

Abstract
Understanding the mechanisms that control processing of the amyloid precursor protein (APP) to produce amyloid-β (Aβ) peptide represents a key area of Alzheimer's disease research. Here, we show that siRNA-mediated loss of calsyntenin-1 in cultured neurons alters APP processing to increase production of Aβ. We also show that calsyntenin-1 is reduced in Alzheimer's disease brains and that the extent of this reduction correlates with increased Aβ levels. Calsyntenin-1 is a ligand for kinesin-1 light chains and APP is transported through axons on kinesin-1 molecular motors. Defects in axonal transport are an early pathological feature in Alzheimer's disease and defective APP transport is known to increase Aβ production. We show that calsyntenin-1 and APP are co-transported through axons and that siRNA-induced loss of calsyntenin-1 markedly disrupts axonal transport of APP. Thus, perturbation to axonal transport of APP on calsyntenin-1 containing carriers induces alterations to APP processing that increase production of Aβ. Together, our findings suggest that disruption of calsyntenin-1-associated axonal transport of APP is a pathogenic mechanism in Alzheimer's disease.
INTRODUCTION
Deposition of amyloid-β (Aβ) within amyloid plaques is a hallmark pathology of Alzheimer's disease. Aβ is an approximate 40 amino acid peptide that is derived by proteolytic cleavage from amyloid precursor protein (APP). Processing of APP to produce Aβ involves cleavage by β-site APP cleaving enzyme-1 (BACE1) and γ-secretase that process APP at the N- and C-termini, respectively, of the Aβ sequence. In addition, APP can be cleaved by α- and γ-secretases and this precludes Aβ production since α-secretase cleaves APP within the Aβ sequence (1).
A large body of evidence suggests that altered production of Aβ is a major pathogenic event in Alzheimer's disease (2). Indeed, some familial forms of Alzheimer's disease are caused by mutations in the APP gene and several of these mutations alter processing of APP and production of Aβ (2). Understanding the molecular mechanisms that control APP processing thus represents a key area of Alzheimer's disease research. Alterations to APP trafficking are acknowledged to be one mechanism for modulating APP processing and Aβ production (3).
Neurons are especially dependent upon correct protein and organelle trafficking since they are polarized with axons and dendrites, and also because transport through axons can involve cargo movement over exceptionally long distances. Moreover, a large body of evidence now implicates defective axonal transport in Alzheimer's disease (reviewed in 4–6). Within neurons, APP is synthesized in cell bodies and then undergoes anterograde axonal transport on kinesin-1 molecular motors (7,8). Most functional kinesin-1 comprises a heterotetramer of two kinesin-1 motor proteins and two kinesin-1 light chains (KLCs). Kinesin-1 contains ATPase activity and uses the chemical energy of ATP to drive conformational changes that generate motile force; in contrast, the KLCs are mainly involved in binding of cargoes (9).
The precise mechanisms by which APP attaches to and is transported by kinesin-1 are not properly understood (reviewed in 10). However, there is evidence that one route may involve calsyntenin-1 (also known as alcadein-α) (11,12). Calsyntenin-1 is a neuronally enriched type-1 membrane-spanning protein that binds directly to KLCs via its intracellular C-terminal domain (13–16). As such, calsyntenin-1 acts as a ligand to mediate transport of a subset of vesicles through axons on kinesin-1 motors. A proportion of APP and calsyntenin-1 co-localize in cells and tissues, and proteomic studies have shown that some calsyntenin-1 containing vesicles also contain APP (11,12). However, such studies are correlational and do not formally demonstrate that calsyntenin-1 is required for movement of APP in neurons. Indeed, other studies suggest that calsyntenin-1 does not normally mediate axonal transport of APP (14). Likewise, the effect of calsyntenin-1 on APP processing and Aβ production is unclear. Some studies indicate that the loss of calsyntenin-1 promotes APP processing, whereas others indicate that overexpression of calsyntenin-1 increases Aβ production (12,14). Notably, the effect of loss of calsyntenin-1 on Aβ production in neurons has not been reported.
To obtain formal evidence on the role of calsyntenin-1 in transport and processing of APP, we monitored how siRNA loss of calsyntenin-1 influenced APP axonal transport and production of endogenous Aβ in living neurons. Here, we show that APP and calsyntenin-1 are co-transported through axons, that the loss of calsyntenin-1 disrupts axonal transport of APP and that calsyntenin-1 loss also leads to altered APP processing and increased production of Aβ. Finally, we demonstrate that calsyntenin-1 expression is reduced and negatively correlates with Aβ burden in Alzheimer's disease brains. Thus, altered axonal transport of APP on calsyntenin-1 carriers may be mechanistic in Alzheimer's disease.
RESULTS
siRNA-mediated loss of calsyntenin-1 increases Aβ production in neurons
We first tested the role of calsyntenin-1 on production of Aβ. To do so, we downregulated calsyntenin-1 expression in rat cortical neurons using siRNAs and monitored production of endogenous Aβ(1–40) and Aβ(1–42) species using ELISAs. Two different siRNAs and a Smartpool mix of four siRNAs all reduced calsyntenin-1 expression to lower than 5% of that seen in control cells (Fig. 1A). Aβ ELISAs revealed that this reduction in calsyntenin-1 expression led to a significant approximate 1.65-fold increase in secretion of both Aβ(1–40) and Aβ(1–42) (Fig. 1B). Since all three siRNAs produced highly similar effects on calsyntenin-1 expression and Aβ production, the Smartpool mix was used in all later experiments.
Figure 1.

siRNA knockdown of calsyntenin-1 increases production of Aβ. (A) Knockdown of calsyntenin-1 in rat cortical neurons. Neurons were treated with control siRNA (Ctrl) or with two different calsyntenin-1 siRNAs (Cstn#1; Cstn#2) or with a pool of four different calsyntenin-1 siRNAs (Cstn-pl). Samples were then probed on immunoblots for calsyntenin-1 (Cstn) or actin as a loading control. Molecular mass markers are shown. (B) ELISA assays for Aβ(1–40) and Aβ(1–42) in conditioned media from rat cortical neurons treated with control siRNA (Ctrl) or with two different calsyntenin-1 siRNAs (Cstn#1; Cstn#2) or with a pool of four different calsyntenin-1 siRNAs (Cstn-pl). Treatment with calsyntenin-1 siRNAs increase the amounts of both Aβ(1–40) and Aβ(1–42) in conditioned media. Statistical significance was determined by one-way ANOVA followed by the Bonferroni post hoc test. n = 6; error bars are SEM; **P < 0.01.
siRNA-mediated loss of calsyntenin-1 increases APP processing at the BACE1 site but does not alter the levels of BACE1, ADAM10, presenilin-1, nicastrin, full-length APP or phosphorylation of APP on threonine-668
To gain insight into the mechanisms by which the loss of calsyntenin-1 increased production of Aβ, we first monitored APP levels and APP processing at the α-secretase and BACE1 sites in neurons treated with control or calsyntenin-1 siRNAs by immunoblotting. Processing of APP by α-secretase and BACE1 generates N-terminal secreted APP fragments (sAPPα and sAPPβ) and the corresponding membrane-associated APP C-terminal domains. These APP C-terminal domains correspond to the two fragments produced by BACE1 processing (99 and 89 amino acids; APP-C99 and APP-C89) and the fragment produced by α-secretase processing (83 amino acids; APP-C83) (1). While the C-terminal APP fragments can be resolved on SDS–PAGE, analysis is complicated since in neurons, a proportion of APP is phosphorylated on threonine-668 (APP-thr668) by c-Jun N-terminal kinase-3 which alters the migration pattern of the different processed species so that some co-migrate (17,18). However, treatment of samples with λ-protein phosphatase prior to analyses facilitates interpretation of the band pattern (18).
