Importantly, some patients displayed a more than 100-fold decrease in viral load and patients treated with high doses of PRO 140 displayed no change in co-receptor tropism and no emergence of PRO 140-resistant viral strains (Jacobson et al., 2010a). Such strong antiviral effects supported the development of subcutaneous formulations of PRO 140. Three weekly subcutaneous injections of PRO 140 led to an antiviral activity similar to that observed with one single intravenous injection (Jacobson et al., 2010b). This phase 2 study provided proof-of-concept for the subcutaneous use of PRO 140 and the subcutaneous PRO 140 was selected for further development on the basis of its potential to be self-administered by patients (Jacobson et al., 2010a,b).
mAbs against CCR4 have been optimized by Kyowa Hakko Kirin Ltd to block the receptor, but also induce antibody-dependent cellular cytotoxicity (ADCC) (Satoh et al., 2006; Ishii et al., 2010). To this end, these mAbs (i.e. KW-0761 and KM2760) have been modified to remove fucose groups from the Fc region of their heavy chains, thereby increasing their affinity for leukocyte receptors FcγRIIIa. Upon binding to CCR4-expressing cells, these mAbs recruit FcγRIIIa-expressing natural killer cells leading to the lysis of CCR4+ tumour cells (Satoh et al., 2006; Ishii et al., 2010). Preclinical studies have shown that the antitumor activity of KW-0761 and KM2760 in adult T-cells leukaemia-lymphoma (ATLL) mouse models is mediated via ADCC (Niwa et al., 2004; Ishii et al., 2010). Furthermore, the clinical application of KW-0761 was demonstrated by its ability to induce ADCC-mediated cytotoxicity of primary ATLL cells ex vivo (Ishii et al., 2010). Phase 1 and 2 clinical trials are currently ongoing to evaluate the therapeutic effect of KW-0761, alone or in combination with multidrug chemotherapy, in patients with T-cell and NK-cell lymphomas (Table 2).
A new class of antibody-based therapeutics has recently joined the family of GPCR-targeting biologicals. VHH antibody fragments, also defined as nanobodies by Ablynx, are immunoglobulin single variable domains of heavy-chain antibodies that occur naturally in the Camelidae family. Due to their small size of approximately 15 kDa, nanobodies can be easily expressed and produced in organisms such as yeasts or bacteria, and are also highly stable and soluble. Furthermore, complementarity-determining regions of nanobodies can penetrate into grooves and cavities of proteins, accessing a new range of non-planar epitopes, such as enzymatic clefts (Ghassabeh et al., 2010). We recently described the first GPCR-specific antagonistic nanobodies against the chemokine receptor CXCR4 (Jähnichen et al., 2010). 238D2 and 238D4 were generated and their epitopes were localized mainly in EL2 but also EL3 loop of CXCR4, with one amino acid common to both different epitopes. Both nanobodies showed high affinity towards CXCR4, with inhibition constants in [125I]-CXCL12 displacement assays, signalling and chemotaxis assays in the nanomolar range. In addition, both nanobodies proved to behave as competitive antagonists. Interestingly, when both 238D2 and 238D4 monovalent nanobodies were linked together, the affinity of the resulting biparatopic nanobody increased almost 30 times compared to its monovalent counterparts, reaching a Ki value of 0.35 nM. The affinity of this biparatopic single domain antibody was 100-fold higher than the CXCR4 benchmark compound AMD3100. Preclinical characterization of the CXCR4 nanobodies also demonstrated their ability to fully inhibit entry of CXCR4-tropic HIV-1 strains in vitro. Finally, a single intravenous injection of biparatopic nanobody resulted in stem cell mobilization in cynomolgus monkeys, to a similar extent as AMD3100 (Jähnichen et al., 2010). Currently, CXCR4 nanobodies are in phase 1 clinical studies for use in stem cell mobilisation (http://www.ablynx.com/wp-content/uploads/2010/11/7_ALX-06511.pdf). It will be interesting to see the potential of these molecules and their therapeutical benefit compared to the AMD3100/Pleraxifor drug currently used in the clinic.
A VHH antibody fragment against the atypical chemokine receptor Duffy antigen receptor for chemokines (DARC) has also been reported. Unlike the CXCR4 nanobody, the DARC VHH fragment has been generated solely against the N-terminal domain of the receptor (Smolarek et al., 2010). The CA52 nanobody binds to DARC with a very high affinity of 0.2 nM. It displaces the endogenous DARC ligand CXCL8 and was able to prevent the infection of red blood cells by Plasmodium vivax. As such, this nanobody may serve as a basis for the development of therapeutics against malaria (Handel and Horuk, 2010; Smolarek et al., 2010).
Crystal structures of CXCR4 and their impact on chemokine receptor-targeted drug design
The determination of the first GPCR crystal structure of bovine rhodopsin (bRho) in 2000 was a milestone in GPCR research (Palczewski et al., 2000). The bRho structure served as template for in silico GPCR homology modelling, including chemokine receptors and structure-based drug design (de Graaf and Rognan, 2009). About 3 years ago the first structures of liganded GPCRs [i.e. ADRB1/2, and adenosine A2A receptor (AA2AR)] were reported (Cherezov et al., 2007; Jaakola et al., 2008; Warne et al., 2008), which were subsequently used as new GPCR modelling templates. While the recently solved DRD3 crystal structure (Chien et al., 2010) gives insights into bioaminergic receptor subtype specificity (by comparison with ADRB1/2), the recently solved CXCR4 chemokine receptor crystal structures (Wu et al., 2010) open up new possibilities for structure-based drug design on the chemokine receptor family. One crystal structure has been elucidated with a large cyclic peptide CVX15 and several crystal structures have been elucidated with the small-molecule antagonist IT1t (Wu et al., 2010).
The GPCR structure features seven TM helices and one intracellular helix (Hx8). Traditionally, the GPCR TM bundle is categorized in two subpockets in which ligands can reside. These are a minor pocket comprised of TMs 1, 2, 3 and 7 (TMS1) (Figure 4E), and a major pocket comprised of TMs 3, 4, 5, 6 and 7 (TMS2) (Surgand et al., 2006) (Figure 4F). While the ligands in the bRho, ADRB1, ADRB2, AA2AR and DRD3 co-crystal structures all occupy TMS2, the CXCR4 crystal structures show for the first time ligand binding to not only TMS2 (CVX15) but also TMS1 (IT1t) (Figure 6A).
Figure 6.
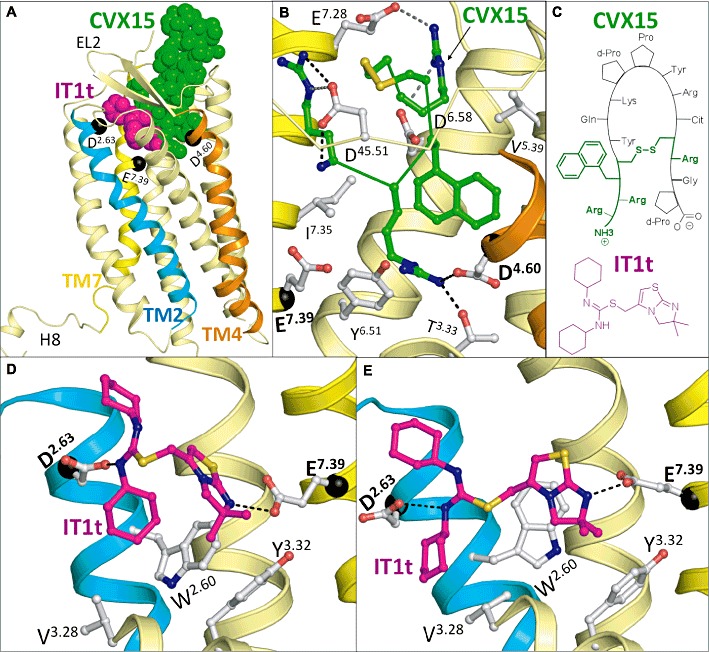
Crystal structures of CXCR4 receptors complexed with ligands. (A) Comparison of the ligand binding modes of the small-molecule antagonist IT1t (magenta, 3odu) and the peptide-like ligand CVX15 (green, 3oe0) in the (IT1t bound) CXCR4 crystal structure. While IT1t binds in the TMS1 (minor pocket) between TM helices 1, 2, 3 and 7, CVX15 binds primarily in TMS2 (major pocket) between TMs 3, 4, 5, 6 and 7. TMs 2, 4 and 7 are coloured cyan, yellow and orange respectively, and Cα atoms of residues D2.63 (TMS1), D4.60 (TMS2) and E7.39 (interface between TMS1 and TMS2) are indicated with black spheres (like in panels B, D and E). Furthermore, the beta sheet of EL2 and the disordered helix 8 are labelled. (B) Interactions between the residues 1–4 and 13–14 of CVX15 (green side chain in ball-and-stick, backbone as ribbon) and CXCR4 (3oe0). Important residues are displayed as ball-and-stick (grey carbon atoms), while CVX15-1T1t H-bond and ionic interactions are indicated with black and grey dashed lines respectively. Nitrogen, oxygen and sulphur atoms are coloured blue, red and yellow respectively. For reasons of clarity the top of TM3 is not shown, while only the first β strand of EL2 is displayed. (C) Chemical structures of IT1t and CVX15 (part of molecule displayed in panel B is coloured green). (D,E) Comparison of the interactions between IT1t (magenta carbon atoms) and CXCR4 in the experimentally determined X-ray crystal structure (3odu, panel D) and the best in silico CXCR4 model in the worldwide GPCR DOCK 2010 competition (panel E) correctly predicting the highest number of IT1t-CXCR4 contacts (prior to release of the CXCR4-IT1t crystal structure). Important residues are displayed as ball-and-stick (grey carbon atoms), while IT1t-CXCR4 H-bonds are indicated with black dashed lines. Colour coding of helices and heteroatoms are the same as defined in panels A and B. For reasons of clarity the top of TM3 is not shown.
