

Summary
The pKa values of internal ionizable groups are usually very different than the normal pKa values of ionizable groups in water. To examine the molecular determinants of pKa values of internal groups, we compared the properties of Lys, Asp and Glu at internal position 38 in staphylococcal nuclease. Lys-38 titrates with a normal or elevated pKa whereas Asp-38 and Glu-38 titrate with elevated pKa values of 7.0 and 7.2, respectively. In the structure of the L38K variant, the buried amino group of the Lys-38 side chain makes an ion pair with Glu-122; whereas, in the structure of the L38E variant, the buried carboxyl group of Glu-38 interacts with two backbone amides and has several nearby carboxyl oxygen atoms. Previously we showed that the pKa of Lys-38 is normal owing to structural reorganization and water penetration concomitant with ionization of the Lys side chain. In contrast, the pKa of Asp-38 and Glu-38 are perturbed significantly owing to an imbalance between favorable polar interactions and unfavorable contributions from dehydration and from Coulomb interactions with surface carboxylic groups. Their ionization is also coupled to subtle structural reorganization. These results illustrate the complex interplay between local polarity, Coulomb interactions and structural reorganization as determinants of pKa values of internal groups in proteins. This study suggests that improvements to computational methods for pKa calculations will require explicit treatment of the conformational reorganization that can occur when internal groups ionize.
Keywords: pKa values, internal ionizable groups, structure-function, energy calculations, electrostatics
Introduction
A small fraction of ionizable residues in proteins are sequestered from water and buried in the protein interior.1-3 These internal ionizable groups are essential for catalysis,4-6 H+/e- transport,7-10 and molecular recognition.11 The pKa values of internal ionizable groups are usually different than the normal pKa values in water,12-19 and are often tuned for specific biological purposes.4 Understanding the determinants of these pKa values is important for quantitative description of the structural basis of function in a large variety of biological processes.
The shift in the pKa of an internal group relative to the normal pKa in water is governed by differences in the polarity and polarizability experienced by the charge in the two environments (ΔGself), and by Coulomb interactions with the charges of other ionizable groups. Structural reorganization of the protein coupled to the ionization of internal groups can also influence their pKa. One of the goals of this study was to examine the relative magnitude of these three determinants of the pKa values of internal groups.
The polarity and polarizability in the protein interior are usually lower than in bulk water; therefore, ΔGself is generally unfavorable for buried ionizable groups. For this reason, the pKa values of internal ionizable groups are usually shifted in the direction that favors the neutral state (i.e. increase in pKa for acidic groups and depression for basic ones).12-18 Surprisingly, the apparent polarity and polarizability in the protein interior reported by internal ionizable groups is not as low as previously thought. 15-17; 20-25 In some cases, hydrogen bonds (i.e. high polarity) can actually compensate fully for the loss of hydration experienced by a charged atom inside a protein.6; 19; 26
Coulomb interactions between surface charges are usually weak because charges are screened effectively by water.27-31 In contrast, the Coulomb interaction of ion pairs sequestered from bulk solvent at protein-protein interfaces can be quite strong (3-5 kcal/mol).32 Coulomb interactions between surface and internal groups in protein active sites have never been studied directly. Surface ionizable group have been shown to have small, but observable effects on enzyme activity.33-35 Even if the effects are small, the sum of many small interactions could lead potentially to a large effect.36; 37 A complete understanding of interactions between internal and surface charges is necessary to understand contributions of surface ionizable residues to the properties of internal groups at active sites and interfaces.4
Staphylococcal nuclease (SNase) is an excellent model system for studying properties of internal ionizable groups systematically, and for dissecting molecular determinants of their pKa values. It has been shown that hyperstable variants of SNase can tolerate substitutions of 25 internal positions with Lys, Asp, Glu and Arg.38 The majority of these internal ionizable groups titrate with pKa values shifted in the direction that promotes the neutral state, some by as much as 5.7 pKa units.15; 16; 22; 23; 25; 26; 39 We have shown previously that, although Lys-38 in staphylococcal nuclease is internal, it titrates with a normal or possibly elevated pKa value. The pKa is not depressed despite the amino group being secluded from bulk water in the crystal structure; water penetration facilitated by structural relaxation ensures hydration of the charged group.39 In contrast, we show here that the pKa of Glu-38 and Asp-38 are shifted significantly. The differences in the ionization behavior of Lys, Glu, and Asp at position 38 in SNase offer opportunities to examine contributions by the reaction field of bulk solvent, local polarity and polarizability, conformational reorganization, and Coulomb interactions to the pKa values of these internal ionizable groups.
Results
Crystal structure of the L38E variant
Two hyperstable variants of SNase were used in this study: PHS and Δ+PHS. The structure of the PHS/L38E variant was solved to 2.0 Å and compared to the structures of PHS nuclease and the PHS/L38K variant.39; 40 Refinement statistics are shown in Table 1 in the Supplementary Information. PHS nuclease was used for crystallographic studies instead of the Δ+PHS form of nuclease that was used for equilibrium thermodynamic and NMR spectroscopy experiments because the PHS/L38E crystallized and Δ+PHS/L38E did not. PHS nuclease contains six residues (44-49) in a dynamic loop and two point mutations (F50G and N51V) that are not present in Δ+PHS nuclease. The structures of Δ+PHS and PHS variants are superimposable.40; 41
The overall structure of the PHS/L38E variant is comparable to the structures of PHS nuclease (Cα RMSD = 0.7 Å) and of the PHS/L38K variant (Cα RMSD = 0.4 Å), even in the region surrounding Glu-38 (Figure 1A). The primary difference between the PHS/L38E and PHS/L38K structures is the position of Glu-122. This residue is in the same position in the structure of PHS and PHS/L38E, whereas, the Cδ of Glu-122 is shifted by 1.6 Å to establish a Lys-38/Glu-122 ion pair in the structure of PHS/L38K.39 Residues 113-115 in the structure of PHS/L38E are in a slightly different conformation than in the other structures owing to the presence of an inhibitor (thymidine-3′,5′-diphosphate) that is present in the structure of PHS/L38E nuclease and absent in the other structures.
Figure 1.
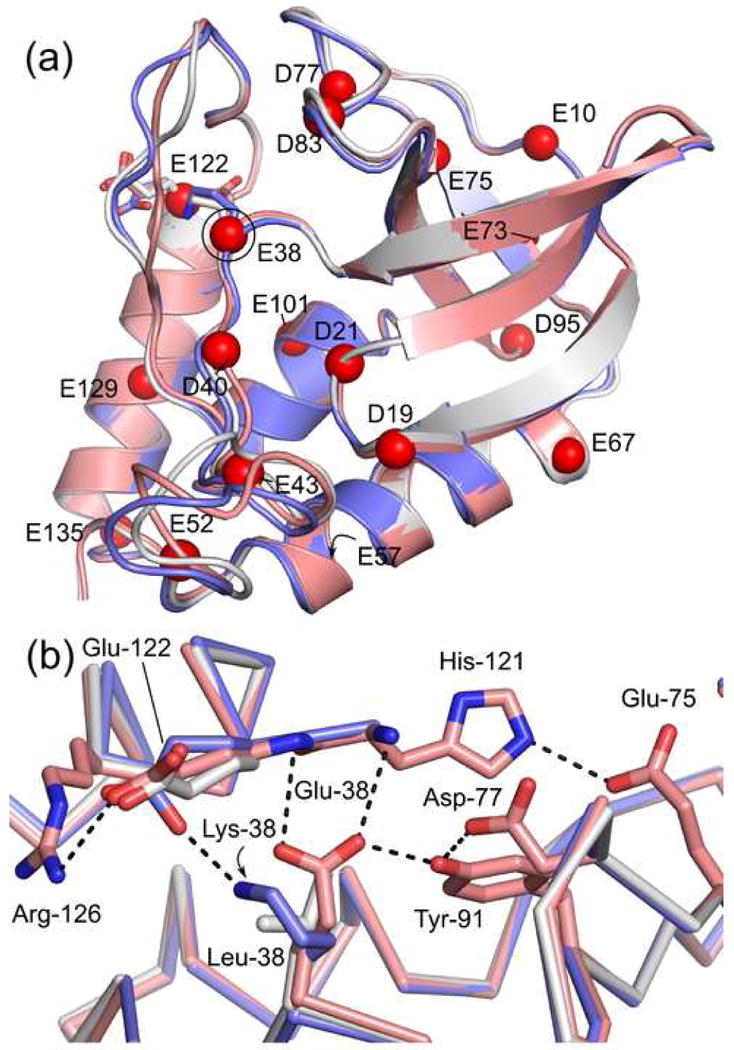
Crystal structure of PHS/L38E (pink, PDB accession code 3D6C) overlaid on the structures of PHS/L38K (blue, PDB accession code 2RKS) and PHS nuclease (white, PDB accession code 1EY8). (A) The global fold of the protein is not perturbed. The Cα atom of Asp and Glu residues are shown as red spheres. Glu-38, Lys-38, and Glu-122 are shown as sticks. (B) Microenvironment of Glu-38 and Lys-38. Ionizable residues within 8.4 Å of Glu-38 are shown in stick, hydrogen bonds as black dashes.
The oxygen atoms of the Glu-38 side chain are completely solvent inaccessible in the structure of PHS/L38E. The nearest crystallographic water molecule is 5.4 Å from the Glu-38 Oε1 atom. Thus far, this is the only crystal structure of a SNase variant with an internal oxygen atom in which the atom is not hydrated by an internal water molecule.16; 42 Interactions with internal water molecules might be precluded by hydrogen bonds between the carboxylic group of Glu-38 and the backbone amides of Thr-120 and His-121, and the hydroxyl group of Tyr-91 (Figure 1B). The hydrogen bond to Tyr-91 directly links Glu-38 into an extensive hydrogen bond network.30; 31; 43; 44
Although SNase is a basic protein, the ionizable residues nearest to Glu-38 in the crystal structure of the PHS/L38E variant are acidic: Asp-77 (4.0 Å) and Glu-122 (5.1 Å). The next nearest ionizable residues are basic: His-121 (6.5 Å) and Arg-126 (6.6 Å) (Figure 1B). The proximity of these residues makes them ideal for direct measurement of Coulomb interactions between surface charges and the carboxylic groups of Asp-38 and Glu-38.
pKa values of Glu-38 and Asp-38
The pKa values of Glu-38 and Asp-38 were measured by analysis of the pH-dependence of protein stability. This method takes advantage of the thermodynamic linkage between proton binding and stability.45 Measurement of the unfolding free energy (ΔG°H2O) of a protein as a function of pH reports on the pKa values of all ionizable residues in the protein. The pKa value of a single group introduced by mutagenesis can be measured by subtracting ΔG°H2O of the background protein (i.e. Δ+PHS nuclease) from ΔG°H2O of the variant protein (i.e. Δ+PHS/L38E). Shifts in the pKa are reflected in the characteristic shape of the pH-dependence of ΔΔG°H2O.15; 23; 25 It was shown previously that the pKa of Lys-38 was ≥ 10.4, comparable to the normal pKa of a Lys in water.39 In contrast, the pKa values of Glu-38 and Asp-38 were 7.0 +/- 0.3 and 6.8 +/-0.3 pH units, respectively (Figure 2). Relative to the normal pKa values of 4.4 and 4.0 for Glu and Asp in water, respectively, this corresponds to shifts in pKa of 2.6 and 2.8 pH units.
