

Sorting of secretory cargo from the Golgi remains an elusive process. Previously a role was identified for cofilin and the Ca2+ATPase SPCA1 in sorting of secretory cargo from the Golgi of mammalian cells. Now it is shown that the yeast orthologues cofilin and Pmr1 are also required for sorting of selective secretory cargo at the Golgi in yeast.
Abstract
The mechanism of cargo sorting at the trans-Golgi network (TGN) for secretion is poorly understood. We previously reported the involvement of the actin-severing protein cofilin and the Ca2+ ATPase secretory pathway calcium ATPase 1 (SPCA1) in the sorting of soluble secretory cargo at the TGN in mammalian cells. Now we report that cofilin in yeast is required for export of selective secretory cargo at the late Golgi membranes. In cofilin mutant (cof1-8) cells, the cell wall protein Bgl2 was secreted at a reduced rate and retained in a late Golgi compartment, whereas the plasma membrane H+ ATPase Pma1, which is transported in the same class of carriers, reached the cell surface. In addition, sorting of carboxypeptidase Y (CPY) to the vacuole was delayed, and CPY was secreted from cof1-8 cells. Loss of the yeast orthologue of SPCA1 (Pmr1) exhibited similar sorting defects and displayed synthetic sickness with cof1-8. In addition, overexpression of PMR1 restored Bgl2 secretion in cof1-8 cells. These findings highlight the conserved role of cofilin and SPCA1/Pmr1 in sorting of the soluble secretory proteins at the TGN/late Golgi membranes in eukaryotes.
INTRODUCTION
Secretory cargoes of eukaryotic cells are transported from the endoplasmic reticulum (ER) to the Golgi membranes by COPII-coated vesicles (Bonifacino and Glick, 2004; Lee et al., 2004). ER-resident KDEL (HDEL in yeast)-containing proteins are transported back to the ER in COPI vesicles by binding the KDEL (HDEL) receptor (Lewis and Pelham, 1990). Within the Golgi apparatus, there are many exit routes, and secretory cargo must be sorted to ensure that it is delivered to the correct destination. In yeast and higher eukaryotes the trafficking and sorting of secreted proteins from the Golgi membranes remain poorly understood. In the late Golgi membranes of higher eukaryotes, mannose-6-phosphate–containing lysosomal hydrolases bind the mannose-6-phosphate receptor (M6PR) and are packed into clathrin-coated vesicles for export to the endosomes/lysosomes (Baranski et al., 1990). Sorting of lysosomal hydrolases in yeast also occurs via endosomes and clathrin-coated vesicles, and the best-characterized example is carboxypeptidase Y (CPY), which is sorted by direct interaction with its receptor Vps10 (Cooper and Stevens, 1996). A distant M6PR orthologue does exist in yeast, Mrl1, which affects transport of some soluble hydrolases, but it is unclear whether this is through a glycan-based interaction as in mammalian cells or through a peptide-based interaction as observed for CPY (Whyte and Munro, 2001).
In yeast, there are at least two transport routes from the late Golgi compartment to the cell surface, which have been classified based on vesicle densities, cargoes, and some of the molecular requirements for their biogenesis (Harsay and Bretscher, 1995; David et al., 1998). The low-density secretory vesicles (LDSVs) are transported directly to the cell surface and contain the soluble cargo Bgl2 and the plasma membrane H+ ATPase Pma1, whereas the high-density secretory vesicles (HDSVs) transit through the endosomal system, require clathrin and the dynamin-like GTPase Vps1, and contain invertase and acid phosphatase as cargoes (Gurunathan et al., 2002; Harsay and Schekman, 2002). No receptors for secretory cargo in the Golgi compartments have been identified in yeast or mammals.
More recent studies revealed the involvement of calcium and pH in cargo sorting from the trans-Golgi network (TGN). The sorting of the LDSV-specific integral membrane protein Pma1 depends on the pH of the late Golgi membranes in yeast (Huang and Chang, 2011). Calcium homeostasis in the TGN and its role in secretory cargo sorting require the actin filament–severing protein cofilin and the Ca2+ ATPase secretory pathway calcium ATPase 1 (SPCA1) in mammalian cells (von Blume et al., 2009, 2011). A defect in the activity of these proteins causes retention of some of the secretory cargoes in the TGN, whereas others are exported at normal or higher rates. Of interest, under such conditions, a soluble TGN resident protein, Cab45, and the lysosomal protease cathepsin D are also secreted from the cells (von Blume et al., 2009, 2011). These findings are beginning to reveal components of the secretory cargo-sorting machinery from the TGN. However, one key issue is whether cofilin and SPCA1 have a similar role in the sorting of secretory cargo in other organisms. In addition, the yeast Golgi membranes, unlike the mammalian Golgi apparatus, are not stacked, thus raising the question of whether cargo sorting for trafficking to the cell surface in yeast is by a mechanism similar to that in mammalian cells.