Loss of calsyntenin-1 did not alter the total levels of full-length APP (Fig. 2A). However, analyses of APP C-terminal processed fragments (APP-CTFs) in cell lysate samples showed that the loss of calsyntenin-1 decreased the level of APP-C83 and increased the levels of both APP-C89 and APP-C99 fragments (Fig. 2A). These changes were most easily observed in the λ-protein phosphatase-treated samples (Fig. 2A). To complement these analyses, we also monitored APP processing by immunoblotting for APP-secreted fragments in conditioned media. Probing with antibody 22C11 (that detects total sAPP) and antibodies that specifically detect sAPPα and sAPPβ revealed that while the loss of calsyntenin-1 did not alter the total sAPP levels, it induced a decrease in sAPPα and a corresponding increase in sAPPβ levels (Fig. 2B).
Figure 2.

siRNA knockdown of calsyntenin-1 alters APP processing in rat cortical neurons without affecting expression of APP or APP secretase proteins. (A). Neurons were treated with control (Ctrl) or calsyntenin-1 (Cstn) siRNAs (pooled mix of four different siRNAs) and samples then probed on immunoblots for full-length APP (APP), full-length APP phosphorylated on threonine-668 (APP-p), APP C-terminal processed fragments (APP-C), APP C-terminal processed fragments phosphorylated on threonine-668 (APP-Cp) or APP C-terminal processed fragments following treatment with λ-protein phosphatase (APP-C λpp). Immunoblots showing APP C-terminal processed fragments are marked to indicate APP species generated following cleavage by BACE1 (C99 and C89) or α-secretase (C83), and their phosphorylated variants (pC99, pC89, pC83). Bar graphs show relative levels of APP C-terminal fragments (APP-C83; APP-C89 and APP-C99) in the different samples following treatment with λ-protein phosphatase. Statistical significance was determined by Student's t-test. n = 5; error bars are SEM. *P < 0.05; **P < 0.01. No significant changes in the levels of full-length APP or full-length APP phosphorylated on threonine-668 were detected in the same samples. (B) sAPP fragments in conditioned media of rat cortical neurons treated with control (Ctrl) or calsyntenin-1 (Cstn) siRNAs. Samples were probed on immunoblots for total sAPP (sAPP), sAPPα and sAPPβ. Bar graph shows relative levels of sAPPα and sAPPβ in the different samples; total sAPP levels were unchanged. Statistical significance was determined by Student's t-test. n = 4; error bars are SEM. *P < 0.05; **P < 0.01. (C) siRNA knockdown of calsyntenin-1 does not affect expression of BACE1, α- or γ-secretase proteins or X11β. Neurons were treated with control (Ctrl) or calsyntenin-1 siRNAs (Cstn) and samples (n = 4) then probed on immunoblots for BACE1, ADAM10, N- and C-terminal presenilin-1 (PS1) fragments, nicastrin and X11β as indicated. Molecular mass markers are shown.
APP-thr668 phosphorylation has been linked to axonal transport of APP and Aβ production (19–21). We therefore monitored the effect of calsyntenin-1 knockdown on APP-thr668 phosphorylation. Loss of calsyntenin-1 did not alter the levels of phosphorylated full-length APP (Fig. 2A). Moreover, changes in phosphorylation of APP-C99, APP-C89 and APP-C83 were in line with the relative changes in the total amounts of these processed fragments (Fig. 2A).
We also monitored whether the loss of calsyntenin-1 altered the levels of BACE1, the α-secretase ADAM10, the γ-secretase components presenilin-1 and nicastrin (1) and X11β by immunoblotting. X11β (also known as X11-like or munc-18 interacting protein-2) binds to both APP and calsyntenin-1 (22,23). Knockdown of calsyntenin-1 did not change the levels of any of these secretases or X11β (Fig. 2C). Thus, the increased production of Aβ associated with the loss of calsyntenin-1 is accompanied by a decrease in processing of APP at the α-secretase site and a corresponding increase in processing at the BACE1 sites. However, these changes do not involve alterations to the levels of full-length APP, APP-thr668 phosphorylation, APP secretases or X11β.
A significant proportion of APP and calsyntenin-1 co-localize in axons and APP-mCherry and enhanced green fluorescent protein-calsyntenin-1 are co-transported through living axons
Since calsyntenin-1 functions as a ligand for kinesin-1 and APP is transported on kinesin-1 motors, the effect of calsyntenin-1 loss on APP processing and Aβ production may be due to changes in axonal transport of APP. Indeed, disruption of kinesin-1-mediated APP axonal transport alters APP processing to increase Aβ production (8). To begin to test this possibility, we first monitored co-localization of APP and calsyntenin-1 in neurons. Both APP and calsyntenin-1 are enriched in the Golgi and calsyntenin-1 has been shown to mediate kinesin-1 transport of cargoes on post-Golgi carriers (12,15). In line with these findings, a proportion of calsyntenin-1 co-localized with the trans-Golgi network (TGN) marker TGN-38 and proportions of both calsyntenin-1 and APP co-localized in neuronal cell bodies (Fig. 3A). Some APP and calsyntenin-1 also co-localized in vesicular structures in axons (Fig. 3B). Quantification of this axonal co-localization revealed that 41% of APP vesicles co-localized with calsyntenin-1. These findings are in broad agreement with previous studies which showed that in sciatic nerve and in axons of cultured hippocampal neurons, ∼30–45% of APP and calsyntenin-1 co-localize (11,14). We also monitored the axonal distributions of transfected enhanced green fluorescent protein (EGFP) -calsyntenin-1 and APP-mCherry in neurons. High levels of co-localization of these transfected proteins were detected in axons (Fig. 3C).
Figure 3.