Figure 2.
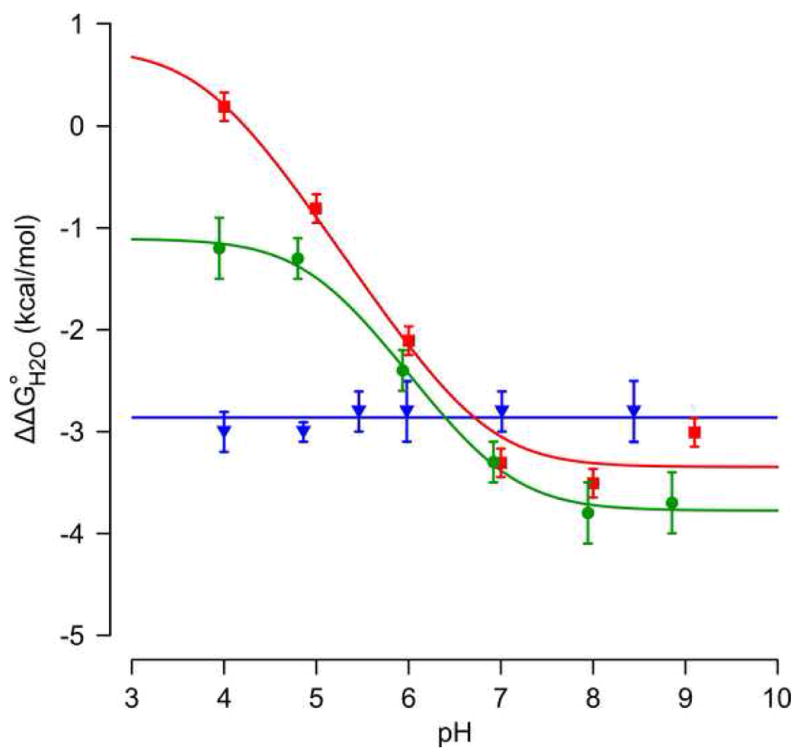
pH-dependence of ΔΔG°H2O for the Δ+PHS/L38D (
 ), Δ+PHS/L38E (
), Δ+PHS/L38E (
 ), and Δ+PHS/L38K (
), and Δ+PHS/L38K (
 ) variants. Solid lines are fits to the data (see methods). Error bars are propagated from GdnHCl denaturation fit errors.
) variants. Solid lines are fits to the data (see methods). Error bars are propagated from GdnHCl denaturation fit errors.
The measurement of pKa values by analysis of the pH dependence of stability is too imprecise for detailed investigation of the contribution of Coulomb interactions to the observed pKa value. An attempt was made to measure the pKa of Glu-38 with NMR spectroscopy using the pH-dependence of the Glu-38 Cδ resonance.41 Although the Cδ resonance could be assigned at low pH, the peak entered intermediate exchange above pH 5.6 and could not be followed at higher pH values. Resonances corresponding to the Cγ/Cδ atoms of Glu-73, Glu-75, Asp-77, Asp-83, and Glu-122 all showed a secondary apparent titration centered at pH 7.0. At positions 75 and 77, the magnitude of the secondary transition was greater than 0.5 ppm. All of the data from NMR spectroscopy are consistent with a pKa of 7.0 for Glu-38.
The pKa of Glu-38 was also obtained by performing a global fit to the pH titrations of multiple resonances.46; 47 Specifically, the pKa was obtained by analysis of the pH-dependence of the 1H chemical shift of six backbone amides (Thr-33, Phe-34, Arg-35, Glu-75, Gly-88, and Leu-89). The titration events monitored by these amide backbone atoms in Δ+PHS nuclease are shown in Figure 3A. No changes larger than 0.06 ppm were observed over the pH range studied. A small transition centered at pH 6.3 +/- 0.3 is visible for positions 34, 35, 75, and 89, most likely reflecting the titration of His-8 or Asp-21, whose pKa values are both 6.5 in Δ+PHS nuclease.41 In contrast, the pH-dependence of the 1H chemical shift of the same six amides in the Δ+PHS/L38E variant (Figure 3B) reflects a large transition. A global fit of the modified Hill equation to this transition yielded a pKa value of 7.0 +/- 0.1, in excellent agreement with the pKa of Glu-38 determined using linkage thermodynamics and the value inferred from the titration of carboxylic acids. A similar analysis of the L38D variant showed that Asp-38 has a pKa of 7.2 +/- 0.1, which is also in agreement with the value of 6.8 +/- 0.3 obtained by analysis of the pH dependence of stability of the Δ+PHS/L38D variant. The pKa values extracted by global fit of NMR spectroscopy data and by linkage analysis are summarized in Table 1.
Figure 3.
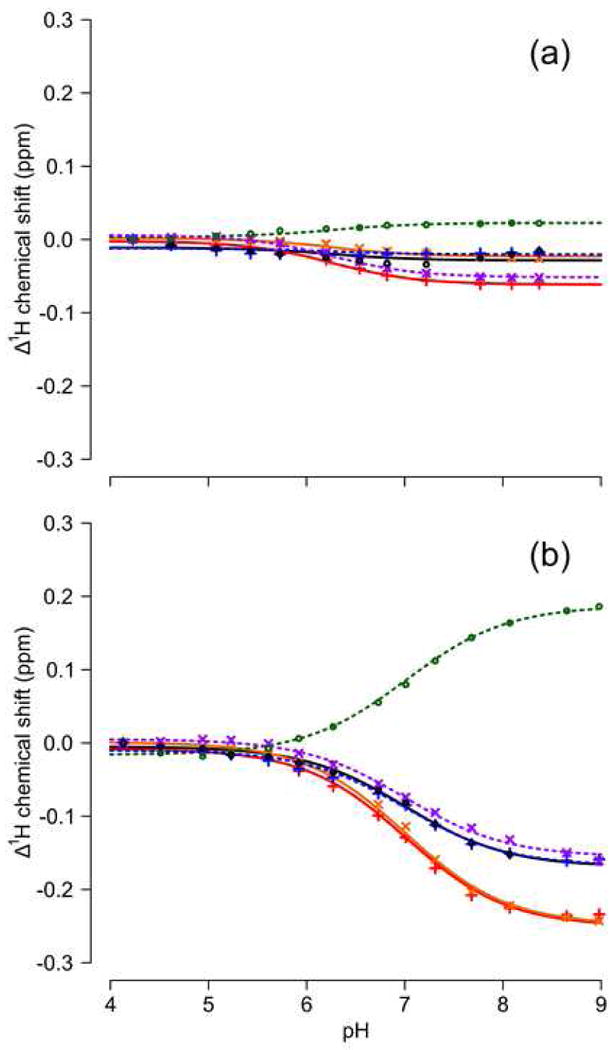
Apparent titration of 1H backbone resonances in Δ+PHS nuclease (A) and the Δ+PHS/L38E variant (B). Series are Thr-33 (•), Phe-34 (+), Arg-35 (×), Glu-75 (
), Gly-88 (+), and Leu-89 (×). Lines indicate a global fit to the apparent titration of all residues.
Table 1. Comparison of pKa values determined by linkage analysis and NMR spectroscopy.
| linkage analysis | NMR | ||||
|---|---|---|---|---|---|
| Variant | residue | pKa | +/- | pKa | +/- |
| Δ+PHS/L38E | Glu-38 | 7.0 | 0.3 | 7.0 | 0.1 |
| Δ+PHS/L38E/E122Q | Glu-38 | --- | --- | 6.2 | 0.1 |
| Δ+PHS/L38D | Asp-38 | 6.8 | 0.3 | 7.2 | 0.1 |
| Δ+PHS/L38D/E122Q | Asp-38 | 6.9 | 0.3 | 6.6 | 0.1 |
pKa could not be determined using linkage analysis because variant unfolds in an apparent 3-state manner
The agreement between the pKa values measured from equilibrium thermodynamic data and from the global fit of titrations of backbone amide resonances suggests that the values obtained by NMR are accurate. However, the NMR experiment does not follow the amino acid of interest directly. Other groups could be responsible for the observed transition. To examine this possibility, the pKa values of all residues that titrate between pH 4.6 and 8.5 in Δ+PHS nuclease were measured in the Δ+PHS/L38E variant. The pKa values of His-8, His-121, and Asp-21 were found to be 6.5, 5.7, and 6.5, respectively in the Δ+PHS/L38E variant (Tables 2 and 3). This demonstrates that none of these groups are responsible for the apparent titration near pH 7 monitored with NMR spectroscopy. The 1H chemical shifts of multiple groups appear to be reporting on the proton titration of Asp-38 or Glu-38.