Cofilin and actin dynamics clearly regulate endocytosis in yeast (Engqvist-Goldstein and Drubin, 2003). The requirement of cofilin in protein transport from the Golgi apparatus of yeast is not known. Actin, on the other hand, has been implicated in secretion; however, its exact role in this process, or more specifically at the Golgi membranes, is not well understood (Novick and Botstein, 1985; Mulholland et al., 1997; Karpova et al., 2000). Cofilin functions by binding monomeric or filamentous actin and depolymerizing actin filaments and as such regulates the organization of the actin cytoskeleton and cortical actin patches, which are proposed sites of endocytosis in yeast (Carlier et al., 1997; Lappalainen et al., 1997). The regulation of cofilin has similarities in yeast and higher eukaryotes, such as regulation by pH and phosphatidylinositol 4,5-bisphosphate (PIP2) binding (Van Troys et al., 2008). Regulation by phosphorylation is an important mechanism of regulation in mammalian cells, although no cofilin kinases or phosphatases have been identified in yeast (Morgan et al., 1993; Agnew et al., 1995). However, this may be a conserved mode of regulation, since mutation of the conserved serine residue to glutamate, which would constitutively inactive cofilin, is lethal (Lappalainen et al., 1997).
We tested the requirement of cofilin in cargo sorting and export from the Golgi apparatus in Saccharomyces cerevisiae. Our findings reveal that cofilin is required for the sorting of select cargo at the late Golgi membranes. The LDSV cargo Bgl2 was found to accumulate in the Golgi membranes of cofilin-mutant cells, whereas Pma1 transport was unaffected. The trafficking of HDSV-specific cargo invertase was mildly affected, whereas the rate of CPY transport to the vacuole was reduced and CPY was secreted from cofilin-mutant cells. Similar sorting defects are described for cells lacking the Golgi apparatus–localized Ca2+ ATPase Pmr1, and we report here that a cofilin mutant interacts genetically with loss of PMR1. Moreover, exogenous expression of PMR1 restored the Bgl2 secretion in cofilin-mutant cells. Together these data indicate that cofilin and actin dynamics regulate protein sorting in the yeast Golgi apparatus and likely do so through Pmr1 as in mammalian cells.
RESULTS
Bgl2 secretion is impaired in cofilin mutants
S. cerevisiae contains a single cofilin gene, which is essential for viability (Lappalainen et al., 1997). A number of mutant alleles of cofilin have been generated, and these have facilitated the analysis of cofilin's role in regulation of actin dynamics through the activity of actin filament depolymerization. Many of these cofilin mutants have been generated as recombinant proteins and examined in vitro for various functions such as F- or G-actin binding, actin filament depolymerization, and PIP2 binding (Lappalainen and Drubin, 1997; Lappalainen et al., 1997; Okreglak and Drubin, 2007; Lin et al., 2010). Drubin and colleagues generated three cofilin temperature-sensitive (ts) alleles: cof1-5, cof1-8, and cof1-22. The cof1-22–mutant protein is defective in F-actin binding, the mutant cof1-5 protein is defective in G-actin binding, and the cof1-8–mutant protein was not characterized for these activities but was later shown to be defective in PIP2 binding (Lappalainen and Drubin, 1997; Lappalainen et al., 1997). Clearly these mutants