The cyclic peptide CVX15 resides in TMS2 and, due to its size, points out of the TM domain towards the extracellular side of the protein (Figure 6B). The peptide makes ionic interactions with D1714.60 and D2626.58 similar to other CXCR4 ligands that bind to TMS2 (Table 1, Figure 4F), and makes additional interactions with D18745.51, D19345.57 and E2777.28 in the extracellular region (Figure 6B). The CXCR4 crystal structures with the antagonist IT1t are unique in the sense that they are the first to portray a ligand binding to TMS1 (Figures 4E and 6D). It forms ionic interactions with D972.63 and E2887.39, the latter being a highly conserved binding partner in other chemokine receptors (Figure 5). The CXCR4 crystal structures as well as site-directed mutagenesis data of other chemokine receptors and their ligands (i.e. TAK-779 and AMD11070, Table 1) show that both pockets (TMS1 and TMS2) are interconnected. The existence of different ligand-binding sites makes the structure-based design of small-molecule ligands for chemokine receptors challenging.
Figure 5.
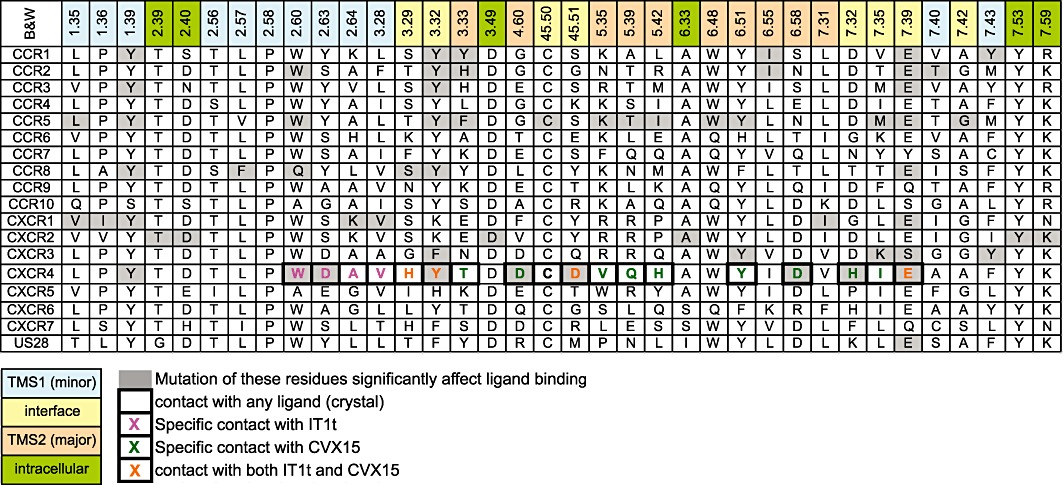
Alignment of important amino acid residues of TM domains and EL2. The TM residues are shown using the Ballesteros–Weinstein (B&W) numbering scheme (Ballesteros and Weinstein, 1995). An adapted version is also used for the EL2 residues (45.50 and 45.51) indicating residues in the loop between TM4 and TM5, using the conserved cysteine as reference: 45.50 (de Graaf et al., 2008). Mutation of residues highlighted in gray significantly affected ligand binding. Also the TXP motif is shown (2.56–2.58), which is conserved among chemokine receptors (see text for details). The residues are coloured blue in case of TMS1 residues (minor pocket), orange for TMS2 residues (major pocket), yellow for interface residues between TMS1 and TMS2, and green for intracellular residues. For CXCR4, contacts from the crystal structures of the receptor with ligands are indicated with a thick black border. Magenta indicates contacts for the small-molecule IT1t, green for the peptide CVX15, and orange for both ligands. For more detailed information on the residues, ligands and associated literature references, see Table 1.
Next to the novel ligand-binding modes, the CXCR4 crystal structures portray several other differences to those of previously resolved GPCRs. First, TM2 of the chemokine receptor family possesses a unique S/TXP motif (Figure 5: residues 2.56–2.58) which induces a unique helical kink to position the two ligand-binding residues W942.60 (Q in CCR8) and D972.63 (S or Y in other chemokine receptors) into TMS1 instead of towards the membrane layer as in the bRho, ADRB2, DRD3 and AA2AR crystal structures (Palczewski et al., 2000; Cherezov et al., 2007; Jaakola et al., 2008; Chien et al., 2010). This alternative kink of TM2 is supported by site-directed mutagenesis data probing the TM2–TM3 interface (Govaerts et al., 2001) and receptor–ligand interactions (Table 1) and is in line with earlier predictions of TM flexibility (Deupi et al., 2004).
Secondly, the crystal structures of ligand IT1t show a disordered Hx8 (Wu et al., 2010). The impact of this distortion on future modelling attempts based on the CXCR4 crystal structures is that models for ligands that bind to a putative intracellular pocket (Figure 4D) will be difficult to construct since they contact TM7 (e.g. SCH-527123, SB265610 in CXCR2, see section on intracellular binding sites and Table 1).
Thirdly, EL2 of CXCR4 contains the same β strand in the crystal structure as observed in rhodopsin, but is oriented outward (Figure 6A), to accommodate the large peptide ligand or Hx8 of the CXCR4 monomer, demonstrating the plasticity of this loop to fold in an open or closed conformation.
Before the release of CXCR4-CVX15 and CXCR4-IT1t structures (Wu et al., 2010), the scientific community was invited to submit a structural model of CXCR4 in complex with either CVX15 or IT1t in the open challenge GPCRDOCK2010 (Kufareva et al., 2011). Given the differences with previously released GPCR crystal structures and the lack of conclusive experimental data to guide the modelling procedure, correct prediction of the CXCR4 fold and CXCR4–ligand interactions was shown to be highly challenging. Our model of the CXCR4–IT1t complex (Figure 6E) was in fact the only model out of 103 models (from 25 different groups) which correctly captured both ionic interactions with D972.63 and E2887.39, placing the dicyclohexylthiourea between TM2 (W942.60, ionic interaction with D972.63), TM3 (V1123.28) and EL2 (I19045.54), and the imidazothiazole ring between TM2 (W942.60) and TM3 (cation–π interaction with Y1163.32), and TM7 (ionic interaction with E2887.39). Although the overall position of the ligand was more shifted towards TM2 and TM3, the model was the only model that identified more than 20% of the ligand–receptor atomic contacts in the crystal structure (Figure 6D,E) (Kufareva et al., 2011). The TM bundle of our CXCR4 model was constructed based on the ADRB2 crystal structure (Cherezov et al., 2007) (next to ADRB1, the structural template with the highest resolution to CXCR4) and TM2 was modelled in an alternative kink, stabilized by the chemokine receptor specific TXP motif (Govaerts et al., 2001; Kellenberger et al., 2007; Shamovsky et al., 2009) and orienting W942.60 and D972.63 towards the ligand-binding pocket. This modelling procedure was guided by site-directed mutagenesis data probing the TM2–TM3 interface (Govaerts et al., 2001) and receptor–ligand interactions (Table 1), including the experimentally determined involvement of D972.63 in the binding of other CXCR4 ligands (Wong et al., 2008). We docked IT1t into the CXCR4 TM bundle in line with ligand-binding mode hypotheses (de Graaf and Rognan, 2009) matching the essential positively ionizable thiorea and imidathiazole groups of IT1t (Thoma et al., 2008) with the negatively charged carboxylate groups of D972.63, D1714.60, D2626.58 and E2887.39 (Wong et al., 2008). Furthermore, the steep structure–activity relationship around the dimethyl-imidathiazole group (Thoma et al., 2008) supported the tight cation–π stacking interaction with Y1163.32 at the bottom of TMS1 (Figure 6D,E). It should be noted that none of the CXCR4-CVX15 models predicted the important interactions in the crystal structure, indicating that accurate prediction of flexible peptide ligands in large receptor-binding pockets (without the availability of experimentally supported receptor–ligand interaction anchors) is currently beyond the reach of molecular modelling approaches. The low accuracy of the loop predictions in the GPCRdock challenge, compared to the reasonable accuracy of the predicted fold of the TM helices (Kufareva et al., 2011), is furthermore in line with a previous evaluation of the implication of EL2 modelling on structure-based virtual screening (de Graaf et al., 2008).
