

Abstract
Agonists acting on µ-opioid receptors (MOR) are very effective analgesics but cause tolerance during long-term or repeated exposure. Intensive efforts have been made to find novel opioid agonists that are efficacious analgesics but can elude the signalling events that cause tolerance. µ-Opioid agonists differentially couple to downstream signalling mechanisms. Some agonists, such as enkephalins, d-Ala(2),N-Me-Phe(4),Gly(5)-ol]-enkephalin (DAMGO), methadone and sufentanyl are efficacious at mediating G-protein and effector coupling, as well as triggering MOR regulatory events that include MOR phosphorylation, β-arrestin binding, receptor endocytosis and recycling. By contrast, morphine and closely related alkaloids can mediate efficacious MOR–effector coupling but poorly trigger receptor regulation. Several models have been proposed to relate differential MOR regulation by different opioids with their propensity to cause tolerance. Most are based on dogma that β-arrestin-2 (βarr-2) binding causes MOR desensitization and/or that MOR endocytosis and recycling are required for receptor resensitization. This review will examine some of these notions in light of recent evidence establishing that MOR dephosphorylation and resensitization do not require endocytosis. Recent evidence from opioid-treated animals also suggests that impaired MOR–effector coupling is driven, at least in part, by enhanced desensitization, as well as impaired resensitization that appears to be βarr-2 dependent. Better understanding of how chronic exposure to opioids alters receptor regulatory mechanisms may facilitate the development of effective analgesics that produce limited tolerance.
LINKED ARTICLES
This article is part of a themed section on the Molecular Pharmacology of G Protein-Coupled Receptors (GPCRs). To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.165.issue-6. To view the 2010 themed section on the same topic visit http://onlinelibrary.wiley.com/doi/10.1111/bph.2010.159.issue-5/issuetoc
Keywords: opioid, morphine, opioid signalling, opioid efficacy, mu-receptor, β-arrestin, protein kinase, desensitization, endocytosis, receptor recycling
Introduction
Opioids are potent and effective analgesics. It is well established that nearly all clinically used opioids mediate their analgesic effects by activating the µ-opioid receptor (MOR; Kieffer and Gaveriaux-Ruff, 2002). However, long-term use of µ-opioid agonists produces adverse effects that include the development of tolerance and addiction, limiting their clinical utility (Williams et al., 2001; Christie, 2008; Morgan and Christie, 2011). Qualitatively, all MOR agonists produce tolerance in vivo although there are differences in the extent of tolerance (Morgan and Christie, 2011), suggesting that opioid analgesics resistant to tolerance could be developed. Recent promising approaches to limit tolerance have been extensively reviewed and include simultaneous activation of more than one opioid receptor type (e.g. MOR and DOR receptors), selective targeting of heteromultimers or opioids that differentially activate distinct intracellular signalling cascades, possibly involving differential activation of Gα subtypes (Pineyro and Archer-Lahlou, 2007), and particularly differential G-protein activation versus endocytosis (e.g.Martini and Whistler, 2007; Christie, 2008; Koch and Hollt, 2008; Berger and Whistler, 2010; von Zastrow, 2010).
The molecular mechanisms mediating opioid tolerance in vivo remain uncertain, but there is accumulating evidence linking mechanisms of MOR desensitization receptor phosphorylation, arrestin association, endocytosis and recycling to tolerance development. For example, knockout (k.o.) mice lacking key MOR regulatory proteins, including β-arrestin-2 k.o. (βarr-2; Bohn et al., 2000; 2002), exhibited enhanced morphine analgesic responses, while development of morphine analgesic tolerance was attenuated. Impetus to investigate MOR regulatory mechanisms of tolerance also came from the finding that different agonists can differentially engage these mechanisms (Keith et al., 1996; Whistler and von Zastrow, 1998; Alvarez et al., 2002). There are many reviews on the topic (see von Zastrow et al., 2003; Connor et al., 2004; Martini and Whistler, 2007; Koch and Hollt, 2008; Berger and Whistler, 2010; von Zastrow, 2010). Notably, morphine and closely related alkaloid agonists were found to quite efficaciously activate G-protein signalling but poorly mediate endocytosis, whereas most efficacious peptides and some small molecule agonists efficiently engage both processes.
The process of MOR regulation is thought to resemble that of the well-characterized β2-adrenoceptor. Briefly, G-protein receptor kinase-2 (GRK2) phosphorylation of the agonist-bound β2-adrenoceptor enhances its affinity for βarr-2 binding, triggering receptor endocytosis via clathrin-dynamin-dependent mechanisms (Gainetdinov et al., 2004). MOR is also predominantly phosphorylated by GRK2 (Wang, 2000; Li and Wang, 2001), GRK3 and perhaps weakly by GRK5 (Kovoor et al., 1998; Terman et al., 2004). To the extent that they have been studied, other GRK isoforms do not contribute to acute MOR desensitization (Johnson et al., 2006a). Like the β2-adrenoceptor, MOR interacts predominantly with βarr-2 (Cheng et al., 1998), although it has been suggested that MOR can interact with βarr-1 in the absence of βarr-2 (but not with morphine activation; Bohn et al., 2004). Following endocytosis, MOR is sorted for recycling back to the surface membrane (von Zastrow et al., 2003). Like the β2-adrenoceptor, βarr-2-dependent endocytosis and recycling are thought to be essential for MOR resensitization (Law et al., 2000; Koch et al., 2005), although more recent evidence strongly challenges this assumption (Arttamangkul et al., 2006; Dang et al., 2011; Doll et al., 2011).
Currently, there are varying and apparently incompatible hypotheses for the involvement of MOR regulatory processes in opioid tolerance (see Bohn et al., 2004; Koch and Hollt, 2008; Berger and Whistler, 2010). It is widely thought that opioid agonists that differentially engage MOR-signalling and receptor regulatory processes have different propensity to cause tolerance. Supporting evidence for this notion revolves around the accepted dogma that MOR endocytosis and recycling are required for receptor dephosphorylation and resensitization. This review considers some of these assumptions and focuses on how the process of MOR regulation contributes to the development opioid tolerance at the receptor level in light of recent findings that strongly suggest that MOR desensitization does not require receptor endocytosis and recycling but tolerance does involve MOR desensitization, as well as resensitization mechanisms that are arrestin-dependent.
Definitions of tolerance and desensitization
The terms tolerance and desensitization are often used to describe very different processes that may be mediated by distinct mechanisms. Drug tolerance is defined as a loss of responsiveness to an agonist after continued exposure and is best quantified by the rightward shift in the dose–response curve that may be associated with a reduction in the maximum response in whole animals or similar shifts to concentration–response curves in isolated systems. However, common use of the term in different experimental contexts can be confusing because the mechanisms regulating MOR function during short-term agonist exposure may differ from that during or after long-term agonist exposure. Whilst most studies examine tolerance following long-term drug exposure of days to weeks, others ascribe tolerance to a very short-term loss of MOR responsiveness that occurs in the minutes to several hours after acute agonist exposure either in vivo or in vitro. Although the latter is correct usage, it can be confusing because loss of MOR responsiveness during short-term, sustained exposure is closely linked to mechanisms of rapid MOR desensitization that may include receptor endocytosis rather than long-term regulation of MOR function. As discussed in detail below, the process of rapid MOR desensitization certainly contributes to long-term MOR regulation and tolerance, but the two are not equivalent. As such, we restrict the usage of the term ‘tolerance’ to phenomena observed after long-term exposure (several days to weeks) to opioids and describe short-term studies, where relevant, as ‘acute tolerance’.
Usage of the term ‘desensitization’ can also be confusing because it is, like tolerance, an operational definition for loss of receptor function that can be applied to very different phenomena. Here we adopt the most common usage ascribed to the rapid loss of MOR–effector coupling that occurs during sustained exposure to agonists, usually in vitro, that occurs within seconds to several minutes. The same term, however, has been applied correctly to measurements of MOR–effector coupling in vitro after long-term opioid exposure (e.g. Bohn et al., 2000; 2002), which does not actually measure the loss of receptor function during sustained agonist application. Loss of sensitivity or what has been described as ‘desensitization’ in this context is equivalent to tolerance at the cellular or molecular level. We avoid use of the term ‘desensitization’ in this context because it confounds the distinction between loss of MOR function that occurs within seconds to minutes during agonist application (usually defined as desensitization) and the tolerance (as defined above) that develops over days and weeks.