Table 2. pKa values and hill coefficients of His residues measured using NMR spectroscopy.
| His-8 | His-121 | |||
|---|---|---|---|---|
| variant | pKa* | n | pKa* | n |
| Δ+PHS | 6.6 | 1.0 | 5.4 | 0.8 |
| Δ+PHS/E122Q | 6.5 | 1.0 | 5.3 | 0.9 |
| Δ+PHS/L38K | 6.5 | 1.0 | 5.6 | 1.0 |
| Δ+PHS/L38E | 6.5 | 1.0 | 5.7 | 0.9 |
| Δ+PHS/L38E/E122Q | 6.4 | 1.1 | 5.7 | 0.9 |
| Δ+PHS/L38D | 6.5 | 1.0 | 5.7 | 0.9 |
| Δ+PHS/L38D/E122Q | 6.5 | 1.1 | 5.8 | 0.9 |
uncertainty in pKa value is +/- 0.1
Table 3. pKa values of Asp and Glu residues measured using NMR spectroscopy.
| Δ+PHS ‡ | Δ+PHS/L38E | Δ+PHS/L38K | |||
|---|---|---|---|---|---|
| position | pKa* | pKa* | ΔpKa† | pKa* | ΔpKa† |
| D19 | 2.2 | < 2.5 | --- | < 2.5 | --- |
| D21 | 6.5 | 6.6 | 0.0 | 6.5 | 0.0 |
| D40 | 3.9 | 3.9 | 0.0 | 3.8 | -0.1 |
| D77 | < 2.5 | < 2.5 | --- | --- | --- |
| D83 | < 2.5 | < 2.5 | --- | < 2.5 | --- |
| D95 | 2.2 | 2.3 | 0.1 | 2.1$ | 0.0 |
| D143 | 3.8 | 3.8 | 0.0 | 4.0 | 0.2 |
| D146 | 3.9 | 3.9 | 0.0 | 4.0 | 0.1 |
| E10 | 2.8 | 3.1 | 0.2 | 3.1$ | 0.2 |
| E43 | 4.3 | 4.4 | 0.0 | 4.5 | 0.2 |
| E52 | 3.9 | 4.0 | 0.1 | 4.1 | 0.2 |
| E57 | 3.5 | 3.6 | 0.1 | 3.6 | 0.1 |
| E67 | 3.8 | 3.8 | 0.1 | 3.9 | 0.1 |
| E73 | 3.3 | 3.4 | 0.1 | 3.3$ | 0.0 |
| E75 | 3.3 | 3.3 | 0.1 | --- | --- |
| E101 | 3.8 | 3.9 | 0.1 | 3.9 | 0.0 |
| E122 | 3.9 | 3.8 | -0.1 | 3.9 | 0.0 |
| E129 | 3.8 | 3.9 | 0.1 | 3.8 | 0.0 |
| E135 | 3.8 | 3.8 | 0.1 | 3.9 | 0.1 |
| E142 | 4.5 | 4.5 | 0.0 | 4.6 | 0.1 |
uncertainty in pKa value is +/- 0.1
uncertainty in ΔpKa value is +/- 0.1
C. Castañeda et al.41
Acid baseline fixed to Δ+PHS value
Spectroscopic probes of structural rearrangement
Structural reorganization associated with the substitution of Leu-38 with Asp or Glu, or with the ionization of Asp, Glu and Lys at position 38 was probed by CD, Trp fluorescence, and NMR spectroscopy. The intrinsic fluorescence of Trp-140, which caps the C-terminal end of helix 3, has been shown to be a robust reporter of the global integrity of SNase.48 Trp fluorescence at neutral pH was insensitive to the substitution of Leu-38 with ionizable groups (data not shown). Circular dichroism spectra of all variants in the far-UV range were also indistinguishable from one another at pH 4, 7, and 10 (Figure 4), suggesting that none of the substitutions altered the secondary structure of the protein significantly, even when the internal Lys, Asp or Glu groups were charged. Similarly, 70 to 90% of the 131 peaks in the 15N-1H HSQC spectrum of Δ+PHS nuclease at pH 4.5 were identifiable by visual inspection on the spectrum of each variant (Figure 5).41 Overall, the spectroscopic probes suggest that the substitutions did not affect the structure of the protein over a wide range of pH.
Figure 4.
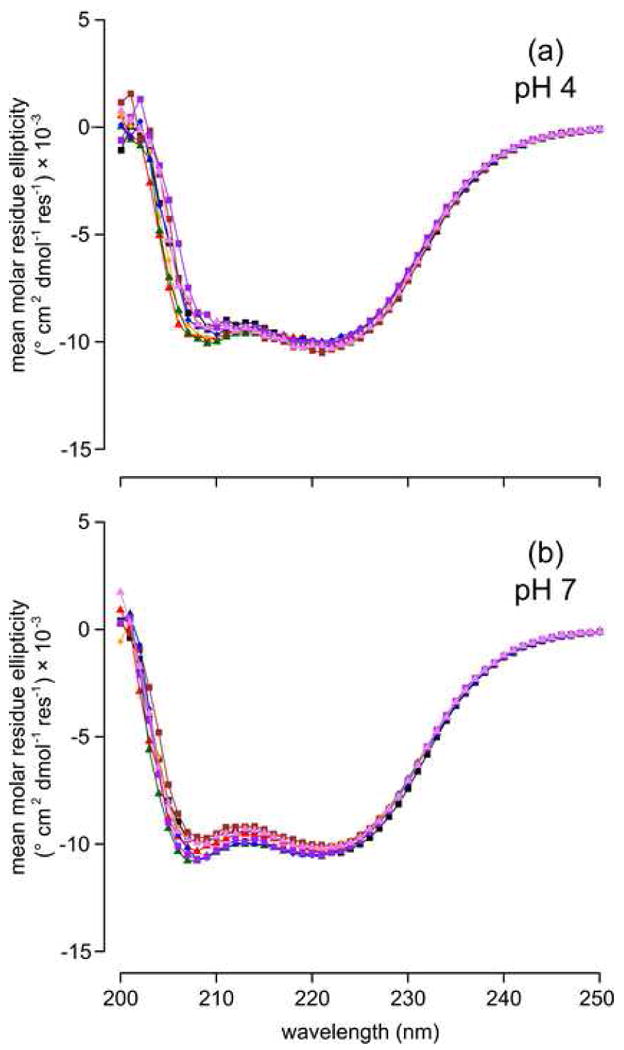
Far-UV CD spectra of at pH 4 (A) and pH 7 (B). Series are Δ+PHS (▾), Δ+PHS/L38E (
 ), Δ+PHS/L38E/D77N (
), Δ+PHS/L38E/D77N (
 ), Δ+PHS/L38E/E122Q (
), Δ+PHS/L38E/E122Q (
 ), Δ+PHS/L38E/E122D (
), Δ+PHS/L38E/R126Q (
), Δ+PHS/L38E/E122D (
), Δ+PHS/L38E/R126Q (
 ), Δ+PHS/L38D (
), Δ+PHS/L38D (
 ), and Δ+PHS/L38D/E122Q (
), and Δ+PHS/L38D/E122Q (
 ).
).
Figure 5.
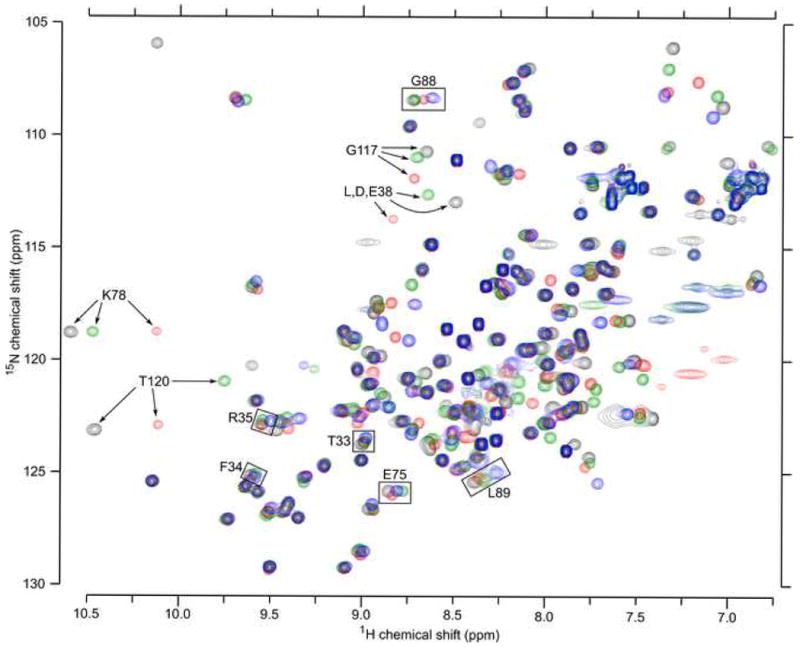
HSQC spectra of the Δ+PHS (black), Δ+PHS/L38D (red), Δ+PHS/L38E (green), and Δ+PHS/L38K variants (blue) at pH 4.6-4.7. The four spectra are overall quite similar. Arrows highlight 4 of the 12 residues that are seen in every spectrum except that of Δ+PHS/L38K. Boxes identify residues used in global fits to extract pKa values of groups at position 38.
Although the global structure of the proteins remained intact, the previous investigation of the Δ+PHS/L38K variant suggested that flexibility of the loop containing residues 113-119 allowed water to penetrate the protein to solvate the charged moiety of the side chain of Lys-38. To probe the conformation of this loop in these variants, an HNN experiment was used to assign all backbone 15N-1H peaks in the HSQC spectra of the Δ+PHS/L38E and Δ+PHS/L38K variants at pH 4.6.49 These spectra were compared to the spectra of Δ+PHS collected previously,41 at pH values between 2.8 and 9.0 in steps of ∼0.4 pH units.
At pH 4.5, 137 peaks were evident for Δ+PHS nuclease, 138 for the Δ+PHS/L38E variant, and only 125 for the Δ+PHS/L38K variant. In the spectrum of Δ+PHS nuclease, only three peaks in the region of interest entered intermediate exchange (i.e. millisecond timescale) with increasing pH: Tyr-113 disappeared above pH 5.7; Lys-116 and Gly-117 disappeared above pH 7.2. Larger changes were observed in the spectrum of the Δ+PHS/L38E variant. The peaks for seven residues (78, 80, 114, 117-120) all entered exchange above pH 6.3, concomitant with ionization of Glu-38. Without further information, it cannot be determined if this is due to structural relaxation or due to the change in the electrostatic environment of the groups owing to the ionization of Glu-38. Changes in proton chemical shift as large as 0.4 ppm were also observed in helix 3 (residues 123-130), around Tyr-91 (residues 88-92), and in the residues adjacent in sequence to position 38 (residues 34-39).
Unlike Glu-38 and Asp-38, Lys-38 was ionized over the entire pH-range under investigation. Twelve peaks are missing in this spectrum at all pH values studied. Of these, 9 peaks correspond to a contiguous stretch from Tyr-113 to Glu-122 (Figure 4). The remaining three missing peaks are Lys-38, Lys-78, and Gln-80. Changes in 15N-1H chemical shifts should not be overinterpreted;50 however, the absence of these peaks is consistent with increased exchange of the amide protons with solvent. This interpretation is also consistent with the previous investigation of the L38K variant and with the measured pKa value of His-121 (see next section).39 Overall, the changes in chemical shift are smaller in the Lys-38 variant than in the Glu-38 variant, having a maximum shift of 0.2 ppm. The largest changes are limited to the N-terminal end of helix 3.