are severely defective in cofilin function in vivo at the nonpermissive temperature of 37°C, as indicated by dramatic changes in actin organization and defective endocytosis. We tested these alleles for their ability to affect secretion of the cell wall protein Bgl2. Cells were grown to logarithmic phase at 25°C and shifted to 37°C for 2 h before removal of the cell wall and determination of intracellular Bgl2. Bgl2 is secreted rapidly postsynthesis and nearly undetectable in the spheroplasts of wild-type cells. All three cofilin ts alleles accumulated Bgl2 to similar levels as a sec14-1 ts strain known to block transport of Bgl2 (Figure 1A; Curwin et al., 2009). We also tested these cofilin mutants at permissive temperatures of 25 and 30°C. The sec14-1 control only accumulated Bgl2 at the nonpermissive temperature of 37°C, whereas all cofilin ts alleles exhibited a Bgl2 transport defect regardless of temperature (Figure 1A). To discard the possibility of differences in protein translation, we also examined total and extracellular Bgl2. There was no reduction in the extracellular levels of Bgl2 in cofilin mutants or the sec14-1 control, as Bgl2 is highly abundant in the cell wall, and a complete block in transport over a long period of time would be required to see reduction in the steady-state extracellular levels (Supplemental Figure S1A). Total Bgl2 was determined in nonspheroplasted cells, and overall levels of Bgl2 were virtually unaltered. A slight increase in total Bgl2 compared with controls was observed only at 25°C for cof1-8, cof1-5, and cof1-22, but this did not account for the accumulated internal Bgl2 (Supplemental Figure S1A). Other non–temperature-sensitive alleles, which affect the function of cofilin to a lesser extent, were also tested. For example, two alleles cof1-18 and cof1-19 that do not affect in vivo actin organization or growth (Lappalainen et al., 1997) also did not affect Bgl2 transport (Supplemental Figure S1B). It has been shown the cof1-19 allele affects actin patch mobility in vivo, and the mutant protein has defects in PIP2 and G-actin binding in vitro, whereas the cof1-18 mutant protein has no characterized defects (Lappalainen et al., 1997; Ojala et al., 2001; Lin et al., 2010). One allele, cof1-4, which does not affect growth but affects actin organization (to lesser extent than the ts alleles; Lappalainen et al., 1997), did accumulate Bgl2 (Supplemental Figure S1B). The cof1-4 allele contains a serine-to-alanine mutation in the conserved serine residue known to regulate cofilin function in higher eukaryotes. Together these data indicate that cofilin activity is required for Bgl2 transport. Mildly perturbing cofilin function was not sufficient to result in Bgl2 accumulation, and this phenotype was only observed in those alleles severe enough to lead to noticeable changes in the actin cytoskeleton (Figure 1 and Supplemental Figure S1). We chose to focus on the cof1-8 allele at 30°C for further analysis to maintain a background with relatively high reduction in cofilin activity but less effect on endocytosis compared with cof1-5 and cof1-22, which reportedly exhibit more endocytic problems, even at permissive temperatures (Lappalainen and Drubin, 1997; Okreglak and Drubin, 2007). We first determined that the defect in Bgl2 transport in cof1-8 cells could be restored by exogenous expression of wild-type COF1 (Figure 1B).