As introduced above, the process of MOR regulation involves multiple processes, including phosphorylation, arrestin binding, endocytosis and resensitization (dephosphorylation); which may not require receptor endocytosis and recycling. It is therefore important to note that measurements of MOR desensitization as defined here can encompass multiple components of the MOR regulatory processes, depending on the temporal resolution of the assay. Thus, measurements of desensitization may include loss of MOR–effector coupling prior to endocytosis (seconds to several minutes), desensitization due to endocytosis (which produces loss of MOR–effector coupling by removing receptors from the surface membrane; usually 2–30 min) or resensitization and recycling (which slowly reverses desensitization; usually 20 min–1 h). Assays of MOR activation of G-proteins employing methods such as activity of G-protein-modulated ion channels (e.g. Dang et al., 2009) or resonance energy transfer (RET) methods (e.g. Molinari et al., 2010) continuously monitor MOR signalling over time scales of seconds to minutes during and shortly after induction of desensitization, so they can easily distinguish these components. However, biochemical assays for desensitization, such as inhibition of adenylate cyclase, that require more than 5 min of sustained opioid exposure (most assays take 10–20 min, e.g. Law et al., 2000, or longer, e.g. Koch et al., 2005) may be measuring the combined effects of rapid desensitization at the cell surface plus endocytosis or resensitization (Connor et al., 2004). Therefore, attempts to compare kinetics and mechanisms regulating MOR signalling across studies can be confusing. Careful consideration should be given to the duration of agonist treatment and the time required to measure MOR–effector coupling when comparing mechanisms of MOR regulation across assays because multiple components may be involved.
Differential MOR desensitization, endocytosis and tolerance
Differential agonist efficacy for G-protein signalling and endocytosis
The discovery that different opioid agonists have different efficacies for G-protein signalling and mediation of receptor endocytosis has provided impetus to determine whether MOR regulatory mechanisms contribute to tolerance, which could explain why MOR function is lost in the absence of reduced MOR expression. It has been hypothesized that the poor ability of morphine to initiate efficient MOR endocytosis gives morphine high liability for causing tolerance. Many studies have established that morphine activates MOR but poorly induces endocytosis (Arden et al., 1995; Keith et al., 1996; Sternini et al., 1996; Whistler and von Zastrow, 1998; Borgland et al., 2003), as widely reviewed (see von Zastrow et al., 2003; Connor et al., 2004; Martini and Whistler, 2007; Koch and Hollt, 2008; Berger and Whistler, 2010; von Zastrow, 2010). Most quantitative studies of signalling efficacy have concluded that intrinsic efficacy to activate G-proteins versus endocytosis or βarr-2 association with MOR are not linearly related (Borgland et al., 2003; Molinari et al., 2010; but also see McPherson et al., 2010). Molinari et al. (2010) using RET methods, reported a hyperbolic relationship between intrinsic activity for G-protein and βarr-2, consistent with earlier studies. By contrast, McPherson et al. (2010) found a more linear relationship for both βarr-2 recruitment and endocytosis, with some outliers (but not morphine). This discrepancy could be due to the methods used to determine G-protein activation (GTPγS binding for 2 h by McPherson et al., 2010 and RET methods by Molinari et al., 2010), the expression of different densities of RET donors and acceptors in the two studies or the use an operational model by McPherson et al. (2010) but not Molinari et al. (2010). It is well established that overexpression of GRKs or arrestins can profoundly enhance induction of endocytosis by morphine (e.g. Whistler and von Zastrow, 1998; Bohn et al., 2004). Morphine also fails to induce MOR endocytosis in spinal cord in vivo (Trafton et al., 2000), but it efficiently induces endocytosis in medium spiny striatal neurons (Haberstock-Debic et al., 2003; Yu et al., 2009).
Do strongly internalizing opioid agonists produce less tolerance than weakly internalizing agonists?
Morphine produces more behavioural tolerance than strongly internalizing agonists. This finding has been widely cited to support the notion that MOR recycling influences tolerance. Morphine, for instance, produced greater opioid tolerance when compared with agonists like DAMGO, sufentanyl or etorphine, when equivalent induction doses and continuous infusions were used to control for pharmacokinetic differences (Stevens and Yaksh, 1989; Duttaroy and Yoburn, 1995; Madia et al., 2009). Whilst this seems to support the notion that strongly internalizing agonists produce less tolerance than weakly internalizing agonists, the interpretation is seriously confounded by large differences in intrinsic efficacy for G-protein activation among these agonists. Etorphine, sufentanyl and DAMGO all exhibit much higher intrinsic efficacy for G-protein activation than morphine (Traynor and Nahorski, 1995; Emmerson et al., 1996; Selley et al., 1998; McPherson et al., 2010; Molinari et al., 2010), although some studies using GTPγS binding have reported that the intrinsic efficacy of sufentanyl is comparable with morphine (Emmerson et al., 1996; Selley et al., 1998). Low intrinsic efficacy agonists usually produce larger rightward shifts in concentration-response curves than high efficacy agonists. This occurs when MOR–effector coupling is impaired either by irreversible antagonists or chronic drug treatment presumably because low intrinsic efficacy agonists such as morphine must occupy a greater fraction of the total receptor population to produce a given level of effect, due to lesser receptor reserve (e.g. Christie et al., 1987; Stevens and Yaksh, 1989; Mjanger and Yaksh, 1991; Connor et al., 1999).
To properly test the notion that strongly versus weakly internalizing opioids produce differential tolerance would therefore require direct comparison of the extent of tolerance produced by morphine with opioids that exhibit comparable intrinsic efficacy for G-protein activation but much higher efficacy for endocytosis than morphine, while ensuring equivalent receptor stimulation and duration of action. Methadone and endomorphins have been considered good candidates because their intrinsic efficacies for G-protein activation appear similar to morphine and both efficiently induce MOR endocytosis. However, the intrinsic efficacy of methadone is more similar to DAMGO than morphine in GTPγS assays (Selley et al., 1998; McPherson et al., 2010) and in vivo (Adams et al., 1990). The apparently low efficacy of methadone in electrophysiological studies is caused by non-MOR actions on ion channels (Rodriguez-Martin et al., 2008). Methadone and endomorphins also have very different pharmacokinetic properties and toxicity compared with morphine that can further complicate interpretations. Although He and Whistler (2005) did examine this issue using methadone and morphine, the results are very difficult to interpret because i.c.v. dose equivalence was not established. By contrast, Soignier et al. (2004) reported comparable rates of tolerance development and completely symmetrical cross-tolerance during continuous i.c.v. infusion of morphine, endomorphin-1 and endomorphin-2, suggesting tolerance may not be different between strongly and weakly internalizing agonists when intrinsic efficacy is matched. Furthermore, there is no clear evidence that strongly internalizing agonists produce differential tolerance compared with weakly internalizing opioids in humans (Morgan and Christie, 2011). For example, comparison of tolerance development in pain patients during continuous administration of transdermal fentanyl (high efficacy, moderate endocytosis) versus buprenorphine (low efficacy, non-internalizing) found fentanyl produced greater tolerance (Sittl et al., 2006). Therefore, it remains uncertain whether or not strongly internalizing agonists produce less tolerance than weakly internalizing agonists.
Decreased MOR–effector coupling contributes to opioid tolerance
Chronic exposure to opioids can cause profound tolerance in both animals and humans (Christie, 2008; Morgan and Christie, 2011).Tolerance measured in whole animals is mediated by multiple adaptive mechanisms ranging from molecular mechanisms of MOR–effector coupling in neurons, second messenger systems in opioid sensitive cells, non-neuronal cells (including glia) and neural networks interacting with opioid sensitive neurons, to learned behaviour in animals (see Christie, 2008). Nonetheless, there is very solid evidence that impaired MOR–effector coupling contributes to tolerance in vivo.
Opioid tolerance has been extensively quantified in isolated tissues, neurons and membrane preparations from morphine tolerant animals, as well as in cell culture models. Functional measurements of impaired MOR–effector coupling in isolated tissues and cells after chronic morphine treatment consistently show a loss of functional receptors without consistent changes in MOR binding density (down-regulation, reviewed by Christie, 2008; Koch and Hollt, 2008). Agonists that strongly promote MOR endocytosis, such as etorphine, are an exception because they do induce receptor down-regulation (Stafford et al., 2001), presumably because a small proportion of endocytosed MOR is degraded during each internalization cycle (Whistler et al., 2002).