The binding modes of ligands in earlier ADRB1/2 homology models generally resemble the binding orientations of ligands in the respective crystal structures (Cherezov et al., 2007; 2010; de Graaf and Rognan, 2009), and the recently solved DRD3-ligand co-crystal structure could be correctly predicted based on the closely related ADRB1/2 crystal structures (Kufareva et al., 2011). The use of experimental anchors to guide the construction of a AA2AR-ligand crystal structure complex were, however, somewhat misleading (de Graaf and Rognan, 2009), while the spatial distribution of experimentally determined interaction partners in CXCR4 (negatively ionizable residues D972.63, D1714.60, D2626.58 and E2887.39) (Wong et al., 2008) and its ligand (two positively ionizable groups) allows the definition of several alternative binding modes. The success of our customized CXCR4 modelling approach demonstrates that a careful consideration of experimental data (site-directed mutagenesis data in combination with structure–activity relationships) is essential in predicting protein structure and protein–ligand interactions (de Graaf and Rognan, 2009). Finally it should be noted that customized chemokine receptor models based on bRho and ADRB2 crystal structures have already been successfully used to identify new ligands for CCR2 (Kim et al., 2011), CCR3 (Becker et al., 2004), CCR4 (Bayry et al., 2008; Davies et al., 2009), CCR5 (Kellenberger et al., 2007; Liu et al., 2008), CXCR4 (Perez-Nueno et al., 2009) and CXCR7 (Jones et al., 2006) in the past. Therefore, it is expected that the novel crystallographic data on CXCR4 will improve the resolution of in silico models and aid the structure-based development of future drugs for targets belonging to the chemokine receptor family.
Cross-modulation within chemokine receptor oligomers
Although GPCRs can function as monomeric signalling units by coupling of intracellular heterotrimeric G proteins or β-arrestins upon agonist binding to their extracellular surface in a 1:1:1 stoichiometry (Whorton et al., 2007; Kuszak et al., 2009; Arcemisbehere et al., 2010; Tsukamoto et al., 2010), accumulating evidence suggest that GPCRs are assembled in homo- and/or heteromeric complexes for at least part of their lifetime (Pin et al., 2007; Milligan, 2009; Lohse, 2010). Several examples of homo- and heteromeric interactions between chemokine receptors, but also between chemokine receptors and other GPCR subtypes have been reported in the last decade (Figure 7).
Figure 7.
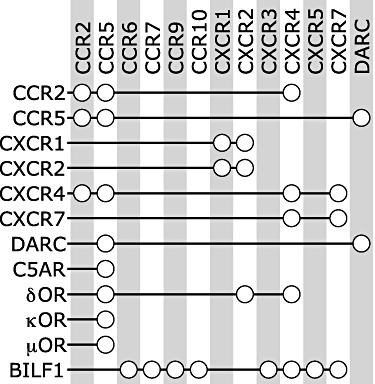
GPCR dimerization partners of chemokine receptors. The open circles connect the chemokine receptors with their suggested dimerization partners.
Initial cross-linking experiments suggested that chemokines induce homo- and/or heteromerization of CCR2, CCR5 and CXCR4 (Rodriguez-Frade et al., 1999; Vila-Coro et al., 1999; 2000; Mellado et al., 2001; Hernanz-Falcon et al., 2004; Rodriguez-Frade et al., 2004). However, more recent co-immunoprecipitation, resonance energy transfer (RET) and protein complementation assay (PCA)-based studies revealed that all tested chemokine receptors oligomerize in a ligand-independent manner (Issafras et al., 2002; Babcock et al., 2003; El-Asmar et al., 2005; Wilson et al., 2005; Sohy et al., 2009). The latter fits well with the current dogma that GPCR oligomers are formed during protein biosynthesis and maturation in the endoplasmic reticulum (ER) to facilitate appropriate protein folding and cell surface targeting (Milligan, 2010). On the other hand, fusion of an ER-retention motif to the C-tail of CXCR2 not only impaired its own trafficking to the cell surface, but also of co-expressed wild-type CXCR1 and CXCR2 through the formation of heteromeric complexes (Wilson et al., 2005). Similar entrapment of wild-type CCR5 by the dominant negative CCR5Δ32 truncation mutant was proposed to explain the delayed progression of HIV-1 infection in heterozygous individuals (Benkirane et al., 1997), although others raised scepticism on the dominant negative nature of this observation (Venkatesan et al., 2002).
Recent studies using fluorescence recovery after photobleaching (FRAP) and total internal reflection fluorescence (TIRF) microscopy revealed that some GPCR subtypes are engaged in short-living transient homodimers that are formed and fall apart within seconds in a ligand-independent manner, whereas others are assembled in stable higher order oligomers at the cell surface (Dorsch et al., 2009; Fonseca and Lambert, 2009; Hern et al., 2010). Similar FRAP or TIRF microscopy evaluation of chemokine receptor oligomer stability remains to be performed. However, CXCR4 and δ-opioid receptors were proposed to exist in a ligand-dependent dynamic equilibrium between homo- and heteromers (Pello et al., 2008). Stimulation with either CXCL12 or [D-Pen2, D-Pen5]-enkephalin shifted this equilibrium towards signalling homomers, whereas simultaneous addition of both agonists induced the formation of signalling-deficient heteromers.
Heteromerization of the non-G protein-signalling chemokine receptors CXCR7 and DARC with CXCR4 and CCR5, respectively, blunted chemokine-induced G protein signalling by the latter two receptors (Chakera et al., 2008; Levoye et al., 2009). On the other hand, heteromerization of CCR5 with CCR2 or CXCR4 shifted G protein coupling from Gi- towards Gq-mediated signalling pathways resulting in cell adhesion rather than chemotaxis (Mellado et al., 2001; Molon et al., 2005; Contento et al., 2008). Recruitment of CCR5/CXCR4 heteromers to the immunological synapse stabilizes the interaction of T cell with antigen-presenting cells in response to chemokine secretion by the latter (Mellado et al., 2001; Molon et al., 2005; Contento et al., 2008).
Chemokines can induce changes in basal RET and/or PCA signals, which are generally interpreted as conformational rearrangements within existing chemokine receptor oligomers, rather than de novo formation or dissociation of oligomers (Toth et al., 2004; Percherancier et al., 2005; Sohy et al., 2007; Chakera et al., 2008; Isik et al., 2008; Levoye et al., 2009; Luker et al., 2009). These conformational rearrangements result in allosterism between individual chemokine receptors within oligomers. Negative chemokine binding cooperativity has been observed within CCR2, CCR5 and CXCR4 heteromers in equilibrium (competition) binding and ‘infinite’ radioligand dilution experiments (El-Asmar et al., 2005; Springael et al., 2006; Sohy et al., 2007; 2009). Assuming that chemokine affinities for their cognate receptors are within the same order of magnitude (Murphy et al., 2011), these heteromeric chemokine receptor configurations allow cells to respond to the highest chemokine (gradient) concentration at the expense of other chemokine subtypes by allosterically inhibiting their interaction to partnering receptors. Moreover, low-molecular-weight (allosteric) antagonists acting at one receptor can cross-inhibit in vitro and in vivo chemokine-induced immune cell recruitment mediated through the other chemokine receptor within the heteromer (Sohy et al., 2007; 2009). In contrast, however, the CXCR2 antagonist SB225002 increased signalling of CXCR2/δOR heteromers in response to opioid agonists (Parenty et al., 2008). Hence, this allosteric cross-modulation should be kept in mind when screening for ligands against a particular chemokine receptor to avoid side effects through heteromerized receptor partners. On the other hand, one can also take advantage of allosteric modulation by targeting a widely abundant receptor in a more cell type-specific manner through its less widely expressed heteromeric partner. Moreover, future development of chemokine receptor heteromer-selective (bivalent) ligands may also contribute to the specific targeting of a smaller subset of cells (Waldhoer et al., 2005; Jähnichen et al., 2010; Tanaka et al., 2010; Zhang et al., 2010).