FIGURE 1:
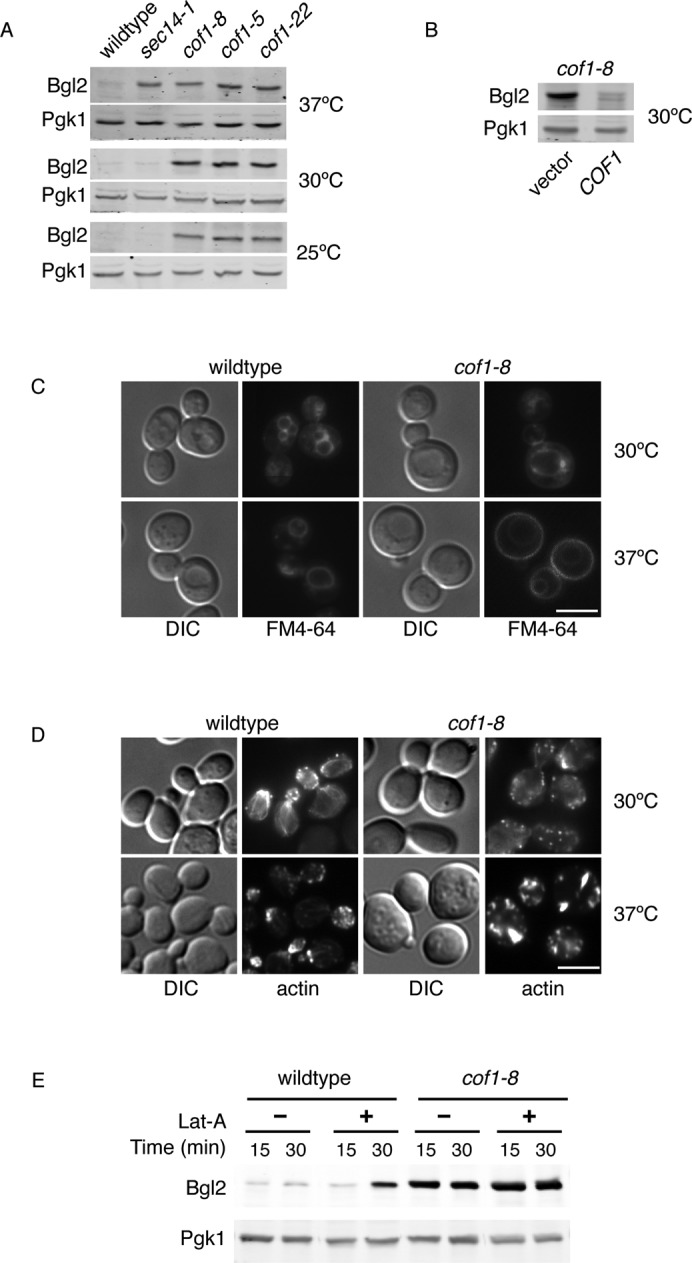
Cofilin and actin are required for proper secretion of Bgl2. (A) Wild-type, sec14-1, and the indicated cofilin mutant cells were grown to logarithmic phase at 25 or 30°C. Cells grown at 25°C were also shifted to 37°C for 2 h. Equal cell numbers were treated to generate spheroplasts, washed to remove the cell wall, and analyzed by Western blot to determine the amount of intracellular Bgl2. (B) Wild-type and cof1-8 cells expressing empty vector or COF1 were grown at 30°C and treated as in A to determine the intracellular levels of Bgl2. (C) FM4-64 transport was determined in live cells grown at 30°C or preshifted to 37°C for 1 h. Cells were labeled with 40 μM FM4-64 for 15 min (at 30 or 37°C, respectively). Cells were washed and resuspended in fresh, prewarmed YPD and further incubated at 30 or 37°C for 45 min. Scale bar, 5 μm. (D) Actin organization was examined by staining with phalloidin. Cells were grown at 30°C to logarithmic phase and either fixed directly in 3.7% formaldehyde or shifted to 37°C for 1 h before fixation. Scale bar, 5 μm. (E) Wild-type and cof1-8 cells were grown to 30°C and treated with 100 μM latrunculin A (Lat-A) or dimethyl sulfoxide (DMSO). Equal cell numbers were taken at the indicated times, and spheroplasts were generated and analyzed by Western blot for detection of Bgl2.
To examine the effect of cof1-8 in endocytosis at the permissive temperature of 30°C, we analyzed the trafficking of the lipophilic dye FM4-64 from the plasma membrane to the vacuole via endosomes. Cells were incubated with FM4-64 for 15 min, washed, and incubated for 45 min to examine the rate of transport to the vacuole via the endosomes. As expected, at the nonpermissive temperature of 37°C, the cof1-8 mutant failed to internalize the majority of FM-464. However there was no defect in the internalization or the rate of FM-464 transport at the permissive temperature of 30°C (Figure 1C). Next we determined the organization of the actin cytoskeleton in cof1-8 cells compared with wild type at 30 and 37°C by staining fixed cells with phalloidin. Wild-type cells, regardless of temperature, contained actin patches mostly in the daughter (budding) cell; filamentous actin cables were often observed traversing the mother–bud axis; however, these were less pronounced at 37°C. In cof1-8 cells at 37°C, the actin cytoskeleton was clearly perturbed, with fewer actin patches that were much larger and depolarized. No actin cables were visible, but instead thick actin structures were observed in nearly all cells. In cof1-8 cells grown at 30°C, the actin cytoskeleton was much less affected than at 37°C, and although depolarized, it was fairly normal in overall appearance. (Figure 1D). Latrunculin A (Lat-A) blocks actin polymerization and therefore counteracts the activity of cofilin. There was no change in intracellular accumulation of Bgl2 in cof1-8 cells treated with Lat-A (Figure 1E). On the other hand, 30 min of treatment with Lat-A treatment in wild-type cells resulted in Bgl2 accumulation, indicating that general perturbations in actin dynamics affect Bgl2 export from the Golgi membranes. This is not surprising, as Lat-A treatment has been shown to result in the intracellular accumulation of invertase and post–Golgi compartment vesicles (Novick and Botstein, 1985; Karpova et al., 2000).