Operational models (or Fuchgott analysis) used to quantify the loss of functional MOR–effector coupling in isolated systems (e.g. Chavkin and Goldstein, 1984; Christie et al., 1987; Bailey et al., 2009a) after chronic morphine have calculated a loss of approximately 80% of functional surface MOR is required to account for the observed shift in agonist concentration–response curves. Studies using physiological end-points (direct Gγβ interactions with ion channels) in single opioid sensitive neurons have reported impaired MOR–effector coupling in a range of neuronal cell types from animals that have been chronically treated with morphine in vivo (except Ingram et al., 2008), including rat and mouse periaqueductal grey (PAG; Bagley et al., 2005), rat and mouse locus coeruleus (LC; Christie et al., 1987; Connor et al., 1999; Dang and Williams, 2004; Bailey et al., 2009a; Dang et al., 2011; Quillinan et al., 2011) and mouse trigeminal ganglion neurons (Johnson et al., 2006b). Similar results were also reported for inhibition of GABAergic synaptic transmission in nerve terminals in PAG (Hack et al., 2003; Fyfe et al., 2010). These findings are consistent with those examining MOR-activated GTPγS binding in brainstem in parallel with MOR binding density (Bohn et al., 2000) and GTPγS binding in some brain regions but not others (Sim et al., 1996; Kim et al., 2008). Taken together, these results are consistent with earlier reports in cultured cells showing that chronic morphine exposure impaired MOR–effector coupling (GTPγS binding) without greatly affecting MOR binding density (Puttfarcken et al., 1988; Puttfarcken and Cox, 1989).
β-Arrestin-2 and endocytic mechanisms are involved in opioid tolerance
Although the phenomenon that chronic morphine impairs MOR–effector coupling without much effect on MOR binding density has been known for more than 20 years, the mechanisms responsible are still uncertain and controversial. There is, however, accumulating evidence that the MOR regulatory mechanisms involved in acute desensitization, including association with βarr-2 and endocytosis, are intimately involved in the development of opioid tolerance.
Bohn et al. (2000; 2002; 2004) established that development of morphine anti-nociceptive tolerance (but not withdrawal) is blunted in βarr-2 k.o. mice. Concurrently, GTPγS assays in brainstem membranes from the k.o. mice also showed a blunted shift in the concentration–response curve (tolerance). Acute anti-nociceptive responses to morphine (but not etorphine, fentanyl or methadone; Bohn et al., 2004; or other opioid actions of morphine, Raehal et al., 2005) were also enhanced in the βarr-2 k.o. It was suggested that MOR is resistant to desensitization in the absence of βarr-2 (see contrary evidence; Bradaia et al., 2005; Walwyn et al., 2007; Dang et al., 2009; 2011). Additional support for the involvement of MOR regulatory processes in the development of opioid tolerance comes from study using GRK3 k.o. mice. This study showed MOR tolerance was reduced in hippocampal neurons from GRK3 k.o. mice (Terman et al., 2004). Development of behavioural tolerance to fentanyl was attenuated; however, there was no effect on morphine tolerance. Together, these studies suggest that arrestin-dependent MOR regulation is linked to morphine tolerance.
These studies suggest that blocking MOR endocytosis, which is presumably impaired in the βarr-2 k.o. (but see Arttamangkul et al., 2008; Quillinan et al., 2011), attenuates tolerance but others have provided seemingly contradictory evidence that induction of MOR endocytosis and recycling limits morphine tolerance, and suppression of endocytosis or recycling enhances it. He et al. (2002) reported that inclusion of an extremely low dose of a strongly-internalizing agonist, DAMGO (that had no anti-nociceptive effect on its own), with constantly infused i.t. morphine limited the development of tolerance and also stimulated MOR endocytosis in spinal cord and cultured cells (but see contrary evidence; Bailey et al., 2003; Koch et al., 2005). This was not observed with either drug alone at the doses used. The authors hypothesized that a very low concentration of DAMGO, which does not induce detectable endocytosis by itself, can stimulate endocytosis of morphine-occupied MOR and thereby reduces tolerance, perhaps via interaction with homomultimers of MOR. Similarly, Kim et al. (2008) studied a transgenic MOR mouse, in which part of the C-terminal region of the DOR is substituted into MOR (rMOR). This conferred the ability of morphine to efficiently mediate MOR endocytosis and recycling. The rMOR mice showed similar anti-nociceptive sensitivity to morphine as wild-types but developed less morphine anti-nociceptive tolerance, as well as less reduction in MOR-activated GTPγS binding in brainstem membranes. Consistent with these studies, the converse has also been reported in spinophilin k.o. mice (Charlton et al., 2008); development of morphine tolerance was enhanced in spinophilin k.o. mice. Spinophilin is a neuronal scaffolding protein that facilitates MOR endocytosis, so endocytosis should be impaired in the k.o., although other regulatory actions of spinophilin cannot be ruled out. Taken together, these studies suggest that MOR endocytosis limits tolerance and, therefore, opioids that do not promote receptor endocytosis should produce greater tolerance than agonists that do promote MOR endocytosis (but see above for lack of direct behavioural evidence that this is the case).
The findings described above appear contradictory in terms of the relationship between endocytosis and tolerance. On the one hand, blocking βarr-2 association with MOR (which should impair endocytosis) inhibits morphine tolerance and, on the other hand, manipulations that enhance MOR endocytosis (and vice versa) impair development of morphine tolerance. Various explanations have been proposed to account for these disparate findings. In the case of manipulations that prevent βarr-2 binding, it was proposed that βarr-2 association is necessary for, or facilitates MOR desensitization (Bohn et al., 2002; 2004, but see below). However, desensitization (as defined above) was not directly examined in those studies. But examination of MOR desensitization in both sensory and LC neurons show that it is unaffected by βarr2- deletion. It is therefore unclear how βarr-2 deletion can account for blunted tolerance in the k.o. mice. Two general interpretations (not mutually exclusive) for the inhibition of tolerance were developed by Whistler and co-workers that are in line with findings from other groups (e.g. Berger and Whistler, 2010). One interpretation is that strongly internalizing agonists produce less tolerance because the cycles of endocytosis promote dephosphorylation of MOR in endosomes and resensitized receptors are then recycled to the cell surface. Because morphine poorly stimulates endocytosis of phosphorylated and desensitized MOR (whether or not MOR associated with arrestins) the desensitized receptors accumulate at the cell surface causing tolerance. The other interpretation is that morphine causes persistent signalling that contributes to secondary adaptations involved in tolerance in vivo, whereas endocytosis terminates persistent signalling, limiting downstream adaptations and tolerance. These concepts are summarized in Figure 1. These authors have also provided extensive evidence that such secondary adaptations are more pronounced following chronic morphine stimulation of wild-type MOR compared with chimeric MOR that can undergo endocytosis and recycling when stimulated by morphine (ibid.).
Figure 1.
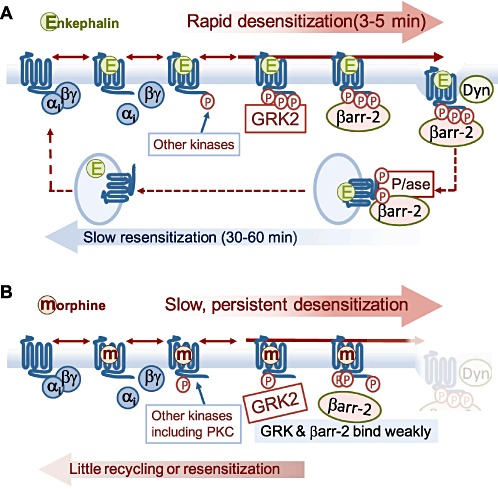
Previous models to explain how strongly internalizing opioid agonists can proiduc less tolerance than weakly internalizing agonists. (A) Strongly internalizing agonists induce rapid desenisitization of MOR coupling. GRK2-mediated phosphorylation is pivotal for βarr-2 binding and endocytosis, both process that were considered ireversible at the cell surface, so MOR slowly resensitizes over the time course of endocytosis and recycling. (B) With weakly internalizing agonists, MOR desensitizes slowly (accelerated by PKC activity) but accumulates in a phosphorylated desensitized state at the cell surface because it stimulates GRK2 and βarr-2 binding very weakly, so cannot resensitize causing tolerance. As discussed in the text, the crucial assumption that endocytosis (and recycling) is necessary for resensitization is incorrect.