Structural reorganization probed with His-121
The properties of His residues of SNase have been characterized extensively.30; 31 Changes in the microenvironments of His residues can be probed by measuring their pKa values by 1D 1H NMR, a method that has high accuracy and precision > 0.1 pH units. The Δ+PHS variant of nuclease only contains two of the four His residues normally present in wildtype SNase: His-8 and His-121. In Δ+PHS nuclease, His-8 and His-121 titrate with pKa values of 6.6 and 5.4, respectively. The pKa of His-8 was entirely insensitive to the presence and ionization of Asp, Glu, or Lys at position 38 (Table 2). This was expected because His-8 is 17 Å from position 38.
It has been shown previously that the pKa of His-121 is depressed owing primarily to dehydration in a partially buried configuration.31 His-121 is 6 Å from the Oε1 of Glu-38 and 9 Å from the Nζ atom of Lys-38 (Figure 1B). In both the L38D and L38E variants, the pKa value of His-121 was elevated from 5.4 to 5.7 (Table 2). This cannot be due to a Coulomb interaction because His-121 and the internal carboxylic groups do not ionize in the same range of pH. It has been observed previously that perturbations to the hydrogen bonding network centered around His-121 almost always cause the pKa value of His-121 to increase relative to its pKa in Δ+PHS.31 Thus, it is likely that Glu-38 and Asp-38 perturb the hydrogen-bond network, possibly allowing more water to reach His-121 than in Δ+PHS nuclease.
Lys-38 is fully charged in the range of pH where His-121 titrates; therefore, a Coulomb interaction between these residues is possible. An unfavorable Coulomb interaction would further depress the pKa of His-121. This was not observed. Instead, as in the L38D and L38E variants, the pKa of His-121 was elevated to 5.6 in the presence of Lys-38. This small change is consistent with slight structural relaxation induced by the substitution of Leu-38 with Lys, or by the ionization of Lys-38. This is fully consistent with the conclusion of our previous study of Lys-38 and with the evidence from NMR spectroscopy of slight structural reorganization in the L38K variant.39
Coulomb interactions between surface and internal ionizable groups
Δ+PHS nuclease has 20 Asp and Glu residues. The pKa values of these groups are known.41 With the exception of Asp-21, which has an elevated pKa, all carboxylic groups titrate with depressed or normal pKa values. The pKa values of surface Asp and Glu residues in the Δ+PHS/L38E and Δ+PHS/L38K variants were measured with NMR spectroscopy (Table 3).
Substitution of Leu-38 with Glu did not alter the pKa values of any of the Asp or Glu residues (Table 3). This does not imply that Glu-38 does not interact with these groups. Glu-38 titrates with a pKa of 7.0, while most other acidic groups titrate with pKa values near 4.0; therefore, Glu-38 is neutral during the titration of the other Asp and Glu residues and cannot affect their pKa values by Coulomb interaction. These data also show that the L38E substitution has no detectable impact on the structure of the protein and thus on the electrostatic environments of the surface Asp and Glu residues. If Asp-21 had a significant Coulomb interaction with Glu-38, its pKa of 6.5 would be affected. The lack of any detectable shift indicates that any Coulomb interaction between these two carboxylic groups is < 0.1 kcal/mol. Overall, the absence of any measurable impact of substitutions of Leu-38 on the pKa values of surface residues corroborates the results from spectroscopic experiments showing that the structure of the variants are very similar if not identical to the structures of the background protein.
To probe interactions between Asp-38 and Glu-38 with neighboring ionizable residues directly, five double mutants were made: L38E/D77N, L38E/E122Q, L38E/E122D, L38E/R126Q, and L38D/E122Q. The pKa value of the internal Glu-38 or Asp-38 was measured by global fit of the pH dependence of 1HN chemical shifts measured with NMR spectroscopy. The measured pKa values are listed in Table 4. These pKa values can be readily converted to apparent ΔGij values by multiplying the difference in pKa between the background protein and the variants with neutral substitutions by RTln(10). To measure the interaction between Glu-38 and Asp-122, the ΔpKa was measured using the L38E/E122Q variant as a background. The observed ΔGij values were strongly distance-dependent and ranged from 0 to 1.5 kcal/mol (Table 5).
Table 4. pKa values of Asp-38 and Glu-38 measured using global fit to 1HN chemical shift data.
| variant | residue | n | pKa* | ΔpKa |
|---|---|---|---|---|
| Δ+PHS/L38E | Glu-38 | 0.8 | 7.0 | 0.0 |
| Δ+PHS/L38E/D77N | Glu-38 | 0.7 | 6.1 | -0.9 |
| Δ+PHS/L38E/E122Q | Glu-38 | 0.9 | 6.2 | -0.8 |
| Δ+PHS/L38E/E122D | Glu-38 | 0.8 | 7.4 | 0.4 |
| Δ+PHS/L38E/R126Q | Glu-38 | 0.9 | 7.2 | 0.2 |
| Δ+PHS/L38D | Asp-38 | 0.9 | 7.2 | 0.0 |
| Δ+PHS/L38D/E122Q | Asp-38 | 1.0 | 6.6 | -0.6 |
uncertainty in pKa value is +/- 0.1
Table 5. Apparent Coulomb interactions between Glu-38 and other ionizable residues.
| interaction | ΔGij * (kcal/mol) |
rij (Å) |
interaction | ΔGij* (kcal/mol) |
rij (Å) |
|---|---|---|---|---|---|
| Glu-38/Asp-122‡ | 1.5 | 3.6† | --- | --- | -- |
| Glu-38/Asp-77 | 1.2 | 4.0 | --- | --- | --- |
| Glu-38/Glu-122 | 1.1 | 5.1 | Asp-38/Glu-122 | 0.8 | 5.3† |
| Glu-38/Arg-126 | -0.3 | 6.6 | --- | --- | --- |
| Glu-38/Asp-21 | 0.0 | 13.1 | --- | --- | --- |
| Glu-38/His-8 | 0.0 | 15.2 | Asp-38/His-8 | 0.0 | 18.9† |
uncertainty is 0.1 kcal/mol
distance estimated using structural model
ΔpKa estimated using L38E/E122Q variant
Unlike Asp-38 or Glu-38, which are neutral at low pH, Lys-38 is ionized below pH 10. Any Coulomb interaction with Lys-38 should therefore alter the pKa values of surface carboxylic groups. In particular, Lys-38 is involved in a 2.7 Å ion pair with Glu-122 in the crystal structure of the L38K variant. A conservative estimate of the possible pKa shift can be made using Coulomb's law using the dielectric constant of pure water. This indicates that an interaction of 1.5 kcal/mol between Lys-38 and Glu-122 is possible. This would shift the pKa of Glu-122 by 1.1 units. The measured pKa values of the carboxylic groups in the Δ+PHS/L38K variant are shown in Table 3. Surprisingly, no shifts in pKa value were observed. This experiment demonstrates conclusively that the ion pair between Lys-38 and Glu-122 observed in the crystal structure is not present in solution, and that Lys-38 does not interact with any carboxylic acid residues in the protein. This behavior is fully consistent with a structural rearrangement leading to hydration of the charged side chain of Lys-38.
Structure-based pKa calculations
Reproducing the shifts in pKa values in multiple sites in a protein is still a difficult challenge for structure-based electrostatics calculations. To test the ability of computational methods to calculate Coulomb interactions accurately, five computational methods were used to calculate the pKa of Glu-38 in the L38E, L38E/D77N, L38E/E122Q, L38E/E122D, and L38E/R126Q variants. The pKa values of Asp-38 (in the L38D variant), His-8, and His-121 were also calculated. Overall, the calculations attempted to reproduce 12 unique pKa values: Glu-38 in the five variants listed above (Table 4), Asp-38 in the L38D variant (Table 4), and His-8 and His-121 in the L38E, L38E/E122Q, and L38D variants (Table 2). A variety of different computational methods were tested: PROPKA,51 the single site (S/FDPB) and full-site PARSE (F/FDPB) implementation of finite difference Poisson-Boltzmann electrostatics,52-54 the pH-adapted conformer FDPB method (PAC),55; 56 and multi-conformer continuum electrostatics (MCCE).57; 58
PROPKA uses a set of empirical rules to estimate pKa values from structure. The pKa of Glu-38 calculated with PROPKA was 5.3. The group was classified as buried and experienced an unfavorable dehydration energy (2.1 kcal/mol) offset by favorable polar interactions with the Tyr-91 side chain and His-121/Glu-122 backbone (-1.3 kcal/mol). No Coulomb interactions were calculated because PROPKA only treats Coulomb interactions between two residues if they are both classified as buried and are within 7 Å of one another. Only His-121 meets both criteria, but no interaction energy was calculated because the calculated pKa of His-121 (5.1) is below that of Glu-38; the PROPKA algorithm excludes possible interaction because the groups are never charged simultaneously. The lack of explicit Coulomb interactions causes PROPKA to be entirely insensitive to substitution of residues at positions 77, 122, or 126 with uncharged analogs. The overall RMS error for the calculated versus experimental pKa values was 1.0 (Table 6).