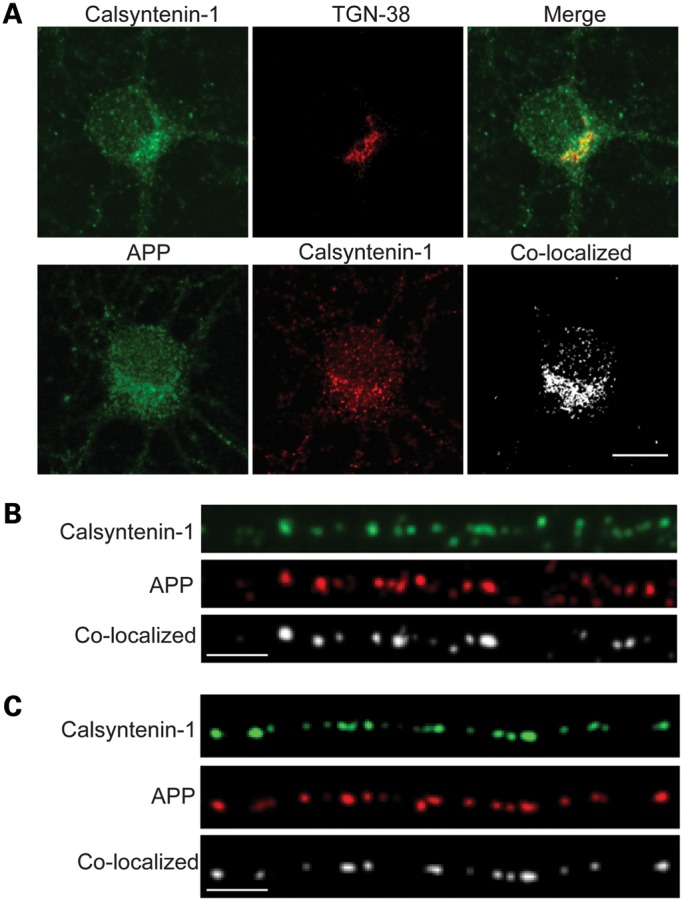
Calsyntenin-1 is present in the TGN and APP and calsyntenin-1 co-localize in cell bodies and in axons of cultured rat cortical neurons. (A and B) Neurons were co-stained for calsyntenin-1 and either TGN-38 or APP as indicated. (A) Confocal slices through cell bodies with merge (calsyntenin-1 and TGN-38) or co-localized pixels (calsyntenin-1 and APP). (B) Endogenous calsyntenin-1 and APP in axons with co-localized puncta. (C) EGFP-calsyntenin-1 and APP-mCherry in axons with co-localized puncta. Scale bars are 10 μm (A) and 5 μm (B and C).
The co-localization of APP and calsyntenin-1 in axons suggest that they are co-transported. We therefore monitored axonal transport of APP-mCherry and EGFP-calsyntenin-1 by dual imaging time-lapse microscopy in co-transfected living rat cortical neurons. In line with our previous work (15,24), we chose cells expressing low levels of transfected proteins (as judged by the fluorescent protein signal) for analyses so as to avoid any possible artefacts produced by high levels of expression. Co-ordinate movement of APP-mCherry and EGFP-calsyntenin-1 in both anterograde and retrograde directions was observed through axons (Fig. 4A). Movement was predominantly anterograde and mean speeds were 1.6 μm/s (anterograde) and 1.1 μm/s (retrograde). These velocities and the bias towards anterograde movement are similar to those described previously for calsyntenin-1 (13–15). APP-YFP/EGFP also moves in a predominantly anterograde direction with reported speeds of 0.8–9 μm/s which may depend to some extent upon the cell type utilized (7,14,25–27). Notably, ∼65% of APP-EGFP is transported at speed of 1–2 μm/s in mouse cortical neurons which are similar to the speeds reported here in rat cortical neurons (26).
Figure 4.

EGFP-calsyntenin-1 and APP-mCherry are co-transported through axons of rat cortical neurons and siRNA loss of calsyntenin-1 disrupts axonal transport of APP-EGFP. (A) Kymograph showing co-transport of EGFP-calsyntenin-1 (Cstn) and APP-mCherry (APP). Merge and co-transported (Co-tr) EGFP-calsyntenin-1/APP-mCherry particles are shown. (B and C) Representative kymographs showing axonal transport of APP-EGFP in control (B) and calsyntenin-1 (C) siRNA-treated neurons. (D) Flux rates for APP-EGFP vesicle movement in control and calsyntenin-1 siRNA-treated neurons. Loss of calsyntenin-1 disrupts APP-EGFP transport in both anterograde and retrograde directions. Statistical significance was determined by Student's t-test. n = 33 control and 33 calsyntenin-1 siRNA-treated cells; error bars are SEM; *P < 0.05; ***P < 0.001.
siRNA loss of calsyntenin-1 decreases axonal transport of APP
The above co-localization and live imaging studies suggest that a significant proportion of APP is co-transported with calsyntenin-1 through axons. To test further this possibility, we quantified APP-EGFP axonal transport using time-lapse microscopy in transfected living rat cortical neurons following siRNA knockdown of calsyntenin-1. APP-EGFP movement was again predominantly anterograde in these neurons. However, analyses of the flux rates for APP-EGFP transport revealed that the loss of calsyntenin-1 markedly reduced the numbers of APP-EGFP moving cargoes (Fig. 4B–D). This reduction was seen in both anterograde (64.3% reduction) and retrograde (46.6% reduction) directions (Fig. 4D). Thus, APP and calsyntenin-1 co-localize in axons, APP-mCherry and EGFP-calsyntenin-1 are co-transported through axons, and the loss of calsyntenin-1 markedly disrupts axonal transport of APP.
siRNA loss of calsyntenin-1 increases APP levels in the TGN
Calsyntenin-1 is enriched in the Golgi and mediates trans-Golgi exit of APP (12). The reduced axonal transport of APP-EGFP seen in calsyntenin-1 knockdown neurons suggests that APP may be retained in the cell body and accumulate in the TGN. To test this possibility, we quantified the APP fluorescent signals in the TGN in control and calsyntenin-1 siRNA-treated neurons. Loss of calsyntenin-1 induced a significant 54.7% increase in APP signals in the TGN (Fig. 5).
Figure 5.