In essence, both types of study described above include the notion that arrestin-dependent endocytosis and recycling is necessary for MOR resensitization to occur. This notion is based largely on the model established for β2-adrenoceptor recycling (Gainetdinov et al., 2004). Although some studies appear to support this for MOR (Koch et al., 1998; 2001; Law et al., 2000; Qiu et al., 2003), more recent findings discussed below clearly establish that MOR dephosphorylation proceeds efficiently at the cell surface, as does resensitization in the βarr-2 k.o. or when endocytosis is blocked.
MOR desensitization does not require βarrestin and is distinct from endocytosis
Two lines of evidence directly establish that MOR desensitization does not require endocytosis. Arttamangkul et al. (2006) directly studied desensitization and endocytosis of MOR in cultured LC neurons in parallel with a transgenic FLAG-tagged MOR mouse. The lectin concanavalin-A completely blocked endocytosis induced by met-enkephalin but did not affect desensitization. Similarly, Dang et al. (2009) reported that inhibition of dynamin-dependent endocytosis had no effect on the rate or extent of MOR desensitization induced by met-enkephalin in mouse LC neurons. Whilst endocytosis could produce desensitization by removing MOR from the surface membrane, it is clear from these studies that functional desensitization of MOR–effector coupling does not require it.
Kinetics of desensitization and endocytosis
MOR desensitization and endocytosis can also be distinguished on the basis of time course and differential efficacies of opioid agonists. Kinetically, rapid desensitization of MOR largely precedes endocytosis for high efficacy peptide agonists. The time constants for rapid desensitization in neurons and cultured cells are of the order of 1–3 min at 33–37° (Bailey et al., 2004; Dang and Williams, 2004; Arttamangkul et al., 2006; Dang et al., 2009), when measured during stimulation by high-efficacy, strongly internalizing agonists (e.g. met-enkephalin, DAMGO) and other methods that provide reliable data in the second to minute range (Connor et al., 2004), after which the process reaches steady state in less than 10 min. The time course of MOR desensitization is also similar to that reported for βarr-2 association (Oakley et al., 2000; McPherson et al., 2010; Molinari et al., 2010) and phosphorylation of residues in the C-terminal region of MOR (T370 and S375; Doll et al., 2011), all of which saturated within 2–3 min when stimulated by efficacious peptide agonists. Endocytosis induced by the same agonists is somewhat slower, with time constants generally in the order of ≥5 min and reaching steady state in less than 30 min (Law et al., 2000; Borgland et al., 2003; Tanowitz and von Zastrow, 2003; Arttamangkul et al., 2006; 2008; Johnson et al., 2006a; Tanowitz et al., 2008). This suggests that MOR desensitization and endocytosis may occur as separate or sequential processes with some temporal overlap.
Differential efficacy of opioids for desensitization and endocytosis
In some studies (but not others), opioids differentially couple to desensitization and endocytosis. In heterologous expression systems, a range of opioid agonists, including morphine, can cause desensitization (Borgland et al., 2003; Johnson et al., 2006a; Chu et al., 2010). Intrinsic efficacies of several opioids to cause G-protein activation are highly correlated with their efficacy to produce rapid desensitization. It should be noted that much higher levels of receptor occupancy are required for the latter; that is, the coupling efficacy is approximately 10-fold lower for desensitization than G-protein activation (Borgland et al., 2003). However, this was not the case for endocytosis when the same agonists are used; morphine displayed distinctly lower efficacy than expected from either G-protein activation or desensitization. Earlier studies that claimed a strong correlation between the intrinsic efficacy for desensitization and endocytosis in cultured cells (Koch et al., 2005) could have been confounded because the duration of the desensitization assays used (inhibition of cAMP formation) encompassed both phenomena.
By contrast with cultured cells, morphine produces little desensitization in native LC neurons (Dang and Williams, 2005; Virk and Williams, 2008; Bailey et al., 2009b). In these cases, desensitization appears better correlated with capacity to induce MOR endocytosis than G-protein activation. Bailey and co-workers have provided a potential explanation for the differences between some cultured cells and this has been confirmed by others (Chu et al., 2010). In cultured cells (HEK293), morphine- but not DAMGO-induced desensitization is blocked by protein kinase C (PKC) inhibition (Johnson et al., 2006a). Conversely, in LC neurons, where morphine induces little desensitization, PKC activation enhances morphine-induced (but not DAMGO) desensitization (Bailey et al., 2009b). This provides clear evidence of agonist-dependent differential desensitization, with morphine but not DAMGO being PKC-dependent. The same studies suggested the reverse sensitivity for GRK inhibition, with DAMGO-induced desensitization being more sensitive to disruption of GRK than morphine (but see lack of effect of GRK inhibition alone in Dang et al., 2009).
Differential MOR phosphorylation and desensitization
Opioid agonists can differentially phosphorylate MOR (Johnson et al., 2006a). This could provide a plausible explanation for differential desensitization between morphine and DAMGO and its dependence on PKC phosphorylation in some cell types. Serial phosphorylation of up to 20 potential sites in the intracellular regions of MOR contribute to receptor desensitization and endocytosis, particularly GRK substrates near the C-terminal (see Connor et al., 2004; Koch and Hollt, 2008 for review). Some of these sites are essential for GRK phosphorylation and arrestin-dependent endocytosis (ibid.). Mutation of several residues in the C-terminal of MOR (S363, T370 or S375 to A) impairs DAMGO-mediated receptor phosphorylation and endocytosis (El Kouhen et al., 2001; Schulz et al., 2004). Until recently, only S375 has been shown to undergo agonist specific phosphorylation by both DAMGO and (more slowly) morphine using phosphosite-specific antibodies (Schulz et al., 2004).
More recently, Doll et al. (2011) have produced phosphosite-specific antibodies for S363, T370 and S375 of mouse MOR. They showed that S363 was constitutively phosphorylated in HEK293 cells. They confirmed that both DAMGO and morphine (less efficiently) induced phosphorylation of S375. DAMGO also induced efficient phosphorylation of T370, but morphine did not. Importantly, PKC stimulation directly phosphorylated at T370 in an agonist-independent manner. This appears to provide a nice explanation of the PKC dependence of morphine- but not DAMGO-induced desensitization. If efficient desensitization requires phosphorylation of bothT370 and S375, then morphine-induced desensitization would require PKC activation but DAMGO would not. PKC activation might therefore be sufficient to induce desensitization when S375 (and perhaps other unidentified sites) is also phosphorylated. It is conceivable that in addition to phosphorylation of both T370 and S375, additional phosphorylation events are required to facilitate βarr-2 binding, so phosphorylation of T370 and S375 may be necessary (El Kouhen et al., 2001) but not be sufficient to induce effective βarr-2-dependent endocytosis when morphine is the agonist.
βarr-2 is not necessary for desensitization of MOR
A possible interpretation of the effects of the βarr-2 k.o. on development of morphine tolerance is that βarr-2 association with MOR is necessary for desensitization (see above). Although this may be correct for some neurons, it is clearly not the case in neurons studied to date, or HEK293 cells where morphine efficiently induces desensitization but not βarr-2-dependent endocytosis. Walwyn et al. (2007) showed that DAMGO-induced desensitization of MOR coupling (Gβγ mediated) to voltage-gated calcium current inhibition in sensory neurons was unaffected in the βarr-2 k.o., and this was substantiated by (Arttamangkul et al., 2008) in LC neurons. More recent studies established in LC that desensitization induced by met-enkephalin can be mediated by at least two distinct mechanisms independently involving ERK1/2 activity and GRK2-βarr-2 (Dang et al., 2009). Blocking either mechanism alone was not sufficient to inhibit desensitization. The specific process for the ERK1/2-dependent mechanism is not yet known but MOR desensitization, internalization and phosphorylation have all been reported to be prevented by ERK1/2 inhibition in some heterologous expression systems (Polakiewicz et al., 1998; Schmidt et al., 2000). As is likely for the initial events of the GRK-βarr-2 interaction, signalling by ERK1/2 may therefore prevent coupling of MOR to effectors by phosphorylating MOR at sites not occupied by Gα-subunits (Schmidt et al., 2000). Alternatively, ERK1/2 may act indirectly to mediate desensitization via phosphorylation of Gα-interacting protein (GAIP), a regulator of G-protein signalling (RGS), by potentiating the rate of GTP hydrolysis, as has been reported in some cell types (Ogier-Denis et al., 2000). There are many other possible mechanisms involved in MOR desensitization and endocytosis that could be differentially affected by morphine-like opioids (Koch and Hollt, 2008), including facilitating translocation of MOR from lipid raft domains (Zheng et al., 2008) or activation of phospholipase D2 (Koch et al., 2006), both of which are poorly induced by morphine. Acute tolerance to morphine in vivo is also differentially sensitive to inhibition of c-Jun N-terminal kinase compared with strongly-internalizing agonists such as fentanyl (Melief et al., 2010).