Table 6. Comparison of calculated and measured pKa values.
| Experimental | FDPB | ||||||
|---|---|---|---|---|---|---|---|
| residue | variant | By NMR | PROPKA | S | F | PAC | MCCE |
| L38E | 7.0 | 5.3 | 7.0 | 7.1 | 4.9 | 8.5 | |
| L38E/D77N | 6.1 | 5.3 | 5.9 | 5.7 | 0.0 | 5.4 | |
| Glu-38 | L38E/E122Q | 6.2 | 5.3 | 5.6 | 6.0 | 2.7 | 7.2 |
| L38E/E122D | 7.4 | 5.2 | 8.2 | 7.5 | 6.5 | 9.0 | |
| L38E/R126Q | 7.2 | 5.2 | 7.5 | 8.1 | 5.3 | 9.0 | |
| Asp-38 | L38D | 7.2 | 5.0 | 6.7 | 7.4 | 16.2 | 8.7 |
| L38E | 6.5 | 6.5 | 6.8 | 6.8 | 6.5 | 6.7 | |
| His-8 | L38E/E122Q | 6.4 | 6.5 | 6.7 | 6.8 | 6.6 | 6.7 |
| L38D | 6.5 | 6.5 | 6.7 | 6.8 | 6.6 | 6.7 | |
| L38E | 5.7 | 5.1 | 8.0 | 10.2 | 8.3 | 7.6 | |
| His-121 | L38E/E122Q | 5.7 | 5.1 | 8.0 | 10.0 | 8.1 | 7.7 |
| L38D | 5.7 | 5.3 | 7.9 | 9.6 | 5.9 | 7.7 | |
| RMS | --- | 1.0 | 1.2 | 2.1 | 3.4 | 1.4 | |
The results of FDPB calculations were highly dependent on the choice of protein dielectric constant. Using S/FDPB, the calculated pKa of Glu-38 ranged from 19.2 when εp = 4, to 5.4 when εp = 20. Likewise, the pKa of Glu-38 calculated using F/FDPB ranges from 19.2 when εp = 4, to 5.2 when εp = 20. The pKa of 7.0 of Glu-38 was reproduced by dielectric constants of 12 and 11 with the S/FDPB and F/FDPB methods, respectively. The dielectric constant that reproduced the experimental pKa value (εapp) was used for all further calculations. In both types of calculations, the pKa at εapp was governed by unfavorable dehydration energy (4 kcal/mol), net favorable polar interactions (-3 kcal/mol), and net unfavorable Coulomb interactions (2.5 kcal/mol). The overall RMS for all 13 measured pKa values was 1.2 and 2.1 for S/FDPB and F/FDPB respectively (Table 6). The relatively large RMS value is due to a large error in the calculated pKa value of His-121 (8.0 and 10.2). If this is excluded from the RMS calculation, both methods have RMS errors of 0.4. S/FDPB overestimated the apparent Coulomb interactions by 75 % and F/FDPB by 25 %.
The PAC method generates ensembles of possible side chain positions at pH extremes, calculates electrostatic potentials of each configuration using FDPB, then Boltzmann weights these ensembles as a function of pH. To test the best-case scenario for PAC calculations, a variety of parameter combinations were used to maximize agreement with experiment. For PAC, it was found that minimizing side chain positions was the most important user-adjustable parameter, lowering the RMS error by 20 %. Even so, the calculations performed poorly. The calculated pKa of Glu-38 was 4.9 and the overall RMS error the pKa values was 3.4 pH units (Table 6). On average, the apparent coulomb interactions calculated using this method were 3.7 times higher than experiment.
MCCE couples conventional FDPB calculations to side chain rotamer sampling with a Monte Carlo method. MCCE does not allow for arbitrary adjustment of the protein dielectric constant without extensive reparameterization; therefore, these calculations used the default εp values of 4 or 8. The calculations with εp = 4 failed to converge, giving pKa values for Glu-38 that were > 14. Only calculations using εp = 8 are reported here. The calculated pKa value of Glu-38 was 8.5 and the overall RMS error was 1.4 pH units (Table 6). The apparent Coulomb interactions reported by MCCE were, on average, twice as strong as observed experimentally. A correlation plot of the experimental and calculated apparent Coulomb interactions for each calculation type is shown in Supplementary Figure 1.
Discussion
Difference in ionization properties of Lys, Glu, and Asp at position 38
Previous experimental and computational studies with the L38K variant suggested that the pKa of Lys-38 was governed by structural reorganization and water penetration, with little or no contribution from the buried ion pair between Lys-38 and Glu-122 observed in the crystal structure.39 We have now confirmed this with NMR spectroscopy. The HSQC spectrum of this variant shows no peaks for residues 113-123 at any pH value studied. Although chemical shifts are sensitive to factors other than structural relaxation, the data are consistent with reorganization of this segment of the protein and exchange with solvent. The shift in the pKa of His-121 from a depressed value towards a more normal value is opposite from the shift expected from Coulomb interactions between His-121 and Lys-38. However, it is fully consistent with enhanced hydration of His-121 owing to structural relaxation in this region of the protein. Finally, Lys-38 does not shift the pKa value of any acidic residue in the protein. This is particularly striking for Glu-122, which is part of a 2.7 Å ion pair in the crystal structure of the L38K variant, but apparently insensitive to the charge in Lys-38. This can only be explained if Lys-38 is fully hydrated when charged, and if the structure of the L38K variant in solution is different than the crystal structure.
In contrast to the behavior of Lys-38, the pKa values of Glu-38 and Asp-38 are higher than normal pKa values by 2.6 and 3.2 pH units, respectively. These two carboxylic groups experience substantial Coulomb interactions with multiple neighboring groups. There are several possible explanations for this difference in behavior between Lys-38 and the carboxylic groups at this same position. The first is proximity to solvent. Although Lys-38 and Glu-38 are internal in the crystal structures of the PHS/L38K and PHS/L38E variants, the side chains point towards the protein-water interface and they are much closer to bulk water than the internal ionizable groups of acidic and basic residues at internal positions 66 and 92 studied previously.15-17; 21-23; 59 The shorter side chain of Glu-38 is further from bulk water than the longer Lys-38 side chain, and therefore will experience the reaction field of bulk water more weakly. This would contribute to the shift in its pKa. Consistent with expectation, the shifts in pKa correlate inversely with side chain length. We also suspect that the short length of the side chain of Asp-38 is responsible for its not being fully dehydrated. A model of the L38D variant made from the crystal structure of the PHS/L38E variant (see Methods) showed that Asp-38 forms a single hydrogen bond with its own backbone, but is too short to reach any other hydrogen bonding partners without substantial structural rearrangement. Water molecules have been found in association with the carboxylic oxygen atoms of internal Asp and Glu in all previous structures of SNase variants with internal Glu or Asp.16; 42 To examine the possibility that internal water molecules can satisfy the hydrogen bonding potential of the side chain of Asp-38, the DOWSER algorithm60, which places internal water molecules in polar cavities in crystal structures, was applied to the model of the L38D variant. DOWSER identified two potential water molecules in the region between Asp-38 and the 113-119 loop, which make hydrogen bonds with Asp-38 thereby linking this residue to the backbones of Asn-118 and Glu-122.
The difference in the pKa of Lys-38 versus Glu-38 or Asp-38 is also consistent with the ability of carboxylic residues to form better hydrogen bonds than internal Lys residues. In the crystal structure, the amino group of Lys-38 makes an ion pair with the carboxylic side chain of Glu-122, and one bond to the backbone of Ala-112. In contrast, Glu-38 forms hydrogen bonds to the side chain of Tyr-91 and to the backbone amides of Thr-120 and His-121. If the predictions with the DOWSER method are correct, Asp-38 forms a hydrogen bond to its own backbone and two hydrogen bonds to internal water molecules. The polar network that Asp-38 and Glu-38 establish might rigidify this part of the protein and thereby prevent these groups from being solvated through water penetration. This would contribute to the shift in pKa values in the direction that was observed.
The differences in the properties of Lys-38 and Glu-38 or Asp-38 probably also reflect differences in the pH dependence of stability of the different variant proteins. The stability of the L38K variant near pH 10, where Lys normally titrates, is lower than the stability of the L38D and L38E variants near pH 4, where Asp and Glu normally titrate. At pH 10, the local stability of the loop comprised of residues 113-119 does not appear to be sufficiently high to maintain Lys-38 buried in the neutral state. Instead, the loop appears to adopt alternative, more open conformations that allow Lys-38 to be charged, hydrated, and probably only partially buried. We showed previously that a subtle structural reorganization is sufficient to promote water penetration and solvation of the charged side chain of Lys-38.39 Near pH 4, the local stability of the 113-119 loop is sufficiently high to maintain Glu-38 and Asp-38 buried in the neutral state; therefore, the pKa values of these internal groups are shifted upwards. Because the stability of the L38E and L38D variants decrease with increasing pH, this loop might shift towards a more open, alternative conformation when Asp-38 and Glu-38 titrate near pH 7. This is analogous to what was observed in the L38K variant. Consistent with this hypothesis, several resonances in the HSQC spectra of the L38E variant corresponding to this loop disappear when Glu-38 ionizes. The disappearance of these peaks is somewhat difficult to interpret; it could reflect structural reorganization but it could also be due by the electrostatic effects of the charge of Glu-38 on the amides.50
Coulomb interactions between internal and surface ionizable residues
How do the observed Coulomb interaction energies between internal and surface groups compare to previously measured interactions between surface ionizable moieties? The distance-dependence of ΔGij between internal and surface charges is plotted in Figure 6 and compared to previously measured long-range interactions between surface charges.30; 31 A single ΔGij vs rij plot is sufficient to describe all data. This indicates that the interactions between internal and surface groups are as weak as those between surface groups, at least for these internal groups, which are close to the protein-water interface. The effective dielectric constant obtained by fitting Coulomb's law with a Debye-Hückel parameter to the ΔGij obtained between Glu-38 and nearby surface residues is 35 +/- 3, which is almost as high as the value of 46 +/- 8 obtained by including ΔGij from interactions between surface groups, measured previously.30; 31 The high effective dielectric constant is fully consistent with the high local polarity in the microenvironment of Glu-38 and subtle structural reorganization coupled to its ionization.61
Figure 6.
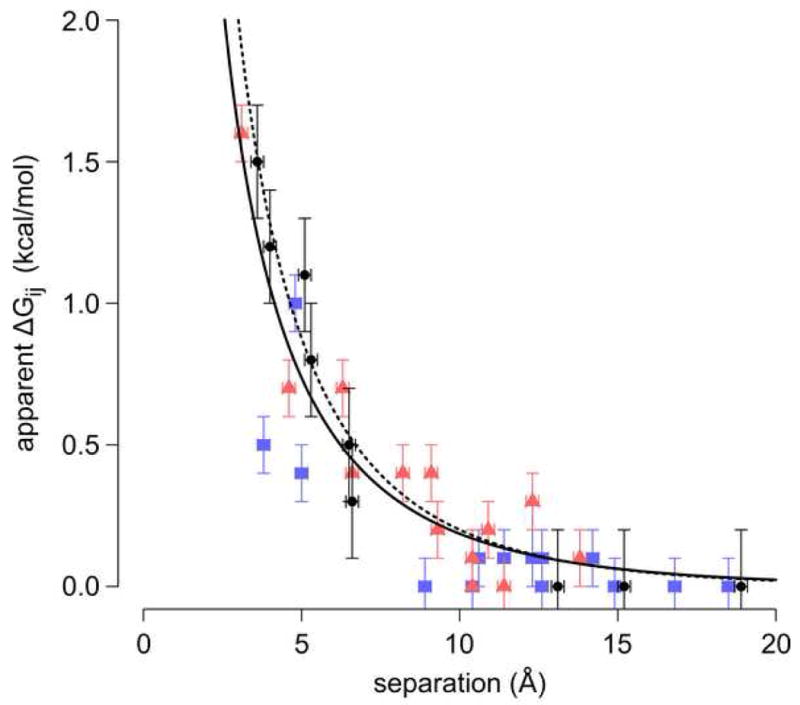
Distance dependence of apparent Coulomb interactions for the current study (•), and for data published previously by Lee et al. (
 ),30 and Baran et al. (
),30 and Baran et al. (
 ).31 The dashed line is a fit to the data from the current study only, the solid line a fit to all available data.