Summary
In summary, the family of chemokines receptors is a perfect showcase for the GPCR family to illustrate the effectiveness of GPCR targeting with small-molecule allosteric modulators and/or biologicals. This is underscored by the recent FDA approval of two small molecules targeting CXCR4 and CCR5. Whereas most biologicals are considered to target extracellular parts of the receptor, the present data on binding modes of the small-molecule antagonists and chemokines suggests that competing with or blocking the entrance of the N-terminus of a chemokine to TM residues poses a mechanism of allosteric (or partially competitive) inhibition of chemokine-mediated receptor activation for many of the chemokine receptor antagonists binding to the classical TMS1- and TMS2-binding pockets (Table 1, Figure 4). As this region is generally not important for chemokine binding, using the term allosterism is, in our opinion, justified. Whether these interactions are purely allosteric or partially competitive largely depends on the used chemokine probe and its specific receptor interactions. There is no discussion needed on the allosteric nature of chemokine receptor antagonists suggested to bind to intracellular binding sites of CXCR2 or CCR4. It remains to be established whether other chemokine receptors or other members of the large GPCR family can be modulated in a similar manner, but the two examples are intriguing. Similarly intriguing is the possibility to specifically target chemokine receptor heterodimers. We would like to stress though, that the evidence for such a mechanism of action of small-molecule modulator still remains to be established.
Targeting chemokine receptors in a functionally selective manner, as suggested to be feasible for CCR5 and CXCR4 (Sachpatzidis et al., 2003; Watson et al., 2005; Saita et al., 2006), is a further promise for future drug discovery. The association of specific signalling pathways with disease or adverse drug effects is starting to emerge, and the overall challenge remains to identify what signalling pathway to target in a particular disease. On the other hand, insights in the structure activity relationships governing functional selectivity is needed, and in vivo studies will have to shed more light on the potential of functional selective ligands in the treatment of chemokine-related disease.
Finally, the recent breakthrough of the CXCR4 crystal structure will give a strong impetus to additional receptor crystallization, mutagenesis, modelling and pharmacological studies, which will be essential to delineate the mechanism of action of the various small-molecule allosteric modulators and/or biologicals.
Acknowledgments
This review was (in part) written within the framework of Top Institute Pharma, project number D1-105 (GPCR forum) and T1-103-1 (Chemokine Receptors).
Glossary
- 7TM
seven-transmembrane
- AA2AR
adenosine A2A receptor
- ADCC
antibody-dependent cellular cytotoxicity
- ADRB
β-adrenoceptor
- ATLL
adult T-cells leukaemia-lymphoma
- bRho
bovine rhodopsin
- CCL
CC chemokine ligand
- CCR
CC chemokine receptor
- CRS
chemokine recognition site
- CXCL
CXC chemokine ligand
- CXCR
CXC chemokine receptor
- DRD3
dopamine D3 receptor
- EL
extracellular loop
- FRAP
fluorescence recovery after photobleaching
- GRK
GPCR kinase
- mAb
monoclonal antibody
- PCA
protein complementation assay
- RET
resonance energy transfer
- TIRF
total internal reflection fluorescence
- TM
transmembrane
- TMS
transmembrane site
- VHH
variable domain of heavy-chain antibody
Conflicts of interest
None.
References
- Ahuja S, Lee J, Murphy P. CXC chemokines bind to unique sets of selectivity determinants that can function independently and are broadly distributed on multiple domains of human interleukin-8 receptor B. Determinants of high affinity binding and receptor activation are distinct. J Biol Chem. 1996;271:225–232. doi: 10.1074/jbc.271.1.225. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Bertini R, Cesta M, Bizzarri C, Di Bitondo R, Di Cioccio V, et al. 2-Arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncompetitive CXCL8 inhibitors. J Med Chem. 2005;48:4312–4331. doi: 10.1021/jm049082i. [DOI] [PubMed] [Google Scholar]
- Allen S, Crown S, Handel T. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Andrews G, Jones C, Wreggett KA. An intracellular allosteric site for a specific class of antagonists of the CC chemokine G protein-coupled receptors CCR4 and CCR5. Mol Pharmacol. 2008;73:855–867. doi: 10.1124/mol.107.039321. [DOI] [PubMed] [Google Scholar]
- Arcemisbehere L, Sen T, Boudier L, Balestre MN, Gaibelet G, Detouillon E, et al. Leukotriene BLT2 receptor monomers activate the G(i2) GTP-binding protein more efficiently than dimers. J Biol Chem. 2010;285:6337–6347. doi: 10.1074/jbc.M109.083477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA. 1999;96:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. 2003;278:3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Stuart CS, editor. Methods in Neurosciences. Vol. 25. New York: Academic Press; 1995. pp. 366–428. [Google Scholar]
- Bayry J, Tchilian EZ, Davies MN, Forbes EK, Draper SJ, Kaveri SV, et al. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc Natl Acad Sci USA. 2008;105:10221–10226. doi: 10.1073/pnas.0803453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, et al. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci USA. 2004;101:11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272:30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- Berchiche YA, Gravel S, Pelletier M-E, St-Onge G, Heveker N. Different effects of the different natural CC-chemokine receptor 2B (CCR2B) ligands on {beta}-arrestin recruitment, G{alpha}i signalling, and receptor internalization. Mol Pharmacol. 2010;79:488–498. doi: 10.1124/mol.110.068486. [DOI] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Berkhout T, Blaney F, Bridges A, Cooper D, Forbes I, Gribble A, et al. CCR2: characterization of the antagonist binding site from a combined receptor modeling/mutagenesis approach. J Med Chem. 2003;46:4070–4086. doi: 10.1021/jm030862l. [DOI] [PubMed] [Google Scholar]
- Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci USA. 2004;101:11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Doranz B, Bondue A, Govaerts C, De Leener A, Vassart G, et al. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J Biol Chem. 2003;278:5179–5187. doi: 10.1074/jbc.M205684200. [DOI] [PubMed] [Google Scholar]
- Bongers G, Maussang D, Muniz LR, Noriega VM, Fraile-Ramos A, Barker N, et al. The cytomegalovirus-encoded chemokine receptor US28 promotes intestinal neoplasia in transgenic mice. J Clin Invest. 2010;120:3969–3978. doi: 10.1172/JCI42563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley ME, Bond ME, Manini J, Brown Z, Charlton SJ. SB265610 is an allosteric, inverse agonist at the human CXCR2 receptor. Br J Pharmacol. 2009;158:328–338. doi: 10.1111/j.1476-5381.2009.00182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelot A, Heveker N, Montes M, Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem. 2000;275:23736–23744. doi: 10.1074/jbc.M000776200. [DOI] [PubMed] [Google Scholar]
- Buntinx M, Hermans B, Goossens J, Moechars D, Gilissen RAHJ, Doyon J, et al. Pharmacological profile of JNJ-27141491 [(S)-3-[3,4-difluorophenyl)-propyl]-5-isoxazol-5-yl-2-thioxo-2,3-dihydro-1H-imidazole-4-carboxyl acid methyl ester], as a noncompetitive and orally active antagonist of the human chemokine receptor CCR2. J Pharmacol Exp Ther. 2008;327:1–9. doi: 10.1124/jpet.108.140723. [DOI] [PubMed] [Google Scholar]
- Casarosa P, Menge WM, Minisini R, Otto C, van Heteren J, Jongejan A, et al. Identification of the first nonpeptidergic inverse agonist for a constitutively active viral-encoded G protein-coupled receptor. J Biol Chem. 2003;278:5172–5178. doi: 10.1074/jbc.M210033200. [DOI] [PubMed] [Google Scholar]
- Casarosa P, Waldhoer M, LiWang PJ, Vischer HF, Kledal T, Timmerman H, et al. CC and CX3C chemokines differentially interact with the N terminus of the human cytomegalovirus-encoded US28 receptor. J Biol Chem. 2005;280:3275–3285. doi: 10.1074/jbc.M407536200. [DOI] [PubMed] [Google Scholar]
- Chakera A, Seeber RM, John AE, Eidne KA, Greaves DR. The duffy antigen/receptor for chemokines exists in an oligomeric form in living cells and functionally antagonizes CCR5 signaling through hetero-oligomerization. Mol Pharmacol. 2008;73:1362–1370. doi: 10.1124/mol.107.040915. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum D, Hanson M, Rasmussen S, Thian F, Kobilka T, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Abola E, Stevens RC. Recent progress in the structure determination of GPCRs, a membrane protein family with high potential as pharmaceutical targets. Methods Mol Biol. 2010;654:141–168. doi: 10.1007/978-1-60761-762-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney RJ, Nelson DJ, Lo YC, Yang G, Scherle PA, Jezak H, et al. Synthesis and evaluation of cis-3,4-disubstituted piperidines as potent CC chemokine receptor 2 (CCR2) antagonists. Bioorg Med Chem Lett. 2008;18:5063–5065. doi: 10.1016/j.bmcl.2008.07.123. [DOI] [PubMed] [Google Scholar]
- Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Mattioli I, Gong J, Loetscher P. Structure-function relationship between the human chemokine receptor CXCR3 and its ligands. J Biol Chem. 2003;278:289–295. doi: 10.1074/jbc.M209470200. [DOI] [PubMed] [Google Scholar]
- Colvin R, Campanella G, Manice L, Luster A. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26:5838–5849. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, et al. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci USA. 2008;105:10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MA, Jenh CH, Gonsiorek W, Fine J, Narula SK, Zavodny PJ, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- Davies MN, Bayry J, Tchilian EZ, Vani J, Shaila MS, Forbes EK, et al. Toward the discovery of vaccine adjuvants: coupling in silico screening and in vitro analysis of antagonist binding to human and mouse CCR4 receptors. PLoS ONE. 2009;4:e8084. doi: 10.1371/journal.pone.0008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil™) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. 2010;128:509–518. doi: 10.1016/j.pharmthera.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Deupi X, Olivella M, Govaerts C, Ballesteros J, Campillo M, Pardo L. Ser and Thr residues modulate the conformation of pro-kinked transmembrane {alpha}-helices. Biophys J. 2004;86:105–115. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bunemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat Methods. 2009;6:225–230. doi: 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Dragic T, Trkola A, Thompson D, Cormier E, Kajumo F, Maxwell E, et al. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc Natl Acad Sci USA. 2000;97:5639–5644. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67:460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol. 2002;42:469–499. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- Fonseca JM, Lambert NA. Instability of a class a G protein-coupled receptor oligomer interface. Mol Pharmacol. 2009;75:1296–1299. doi: 10.1124/mol.108.053876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Gerlach L, Skerlj R, Bridger G, Schwartz T. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem. 2001;276:14153–14160. doi: 10.1074/jbc.M010429200. [DOI] [PubMed] [Google Scholar]
- Ghassabeh GH, Muyldermans S, Saerens D. Nanobodies, single-domain antigen-binding fragments of camelid heavy-chain antibodies. In: Shire SJ, et al., editors. Current Trends in Monoclonal Antibody Development and Manufacturing. New York: Springer; 2010. pp. 29–48. [Google Scholar]
- Gilbert J, Lekstrom-Himes J, Donaldson D, Lee Y, Hu M, Xu J, et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol. 2011;107:906–911. doi: 10.1016/j.amjcard.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Gong J, Clark-Lewis I. Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal residues. J Exp Med. 1995;181:631–640. doi: 10.1084/jem.181.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsiorek W, Fan X, Hesk D, Fossetta J, Qiu H, Jakway J, et al. Pharmacological characterization of 2-Hydroxy-N,N- dimethyl-3-{2-[-[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4- dioxo-cyclobut-1-enylamino}-benzamide (Sch527123), a potent allosteric CXCR1/CXCR2 antagonist. J Pharmacol Exp Ther. 2007;322:477–485. doi: 10.1124/jpet.106.118927. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Zhang C, Wu S, Kunstman K, Trachtenberg E, Phair J, et al. Persistence of dual-tropic HIV-1 in an individual homozygous for the CCR5 delta 32 allele. Lancet. 2002;359:1832–1834. doi: 10.1016/S0140-6736(02)08681-6. [DOI] [PubMed] [Google Scholar]
- Govaerts C, Blanpain C, Deupi X, Ballet S, Ballesteros J, Wodak S, et al. The TXP motif in the second transmembrane helix of CCR5. A structural determinant of chemokine-induced activation. J Biol Chem. 2001;276:13217–13225. doi: 10.1074/jbc.M011670200. [DOI] [PubMed] [Google Scholar]
- Govaerts C, Bondue A, Springael J, Olivella M, Deupi X, Le Poul E, et al. Activation of CCR5 by chemokines involves an aromatic cluster between transmembrane helices 2 and 3. J Biol Chem. 2003;278:1892–1903. doi: 10.1074/jbc.M205685200. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Rognan D. Customizing G Protein-coupled receptor models for structure-based virtual screening. Curr Pharm Des. 2009;15:4026–4048. doi: 10.2174/138161209789824786. [DOI] [PubMed] [Google Scholar]
- de Graaf C, Foata N, Engkvist O, Rognan D. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins. 2008;71:599–620. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- Gupta S, Pillarisetti K, Thomas R, Aiyar N. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1alpha: implications for development of selective CXCR4 antagonists. Immunol Lett. 2001;78:29–34. doi: 10.1016/s0165-2478(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Hall SE, Mao A, Nicolaidou V, Finelli M, Wise EL, Nedjai B, et al. Elucidation of binding sites of dual antagonists in the human chemokine receptors CCR2 and CCR5. Mol Pharmacol. 2009;75:1325–1336. doi: 10.1124/mol.108.053470. [DOI] [PubMed] [Google Scholar]
- Handel TM, Horuk R. Duffy antigen inhibitors: useful therapeutics for malaria? Trends Parasitol. 2010;26:329–333. doi: 10.1016/j.pt.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Haskell C, Horuk R, Liang M, Rosser M, Dunning L, Islam I, et al. Identification and characterization of a potent, selective nonpeptide agonist of the CC chemokine receptor CCR8. Mol Pharmacol. 2006;69:309–316. doi: 10.1124/mol.105.014779. [DOI] [PubMed] [Google Scholar]
- Hatse S, Huskens D, Princen K, Vermeire K, Bridger G, de Clercq E, et al. Modest human immunodeficiency virus coreceptor function of CXCR3 is strongly enhanced by mimicking the CXCR4 ligand binding pocket in the CXCR3 receptor. J Virol. 2007;81:3632–3639. doi: 10.1128/JVI.01941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatse S, Princen K, Vermeire K, Gerlach L-O, Rosenkilde MM, Schwartz TW, et al. Mutations at the CXCR4 interaction sites for AMD3100 influence anti-CXCR4 antibody binding and HIV-1 entry. FEBS Lett. 2003;546:300–306. doi: 10.1016/s0014-5793(03)00609-4. [DOI] [PubMed] [Google Scholar]
- Haworth B, Lin H, Fidock M, Dorr P, Strange P. Allosteric effects of antagonists on signalling by the chemokine receptor CCR5. Biochem Pharmacol. 2007;74:891–897. doi: 10.1016/j.bcp.2007.06.032. [DOI] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc Natl Acad Sci USA. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz-Falcon P, Rodriguez-Frade JM, Serrano A, Juan D, del Sol A, Soriano SF, et al. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat Immunol. 2004;5:216–223. doi: 10.1038/ni1027. [DOI] [PubMed] [Google Scholar]
- Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs. 2010;2:594–606. doi: 10.4161/mabs.2.6.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. 2010;16:1520–1531. doi: 10.1158/1078-0432.CCR-09-2697. [DOI] [PubMed] [Google Scholar]
- Isik N, Hereld D, Jin T. Fluorescence resonance energy transfer imaging reveals that chemokine-binding modulates heterodimers of CXCR4 and CCR5 receptors. PLoS ONE. 2008;3:e3424. doi: 10.1371/journal.pone.0003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issafras H, Angers S, Bulenger S, Blanpain C, Parmentier M, Labbe-Jullie C, et al. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J Biol Chem. 2002;277:34666–34673. doi: 10.1074/jbc.M202386200. [DOI] [PubMed] [Google Scholar]
- Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JM, Saag MS, Thompson MA, Fischl MA, Liporace R, Reichman RC, et al. Antiviral activity of single-dose PRO 140, a CCR5 monoclonal antibody, in HIV-infected adults. J Infect Dis. 2008;198:1345–1352. doi: 10.1086/592169. [DOI] [PubMed] [Google Scholar]
- Jacobson JM, Lalezari JP, Thompson MA, Fichtenbaum CJ, Saag MS, Zingman BS, et al. Phase 2a study of the CCR5 monoclonal antibody PRO 140 administered intravenously to HIV-infected adults. Antimicrob Agents Chemother. 2010a;54:4137–4142. doi: 10.1128/AAC.00086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JM, Thompson MA, Lalezari JP, Saag MS, Zingman BS, D'Ambrosio P, et al. Anti-HIV-1 activity of weekly or biweekly treatment with subcutaneous PRO 140, a CCR5 monoclonal antibody. J Infect Dis. 2010b;201:1481–1487. doi: 10.1086/652190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jähnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, et al. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci USA. 2010;107:20565–20570. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekle A, Keppler OT, De Clercq E, Schols D, Weinstein M, Goldsmith MA. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J Virol. 2003;77:5846–5854. doi: 10.1128/JVI.77.10.5846-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PC, Nygaard R, Thiele S, Elder A, Zhu G, Kolbeck R, et al. Molecular interaction of a potent nonpeptide agonist with the chemokine receptor CCR8. Mol Pharmacol. 2007;72:327–340. doi: 10.1124/mol.106.035543. [DOI] [PubMed] [Google Scholar]