These findings also underscore the possibility that desensitization may be mediated by multiple mechanisms in different cell types or cellular compartments. LC neurons display strong ERK1/2 activation (Eitan et al., 2003; Dang et al., 2009) after opioid administration, but many other neurons (and cell types) do not (Eitan et al., 2003). Therefore, desensitization may be mediated primarily by GRK- βarr2-dependent mechanisms in some neuronal types (e.g. Li and Wang, 2001) but can be initiated by other redundant mechanisms in other cells (see Koch and Hollt, 2008). Other differences between cell types can influence MOR desensitization; for example, the capacity of opioids to induce endocytosis can be strongly influenced by co-expression of other GPCRs such the NK1 receptor in the same cell (Yu et al., 2009). It is also likely that the mechanisms of desensitization are distinct in different cellular compartments. Fyfe et al. (2010) reported that no desensitization of MOR-induced presynaptic GABAergic inhibition, during superfusion of morphine, met-enkephalin or DAMGO for up to 30 min in rat PAG neurons; even when a fraction of receptors had been inactivated with an irreversible MOR antagonist to rule out the potential confound of large receptor reserve.
Morphine tolerance is associated with enhanced MOR desensitization
Desensitization induced by met-enkephalin, DAMGO and morphine (and methadone; Quillinan et al., 2011) are all more pronounced in LC (Dang and Williams, 2004; 2005), as well as PAG neurons (Ingram et al., 2008) after chronic exposure to morphine. Enhanced desensitization would be expected to contribute to opioid tolerance by more prominently reducing functional MOR on cell surface during episodes of agonist administration. There are many possible adaptations caused by chronic morphine that could be responsible for this observation, but enhanced endocytosis does not appear to be responsible. Enhanced desensitization after chronic morphine treatment was associated with reduced endocytosis (Quillinan et al., 2011). Other adaptive mechanisms could include those directly involved with MOR phosphorylation such as ERK1/2, GRKs (but GRK2 is decreased; Fan et al., 2002) or arrestins (but βarr-2 is decreased in PAG; Fan et al., 2003) or others such as RGS proteins (Gold et al., 2003), phospholipase D2 (Koch et al., 2006) or spinophilin (Charlton et al., 2008).
Resensitization and dephosphorylation of MOR do not require endocytosis and recycling
The models of differential tolerance between strongly and weakly internalizing agonists introduced above generally require endocytosis and recycling to resensitize MOR. Morphine and similar agonists, by failing to induce endocytosis, are thought to produce accumulation of desensitized MOR at the cell surface, thereby producing tolerance. More recent evidence discussed below establishes that MOR dephosphorylates and resensitizes efficiently at the cell surface regardless of whether strongly or weakly internalizing agonists are examined, so other explanations for the involvement of MOR regulatory mechanisms in tolerance are required.
Schulz et al. (2004) provided evidence that recycling may be required to dephosphorylate MOR at S375. Briefly, phosphorylation of S375 persisted long after removal of morphine from cells but was readily reversible using the strongly internalizing agonist, DAMGO (but see below for strong evidence to the contrary). Functional studies using inhibition of cAMP formation as an endpoint showed monensin (to inhibit endosomal recycling), truncated MOR mutants (Qiu et al., 2003) or MOR splice variants (Koch et al., 2001; Tanowitz and von Zastrow, 2003; Tanowitz et al., 2008) all reduced both recycling and resensitization of endocytosed MOR. Although these appear to support a requirement for endocytosis and recycling to resensitize MOR, assays of MOR function were performed over time scales greatly exceeding acute desensitization of G-protein coupling to MOR (see above), βarr-2 binding (Oakley et al., 2000), endocytosis (Tanowitz and von Zastrow, 2003; Arttamangkul et al., 2006, 2008) and often recycling (Koch et al., 2001; Tanowitz and von Zastrow, 2003; Arttamangkul et al., 2008; Tanowitz et al., 2008) Therefore, such methods cannot distinguish recovery of functional MOR at the cell surface from the increased MOR surface density (and therefore function) resulting from recycling (Connor et al., 2004).
More recent studies have established conclusively that endocytosis is not necessary for either resensitization or dephosphorylation of MOR. Using met-enkephalin in cultured LC neurons, Arttamangkul et al. (2006) showed directly that concanavalin-A blocks endocytosis of FLAG-tagged MOR but does not affect resensitization. Doll et al. (2011) have shown conclusively that dephosphorylation of S375 is rapid using both DAMGO and morphine as agonists. This contradicts the earlier study of Schulz et al. (2004), but the explanation may be that morphine did not wash effectively from the cell preparations in the earlier study because in Doll et al. (2011) dephosphorylation for several agonists was enhanced by a brief rinse with low pH which presumably facilitates agonist removal from the preparation. More importantly, Doll et al. (2011) showed that after DAMGO exposure, dephosphorylation of both S375 and T370 were just as rapid in cells incubated in concanavalin-A, which completely blocked endocytosis. Although these findings may not generalise to the many other phosphorylation sites on MOR (see Koch and Hollt, 2008) they do establish that sites involved in βarr-2 binding dephosphorylate just as efficiently when endocytosis is blocked.
Recent studies of MOR resensitization in LC neurons from βarr-2 k.o. and wild-type mice (Dang et al., 2011; Quillinan et al., 2011) are consistent with the study of Doll et al. (2011). If arrestin-dependent endocytosis is required for MOR resensitization, then recovery from desensitization induced by a strongly internalizing agonist should be impaired but the opposite was found. In wild-type mice, MOR resensitized slowly after met-enkephalin induced desensitization (approximately 60 min), similar to that reported earlier for LC neurons from rat (Osborne and Williams, 1995; Dang and Williams, 2004) and similar to the rate of MOR recycling reported in cultured cells (Koch et al., 2001; Tanowitz and von Zastrow, 2003). In LC neurons from βarr-2 k.o. mice, MOR resensitization was accelerated, being nearly complete within 20 min (Dang et al., 2011; Quillinan et al., 2011). Accelerated resensitization in the βarr-2 k.o. was mimicked in wild-type LC by manipulations that should block arrestin association upstream (an intracellular GRK inhibitor) or endocytosis downstream of arrestin association (an intracellular dynamin inhibitor; Dang et al., 2011). Conversely, resensitization was slowed by a phosphatase inhibitor under conditions of impaired arrestin association (βarr-2 k.o. plus GRK inhibitor, Dang et al., 2011). This shows that MOR resensitization is rapid when endocytosis is blocked and the time course is quite consistent with the dephosphorylation rate reported by Doll et al. (2011). The slow resensitization in wild-type LC is almost certainly due to the fact that once receptors are endocytosed, relatively slow receptor recycling (Koch et al., 2001; Tanowitz and von Zastrow, 2003) is necessary for recovery of MOR localization and signalling at the surface membrane. It should be noted that such resensitization rates may differ in different neurons because the three most abundant splice variants recycle at different rates (MOR1, MOR1A and MOR1B; Oldfield et al., 2008).
The necessity for endocytosis and recycling to resensitize some GPCRs presumably depends on the affinity of arrestins for the agonist occupied receptor (Oakley et al., 1999; 2000; Gainetdinov et al., 2004). The rapid resensitization and dephosphorylation of MOR at the cell surface suggests that the affinity of the βarr-2 association is relatively weak (Oakley et al., 2000), so that it can dissociate rapidly prior to endocytosis thereby exposing the phosphorylated C-terminal residues (S375, T370 and presumably others) to phosphatases. The very rapid reversal of MOR-βarr-2 RET signals upon agonist washout reported by McPherson et al. (2010) for most strongly and weakly internalizing opioids (except etorphine, which has extremely high affinity for MOR) is consistent with this possibility.