).31 The dashed line is a fit to the data from the current study only, the solid line a fit to all available data.
Another interesting feature of the measured Coulomb interactions is the asymmetry of the Glu-38/Asp-122 and Asp-38/Glu-122 interactions. The apparent Coulomb interaction of Glu-38/Asp-122 is 1.5 kcal/mol, whereas the apparent Coulomb interaction of Asp-38/Glu-122 is 0.8 kcal/mol. This is surprising: how could simply reversing the placement of the charges significantly alter the observed energy of interaction? Simple structural models of the L38D and L38E/E122D variants suggest an explanation. In these models, the nearest side chain oxygen atoms of the Glu-38/Asp-122 pair were 5.3 Å apart and the Asp-38/Glu-122 atoms were 3.6 Å apart. Due to their burial, neither Asp-38 nor Glu-38 can change position without a steric clash. Asp-122 and Glu-122, however, are on the surface and therefore not tethered in a fix position. Glu, with its extra methylene carbon, is simply longer and better able to respond to the repulsive interaction with Asp-38.
Implications for structure-based pKa calculations
The pKa values of internal ionizable residues are especially useful to test models for structure-based calculation of electrostatic energies and pKa values.61; 62 In general, it is not sufficient to test the validity of these methods simply by reproducing pKa values, especially when the methods include ad hoc modifications to improve their performance.52; 53; 63-65 In these cases the experimental pKa values can be reproduced accurately but for entirely incorrect physical reasons.62 The utility of the data in this study is that it contributes additional constraints, such as the magnitude of pair-wise Coulomb interactions, that must be satisfied to ensure that pKa values are reproduced for the right physical reasons. To reproduce these data, a computational method must correctly capture both the self-energy and Coulomb interaction energies experienced by ionizable residues at a single position, and it must also reproduce the differences in the ionization properties of Lys-38 and Asp-38 or Glu-38.
Our experimental data expose several weaknesses in current computational methods. For example, the attempts to reproduce the experimental pKa values with PROPKA revealed the importance of medium-range electrostatics interactions, which can be an important determinant of pKa values. These interactions are neglected in PROPKA, thus it fails to reproduce the experimental pKa values (Table 6). Inclusion of this term would likely bring the calculated pKa from 5.3 closer to the experimentally measured value. On the other hand, all of the continuum calculations overestimated the strength of Coulomb interactions, especially the PAC and MCCE methods. This reflects the fact that because the PAC and MCCE methods attempt to model side chain relaxation explicitly, lower protein dielectric constants are used. The failure of these methods demonstrates that these models either neglect important energetic terms in their Boltzmann-weighted ensembles, or that relaxation involving the protein backbone occurs on the timescale of the experiments, which is not treated by PAC or MCCE.
Conventional FDPB calculations did better than the PAC and MCCE methods, however, it is important to emphasize that in these calculations the protein dielectric constant was used as a tunable variable to maximize agreement between calculated and measured pKa values. FDPB calculations with a static structure cannot predict pKa values of internal ionizable groups. Both the S/FDPB and F/FDPB calculations slightly overestimated the strength of unfavorable Coulomb interactions. As the calculation reproduced the pKa of Glu-38, the strength of the favorable polar interactions is either overestimated, the energetic penalty of dehydration is underestimated, or both. If slight relaxation of the structure occurs in solution, the strength of Coulomb interactions, polar interactions, and the energetic penalty of dehydration would all decrease. Such a process would explain the discrepancy between the experimental and computational results.
Our study demonstrates that very different factors can govern the pKa values of different types of ionizable residues buried at the same location in a protein. Water penetration and structural reorganization are the primary determinants of the pKa of Lys-38. In contrast, favorable polar interactions and unfavorable dehydration and Coulomb interactions determine the pKa values of Asp-38 and Glu-38. Some amount of structural reorganization also influences the pKa values of Asp-38 and Glu-38, as demonstrated by the fact that relatively high protein dielectric constants were needed to reproduce their pKa values in structure-based calculations with static structures.
If structure-based electrostatics calculations are going to be useful predictors of structure/function relationships, they must be able to reproduce the pKa values of internal ionizable groups. To this end, they must be able of identify a priori the cases in which the ionization of an internal group triggers structural reorganization. They must also be able to distinguish these cases from cases in which the ionization of an internal group does not promote structural reorganization. Because the probability that a protein undergoes local structural reorganization coupled to the ionization of an internal group is governed by the free energies of the folded protein and of the low lying excited states, improved methods for structure-based pKa calculations will require methodology for improved conformational sampling and improved ability to estimate the stability of proteins.
Materials and Methods
Staphylococcal nuclease
Two hyperstable variants of SNase, known as PHS and Δ+PHS, were used for crystallography and equilibrium thermodynamic experiments, respectively. Both variants contain three substitutions: P117G, H124L and S128A. Δ+PHS nuclease has two additional mutations (G50F and V51N) and a short deletion (residues 44-49). All genes were engineered into the pET24A+ vector (Novagen, Madison, WI). Substitutions were introduced using QuikChange™ site-directed mutagenesis (Stratagene, La Jolla, CA). All proteins were expressed in BL21(DE3) cells from Stratagene, and purified using the protocol of Shortle and Meeker.66 Uniformly 13C/15N labeled protein was made by growing E. coli in minimal media with 15NH4Cl (1 g/L) and 13C6-D-glucose (4 g/L) (Isotec, Inc.) Purity was determined to be greater than 98% by SDS-PAGE. Protein concentration was determined by absorbance at 280 nm using an extinction coefficient of 0.93 mL mg-1 cm-1.67
Crystallography
PHS/L38E was crystallized in hanging drops using the vapor diffusion method. Three drops containing 4 μL: 4 μL mixture of protein and reservoir solution were suspended over 1 mL of reservoir solution and equilibrated at 4 °C. The protein solution consisted of 16.5 mg/mL protein, 2 milliequivalents of thymidine-3′,5′-diphosphate (THP), and 3 milliequivalents of CaCl2. THP was synthesized in our laboratory, as described previously.68 Reservoir solution consisted of 35 % (v/v) 2-methyl-2,4-pentanediol and 25 mM potassium phosphate at pH 7.0. Diffraction data were collected from a single crystal suspended in mother liquor in a cryoloop flash frozen in liquid nitrogen. Data were collected at 100 K using a Kappa ApexII diffractometer outfitted with a sealed copper tube, multilayer optics, and a CCD detector (Bruker/AXS, Madison, WI). Reflections were indexed and integrated using ApexII software, and merged using XPREP. Structure determination and refinement were performed using the ccp4i interface to the CCP4 program suite.69; 70 Molecular replacement was done with PHASER,71 using the PHS nuclease structure (PDB ID: 1EY8) as a search model.40 Solvent atoms were removed, Leu-38 was truncated to Ala, and all B-factors were set to 20 Å2 prior to molecular replacement. Iterative model building and refinement were performed using COOT and refmac5.72; 73 The sidechain for Glu-38 and the THP ligand were visible after the first round of refinement. Waters, phosphates, and ions were added in later rounds of refinement.
pKa values measured using pH-dependence of stability
GdnHCl denaturation experiments were done using an Aviv Automatic Titrating Flourimeter 105 (Lakewood, NJ) using approach-to-equilibrium, as described previously.20; 22; 39; 74 pKa values were extracted by the single-site linkage relationship to the pH-dependence of ΔΔG°H2O, as described previously.15; 23; 39
NMR data collection
For NMR titration experiments, a 1.4 mL sample of 1 mM protein was prepared in a solution of 100 mM KCl, 0.5 mM NaN3 and 10% D2O (v/v) by buffer exchange using Amicon Ultra-4 centrifugal filter units with a 10,000 MWCO (Millipore Corp., Billerica, MA). This sample was split into two equal fractions, one for titration with acid and one for base. Titrations were performed as described elsewhere.41
NMR experiments were performed on a Bruker Avance II-600 equipped with a cryoprobe. All experiments were conducted at a calibrated temperature of 298 K. NMR spectra were referenced against the position of the HDO peak, which was determined relative to the DSS peak at 0.00 ppm.41; 75 All spectra were processed using NMRPipe76 and peak assignments were completed using Sparky.77
Backbone assignments (15N, 1HN) for the Δ+PHS/L38E and Δ+PHS/L38K variants were collected using the 3D HNN experiment.49 The 15N carrier frequency was set to 117.0 ppm; a total of 42 complex points were collected in each of the t1 and t2 dimensions. Spectral widths of 10000 Hz, 1825 Hz and 1825 Hz were used for the 1H, 15N, and 15N dimensions, respectively.
Assignments for the Cγ and Cδ resonances of Asp and Glu residues in the Δ+PHS/L38E and Δ+PHS/L38K variants were determined primarily by transferring assignments from the CBCGCO spectra of Δ+PHS41 at pH 4.7 to the spectra of the variants. The side chain aliphatic carbon (Cα, Cβ, Cγ) assignments of Glu-38 were collected using the C-C TOCSY (CO)NH experiment.78 Through modification of the 13C-detect HBHGCBCGCO pulse sequence to include a TOCSY element, the Glu-38 Cδ resonance was determined by correlation with its side chain aliphatic carbon assignments (Castañeda and Majumdar, unpublished).