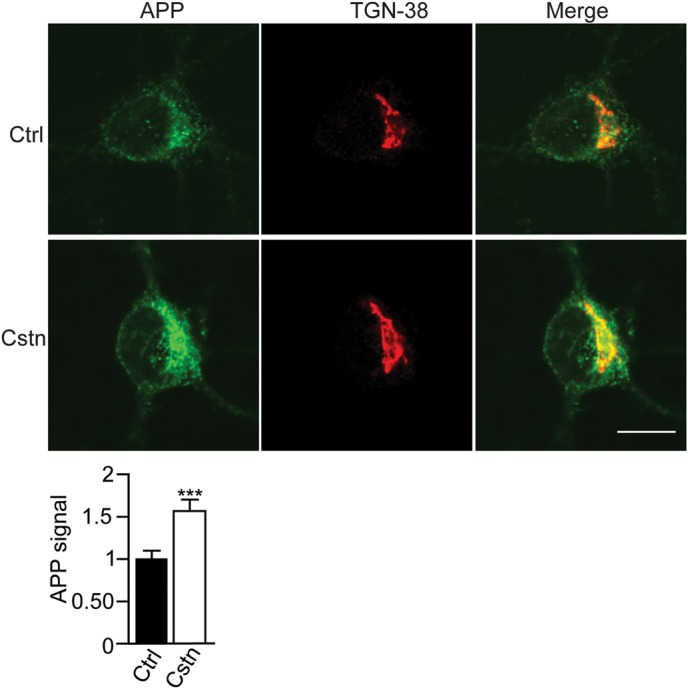
siRNA loss of calsyntenin-1 increases APP levels in the TGN. Neurons were treated with control (Ctrl) or calsyntenin-1 (Cstn) siRNAs and immunostained for APP and TGN-38. Representative cells are shown with merge. Scale bar is 10 μm. Bar chart shows relative APP fluorescent signals in the TGN. Statistical significance was determined by Student's t-test. n = 50 control and 54 calsyntenin-1 siRNA-treated cells; error bars are SEM; ***P < 0.001.
Calsyntenin-1 expression is reduced in Alzheimer's disease brains and the extent of this reduction correlates inversely with Aβ levels
The above studies, which show that the loss of calsyntenin-1 increases Aβ production and that this is associated with a disruption to axonal transport of APP, suggest that defects in calsyntenin-1 metabolism may be mechanistic in Alzheimer's disease. To begin to test this possibility, we monitored calsyntenin-1 expression in Alzheimer's disease brains. Immunoblotting of cortical samples from control and Braak stage V–VI Alzheimer's disease brains revealed that compared with controls, calsyntenin-1 expression was significantly reduced in Alzheimer's disease brains (73.5 ± 6.72% of control) (Fig. 6A). There was no significant correlation between calsyntenin-1 levels and post-mortem delay.
Figure 6.

Calsyntenin-1 is reduced in Alzheimer's disease brains and the extent of this reduction correlates with Aβ levels. (A) Immunoblots of representative samples from control (Ctrl) and Alzheimer's disease (AD) cortex. Samples were probed for calsyntenin-1 (Cstn) and NSE. Bar graph shows relative levels of calsyntenin-1 in the two sample sets following normalization to NSE signals in each sample. Statistical significance was determined by Student's t-test. n = 19 control (Ctrl) and n = 16 Alzheimer's disease samples; error bars are SEM. **P < 0.01. (B) Correlation between cortical calsyntenin-1 amounts and Aβ burden in post-mortem brains. Aβ levels in human tissues were correlated with calsyntenin-1 signals by generating correlation coefficients and significance established by non-parametric, two-tailed, Spearman Rho tests. Graphs show Aβ(1–40) and Aβ(1–42) levels (ng/g brain tissue) plotted against calsyntenin-1 signals (arbitrary units) obtained from immunoblots. Significant negative correlations were observed between both Aβ(1–40) (r2 = −0.396; P < 0.05) and Aβ(1–42) (r2 = −0.441; P < 0.01) and calsyntenin-1 levels (n = 19 control; n = 16 Alzheimer's disease samples).
We also tested whether calsyntenin-1 levels correlated with Aβ burden in these samples. Total Aβ(1–40) and Aβ(1–42) were quantified by ELISAs and correlated with calsyntenin-1 protein levels. Significant negative correlations were observed between both Aβ(1–40) (r2 = −0.396) and Aβ(1–42) (r2 = −0.441), and calsyntenin-1 levels (Fig. 6B).
DISCUSSION
Here, we show that the loss of calsyntenin-1 increases production of endogenous Aβ(1–40) and Aβ(1–42) in rat neurons. We also show that calsyntenin-1 levels are reduced significantly in Alzheimer's disease brains and that the extent of this reduction correlates significantly with increased Aβ levels. Loss of calsyntenin-1 in the cultured neurons was associated with alterations to APP processing involving increased cleavage at the BACE1 sites and decreased cleavage at the α-secretase site.
APP is synthesized in the endoplasmic reticulum (ER) and then trafficked through the Golgi to the cell surface. Here, it can be re-internalized to endocytic and recycling compartments and trafficked back to the cell surface or degraded in the lysosome (3). Calsyntenin-1 is enriched in the TGN and recent evidence suggests that it mediates transport of a subset of cargoes on post-Golgi carriers via kinesin-1 (11,12). Our findings that siRNA loss of calsyntenin-1 reduces axonal transport of APP and increases APP levels in the TGN are thus consistent with a role for calsyntenin-1 in mediating transport of APP on post-Golgi carriers.
The precise intracellular location(s) where APP is processed to produce Aβ in neurons are not clear. BACE1 localizes mainly to the TGN and endosomes, whereas γ-secretase components are found in multiple locations, including ER, Golgi, endosomes and the cell surface (3,28). Thus, some studies suggest that Aβ is produced in endosomes (29–33), whereas others suggest that the TGN is a primary site for Aβ production (34–39). There is also evidence that Aβ is produced in other locations, including autophagosomes (40). Whatever the scenario, disrupting anterograde transport of APP may well promote amyloidogenic processing by targeting APP to sites of cleavage by BACE1 and/or γ-secretase. In particular, our finding that siRNA loss of calsyntenin-1 increases APP levels in the TGN, a favoured site for APP processing by BACE1 (see above) may explain the increased cleavage of APP at the BACE1 sites that we detect in the calsyntenin-1 knockdown neurons.
Despite highly efficient siRNA knockdown of calsyntenin-1, its loss reduced but did not abolish anterograde axonal transport of APP. Other mechanisms for transport of APP by kinesin-1 have been described, including direct attachment of APP to KLCs, or indirect attachment to KLCs via adaptor proteins such as JIPs (C-Jun N-terminal kinase interacting proteins) and PAT1 which bind to both KLC and APP (14,19,41–43). The GTPase Rab3A has also been implicated in APP transport (26). Transport involving these other mechanisms may account for the remaining APP movement seen in the calsyntenin-1 knockdown neurons. It is also possible that the co-ordinate transport of APP-mCherry and EGFP-calsyntenin-1 seen in our dual imaging studies overestimates the proportion of APP that is co-transported with calsyntenin-1 (e.g. overexpression of calsyntenin-1 in these experiments may recruit APP from JIP1/PAT1 carriers). Nevertheless, our observations that siRNA knockdown of calsyntenin-1 markedly inhibits anterograde movement of APP strongly suggests that a significant proportion of APP is transported on calsyntenin-1 carriers. Indeed, using alternative approaches that do not directly assay movement of APP through axons, others have also concluded that an important route for axonal transport of APP by kinesin-1 may involve calsyntenin-1 (11,12).