Arrestin-dependent impairment of MOR resensitization contributes to morphine tolerance
Impairment of the capacity of MOR to rapidly resensitize appears to contribute to morphine tolerance. In addition to enhanced desensitization, MOR resensitization is impaired in LC neurons after chronic morphine (Dang and Williams, 2004), but the mechanisms are still not certain. Dang et al. (2011) and Quillinan et al. (2011) recently confirmed this in mouse LC and further established that the impairment is arrestin-dependent. Impaired MOR resensitization after chronic morphine in wild-type LC neurons was reversed and resembled that in the βarr-2 k.o. either by disrupting GRK2 function or inhibition of dynamin function with intracellular inhibitors. These findings link the impairment of MOR resensitization in LC to adaptations within the process of GRK2-βarr-2-dynamin-dependent MOR regulation.
Dang et al. (2011) and Quillinan et al. (2011) also reported that cellular morphine tolerance in the same population of LC neurons was similar to that previously reported in wild-type neurons (see above) but abolished in the βarr-2 k.o. The finding that morphine treatment failed to produce cellular tolerance in LC neurons from βarr-2 k.o. mice is consistent with the seminal findings that analgesic morphine tolerance, as well as tolerance to DAMGO-stimulated GTPγS binding in brainstem and spinal cord membranes, is attenuated in these animals (Bohn et al., 2000; 2002). These findings suggest that persistence of rapid recovery from desensitization after chronic morphine could contribute to the attenuation of behavioural opioid tolerance in βarr-2 k.o. mice if the mechanism found in LC is found to generalise to analgesia-related neurons. It was proposed that following chronic morphine, βarr-2-dependent regulation of MOR is enhanced, slowing MOR resensitization, thereby shifting the equilibrium between receptor desensitization and resensitization to an accumulation of desensitized MOR that accounts for MOR tolerance (Dang et al., 2011). As such, ablation of βarr-2 in the k.o. mice facilitates resensitization and prevents cellular opioid tolerance in LC neurons. Impaired resensitization could be important for tolerance in vivo if the phenomenon is found to generalize to neurons involved in analgesia.
The mechanisms of enhanced desensitization (see above) and βarr-2-dependent impairment of resensitization during chronic morphine treatment in vivo are still not known. Impaired resensitization was observed after very brief exposure to met-enkephalin and was sensitive to GRK, βarr-2 or dynamin inhibition, suggesting a possibly enhanced rate of GRK phosphorylation after chronic morphine that engages βarr-2 and clathrin-dynamin-dependent processes (Dang et al., 2011). Quillinan et al. (2011) also reported in a GRK2 transgenic that can be blocked by a novel agent (NaPP1) that both impaired resensitization and cellular opioid tolerance in LC neurons were reversed by the GRK2 inhibitor. The dependence of both tolerance and resensitization on GRK, βarr-2 and dynamin would predict the explanation for MOR tolerance (and slow resensitization) may be an enhanced rate of endocytosis after chronic morphine. However, Quillinan et al. (2011) found no difference in the extent of met-enkephalin-induced MOR endocytosis in LC neurons from chronically treated with morphine. Similarly, the extent of endocytosis induced by DAMGO in spinal cord in vivo was also not reduced by chronic morphine treatment (Trafton and Basbaum, 2004). The latter findings seem at odds with the effects of βarr-2 deletion and dynamin -inhibition. The mechanism of impaired resensitization is, therefore, still unclear but a range of adaptations produced by chronic morphine could be responsible. Although untested, it is possible sites other than T370 and S375 are more persistently phosphorylated by chronic morphine to enhance other downstream events that do not increase endocytosis or that post-endocytic trafficking and sorting mechanisms are affected by chronic morphine.
Concluding remarks
The discovery of differential signalling efficacies of opioid agonists for G-protein coupling, desensitization and endocytosis and their potential involvement in the development of opioid tolerance stimulated much research to understand these mechanisms with the hope of developing opioids that can elude or limit tolerance. This idea seems to be substantiated by the consistent findings that greater opioid tolerance develops to agonists with low (morphine and related alkaloids) versus high (enkephalin-related peptides, sufentanyl, etorphine, etc.) differential efficacy for endocytosis. However, that interpretation is much less certain when the direct influence of intrinsic efficacies of these drugs for G-protein signalling on tolerance are taken into account. Nonetheless, the effects on morphine tolerance of genetically ablating trafficking proteins (βarr-2 k.o.) or constructing MOR mutants that recycle efficiently with morphine both strongly suggest MOR desensitization, endocytosis and recycling are important for tolerance. Some of the assumptions underpinning explanations of how this works are incomplete or incorrect. First, βarr-2 binding and endocytosis are not necessary to produce desensitization of MOR. In the absence of βarr-2, other, non-arrestin mechanisms can very efficiently desensitize the receptor. More importantly, there is now very strong evidence that one of the simplest explanations for greater tolerance with weakly internalizing agonists, that phosphorylated and desensitized MOR accumulates at the surface because endocytosis is required for dephosphorylation and resensitization, is incorrect. MOR dephosphorylates and resensitizes as efficiently or more efficiently when endocytosis is blocked, regardless of the agonist used. These findings are outlined in Figure 2. This demands rethinking of models used to explain the effects of transgenics and knockouts.
Figure 2.
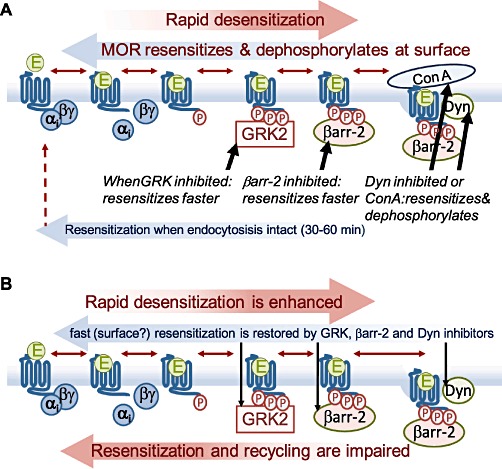
Summary of current evidence for mechanisms of MOR regulation in resensitization and tolerance. (A) Desensitized MOR efficiently resensitizes when GRK2, βarr-2 (k.o.) or dynamin (to block endocytosis directly) are blocked, suggesting that resensitization is very efficient in the absence of endocytosis. Directly blocking endocytosis with concanavalin-A (ConA) does not affect resensitization or dephosphorylation of MOR. (B) After chronic morphine treatment, desensitization is enhanced, and resensitization is blocked. This does not appear to involve changes in endocytosis, but impaired resesnitization is restored to control rates by inhibiting GRK2, βarr-2 or dynamin.