The pKa values of Asp and Glu residues were determined by monitoring the pH dependence of their Cγ and Cδ resonances, respectively, using a 13C-detect version of the 2D HBHGCBCGCO experiment (Castañeda and Majumdar, unpublished). The spectra were collected in 0.4 pH increments between pH 2 and pH 9. For indirect pKa determination of carboxylic residues using 1HN chemical shifts, a series of 15N-1H HSQC spectra were collected in a similar manner. These experiments were performed and processed as previously described.41
For pKa determination of histidine residues in the Δ+PHS variants, samples were prepared in the buffer described above except in 100% D2O. Samples were heated to 330 K for 15 minutes prior to experiments to promote exchange of amide protons. The Hε1 resonance of the imidazole ring was monitored as a function of pH using 1D 1H NMR experiments as described previously.23 The His Hε1 resonances were unambiguously assigned by comparison to the same resonances in Δ+PHS.
pKa values from pH-dependent chemical shifts
pKa values of Asp, Glu and His residues were extracted using the modified Hill equation, as described previously.41 Fitting was done using the nls function within the R 2.7.1 Statistical environment.79
A global fit to the pH-dependence of the 1H chemical shift of six amides (Thr-33, Phe-34, Arg-35, Glu-75, Gly-88, and Leu-89) was used to determine the pKa values of Glu-38 and Asp-38. Three criteria were used to select these resonances: 1) An apparent titration > 0.15 ppm in the variant spectra that was not evident in the Δ+PHS spectra; 2) The resonance could be unambiguously assigned in all variants and did not exhibit exchange over the course of the titration (Figures 2 and 4); 3) The residues were within close spatial proximity of position 38. The fit minimized the root mean squared (RMS) deviation between the calculated and measured chemical shifts:
| 1. |
where N is the number of resonances included in the fit and δi,meas is the experimentally measured chemical shift. δi,calc is described by the modified Hill equation:
| 2. |
where δi,acidic and δi,basic are the acidic and basic baselines of the apparent titration. A single pKa and Hill coefficient (n) were used for all resonances included in the fit. Minimization was done using the optim function inside the R 2.7.1 statistical environment.79 To ensure that the pKa and Hill coefficient did not depend on the resonances chosen, bootstrap sampling of resonances included in the fit was performed. The list of resonances was also expanded to include resonances that only met two of the three inclusion criteria. In all cases, the bootstrap error was less than 0.05 pH units for the pKa value. The errors reported in the paper reflect the maximum possible systematic error in pH measurement rather than the fit error.
Structural models
Models of the PHS/L38D, PHS/L38E/D77N, PHS/L38E/E122Q, PHS/L38E/E122D, and PHS/L38E/R126Q variants were made using the structure of the PHS/L38E variant as a template. The positions of the atoms of the side chain that was mutated in silico were minimized using the CHARMM22 forcefield, as described previously.23 All other atoms were held fixed.
Structure-based electrostatics calculations
PROPKA calculations were performed using PDB2PQR 1.1.2, downloaded from http://pdb2pqr.sourceforge.net/.51; 80 Conventional FDPB calculations were done using the University of Houston Brownian Dynamics (UHBD) package, version 5.1.81 FDPB calculations were done using two parameter sets: single-site (S/FDPB)52; 53 and full-site (F/FDPB) using the PARSE parameter set.54 All calculations were performed at 100 mM ionic strength as described previously.24 Multi-Conformer Continuum Electrostatics (MCCE) calculations were done using MCCE 2.2, downloaded from http://wwwsci.ccny.cuny.edu/∼mcce/.52 ; 58 The parameters distributed with the program were used for the εp = 4 and εp = 8 calculations. Calculations were done at 100 mM ionic strength. PAC calculations were done using the Karlsberg+ web interface (http://agknapp.chemie.fu-berlin.de/karlsberg/).55; 56 Side chain positions were minimized and salt bridges were randomized at high and low pH. Results were found to be insensitive to choice of solvation model. Values reported herein were calculated using the vacuum solvation model.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. Ananya Majumdar for assistance with NMR experiments and for the use of the Johns Hopkins University Biomolecular NMR Facility. We gratefully acknowledge Dr. Dan Isom and Dr. Brian Cannon for making the initial pKa measurements of Glu-38 and Asp-38. This work was supported by National Institutes of Health grant GM-065197 (BGME), and a graduate research fellowship from the National Science Foundation (MJH).
Footnotes
Accession Code: Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3D6C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? Journal of Molecular Biology. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 2.Rashin AA, Honig B. On the Environment of Ionizable Groups in Globular-Proteins. Journal of Molecular Biology. 1984;173:515–521. doi: 10.1016/0022-2836(84)90394-2. [DOI] [PubMed] [Google Scholar]
- 3.Kajander T, Kahn PC, Passila SH, Cohen DC, Lehtio L, Adolfsen W, Warwicker J, Schell U, Goldman A. Buried charged surface in proteins. Structure. 2000;8:1203–1214. doi: 10.1016/s0969-2126(00)00520-7. [DOI] [PubMed] [Google Scholar]
- 4.Harris TK, Turner GJ. Structural basis of perturbed pK(a) values of catalytic groups in enzyme active sites. Iubmb Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. Journal of Molecular Biology. 2002;324:105–21. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 6.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic basis for enzyme catalysis. Chemical Reviews. 2006;106:3210–35. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 7.Adelroth P, Ek MS, Mitchell DM, Gennis RB, Brzezinski P. Glutamate 286 in cytochrome aa(3) from Rhodobacter sphaeroides is involved in proton uptake during the reaction of the fully-reduced enzyme with dioxygen. Biochemistry. 1997;36:13824–13829. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- 8.Cutler RL, Davies AM, Creighton S, Warshel A, Moore GR, Smith M, Mauk AG. Role of arginine-38 in regulation of the cytochrome c oxidation-reduction equilibrium. Biochemistry. 1989;28:3188–97. doi: 10.1021/bi00434a012. [DOI] [PubMed] [Google Scholar]
- 9.Luecke H, Richter HT, Lanyi JK. Proton transfer pathways in bacteriorhodopsin at 2.3 Angstrom resolution. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 10.Jiang YX, Ruta V, Chen JY, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 11.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. Journal of Molecular Biology. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 12.Jeng MF, Dyson HJ. Direct measurement of the aspartic acid 26 pK(a) for reduced Escherichia coli thioredoxin by C-13 NMR. Biochemistry. 1996;35:1–6. doi: 10.1021/bi952404n. [DOI] [PubMed] [Google Scholar]
- 13.Chivers PT, Prehoda KE, Volkman BF, Kim BM, Markley JL, Raines RT. Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry. 1997;36:14985–91. doi: 10.1021/bi970071j. [DOI] [PubMed] [Google Scholar]
- 14.Kaushik JK, Iimura S, Ogasahara K, Yamagata Y, Segawa S, Yutani K. Completely buried, non-ion-paired glutamic acid contributes favorably to the conformational stability of pyrrolidone carboxyl peptidases from hyperthermophiles. Biochemistry. 2006;45:7100–12. doi: 10.1021/bi052610n. [DOI] [PubMed] [Google Scholar]
- 15.Stites WE, Gittis AG, Lattman EE, Shortle D. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. Journal of Molecular Biology. 1991;221:7–14. doi: 10.1016/0022-2836(91)80195-z. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen DM, Leila Reynald R, Gittis AG, Lattman EE. X-ray and thermodynamic studies of staphylococcal nuclease variants I92E and I92K: insights into polarity of the protein interior. Journal of Molecular Biology. 2004;341:565–74. doi: 10.1016/j.jmb.2004.05.066. [DOI] [PubMed] [Google Scholar]
- 17.Karp DA, Gittis AG, Stahley MR, Fitch CA, Stites WE, Garcia-Moreno EB. High apparent dielectric constant inside a protein reflects structural reorganization coupled to the ionization of an internal Asp. Biophysical Journal. 2007;92:2041–53. doi: 10.1529/biophysj.106.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambeir AM, Backmann J, Ruiz-Sanz J, Filimonov V, Nielsen JE, Kursula I, Norledge BV, Wierenga RK. The ionization of a buried glutamic acid is thermodynamically linked to the stability of Leishmania mexicana triose phosphate isomerase. European Journal of Biochemistry. 2000;267:2516–2524. doi: 10.1046/j.1432-1327.2000.01254.x. [DOI] [PubMed] [Google Scholar]
- 19.Giletto A, Pace CN. Buried, charged, non-ion-paired aspartic acid 76 contributes favorably to the conformational stability of ribonuclease T1. Biochemistry. 1999;38:13379–84. doi: 10.1021/bi991422s. [DOI] [PubMed] [Google Scholar]
- 20.García-Moreno B, Dwyer JJ, Gittis AG, Lattman EE, Spencer DS, Stites WE. Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophysical Chemistry. 1997;64:211–24. doi: 10.1016/s0301-4622(96)02238-7. [DOI] [PubMed] [Google Scholar]
- 21.García-Moreno B, Dwyer JJ, Gittis AG, Lattman EE, Spencer DS, Stites WE. Solvent penetration may be responsible for the high dielectric constant inside a protein. Biophysical Journal. 1998;74:A132–A132. [Google Scholar]
- 22.Dwyer JJ, Gittis AG, Karp DA, Lattman EE, Spencer DS, Stites WE, Garcia-Moreno B. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophysical Journal. 2000;79:1610–1620. doi: 10.1016/S0006-3495(00)76411-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitch CA, Karp DA, Lee KK, Stites WE, Lattman EE, García-Moreno EB. Experimental pK(a) values of buried residues: analysis with continuum methods and role of water penetration. Biophysical Journal. 2002;82:3289–304. doi: 10.1016/s0006-3495(02)75670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitch CA, Whitten ST, Hilser VJ, García-Moreno EB. Molecular mechanisms of pH-driven conformational transitions of proteins: insights from continuum electrostatics calculations of acid unfolding. Proteins: Structure, Function, and Bioinformatics. 2006;63:113–26. doi: 10.1002/prot.20797. [DOI] [PubMed] [Google Scholar]
- 25.Isom DG. Proc Natl Acad Sci U S A 2009 [Google Scholar]
- 26.Harms MJ, Schlessman JL, Sue GR, Garcia-Moreno EB. Unusual ability of internal arginine residues to solvate their charged sidechain. In preparation. [Google Scholar]
- 27.Daopin S, Sauer U, Nicholson H, Matthews BW. Contributions of Engineered Surface Salt Bridges to the Stability of T4 Lysozyme Determined by Directed Mutagenesis. Biochemistry. 1991;30:7142–7153. doi: 10.1021/bi00243a015. [DOI] [PubMed] [Google Scholar]