Our findings that the loss of calsyntenin-1 disrupts axonal transport of APP and increases production of Aβ focus attention on the mechanism that controls APP transport on calsyntenin-1 carriers. In particular, the mechanisms that control loading of APP onto calsyntenin-1 containing vesicles and binding of calsyntenin-1 to KLCs represent key regulatory points that could influence APP trafficking.
Although the route(s) by which APP is loaded onto calsyntenin-1-containing vesicles are not clear, both APP and calsyntenin-1 bind to X11β (22,23,44). X11β is a neuronal adaptor protein that is enriched in the Golgi and which regulates APP processing and Aβ production (45–50). There is evidence that X11β functions as a vesicle coat protein; coat proteins function in the selection of cargo for inclusion into particular vesicle sub-types (51). Interestingly, phosphorylation of KLC1 on serine-460 has recently been shown to selectively regulate binding of calsyntenin-1 but not several other ligands to kinesin-1 motors (15). Thus, via its binding to both APP and calsyntenin-1, X11β may function to load APP onto calsyntenin-1-containing vesicles for post-Golgi transport on kinesin-1 motors, and phosphorylation of KLCs may regulate attachment of calsyntenin-1 with associated APP to kinesin-1. Disruption to loading of APP onto calsyntenin-1 vesicles or transport of APP by kinesin-1 on calsyntenin-1 containing vesicles are thus likely to promote amyloidogenic processing of APP. Moreover, the negative correlation we detect between calsyntenin-1 and Aβ levels in Alzheimer's disease brains suggest that the loss of calsyntenin-1 is mechanistic in the disease process.
MATERIALS AND METHODS
Plasmids and siRNAs
EGFP-calsyntenin-1 and APP-EGFP were as described previously (13,52). APP-mCherry was generated by replacement with mCherry sequences in APP-EGFP. Calysntenin-1 and negative control siRNAs were from Dharmacon (Accell range). Calsyntenin-1 siRNAs were #1 5′-GCAAAGAGCAUCAGUAUAA-3′, #2 5′-CCCUUAAGAUGUGUAUAUU-3′, #3 5′-CCAUCACGCUUGCAGUUUU-3′ and #4 5′-GUGUCAGCUUCCUGGUUUU-3′. Negative control siRNA (catalogue number D-001910-03) has been designed, modified and microarray-confirmed by Dharmacon to have minimal targeting of genes in rat cells (see http://www.dharmacon.com/).
Antibodies
Rabbit calsyntenin-1 and APP C-terminal antibodies were as described previously (15,53). Anti-beta-Actin clone AC-15 and anti-Nicastrin rabbit polyclonal antibodies were from Abcam (Cambridge, MA, USA); anti-ADAM10/Kuz and anti-presenilin-1 N-terminus rabbit polyclonal antibodies were from EMD Chemicals (Gibbstown, NJ, USA); anti-presenilin-1 C-terminus clone PS1-loop, anti-APP mouse clone 22C11 and anti-APP mouse clone 2.F2.19B4 were from Millipore Bioscience Research Reagents (Massachusetts, MA, USA); anti-sAPPα clone 2B3 and anti-sAPPβ rabbit polyclonal antibodies were from IBL International (Hamburg, Germany), anti-phospho-threonine-668 APP rabbit polyclonal antibody was from Cell Signaling Technology (Danvers, MA, USA); anti-BACE1 rabbit polyclonal antibody was from Sigma-Aldrich (St Louis, MO, USA); anti-X11β (Mint 2) and TGN-38 mouse monoclonal antibodies were from BD Biosciences (San Jose, CA, USA); neuron-specific enolase (NSE) antibody was from Dako (Cambridge UK).
Cell culture and transfection
Cortical neurons were obtained from embryonic day 18 rat embryos and cultured in Neurobasal medium containing B27 supplement, 100 IU/ml penicillin, 100 μg/ml streptomycin and 2 mm l-glutamine (Invitrogen). For siRNA knockdown studies, neurons at DIV4 were treated with 1 μm siRNAs and analysed at DIV8. For transfection studies, neurons cultured on poly-l-lysine-coated glass cover slips in 12-well plates were transfected using Lipofectamine 2000 (Invitrogen) (1 μg DNA and 0.5 μl Lipofectamine 2000) as described by the manufacturer at DIV7 and analysed on DIV8.
Aβ1-40 and Aβ1-42 ELISAs
Rat/mouse and human Aβ1-40 and Aβ1-42 ELISAs were obtained from Invitrogen. Samples were processed according to the manufacturer's instructions.
SDS–PAGE and immunoblotting
Cells were harvested for SDS–PAGE by washing with phosphate-buffered saline pre-warmed at 37°C and scraping into SDS–PAGE sample buffer and immediately heating to 100°C. λ-Phosphatase treatment was as described previously (54). Samples were separated on either 8 or 10% (w/v) acrylamide gels or for analyses of APP C-terminal processed fragments on 16% (w/v) acrylamide Tris-tricine gels as described previously (55). Separated proteins were then transferred to Protran nitrocellulose membranes or for Tris-tricine gels, to 0.2 μm Optitran nitrocellulose membranes (Sigma) using a Transblot system (BioRad) and then processed for immunoblotting. Signals on immunoblots were quantified using ImageJ (National Institutes of Health) after scanning with an Epson Precision V700 Photo scanner essentially as described by us in previous studies (15,56). To ensure the signals obtained were within the linear range, the mean background-corrected optical density (OD) of each signal was interpolated for an OD calibration curve created using a calibrated OD step tablet (Kodak). Only film exposures that gave OD signals within the linear range of the OD calibration curve were used for the statistical analyses.
Frozen human brain tissue was prepared as a 20% homogenate in ice-cold lysis buffer containing 50 mm Tris–HCl pH 7.4, 0.9% (w/v) NaCl, 0.1% (v/v) Triton X-100, 10 mm sodium fluoride, 1 mm sodium orthovanadate, 2 mm ethylenediamine tetraacetic acid, 1 mm phenylmethylsulfonyl fluoride and complete protease inhibitors (Roche). Following homogenization, samples were centrifuged at 25 000g (av) for 20 min at 4°C and prepared for SDS–PAGE by addition of sample buffer. Signals on immunoblots were simultaneously visualized and the relative amounts quantified using an Odyssey imaging system and associated software (Li-Cor Biosciences, Cambridge, UK). Data were analysed using Student's t-test.