- Jensen PC, Thiele S, Ulven T, Schwartz TW, Rosenkilde MM. Positive versus negative modulation of different endogenous chemokines for CC-chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J Biol Chem. 2008;283:23121–23128. doi: 10.1074/jbc.M803458200. [DOI] [PubMed] [Google Scholar]
- Jin H, Shen X, Baggett BR, Kong X, LiWang PJ. The human CC chemokine MIP-1beta dimer is not competent to bind to the CCR5 receptor. J Biol Chem. 2007;282:27976–27983. doi: 10.1074/jbc.M702654200. [DOI] [PubMed] [Google Scholar]
- Jones SW, Brockbank SM, Mobbs ML, Le Good NJ, Soma-Haddrick S, Heuze AJ, et al. The orphan G-protein coupled receptor RDC1: evidence for a role in chondrocyte hypertrophy and articular cartilage matrix turnover. Osteoarthritis Cartilage. 2006;14:597–608. doi: 10.1016/j.joca.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75:1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- Kehrl JH, Hwang IY, Park C. Chemoattract receptor signaling and its role in lymphocyte motility and trafficking. Curr Top Microbiol Immunol. 2009;334:107–127. doi: 10.1007/978-3-540-93864-4_5. [DOI] [PubMed] [Google Scholar]
- Kellenberger E, Springael J-Y, Parmentier M, Hachet-Haas M, J-l G, Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J Med Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2010a;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Kenakin T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res. 2010b;30:313–321. doi: 10.3109/10799893.2010.503964. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lim JW, Lee SW, Kim K, No KT. Ligand supported homology modeling and docking evaluation of CCR2: docked pose selection by consensus scoring. J Mol Model. 2011 doi: 10.1007/s00894-010-0943-x. DOI: 10.1007/s00894-010-0943-x. [DOI] [PubMed] [Google Scholar]
- Klibanov OM. Vicriviroc, a CCR5 receptor antagonist for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2009;10:845–859. [PubMed] [Google Scholar]
- Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem. 2009;284:35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout T, Nicholas S, Perry S, Reinhart G, Junger S, Struthers R. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, et al. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol. 2008;73:789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- Kouroumalis A, Nibbs R, Aptel H, Wright K, Kolios G, Ward S. The chemokines CXCL9, CXCL10, and CXCL11 differentially stimulate G alpha i-independent signaling and actin responses in human intestinal myofibroblasts. J Immunol. 2005;175:5403–5411. doi: 10.4049/jimmunol.175.8.5403. [DOI] [PubMed] [Google Scholar]
- Kraneveld AD, Braber S, Overbeek S, de Kruijf P, Koelink P, Smit MJ. Chemokine receptors in inflammatory diseases. In: Smit MJ, Lira SA, Leurs R, editors. Chemokine Receptors as Drug Targets. Vol. 46. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2011. pp. 107–150. [Google Scholar]
- de Kruijf P, van Heteren J, Lim H, Conti P, van der Lee M, Bosch L, et al. Nonpeptidergic allosteric antagonists differentially bind to the CXCR2 chemokine receptor. J Pharmacol Exp Ther. 2009;329:783–790. doi: 10.1124/jpet.108.148387. [DOI] [PubMed] [Google Scholar]
- Kufareva I, Rueda M, Katritch V, Participants of GPCR Dock 2010 (Box 1) Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community wide GPCR Dock 2010 assessment. Structure. 2011 doi: 10.1016/j.str.2011.05.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalezari J, Yadavalli GK, Para M, Richmond G, Dejesus E, Brown SJ, et al. Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1-infected patients. J Infect Dis. 2008;197:721–727. doi: 10.1086/527327. [DOI] [PubMed] [Google Scholar]
- Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, Borlat F, et al. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279:22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- Laurence JS, Blanpain C, Burgner JW, Parmentier M, LiWang PJ. CC chemokine MIP-1 beta can function as a monomer and depends on Phe13 for receptor binding. Biochemistry. 2000;39:3401–3409. doi: 10.1021/bi9923196. [DOI] [PubMed] [Google Scholar]
- Leach K, Charlton S, Strange P. Analysis of second messenger pathways stimulated by different chemokines acting at the chemokine receptor CCR5. Biochem Pharmacol. 2007;74:881–890. doi: 10.1016/j.bcp.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou E, Yu K, Zhu J, Zhang Y, Xie X, et al. Discovery of a novel CCR5 antagonist lead compound through fragment assembly. Molecules. 2008;13:2426–2441. doi: 10.3390/molecules13102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ. Dimerization in GPCR mobility and signaling. Curr Opin Pharmacol. 2010;10:53–58. doi: 10.1016/j.coph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Luker KE, Gupta M, Luker GD. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23:823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, et al. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med. 1998;187:1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Das D, Ogata-Aoki H, Nakata H, Miyakawa T, Tojo Y, et al. Structural and molecular interactions of CCR5 inhibitors with CCR5. J Biol Chem. 2006;281:12688–12698. doi: 10.1074/jbc.M512688200. [DOI] [PubMed] [Google Scholar]
- Maeda K, Das D, Yin PD, Tsuchiya K, Ogata-Aoki H, Nakata H, et al. Involvement of the second extracellular loop and transmembrane residues of CCR5 in inhibitor binding and HIV-1 fusion: insights into the mechanism of allosteric inhibition. J Mol Biol. 2008;381:956–974. doi: 10.1016/j.jmb.2008.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, et al. Spirodiketopiperazine-Based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol. 2004;78:8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda DY, Quinn MT, Schepetkin IA, Kirpotina LN, Zebala JA. Nicotinamide glycolates antagonize CXCR2 activity through an intracellular mechanism. J Pharmacol Exp Ther. 2010;332:145–152. doi: 10.1124/jpet.109.159020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20:254–257. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6:907–918. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- Maussang D, Langemeijer E, Fitzsimons CP, Stigter-van Walsum M, Dijkman R, Borg MK, et al. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009;69:2861–2869. doi: 10.1158/0008-5472.CAN-08-2487. [DOI] [PubMed] [Google Scholar]
- May L, Leach K, Sexton P, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de Ana A, Jones DR, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20:2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendonça F, da Fonseca P, Phillips R, Saldanha J, Williams T, Pease J. Site-directed mutagenesis of CC chemokine receptor 1 reveals the mechanism of action of UCB 35625, a small molecule chemokine receptor antagonist. J Biol Chem. 2005;280:4808–4816. doi: 10.1074/jbc.M412267200. [DOI] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. The role of dimerisation in the cellular trafficking of G-protein-coupled receptors. Curr Opin Pharmacol. 2010;10:23–29. doi: 10.1016/j.coph.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Mirzadegan T, Diehl F, Ebi B, Bhakta S, Polsky I, McCarley D, et al. Identification of the binding site for a novel class of CCR2b chemokine receptor antagonists: binding to a common chemokine receptor motif within the helical bundle. J Biol Chem. 2000;275:25562–25571. doi: 10.1074/jbc.M000692200. [DOI] [PubMed] [Google Scholar]
- Molon B, Gri G, Bettella M, Gomez-Mouton C, Lanzavecchia A, Martinez AC, et al. T cell costimulation by chemokine receptors. Nat Immunol. 2005;6:465–471. doi: 10.1038/ni1191. [DOI] [PubMed] [Google Scholar]
- Monteclaro FS, Charo IF. The amino-terminal extracellular domain of the MCP-1 receptor, but not the RANTES/MIP-1alpha receptor, confers chemokine selectivity. Evidence for a two-step mechanism for MCP-1 receptor activation. J Biol Chem. 1996;271:19084–19092. doi: 10.1074/jbc.271.32.19084. [DOI] [PubMed] [Google Scholar]
- Moriconi A, Cesta M, Cervellera M, Aramini A, Coniglio S, Colagioia S, et al. Design of noncompetitive interleukin-8 inhibitors acting on CXCR1 and CXCR2. J Med Chem. 2007;50:3984–4002. doi: 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]
- Muniz-Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, Kazmierski WM, et al. The relative activity of ‘function sparing’ HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol. 2009;75:490–501. doi: 10.1124/mol.108.052555. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Charo IF, Hills R, Horuk R, Matsushima K, Oppenheim JJ. 2011. Chemokine receptors. IUPHAR database (IUPHAR-DB), available at: http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=14.
- Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Nicholls DJ, Tomkinson NP, Wiley KE, Brammall A, Bowers L, Grahames C, et al. Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol Pharmacol. 2008;74:1193–1202. doi: 10.1124/mol.107.044610. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Leurs R, Smit MJ, Vischer HF. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J Biol Chem. 2010;285:29632–29641. doi: 10.1074/jbc.M110.115618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64:2127–2133. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Valentin-Hansen L, Mokrosinski J, Frimurer TM, Schwartz TW. Conserved water-mediated hydrogen bond network between TM-I, -II, -VI, and -VII in 7TM receptor activation. J Biol Chem. 2010;285:19625–19636. doi: 10.1074/jbc.M110.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J. 2008;409:635–649. doi: 10.1042/BJ20071493. [DOI] [PubMed] [Google Scholar]
- Olson WC, Jacobson JM. CCR5 monoclonal antibodies for HIV-1 therapy. Curr Opin HIV AIDS. 2009;4:104–111. doi: 10.1097/COH.0b013e3283224015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson WC, Rabut GE, Nagashima KA, Tran DN, Anselma DJ, Monard SP, et al. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J Virol. 1999;73:4145–4155. doi: 10.1128/jvi.73.5.4145-4155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, Mack M, Proudfoot A, Olbrich H. Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J Biol Chem. 1999;274:8875–8885. doi: 10.1074/jbc.274.13.8875. [DOI] [PubMed] [Google Scholar]
- Ott T, Pahuja A, Nickolls S, Alleva D, Struthers R. Identification of CC chemokine receptor 7 residues important for receptor activation. J Biol Chem. 2004;279:42383–42392. doi: 10.1074/jbc.M401097200. [DOI] [PubMed] [Google Scholar]
- Pakianathan DR, Kuta EG, Artis DR, Skelton NJ, Hebert CA. Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3 and CCR5. Biochemistry. 1997;36:9642–9648. doi: 10.1021/bi970593z. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Parenty G, Appelbe S, Milligan G. CXCR2 chemokine receptor antagonism enhances DOP opioid receptor function via allosteric regulation of the CXCR2-DOP receptor heterodimer. Biochem J. 2008;412:245–256. doi: 10.1042/BJ20071689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J, Wang J, Ponath P, Murphy P. The N-terminal extracellular segments of the chemokine receptors CCR1 and CCR3 are determinants for MIP-1alpha and eotaxin binding, respectively, but a second domain is essential for efficient receptor activation. J Biol Chem. 1998;273:19972–19976. doi: 10.1074/jbc.273.32.19972. [DOI] [PubMed] [Google Scholar]
- Pello OM, Martinez-Munoz L, Parrillas V, Serrano A, Rodriguez-Frade JM, Toro MJ, et al. Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol. 2008;38:537–549. doi: 10.1002/eji.200737630. [DOI] [PubMed] [Google Scholar]
- Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- Perez-Nueno VI, Pettersson S, Ritchie DW, Borrell JI, Teixido J. Discovery of novel HIV entry inhibitors for the CXCR4 receptor by prospective virtual screening. J Chem Inf Model. 2009;49:810–823. doi: 10.1021/ci800468q. [DOI] [PubMed] [Google Scholar]
- Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, et al. International union of basic and clinical pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE. Is CCR2 the right chemokine receptor to target in rheumatoid arthritis? Arthritis Rheum. 2008;58:1889–1891. doi: 10.1002/art.23590. [DOI] [PubMed] [Google Scholar]
- Proudfoot A, Power C, Hoogewerf A, Montjovent M, Borlat F, Offord R, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot AEI, Power CA, Schwarz MK. Anti-chemokine small molecule drugs: a promising future? Expert Opin Investig Drugs. 2010;19:345–355. doi: 10.1517/13543780903535867. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-arrestin- but not G protein-mediated signaling by the ‘decoy’ receptor CXCR7. Proc Natl Acad Sci USA. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan L, Rajarathnam K. Ligand selectivity and affinity of chemokine receptor CXCR1. Role of N-terminal domain. J Biol Chem. 2004;279:30000–30008. doi: 10.1074/jbc.M313883200. [DOI] [PubMed] [Google Scholar]
- Reis RI, Santos EL, Pesquero JB, Oliveira L, Schanstra JP, Bascands JL, et al. Participation of transmembrane proline 82 in angiotensin II AT1 receptor signal transduction. Regul Pept. 2007;140:32–36. doi: 10.1016/j.regpep.2006.11.028. [DOI] [PubMed] [Google Scholar]
- Richter R, Casarosa P, Standker L, Munch J, Springael JY, Nijmeijer S, et al. Significance of N-terminal proteolysis of CCL14a to activity on the chemokine receptors CCR1 and CCR5 and the human cytomegalovirus-encoded chemokine receptor US28. J Immunol. 2009;183:1229–1237. doi: 10.4049/jimmunol.0802145. [DOI] [PubMed] [Google Scholar]
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–2138. doi: 10.1016/j.jacc.2007.02.052. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Frade JM, Vila-Coro AJ, de Ana AM, Albar JP, Martinez AC, Mellado M. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc Natl Acad Sci USA. 1999;96:3628–3633. doi: 10.1073/pnas.96.7.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Frade JM, del Real G, Serrano A, Hernanz-Falcon P, Soriano SF, Vila-Coro AJ, et al. Blocking HIV-1 infection via CCR5 and CXCR4 receptors by acting in trans on the CCR2 chemokine receptor. EMBO J. 2004;23:66–76. doi: 10.1038/sj.emboj.7600020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde M, Gerlach L, Jakobsen J, Skerlj R, Bridger G, Schwartz T. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: transfer of binding site to the CXCR3 receptor. J Biol Chem. 2004;279:3033–3041. doi: 10.1074/jbc.M309546200. [DOI] [PubMed] [Google Scholar]
- Rosenkilde M, Andersen M, Nygaard R, Frimurer T, Schwartz T. Activation of the CXCR3 chemokine receptor through anchoring of a small molecule chelator ligand between TM-III, -IV, and -VI. Mol Pharmacol. 2007a;71:930–941. doi: 10.1124/mol.106.030031. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Gerlach L-O, Hatse S, Skerlj RT, Schols D, Bridger GJ, et al. Molecular mechanism of action of monocyclam versus bicyclam non-peptide antagonists in the CXCR4 chemokine receptor. J Biol Chem. 2007b;282:27354–27365. doi: 10.1074/jbc.M704739200. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Benned-Jensen T, Frimurer TM, Schwartz TW. The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol Sci. 2010;31:567–574. doi: 10.1016/j.tips.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Sabroe I, Peck M, Van Keulen B, Jorritsma A, Simmons G, Clapham P, et al. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J Biol Chem. 2000;275:25985–25992. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- Sachpatzidis A, Benton B, Manfredi J, Wang H, Hamilton A, Dohlman H, et al. Identification of allosteric peptide agonists of CXCR4. J Biol Chem. 2003;278:896–907. doi: 10.1074/jbc.M204667200. [DOI] [PubMed] [Google Scholar]
- Saita Y, Kodama E, Orita M, Kondo M, Miyazaki T, Sudo K, et al. Structural basis for the interaction of CCR5 with a small molecule, functionally selective CCR5 agonist. J Immunol. 2006;177:3116–3122. doi: 10.4049/jimmunol.177.5.3116. [DOI] [PubMed] [Google Scholar]
- Salchow K, Bond ME, Evans SC, Press NJ, Charlton SJ, Hunt PA, et al. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br J Pharmacol. 2010;159:1429–1439. doi: 10.1111/j.1476-5381.2009.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Iida S, Shitara K. Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin Biol Ther. 2006;6:1161–1173. doi: 10.1517/14712598.6.11.1161. [DOI] [PubMed] [Google Scholar]
- Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–159. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- Schulte A, Schulz B, Andrzejewski MG, Hundhausen C, Mletzko S, Achilles J, et al. Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by alpha- and gamma-secretases. Biochem Biophys Res Commun. 2007;358:233–240. doi: 10.1016/j.bbrc.2007.04.100. [DOI] [PubMed] [Google Scholar]
- Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann S, Palani A, et al. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349:41–54. doi: 10.1016/j.virol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Shamovsky I, de Graaf C, Alderin L, Bengtsson M, Bladh H, Borjesson L, et al. Increasing selectivity of CC chemokine receptor 8 antagonists by engineering nondesolvation related interactions with the intended and off-target binding sites. J Med Chem. 2009;52:7706–7723. doi: 10.1021/jm900713y. [DOI] [PubMed] [Google Scholar]
- Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, et al. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci Signal. 2010;3:ra58. doi: 10.1126/scisignal.2001180. [DOI] [PubMed] [Google Scholar]
- Smolarek D, Bertrand O, Czerwinski M, Colin Y, Etchebest C, de Brevern AG. Multiple interests in structural models of DARC transmembrane protein. Transfus Clin Biol. 2010;17:184–196. doi: 10.1016/j.tracli.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Sohy D, Parmentier M, Springael JY. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. 2007;282:30062–30069. doi: 10.1074/jbc.M705302200. [DOI] [PubMed] [Google Scholar]
- Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of ‘selective’ antagonists. J Biol Chem. 2009;284:31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Staudinger R, Wang X, Bandres JC. Allosteric regulation of CCR5 by guanine nucleotides and HIV-1 envelope. Biochem Biophys Res Commun. 2001;286:41–47. doi: 10.1006/bbrc.2001.5345. [DOI] [PubMed] [Google Scholar]
- Stroke I, Cole A, Simhadri S, Brescia M, Desai M, Zhang J, et al. Identification of CXCR3 receptor agonists in combinatorial small-molecule libraries. Biochem Biophys Res Commun. 2006;349:221–228. doi: 10.1016/j.bbrc.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Stupple PA, Batchelor DV, Corless M, Dorr PK, Ellis D, Fenwick DR, et al. An imidazopiperidine series of CCR5 antagonists for the treatment of HIV: the discovery of N-{(1S)-1-(3-Fluorophenyl)-3-[(3-endo)-3-(5-isobutyryl-2-methyl-4,5,6,7-te trahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl] propyl}acetamide (PF-232798) J Med Chem. 2010;54:67–77. doi: 10.1021/jm100978n. [DOI] [PubMed] [Google Scholar]
- Surgand J-S, Rodrigo J, Kellenberger E, Rognan D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J Am Chem Soc. 2010;132:15899–15901. doi: 10.1021/ja107447w. [DOI] [PubMed] [Google Scholar]
- Tchernychev B, Ren Y, Sachdev P, Janz JM, Haggis L, O'Shea A, et al. Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc Natl Acad Sci USA. 2010;107:22255–22259. doi: 10.1073/pnas.1009633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma G, Streiff MB, Kovarik J, Glickman F, Wagner T, Beerli C, et al. Orally bioavailable isothioureas block function of the chemokine receptor CXCR4 in vitro and in vivo. J Med Chem. 2008;51:7915–7920. doi: 10.1021/jm801065q. [DOI] [PubMed] [Google Scholar]
- Tian Y, New D, Yung L, Allen R, Slocombe P, Twomey B, et al. Differential chemokine activation of CC chemokine receptor 1-regulated pathways: ligand selective activation of Galpha 14-coupled pathways. Eur J Immunol. 2004;34:785–795. doi: 10.1002/eji.200324166. [DOI] [PubMed] [Google Scholar]
- Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1alpha and the HIV-1 coat protein gp120: a fluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Ther. 2004;310:8–17. doi: 10.1124/jpet.103.064956. [DOI] [PubMed] [Google Scholar]
- Tsibris A, Kuritzkes D. Chemokine antagonists as therapeutics: focus on HIV-1. Annu Rev Med. 2007;58:445–459. doi: 10.1146/annurev.med.58.080105.102908. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Sinha A, DeWitt M, Farrens DL. Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J Mol Biol. 2010;399:501–511. doi: 10.1016/j.jmb.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidehi N, Schlyer S, Trabanino R, Floriano W, Abrol R, Sharma S, et al. Predictions of CCR1 chemokine receptor structure and BX 471 antagonist binding followed by experimental validation. J Biol Chem. 2006;281:27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, et al. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S, Petrovic A, Van Ryk DI, Locati M, Weissman D, Murphy PM. Reduced cell surface expression of CCR5 in CCR5Delta 32 heterozygotes is mediated by gene dosage, rather than by receptor sequestration. J Biol Chem. 2002;277:2287–2301. doi: 10.1074/jbc.M108321200. [DOI] [PubMed] [Google Scholar]
- Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008;58:1931–1939. doi: 10.1002/art.23591. [DOI] [PubMed] [Google Scholar]
- Verzijl D, Storelli S, Scholten DJ, Bosch L, Reinhart TA, Streblow DN, et al. Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J Pharmacol Exp Ther. 2008;325:544–555. doi: 10.1124/jpet.107.134783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, Moreno-Ortiz MC, Martinez AC, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13:1699–1710. [PubMed] [Google Scholar]
- Vila-Coro AJ, Mellado M, Martin de Ana A, Lucas P, del Real G, Martinez AC, et al. HIV-1 infection through the CCR5 receptor is blocked by receptor dimerization. Proc Natl Acad Sci USA. 2000;97:3388–3393. doi: 10.1073/pnas.050457797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, et al. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci USA. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- Weiss RA, Clapham PR. Hot fusion of HIV. Nature. 1996;381:647–648. doi: 10.1038/381647a0. [DOI] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem. 2005;280:28663–28674. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]
- Wise E, Duchesnes C, da Fonseca P, Allen R, Williams T, Pease J. Small molecule receptor agonists and antagonists of CCR3 provide insight into mechanisms of chemokine receptor activation. J Biol Chem. 2007;282:27935–27943. doi: 10.1074/jbc.M703255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RSY, Bodart V, Metz M, Labrecque J, Bridger G, Fricker SP. Comparison of the potential multiple binding modes of bicyclam, monocylam, and noncyclam small-molecule CXC chemokine receptor 4 inhibitors. Mol Pharmacol. 2008;74:1485–1495. doi: 10.1124/mol.108.049775. [DOI] [PubMed] [Google Scholar]
- Wood A, Armour D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog Med Chem. 2005;43:239–271. doi: 10.1016/S0079-6468(05)43007-6. [DOI] [PubMed] [Google Scholar]
- Wu L, Ruffing N, Shi X, Newman W, Soler D, Mackay CR, et al. Discrete steps in binding and signaling of interleukin-8 with its receptor. J Biol Chem. 1996;271:31202–31209. doi: 10.1074/jbc.271.49.31202. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthou G, Williams T, Pease J. Molecular characterization of the chemokine receptor CXCR3: evidence for the involvement of distinct extracellular domains in a multi-step model of ligand binding and receptor activation. Eur J Immunol. 2003;33:2927–2936. doi: 10.1002/eji.200324235. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, et al. Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J Med Chem. 2010;53:7048–7060. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ben L, Wu Y, Hu W, Ling K, Xin S, et al. Anti-HIV agent trichosanthin enhances the capabilities of chemokines to stimulate chemotaxis and G protein activation, and this is mediated through interaction of trichosanthin and chemokine receptors. J Exp Med. 1999;190:101–111. doi: 10.1084/jem.190.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidar D, Violin J, Whalen E, Lefkowitz R. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA. 2009;106:9649–9654. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]