- 28.Daopin S, Soderlind E, Baase WA, Wozniak JA, Sauer U, Matthews BW. Cumulative Site-Directed Charge-Change Replacements in Bacteriophage-T4 Lysozyme Suggest That Long-Range Electrostatic Interactions Contribute Little to Protein Stability. Journal of Molecular Biology. 1991;221:873–887. doi: 10.1016/0022-2836(91)80181-s. [DOI] [PubMed] [Google Scholar]
- 29.Meeker AK, García-Moreno B, Shortle D. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry. 1996;35:6443–6449. doi: 10.1021/bi960171+. [DOI] [PubMed] [Google Scholar]
- 30.Lee KK, Fitch CA, Garcia-Moreno B. Distance dependence and salt sensitivity of pairwise, coulombic interactions in a protein. Protein Science. 2002;11:1004–1016. doi: 10.1110/ps.4700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baran KL, Chimenti MS, Schlessman JL, Fitch CA, Herbst KJ, Garcia-Moreno BE. Electrostatic effects in a network of polar and ionizable groups in staphylococcal nuclease. Journal of Molecular Biology. 2008;379:1045–62. doi: 10.1016/j.jmb.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Tissot AC, Vuilleumier S, Fersht AR. Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry. 1996;35:6786–6794. doi: 10.1021/bi952930e. [DOI] [PubMed] [Google Scholar]
- 33.Fisher BM, Schultz LW, Raines RT. Coulombic effects of remote subsites on the active site of ribonuclease A. Biochemistry. 1998;37:17386–17401. doi: 10.1021/bi981369s. [DOI] [PubMed] [Google Scholar]
- 34.de Kreij A, van den Burg B, Venema G, Vriend G, Eijsink VGH, Nielsen JE. The effects of modifying the surface charge on the catalytic activity of a thermolysin-like protease. Journal of Biological Chemistry. 2002;277:15432–15438. doi: 10.1074/jbc.M200807200. [DOI] [PubMed] [Google Scholar]
- 35.Jackson SE, Fersht AR. Contribution of Long-Range Electrostatic Interactions to the Stabilization of the Catalytic Transition-State of the Serine-Protease Subtilisin Bpn'. Biochemistry. 1993;32:13909–13916. doi: 10.1021/bi00213a021. [DOI] [PubMed] [Google Scholar]
- 36.Grimsley GR, Shaw KL, Fee LR, Alston RW, Huyghues-Despointes BMP, Thurlkill RL, Scholtz JM, Pace CN. Increasing protein stability by altering long-range coulombic interactions. Protein Science. 1999;8:1843–1849. doi: 10.1110/ps.8.9.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, 3rd, Hilvert D, Houk KN, Stoddard BL, Baker D. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–91. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isom DG, Cannon BR, Castaneda CA, Robinson A, Garcia-Moreno B. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17784–8. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harms MJ, Schlessman JL, Chimenti MS, Sue GR, Damjanovic A, Garcia-Moreno B. A buried lysine that titrates with a normal pKa: role of conformational flexibility at the protein-water interface as a determinant of pKa values. Protein Science. 2008;17:833–45. doi: 10.1110/ps.073397708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J, Lu Z, Sakon J, Stites WE. Increasing the thermostability of staphylococcal nuclease: implications for the origin of protein thermostability. Journal of Molecular Biology. 2000;303:125–30. doi: 10.1006/jmbi.2000.4140. [DOI] [PubMed] [Google Scholar]
- 41.Castaneda C, Fitch CA, Khangulov V, Schlessman JL, Majumdar A, Garcia-Moreno EB. Molecular Determinants of the pKa Values of Asp and Glu Residues in Staphylococcal Nuclease. Proteins: Structure, Function, and Bioinformatics. doi: 10.1002/prot.22470. Submitted. [DOI] [PubMed] [Google Scholar]
- 42.Schlessman JL, Abe C, Gittis A, Karp DA, Dolan MA, Garcia-Moreno BE. Crystallographic study of hydration of an internal cavity in engineered proteins with buried polar or ionizable groups. Biophysical Journal. 2008;94:3208–3216. doi: 10.1529/biophysj.107.122473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leung KW, Liaw YC, Chan SC, Lo HY, Musayev FN, Chen JZW, Fang HJ, Chen HM. Significance of local electrostatic interactions in staphylococcal nuclease studied by site-directed mutagenesis. Journal of Biological Chemistry. 2001;276:46039–46045. doi: 10.1074/jbc.M106620200. [DOI] [PubMed] [Google Scholar]
- 44.Chen HM, Chan SC, Leung KW, Wu JM, Fang HJ, Tsong TY. Local stability identification and the role of key acidic amino acid residues in staphylococcal nuclease unfolding. Febs Journal. 2005;272:3967–3974. doi: 10.1111/j.1742-4658.2005.04816.x. [DOI] [PubMed] [Google Scholar]
- 45.Wyman J., Jr Linked Functions and Reciprocal Effects in Hemoglobin: A Second Look. Advances in Protein Chemistry. 1964;19:223–86. doi: 10.1016/s0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 46.Sorensen MD, Led JJ. Structural Details of Asp(B9) Human Insulin at Low Ph from 2-Dimensional Nmr Titration Studies. Biochemistry. 1994;33:13727–13733. doi: 10.1021/bi00250a025. [DOI] [PubMed] [Google Scholar]
- 47.Ionescu RM, Eftink MR. Global analysis of the acid-induced and urea-induced unfolding of staphylococcal nuclease and two of its variants. Biochemistry. 1997;36:1129–1140. doi: 10.1021/bi9609681. [DOI] [PubMed] [Google Scholar]
- 48.Hirano S, Kamikubo H, Yamazaki Y, Kataoka M. Elucidation of information encoded in tryptophan 140 of staphylococcal nuclease. Proteins: Structure, Function, and Bioinformatics. 2005;58:271–7. doi: 10.1002/prot.20333. [DOI] [PubMed] [Google Scholar]
- 49.Panchal SC, Bhavesh NS, Hosur RV. Improved 3D triple resonance experiments, HNN and HN(C)N, for H-N and N-15 sequential correlations in (C-13, N-15) labeled proteins: Application to unfolded proteins. Journal of Biomolecular Nmr. 2001;20:135–147. doi: 10.1023/a:1011239023422. [DOI] [PubMed] [Google Scholar]
- 50.Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Nuclear Magnetic Resonance of Biologica Macromolecules, Pt A. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 51.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Structure, Function, and Bioinformatics. 2005;61:704–21. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 52.Antosiewicz J, Mccammon JA, Gilson MK. Prediction of pH-Dependent Properties of Proteins. Journal of Molecular Biology. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 53.Antosiewicz J, McCammon JA, Gilson MK. The determinants of pK(a)s in proteins. Biochemistry. 1996;35:7819–7833. doi: 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 54.Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free-Energies Using Macroscopic Solvent Models. Journal of Physical Chemistry. 1994;98:1978–1988. [Google Scholar]
- 55.Kieseritzky G, Knapp EW. Optimizing pK(A) computation in proteins with pH adapted conformations. Proteins: Structure Function and Bioinformatics. 2008;71:1335–1348. doi: 10.1002/prot.21820. [DOI] [PubMed] [Google Scholar]
- 56.Rabenstein B, Knapp EW. Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophysical Journal. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophysical Journal. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophysical Journal. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Damjanovic A, Wu XW, Brooks B, Garcia-Moreno EB. Backbone relaxation triggered by the ionization of internal groups in proteins: A Self-Guided Langevin Dynamics study. Biophysical Journal. 2007:193a–193a. doi: 10.1529/biophysj.108.130906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins: Structure Function and Genetics. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 61.Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 62.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins: Structure, Function, and Bioinformatics. 2001;44:400–17. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 63.Matthew JB, Gurd FRN, García-moreno EB, Flanagan MA, March KL, Shire SJ. Ph-Dependent Processes in Proteins. CRC Critical Reviews in Biochemistry. 1985;18:91–197. doi: 10.3109/10409238509085133. [DOI] [PubMed] [Google Scholar]
- 64.Vijayakumar M, Zhou HX. Salt bridges stabilize the folded structure of barnase. Journal of Physical Chemistry B. 2001;105:7334–7340. [Google Scholar]
- 65.van Vlijmen HW, Schaefer M, Karplus M. Improving the accuracy of protein pKa calculations: conformational averaging versus the average structure. Proteins: Structure, Function, and Genetics. 1998;33:145–58. doi: 10.1002/(sici)1097-0134(19981101)33:2<145::aid-prot1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 66.Shortle D, Meeker AK. Residual structure in large fragments of staphylococcal nuclease: effects of amino acid substitutions. Biochemistry. 1989;28:936–44. doi: 10.1021/bi00429a003. [DOI] [PubMed] [Google Scholar]
- 67.Fuchs S, Cuatreca P, Anfinsen CB. An Improved Method for Purification of Staphylococcal Nuclease. Journal of Biological Chemistry. 1967;242:4768–&. [PubMed] [Google Scholar]
- 68.Tener GM. 2-Cyanoethyl Phosphate and Its Use in Synthesis of Phosphate Esters. Journal of the American Chemical Society. 1961;83:159–&. [Google Scholar]
- 69.The CCP4 suite: programs for protein crystallography. Acta Crystallographica Section D: Biological Crystallography. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 70.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallographica Section D: Biological Crystallography. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 71.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallographica Section D: Biological Crystallography. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 72.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 73.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D: Biological Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 74.Whitten ST, García-Moreno EB. pH dependence of stability of staphylococcal nuclease: evidence of substantial electrostatic interactions in the denatured state. Biochemistry. 2000;39:14292–304. doi: 10.1021/bi001015c. [DOI] [PubMed] [Google Scholar]
- 75.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. H-1, C-13 and N-15 Chemical-Shift Referencing in Biomolecular Nmr. Journal of Biomolecular NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 76.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 77.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 78.Grzesiek S, Anglister J, Bax A. Correlation of Backbone Amide and Aliphatic Side-Chain Resonances in C-13/N-15-Enriched Proteins by Isotropic Mixing of C-13 Magnetization. Journal of Magnetic Resonance Series B. 1993;101:114–119. [Google Scholar]
- 79.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]
- 80.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–7. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davis ME, Madura JD, Luty BA, Mccammon JA. Electrostatics and Diffusion of Molecules in Solution - Simulations with the University-of-Houston-Brownian Dynamics Program. Computer Physics Communications. 1991;62:187–197. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.