Immunofluorescence and time-lapse microscopy
Neurons were prepared for immunostaining by fixing in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min, quenching with 0.05 m NH4Cl for 15 min, washing in PBS and then permeabilizing with 0.2% Triton X-100 in PBS for 5 min. Following blocking with 5% goat serum (Sigma) in PBS for 30 min, the samples were probed with primary antibodies diluted in blocking solution (PBS containing 5% goat serum). Primary antibodies were then detected using goat anti-mouse and anti-rabbit Igs coupled to Alexa Fluor 488 or 546 (Invitrogen). Confocal images were captured using a Zeiss LSM510Meta confocal microscope equipped with a ×100 Plan-Apochromat 1.4 N.A. objective (Carl Zeiss Ltd, Welwyn Garden City, UK). Non-confocal images were captured using a Leica DM5000B microscope with ×63 HCX PL Fluotar phase objective. Co-localization of signals was performed using ImageJ 1.44p with the RG2B co-localization plug-in to determine co-localized pixels. To quantify APP signals in the TGN, TGNs were first marked by immunostaining with TGN-38. APP signals in the delineated areas were then quantified by calculating the average pixel fluorescence intensity using ImageJ. These signals were then normalized by subtracting the background fluorescence signals in each cell which were obtained from measurements in nuclei. Data were analysed using Student's t-test.
Live microscopy of APP-EGFP, APP-mCherry and EGFP-calsyntenin-1 was performed with an Axiovert S100 microscope (Zeiss) equipped with a Lambda LS Xenon-Arc light source (Sutter Instrument Company, Novato, CA, USA), GFP or GFP-mCherry filter set (Chroma Technology Corp., Rockingham, VT, USA), ×100 Plan-Apochromat 1.4 N.A. objective (Zeiss), Lambda 10-3 filter wheel (Sutter Instrument Co.) and a Photometrics Cascade-II 512B High Speed EMCCD camera (Photometrics, Tuscon, AZ, USA). Twenty-four hours post-transfection, the cells were transferred to a Ludin imaging chamber (Life Imaging Systems, Basel, Switzerland) mounted on the stage of the microscope. Neurons were maintained in a HEPES-buffered extracellular neuronal solution (composition in mm: NaCl, 140; KCl, 5; NaHCO3, 5; MgCl2·6H2O, 1; CaCl2, 1.2; Na2HPO4, 1.2; Glucose, 10; Hepes, 20; pH 7.4) at 37°C using an objective heater (IntraCell, Royston, UK) and ‘The Box’ Microscope Temperature Control System (Life Imaging Systems). Vesicle movements were recorded for 5 min with a 1 s time-lapse interval using MetaMorph software (Molecular Devices). A movement was defined as a displacement of a particle of at least 2 μm, without reversal of direction of more than 2 μm or pausing for longer than 5 s. For dual imaging of APP-mCherry and EGFP-calsyntenin-1 transport, co-transfected neurons were imaged using a two-channel simultaneous imaging system (Dual View, Photometrics) and vesicle movements were recorded for 2 min with a 2 s time-lapse interval using MetaMorph software (Molecular Devices). Image analysis was performed with ImageJ. The flux rates for APP-EGFP transport in both anterograde and retrograde directions were determined essentially as described (8,27) by counting the numbers of APP-EGFP particles that crossed a defined line in mid axons per minute. Statistical significance was determined using Student's t-test. Images presented were constructed using Adobe CS5 and have been smoothened in ImageJ.
Human tissues
The post-mortem human frontal cortex was obtained from control and pathologically confirmed cases of Alzheimer's disease (Braak Stage V–VI) (Table 1). All tissue collection and processing were carried out under the regulations and licensing of the Human Tissue Authority and in accordance with the Human Tissue Act, 2004. Calsyntenin-1 signals were normalized against NSE, as neuronal loss or astrogliosis may bias the data toward downregulation of neuronal proteins.
Table 1.
Case data for human post-mortem brain samples
| Stage | Case no. | Sex | Age (years) | Post-mortem delay (h) |
|---|---|---|---|---|
| Control | A042/01 | F | 52 | 44 |
| Control | A278/96 | F | 77 | 29 |
| Control | A239/95 | F | 79 | 38 |
| Control | A155/95 | F | 63 | 34 |
| Control | A094/95 | F | 80 | 31 |
| Control | A047/02 | F | 87 | 22 |
| Control | A170/00 | F | 68 | 9 |
| Control | A322/94 | F | 62 | 81 |
| Control | A346/95 | M | 85 | 16 |
| Control | A135/95 | M | 65 | 24 |
| Control | A134/00 | M | 86 | 6 |
| Control | A401/97 | M | 85 | 42 |
| Control | A223/96 | M | 80 | 11 |
| Control | A133/95 | M | 85 | 48 |
| Control | A149/01 | M | 95 | 44 |
| Control | A077/00 | M | 68 | 53 |
| Control | A066/00 | M | 61 | 53 |
| Control | A330/94 | M | 69 | 52 |
| Control | A261/94 | M | 75 | 85 |
| Braak VI | A157/00 | F | 75 | 9 |
| Braak V | A240/06 | F | 97 | 12 |
| Braak VI | A010/06 | F | 67 | 56 |
| Braak V | A210/05 | F | 84 | 18 |
| Braak V–VI | A169/05 | F | 82 | 43 |
| Braak V–VI | A168/05 | F | 84 | 36 |
| Braak V–VI | A074/05 | F | 89 | 29 |
| Braak V | A203/04 | F | 84 | 37 |
| Braak V | A221/03 | F | 81 | 37 |
| Braak VI | A188/00 | F | 64 | 10 |
| Braak VI | A232/00 | F | 79 | 8 |
| Braak VI | A074/06 | F | 69 | 25 |
| Braak VI | A206/07 | M | 81 | 47 |
| Braak V–VI | A186/04 | M | 71 | 5 |
| Braak VI | A122/04 | M | 86 | 26 |
| Braak VI | A176/01 | M | 71 | 41 |
FUNDING
This work was supported by grants from the Wellcome Trust (http://www.wellcome.ac.uk/078662 to C.C.J.M.), UK Medical Research Council (http://www.mrc.ac.uk/index.htm G0501573 to C.C.J.M.), Alzheimer's Research UK (http://www.alzheimersresearchuk.org/ to C.C.J.M.) and the Royal Society (http://royalsociety.org/ to W.N.). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
ACKNOWLEDGEMENTS
We thank Peter Sonderegger (University of Zurich, Switzerland) for calsyntenin-1 clone. Human post-mortem brain tissues were obtained from the MRC London Brain Bank for Neurodegenerative Diseases, Institute of Psychiatry, King's College London, UK.
Conflict of Interest statement. None declared.