The finding that rapid desensitization of MOR is enhanced and resensitization is impaired in LC neurons after chronic morphine, if widely substantiated in other neurons, may contribute to further developments. If it is confirmed widely through the CNS and in different cellular compartments that enhanced rapid MOR desensitization and arrestin-dependent impairment of resensitization strongly contribute to opioid tolerance, then drugs able to elude these mechanisms might be found to produce less tolerance. The finding that a salvinorin A analogue, herkinorin, efficaciously engages MOR–G-protein signalling but does not induce βarr-2 translocation, even when GRK2 is overexpressed (Groer et al., 2007) confirms the possibility that opioid agonists may be found that would not facilitate arrestin-dependent impairment of resensitization in tolerance. Virk et al. (2009) reported the intriguing finding that met-enkephalin can engage G-protein signalling in the presence of low concentrations of buprenorphine (a low efficacy for G-proteins, non-internalizing agonist) but no longer produces any rapid desensitization. If validated more widely this suggests that opioids, or related drugs could be found to stabilize MOR in conformations that are able to signal to G-proteins but cannot desensitize, which could perhaps limit tolerance.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia
Glossary
- βarr-2
β-arrestin-2 (a.k.a. Arrestin 3)
- DAMGO
d-Ala(2),N-Me-Phe(4),Gly(5)-ol]-enkephalin
- GRK
G-protein receptor kinase
- LC
locus coeruleus; HEK 293, human embryonic kidney 293 cells
- MOR
µ-opioid receptor
- PAG
periaqueductal grey
- PKC
protein kinase C
- RET
resonance energy transfer
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- Adams JU, Paronis CA, Holtzman SG. Assessment of relative intrinsic activity of mu-opioid analgesics in vivo by using beta-funaltrexamine. J Pharmacol Exp Ther. 1990;255:1027–1032. [PubMed] [Google Scholar]
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, von Zastrow M, et al. µ-Opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden JR, Segredo V, Wang Z, Lameh J, Sadee W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged µ-opioid receptor expressed in HEK 293 cells. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT. Separation of µ-opioid receptor desensitization and internalization: endogenous receptors in primary neuronal cultures. J Neurosci. 2006;26:4118–4125. doi: 10.1523/JNEUROSCI.0303-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Quillinan N, Low MJ, von Zastrow M, Pintar J, Williams JT. Differential activation and trafficking of µ-opioid receptors in brain slices. Mol Pharmacol. 2008;74:972–979. doi: 10.1124/mol.108.048512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley EE, Chieng BC, Christie MJ, Connor M. Opioid tolerance in periaqueductal gray neurons isolated from mice chronically treated with morphine. Br J Pharmacol. 2005;146:68–76. doi: 10.1038/sj.bjp.0706315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G. µ-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci. 2003;23:10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitization of µ-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. doi: 10.1124/mol.104.004747. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, et al. Role of protein kinase C and µ-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009a;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, et al. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of µ-opioid receptors in mature brain neurons. Br J Pharmacol. 2009b;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Whistler JL. How to design an opioid drug that causes reduced tolerance and dependence. Ann Neurol. 2010;67:559–569. doi: 10.1002/ana.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. µ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in βarrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of µ-opioid receptors. J Biol Chem. 2003;278:18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- Bradaia A, Berton F, Ferrari S, Luscher C. β-Arrestin2, interacting with phosphodiesterase 4, regulates synaptic release probability and presynaptic inhibition by opioids. Proc Natl Acad Sci USA. 2005;102:3034–3039. doi: 10.1073/pnas.0406632102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton JJ, Allen PB, Psifogeorgou K, Chakravarty S, Gomes I, Neve RL, et al. Multiple actions of spinophilin regulate µ opioid receptor function. Neuron. 2008;58:238–247. doi: 10.1016/j.neuron.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Goldstein A. Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci USA. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZJ, Yu QM, Wu YL, Ma L, Pei G. Selective interference of β-arrestin 1 with kappa and delta but not µ opioid receptor/G protein coupling. J Biol Chem. 1998;273:24328–24333. doi: 10.1074/jbc.273.38.24328. [DOI] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids : tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- Chu J, Zheng H, Zhang Y, Loh HH, Law PY. Agonist-dependent µ-opioid receptor signaling can lead to heterologous desensitization. Cell Signal. 2010;22:684–696. doi: 10.1016/j.cellsig.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Borgland SL, Christie MJ. Continued morphine modulation of calcium channel currents in acutely isolated locus coeruleus neurons from morphine-dependent rats. Br J Pharmacol. 1999;128:1561–1569. doi: 10.1038/sj.bjp.0702922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. µ-opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT. Chronic morphine treatment reduces recovery from opioid desensitization. J Neurosci. 2004;24:7699–7706. doi: 10.1523/JNEUROSCI.2499-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT. Morphine-induced µ-opioid receptor desensitization. Mol Pharmacol. 2005;68:1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- Dang VC, Napier IA, Christie MJ. Two distinct mechanisms mediate acute µ-opioid receptor desensitization in native neurons. J Neurosci. 2009;29:3322–3327. doi: 10.1523/JNEUROSCI.4749-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Chieng B, Azriel Y, Christie MJ. Morphine tolerance produced by βarrestin-2-dependent impairment of µ-opioid receptor resensitization. J Neurosci. 2011;31:7122–7130. doi: 10.1523/JNEUROSCI.5999-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Konietzko J, Koch T, Höllt V, Schulz S. Agonist-selective patterns of µ-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. 2011;164:298–307. doi: 10.1111/j.1476-5381.2011.01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiol. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- Eitan S, Bryant CD, Saliminejad N, Yang YC, Vojdani E, Keith D, et al. Brain region-specific mechanisms for acute morphine-induced mitogen-activated protein kinase modulation and distinct patterns of activation during analgesic tolerance and locomotor sensitization. J Neurosci. 2003;23:8360–8369. doi: 10.1523/JNEUROSCI.23-23-08360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates µ-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the µ opioid receptor. J Pharmacol Exp Ther. 1996;278:1121–1127. [PubMed] [Google Scholar]
- Fan X, Zhang J, Zhang X, Yue W, Ma L. Acute and chronic morphine treatments and morphine withdrawal differentially regulate GRK2 and GRK5 gene expression in rat brain. Neuropharmacology. 2002;43:809–816. doi: 10.1016/s0028-3908(02)00147-8. [DOI] [PubMed] [Google Scholar]
- Fan XL, Zhang JS, Zhang XQ, Yue W, Ma L. Differential regulation of β-arrestin 1 and β-arrestin 2 gene expression in rat brain by morphine. Neuroscience. 2003;117:383–389. doi: 10.1016/s0306-4522(02)00930-2. [DOI] [PubMed] [Google Scholar]
- Fyfe LW, Cleary DR, Macey TA, Morgan MM, Ingram SL. Tolerance to the antinociceptive effect of morphine in the absence of short-term presynaptic desensitization in rat periaqueductal gray neurons. J Pharmacol Exp Ther. 2010;335:674–680. doi: 10.1124/jpet.110.172643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Han MH, Herman AE, Ni YG, Pudiak CM, Aghajanian GK, et al. Regulation of RGS proteins by chronic morphine in rat locus coeruleus. Eur J Neurosci. 2003;17:971–980. doi: 10.1046/j.1460-9568.2003.02529.x. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, et al. An opioid agonist that does not induce µ-opioid receptor–arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, et al. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45:575–584. doi: 10.1016/s0028-3908(03)00205-3. [DOI] [PubMed] [Google Scholar]
- He L, Whistler JL. An opiate cocktail that reduces morphine tolerance and dependence. Curr Biol. 2005;15:1028–1033. doi: 10.1016/j.cub.2005.04.052. [DOI] [PubMed] [Google Scholar]
- He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Macey TA, Fossum EN, Morgan MM. Tolerance to repeated morphine administration is associated with increased potency of opioid agonists. Neuropsychopharmacology. 2008;33:2494–2504. doi: 10.1038/sj.npp.1301634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, et al. Agonist-selective mechanisms of µ-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006a;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Johnson EE, Chieng B, Napier I, Connor M. Decreased µ-opioid receptor signalling and a reduction in calcium current density in sensory neurons from chronically morphine-treated mice. Br J Pharmacol. 2006b;14:947–955. doi: 10.1038/sj.bjp.0706820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, et al. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Hollt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol Ther. 2008;117:199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Schroder H, Wolf R, Raulf E, Hollt V. Carboxyl-terminal splicing of the rat µ opioid receptor modulates agonist-mediated internalization and receptor resensitization. J Biol Chem. 1998;273:13652–13657. doi: 10.1074/jbc.273.22.13652. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Pfeiffer M, Klutzny M, Schroder H, Kahl E, et al. C-terminal splice variants of the mouse µ-opioid receptor differ in morphine-induced internalization and receptor resensitization. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, et al. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Koch T, Wu DF, Yang LQ, Brandenburg LO, Hollt V. Role of phospholipase D2 in the agonist-induced and constitutive endocytosis of G-protein coupled receptors. J Neurochem. 2006;97:365–372. doi: 10.1111/j.1471-4159.2006.03736.x. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of µ-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- Law PY, Erickson LJ, El-Kouhen R, Dicker L, Solberg J, Wang W, et al. Receptor density and recycling affect the rate of agonist-induced desensitization of µ-opioid receptor. Mol Pharmacol. 2000;58:388–398. doi: 10.1124/mol.58.2.388. [DOI] [PubMed] [Google Scholar]