References
- 1.De Strooper B., Vassar R., Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. doi:10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh D.M., Selkoe D.J. A beta oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. doi:10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 3.Thinakaran G., Koo E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. doi:10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stokin G.B., Goldstein L.S. Axonal transport and Alzheimer's disease. Annu. Rev. Biochem. 2006;75:607–627. doi: 10.1146/annurev.biochem.75.103004.142637. doi:10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- 5.De Vos K.J., Grierson A.J., Ackerley S., Miller C.C.J. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. doi:10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 6.Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr, Brown H., Tiwari A., Hayward L., et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. doi:10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaether C., Skehel P., Dotti C.G. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol. Biol. Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. doi:10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 9.Hirokawa N., Noda Y., Tanaka Y., Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. doi:10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 10.Brunholz S., Sisodia S., Lorenzo A., Deyts C., Kins S., Morfini G. Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp. Brain Res. 2012;217:353–364. doi: 10.1007/s00221-011-2870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steuble M., Gerrits B., Ludwig A., Mateos J.M., Diep T.M., Tagaya M., Stephan A., Schatzle P., Kunz B., Streit P., et al. Molecular characterization of a trafficking organelle: dissecting the axonal paths of calsyntenin-1 transport vesicles. Proteomics. 2010;10:3775–3788. doi: 10.1002/pmic.201000384. doi:10.1002/pmic.201000384. [DOI] [PubMed] [Google Scholar]
- 12.Ludwig A., Blume J., Diep T.M., Yuan J., Mateos J.M., Leuthauser K., Steuble M., Streit P., Sonderegger P. Calsyntenins mediate TGN exit of APP in a kinesin-1-dependent manner. Traffic. 2009;10:572–589. doi: 10.1111/j.1600-0854.2009.00886.x. doi:10.1111/j.1600-0854.2009.00886.x. [DOI] [PubMed] [Google Scholar]
- 13.Konecna A., Frischknecht R., Kinter J., Ludwig A., Steuble M., Meskenaite V., Indermuhle M., Engel M., Cen C., Mateos J.M., et al. Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol. Biol. Cell. 2006;17:3651–3663. doi: 10.1091/mbc.E06-02-0112. doi:10.1091/mbc.E06-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Araki Y., Kawano T., Taru H., Saito Y., Wada S., Miyamoto K., Kobayashi H., Ishikawa H.O., Ohsugi Y., Yamamoto T., et al. The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 2007;26:1475–1486. doi: 10.1038/sj.emboj.7601609. doi:10.1038/sj.emboj.7601609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vagnoni A., Rodriguez L., Manser C., De Vos K.J., Miller C.C.J. Phosphorylation of kinesin light chain-1 at serine-460 modulates binding and trafficking of calsyntenin-1. J. Cell Sci. 2011;124:1032–1042. doi: 10.1242/jcs.075168. doi:10.1242/jcs.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dodding M.P., Mitter R., Humphries A.C., Way M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011;30:4523–4538. doi: 10.1038/emboj.2011.326. doi:10.1038/emboj.2011.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Standen C.L., Brownlees J., Grierson A.J., Kesavapany S., Lau K.F., McLoughlin D.M., Miller C.C.J. Phosphorylation of thr668 in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3) J. Neurochem. 2001;76:316–320. doi: 10.1046/j.1471-4159.2001.00102.x. doi:10.1046/j.1471-4159.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- 18.Kimberly W.T., Zheng J.B., Town T., Flavell R.A., Selkoe D.J. Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J. Neurosci. 2005;25:5533–5543. doi: 10.1523/JNEUROSCI.4883-04.2005. doi:10.1523/JNEUROSCI.4883-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muresan Z., Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J. Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. doi:10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ando K., Iijima K.I., Elliott J.I., Kirino Y., Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J. Biol. Chem. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- 21.Lee M.S., Kao S.C., Lemere C.A., Xia W., Tseng H.C., Zhou Y., Neve R., Ahlijanian M.K., Tsai L.H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. doi:10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLoughlin D.M., Miller C.C.J. The intracellular cytoplasmic domain of the Alzheimer's disease amyloid precursor protein interacts with phosphotyrosine binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. doi:10.1016/S0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- 23.Araki Y., Tomita S., Yamaguchi H., Miyagi N., Sumioka A., Kirino Y., Suzuki T. Novel cadherin-related membrane proteins, Alcadeins, enhance the X11-like protein-mediated stabilization of amyloid beta -protein precursor metabolism. J. Biol. Chem. 2003;278:49448–49458. doi: 10.1074/jbc.M306024200. doi:10.1074/jbc.M306024200. [DOI] [PubMed] [Google Scholar]
- 24.Ackerley S., Thornhill P., Grierson A.J., Brownlees J., Anderton B.H., Leigh P.N., Shaw C.E., Miller C.C.J. Neurofilament heavy chain side-arm phosphorylation regulates axonal transport of neurofilaments. J. Cell Biol. 2003;161:489–495. doi: 10.1083/jcb.200303138. doi:10.1083/jcb.200303138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandelkow E.M., Thies E., Trinczek B., Biernat J., Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. doi:10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szodorai A., Kuan Y.H., Hunzelmann S., Engel U., Sakane A., Sasaki T., Takai Y., Kirsch J., Muller U., Beyreuther K., et al. APP anterograde transport tequires Rab3A GTPase activity for assembly of the transport vesicle. J. Neurosci. 2009;29:14534–14544. doi: 10.1523/JNEUROSCI.1546-09.2009. doi:10.1523/JNEUROSCI.1546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldsbury C., Mocanu M.M., Thies E., Kaether C., Haass C., Keller P., Biernat J., Mandelkow E., Mandelkow E.M. Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic. 2006;7:873–888. doi: 10.1111/j.1600-0854.2006.00434.x. doi:10.1111/j.1600-0854.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- 28.Small S.A., Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. doi:10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haass C., Schlossmacher G., Hung A.Y., Vigo-Palfrey C., Mellon A., Ostaszewski B.L., Lieberburg I., Koo E.H., Schenk D., Teplow D.B., et al. Amyloid beta peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. doi:10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 30.Koo E.H., Squazzo S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 31.Sannerud R., Declerck I., Peric A., Raemaekers T., Menendez G., Zhou L., Veerle B., Coen K., Munck S., De Strooper B., et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl Acad. Sci. USA. 2011;108:E559–E568. doi: 10.1073/pnas.1100745108. doi:10.1073/pnas.1100745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J., Petralia R.S., Kurushima H., Patel H., Jung M.Y., Volk L., Chowdhury S., Shepherd J.D., Dehoff M., Li Y., et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell. 2011;147:615–628. doi: 10.1016/j.cell.2011.09.036. doi:10.1016/j.cell.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez R.G., Soriano S., Hayes J.D., Ostaszewski B., Xia W., Selkoe D.J., Chen X., Stokin G.B., Koo E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. doi:10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 34.Baulac S., LaVoie M.J., Kimberly W.T., Strahle J., Wolfe M.S., Selkoe D.J., Xia W. Functional gamma-secretase complex assembly in Golgi/trans-Golgi network: interactions among presenilin, nicastrin, Aph1, Pen-2, and gamma-secretase substrates. Neurobiol. Dis. 2003;14:194–204. doi: 10.1016/s0969-9961(03)00123-2. doi:10.1016/S0969-9961(03)00123-2. [DOI] [PubMed] [Google Scholar]
- 35.Burgos P.V., Mardones G.A., Rojas A.L., daSilva L.L., Prabhu Y., Hurley J.H., Bonifacino J.S. Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell. 2010;18:425–436. doi: 10.1016/j.devcel.2010.01.015. doi:10.1016/j.devcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skovronsky D.M., Moore D.B., Milla M.E., Doms R.W., Lee V.M.Y. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-Golgi network. J. Biol. Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. doi:10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- 37.Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. doi:10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 38.Xia W.M., Ray W.J., Ostaszewski B.L., Rahmati T., Kimberly W.T., Wolfe M.S., Zhang J.M., Goate A.M., Selkoe D.J. Presenilin complexes with the C-terminal fragments of amyloid precursor protein at the sites of amyloid beta-protein generation. Proc. Natl Acad. Sci. USA. 2000;97:9299–9304. doi: 10.1073/pnas.97.16.9299. doi:10.1073/pnas.97.16.9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muresan V., Varvel N.H., Lamb B.T., Muresan Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J. Neurosci. 2009;29:3565–3578. doi: 10.1523/JNEUROSCI.2558-08.2009. doi:10.1523/JNEUROSCI.2558-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nixon R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. doi:10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 41.Kamal A., Stokin G.B., Yang Z.H., Xia C.H., Goldstein L.S.B. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. doi:10.1016/S0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 42.Kuan Y.H., Gruebl T., Soba P., Eggert S., Nesic I., Back S., Kirsch J., Beyreuther K., Kins S. PAT1a modulates intracellular transport and processing of amyloid precursor protein (APP), APLP1, and APLP2. J. Biol. Chem. 2006;281:40114–40123. doi: 10.1074/jbc.M605407200. doi:10.1074/jbc.M605407200. [DOI] [PubMed] [Google Scholar]
- 43.Zheng P., Eastman J., Vande Pol S., Pimplikar S.W. PAT1, a microtubule-interacting protein, recognizes the basolateral sorting signal of amyloid precursor protein. Proc. Natl Acad. Sci. USA. 1998;95:14745–14750. doi: 10.1073/pnas.95.25.14745. doi:10.1073/pnas.95.25.14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borg J.-P., Ooi J., Levy E., Margolis B. The phosphotyrosine interaction domains of X11 and Fe65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J.H., Lau K.F., Perkinton M.S., Standen C.L., Rogelj B., Falinska A., McLoughlin D.M., Miller C.C. The neuronal adaptor protein X11beta reduces Abeta levels and amyloid plaque formation in the brains of transgenic mice. J. Biol. Chem. 2004;279:49099–49104. doi: 10.1074/jbc.M405602200. doi:10.1074/jbc.M405602200. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell J.C., Ariff B.B., Yates D.M., Lau K.F., Perkinton M.S., Rogelj B., Stephenson J.D., Miller C.C., McLoughlin D.M. X11beta rescues memory and long-term potentiation deficits in Alzheimer's disease APPswe Tg2576 mice. Hum. Mol. Genet. 2009;18:4492–4500. doi: 10.1093/hmg/ddp408. doi:10.1093/hmg/ddp408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sano Y., Syuzo-Takabatake A., Nakaya T., Saito Y., Tomita S., Itohara S., Suzuki T. Enhanced amyloidogenic metabolism of APP in the X11L-deficient mouse brain. J. Biol. Chem. 2006;281:37853–37860. doi: 10.1074/jbc.M609312200. doi:10.1074/jbc.M609312200. [DOI] [PubMed] [Google Scholar]
- 48.Tomita S., Ozaki T., Taru H., Oguchi S., Takeda S., Yagi Y., Sakiyama S., Kirino Y., Suzuki T. Interaction of a neuron-specific protein containing PDZ domains with Alzheimer's amyloid precursor protein. J. Biol. Chem. 1999;274:2243–2254. doi: 10.1074/jbc.274.4.2243. doi:10.1074/jbc.274.4.2243. [DOI] [PubMed] [Google Scholar]
- 49.Miller C.C., McLoughlin D.M., Lau K.F., Tennant M.E., Rogelj B. The X11 proteins, Abeta production and Alzheimer's disease. Trends Neurosci. 2006;29:280–285. doi: 10.1016/j.tins.2006.03.001. doi:10.1016/j.tins.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Ho A., Liu X., Sudhof T.C. Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease. J. Neurosci. 2008;28:14392–14400. doi: 10.1523/JNEUROSCI.2481-08.2008. doi:10.1523/JNEUROSCI.2481-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill K., Li Y., Bennett M., McKay M., Zhu X., Shern J., Torre E., Lah J.J., Levey A.I., Kahn R.A. Munc18 interacting proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of beta-Alzheimer's precursor protein. J. Biol. Chem. 2003;278:36032–36040. doi: 10.1074/jbc.M301632200. doi:10.1074/jbc.M301632200. [DOI] [PubMed] [Google Scholar]
- 52.De Vos K.J., Chapman A.L., Tennant M.E., Manser C., Tudor E.L., Lau K.F., Brownlees J., Ackerley S., Shaw P.J., McLoughlin D.M., et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. doi:10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cousins S.L., Hoey S.E., Stephenson F.A., Perkinton M.S. Amyloid precursor protein 695 assocoates with assembled NR2A- and NR2B-containing NMDA receptors to result in the enhancement of their cell surface delivery. J. Neurochem. 2009;111:1501–1513. doi: 10.1111/j.1471-4159.2009.06424.x. doi:10.1111/j.1471-4159.2009.06424.x. [DOI] [PubMed] [Google Scholar]
- 54.Standen C.L., Perkinton M.S., Byers H.L., Kesavapany S., Lau K.F., Ward M., McLoughlin D., Miller C.C. The neuronal adaptor protein Fe65 is phosphorylated by mitogen-activated protein kinase (ERK1/2) Mol. Cell Neurosci. 2003;24:851–857. doi: 10.1016/j.mcn.2003.07.002. doi:10.1016/j.mcn.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Hao C.Y., Perkinton M.S., Chan W.W., Chan H.Y., Miller C.C., Lau K.F. GULP1 is a novel APP-interacting protein that alters APP processing. Biochem. J. 2011;436:631–639. doi: 10.1042/BJ20110145. doi:10.1042/BJ20110145. [DOI] [PubMed] [Google Scholar]
- 56.Lee J.H., Lau K.F., Perkinton M.S., Standen C.L., Shemilt S.J., Mercken L., Cooper J.D., McLoughlin D.M., Miller C.C. The neuronal adaptor protein X11alpha reduces Abeta levels in the brains of Alzheimer's APPswe Tg2576 transgenic mice. J. Biol. Chem. 2003;278:47025–47029. doi: 10.1074/jbc.M300503200. doi:10.1074/jbc.M300503200. [DOI] [PubMed] [Google Scholar]