- Li AH, Wang HL. G protein-coupled receptor kinase 2 mediates µ-opioid receptor desensitization in GABAergic neurons of the nucleus raphe magnus. J Neurochem. 2001;77:435–444. doi: 10.1046/j.1471-4159.2001.00267.x. [DOI] [PubMed] [Google Scholar]
- Madia PA, Dighe SV, Sirohi S, Walker EA, Yoburn BC. Dosing protocol and analgesic efficacy determine opioid tolerance in the mouse. Psychopharmacol. 2009;207:413–422. doi: 10.1007/s00213-009-1673-6. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of µ opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:17556–17564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, et al. µ-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010;78:756–766. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci USA. 2010;107:11608–11613. doi: 10.1073/pnas.1000751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjanger E, Yaksh TL. Characteristics of dose-dependent antagonism by beta-funaltrexamine of the antinociceptive effects of intrathecal mu agonists. J Pharmacol Exp Ther. 1991;258:544–550. [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Gro C, Riitano D, Ambrosio C, et al. Morphine-like opiates selectively antagonize receptor-arrestin interactions. J Biol Chem. 2010;285:12522–12535. doi: 10.1074/jbc.M109.059410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MM, Christie MJ. Analysis of opioid efficacy, tolerance, addiction, and dependence from cell culture to human. Br J Pharmacol. 2011;164:1322–1334. doi: 10.1111/j.1476-5381.2011.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–39095. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- Oldfield S, Braksator E, Rodriguez-Martin I, Bailey CP, Donaldson LF, Henderson G, et al. C-terminal splice variants of the µ-opioid receptor: existence, distribution and functional characteristics. J Neurochem. 2008;104:937–945. doi: 10.1111/j.1471-4159.2007.05057.x. [DOI] [PubMed] [Google Scholar]
- Osborne PB, Williams JT. Characterization of acute homologous desensitization of µ-opioid receptor-induced currents in locus coeruleus neurones. Br J Pharmacol. 1995;115:925–932. doi: 10.1111/j.1476-5381.1995.tb15899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineyro G, Archer-Lahlou E. Ligand-specific receptor states: implications for opiate receptor signalling and regulation. Cell Signal. 2007;19:8–19. doi: 10.1016/j.cellsig.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Polakiewicz RD, Schieferl SM, Dorner LF, Kansra V, Comb MJ. A mitogen-activated protein kinase pathway is required for mu-opioid receptor desensitization. J Biol Chem. 1998;273:12402–12406. doi: 10.1074/jbc.273.20.12402. [DOI] [PubMed] [Google Scholar]
- Puttfarcken PS, Cox BM. Morphine-induced desensitization and down-regulation at µ-receptors in 7315C pituitary tumor cells. Life Sci. 1989;45:1937–1942. doi: 10.1016/0024-3205(89)90548-1. [DOI] [PubMed] [Google Scholar]
- Puttfarcken PS, Werling LL, Cox BM. Effects of chronic morphine exposure on opioid inhibition of adenylyl cyclase in 7315c cell membranes: a useful model for the study of tolerance at µ opioid receptors. Mol Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- Qiu Y, Law PY, Loh HH. µ-opioid receptor desensitization: role of receptor phosphorylation, internalization, and representation. J Biol Chem. 2003;278:36733–36739. doi: 10.1074/jbc.M305857200. [DOI] [PubMed] [Google Scholar]
- Quillinan N, Lau E, Virk M, von Zastrow M, Williams JT. Recovery from Mu-opioid receptor desensitization following chronic treatment with morphine and methadone. J Neurosci. 2011;31:4434–4443. doi: 10.1523/JNEUROSCI.4874-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martin I, Braksator E, Bailey CP, Goodchild S, Marrion NV, Kelly E, et al. Methadone: does it really have low efficacy at µ-opioid receptors? Neuroreport. 2008;19:589–593. doi: 10.1097/WNR.0b013e3282f97b64. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schulz S, Klutzny M, Koch T, Handel M, Hollt V. Involvement of mitogen-activated protein kinase in agonist-induced phosphorylation of the µ-opioid receptor in HEK 293 cells. J Neurochem. 2000;74:414–422. doi: 10.1046/j.1471-4159.2000.0740414.x. [DOI] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal µ-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley DE, Liu Q, Childers SR. Signal transduction correlates of µ opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on µ opioid receptor-stimulated [35S]GTPγS autoradiography in rat brain. J Neurosci. 1996;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Nuijten M, Nautrup BP. Patterns of dosage changes with transdermal buprenorphine and transdermal fentanyl for the treatment of noncancer and cancer pain: a retrospective data analysis in Germany. Clin Therapeut. 2006;28:1144–1154. doi: 10.1016/j.clinthera.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Soignier RD, Vaccarino AL, Fanti KA, Wilson AM, Zadina JE. Analgesic tolerance and cross-tolerance to i.c.v. endomorphin-1, endomorphin-2, and morphine in mice. Neurosci Lett. 2004;366:211–214. doi: 10.1016/j.neulet.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Stafford K, Gomes AB, Shen J, Yoburn BC. µ-Opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol Biochem Behav. 2001;69:233–237. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- Sternini C, Spann M, Anton B, Keith DE, Jr, Bunnett NW, von Zastrow M, et al. Agonist-selective endocytosis of µ opioid receptor by neurons in vivo. Proc Natl Acad Sci USA. 1996;93:9241–9246. doi: 10.1073/pnas.93.17.9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CW, Yaksh TL. Potency of infused spinal antinociceptive agents is inversely related to magnitude of tolerance after continuous infusion. J Pharmacol Exp Ther. 1989;250:1–8. [PubMed] [Google Scholar]
- Tanowitz M, von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem. 2003;278:45978–45986. doi: 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- Tanowitz M, Hislop JN, von Zastrow M. Alternative splicing determines the post-endocytic sorting fate of G-protein-coupled receptors. J Biol Chem. 2008;283:35614–35621. doi: 10.1074/jbc.M806588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, et al. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton JA, Basbaum AI. [d-Ala2,N-MePhe4,Gly-ol5]enkephalin-induced internalization of the µ opioid receptor in the spinal cord of morphine tolerant rats. Neurosci. 2004;125:541–543. doi: 10.1016/j.neuroscience.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marek K, Basbaum AI. Postsynaptic signaling via the [mu]-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci. 2000;20:8578–8584. doi: 10.1523/JNEUROSCI.20-23-08578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor JR, Nahorski SR. Modulation by µ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1995;47:848–854. doi: 10.1016/S0026-895X(25)08634-1. [DOI] [PubMed] [Google Scholar]
- Virk MS, Williams JT. Agonist-specific regulation of µ-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol. 2008;73:1301–1308. doi: 10.1124/mol.107.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Arttamangkul S, Birdsong WT, Williams JT. Buprenorphine is a weak partial agonist that inhibits opioid receptor desensitization. J Neurosci. 2009;29:7341–7348. doi: 10.1523/JNEUROSCI.3723-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. β-arrestin2 and c-Src regulate the constitutive activity and recycling of µ opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL. A cluster of Ser/Thr residues at the C-terminus of µ-opioid receptor is required for G protein-coupled receptor kinase 2-mediated desensitization. Neuropharmacology. 2000;39:353–363. doi: 10.1016/s0028-3908(99)00174-4. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by β-arrestin. Proc Natl Acad Sci USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P, et al. Modulation of postendocytic sorting of G protein-coupled receptors. Science. 2002;297:615–620. doi: 10.1126/science.1073308. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Yu YJ, Arttamangkul S, Evans CJ, Williams JT, von Zastrow M. Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of µ-opioid receptors in CNS neurons. J Neurosci. 2009;29:222–233. doi: 10.1523/JNEUROSCI.4315-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M. Regulation of opioid receptors by endocytic membrane traffic: mechanisms and translational implications. Drug Alcohol Depend. 2010;108:166–171. doi: 10.1016/j.drugalcdep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M, Svingos A, Haberstock-Debic H, Evans C. Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr Opin Neurobiol. 2003;13:348–353. doi: 10.1016/s0959-4388(03)00069-2. [DOI] [PubMed] [Google Scholar]
- Zheng H, Chu J, Qiu Y, Loh HH, Law PY. Agonist-selective signaling is determined by the receptor location within the membrane domains. Proc Natl Acad Sci USA. 2008;105:9421–9426. doi: 10.1073/pnas.0802253105. [DOI] [PMC free article] [PubMed] [Google Scholar]