A reduced rate of endocytosis does not affect Bgl2 export from the Golgi membranes
To further address the connection between cofilin's roles in regulation of endocytosis and secretion, we examined Bgl2 transport in a number of mutants defective in endocytosis. Abp1 and Aip1 are two proteins that function intimately with cofilin in the formation of actin patches and the regulation of endocytosis. Abp1 localizes to sites of endocytosis before cofilin and is required to activate the Arp2/3 complex (Okreglak and Drubin, 2007). Aip1 interacts directly with cofilin and is required to restrict cofilin localization to cortical patches and promote actin filament turnover (Clark and Amberg, 2007; Lin et al., 2010). As before, intracellular accumulation of Bgl2 was determined in spheroplasted cells. Deletion of either of these proteins did not lead to intracellular accumulation of Bgl2 (Figure 2A). We also examined the secretion of Bgl2 in other endocytic mutants that do not interact directly with cofilin at actin patches but are reportedly blocked in all forms of endocytosis, including receptor-mediated endocytosis, fluid-phase endocytosis, and FM4-64 transport (Engqvist-Goldstein and Drubin, 2003). These included endocytic mutants that function directly at sites of endocytosis (Sla1, Sla2, Inp51, and End3) and endocytic mutants that block endocytosis indirectly by affecting ergosterol synthesis (Erg2, Erg3, and Erg6). Sla1 binds actin and End3, and these are required for cortical actin assembly and endocytosis (Tang et al., 1997, 2000; Warren et al., 2002); Sla2 is a transmembrane protein that links actin and clathrin at sites of endocytosis (Wesp et al., 1997; Yang et al., 1999); Inp51 is a PIP2 phosphatase of the synaptojanin family that regulates PIP2 homeostasis at the plasma membrane (PM) and is required for endocytosis (Singer-Kruger et al., 1998); Erg2 is a sterol isomerase catalyzing an intermediate step in ergosterol biosynthesis (Ashman et al., 1991); Erg3 is a sterol desaturase, generating a precursor in ergosterol biosynthesis (Arthington et al., 1991); and Erg6 is a methyltransferase converting zymosterol to fecosterol in the ergosterol biosynthetic pathway and acts upstream of both Erg2 and Erg3 (Lees et al., 1995; Parks et al., 1995). Of these seven endocytic mutants, only two, erg6∆ and sla2∆, exhibited accumulation of Bgl2 (Figure 2A). Of interest, Sla2 is a clathrin adapter, linking clathrin and actin, and sla2∆ cells have been shown to accumulate post–Golgi compartment vesicles (Mulholland et al., 1997). The human Sla2 homologue (Hip1R) has also been implicated in transport from the TGN (Carreno et al., 2004). Erg6 is an enzyme required for ergosterol synthesis, and perturbing ergosterol levels would alter lipid homeostasis in the Golgi membranes, which is known to affect secretion of some cargoes (Proszynski et al., 2005; Klemm et al., 2009). We suggest that the accumulation of Bgl2 in erg6∆ and sla2∆ mutant cells is likely due to direct effects on the function of the Golgi apparatus. Taken together, these data strongly indicate that the defect in Bgl2 secretion in cof1-8 cells at 30°C is not due to a defect in endocytosis.
FIGURE 2:

Endocytosis is not required for proper secretion of Bgl2. (A) The indicated endocytosis-defective strains were grown at 30°C to logarithmic phase, spheroplasts were generated, cell walls were removed, and intracellular Bgl2 was examined by Western blot. (B) Snc1-GFP localization was examined in live cells grown to logarithmic phase at 30°C. Scale bar, 5 μm.
To further assess defects in endocytosis in cof1-8 cells at 30°C, we examined the localization of the vesicle soluble N-ethylmaleimide–sensitive factor attachment protein receptor (v-SNARE) Snc1. Snc1 is packaged into both HDSV and LDSV transport carriers for export from the Golgi apparatus (Gurunathan et al., 2002). After reaching the PM, Snc1 is internalized via endosomes, recycled back to the TGN, and then transported to the PM (Lewis et al., 2000). As expected, at steady state, in wild-type cells Snc1-GFP was visible at the PM of the growing bud and internal structures previously identified as TGN and endosomes (Figure 2B; Robinson et al., 2006). However, in cof1-8 cells at 30°C Snc1-GFP was only visible in the PM, and the localization was no longer polarized to the growing bud but instead was distributed uniformly in the PM of the growing cells (Figure 2B). This implies that the rate of endocytosis and polarized secretion is affected in cof1-8 cells at 30°C. This is not surprising, since the actin cytoskeleton is also depolarized at this temperature (Figure 1D). Snc1–green fluorescent protein (GFP) localization is often used to assess the rate of endocytosis, and a PM-only localization is a hallmark of a defect in endocytosis. However, when we examined the localization of the Snc1-GFP in the other endocytic mutants described, we observed varying phenotypes. Snc1-GFP localization was similar to cof1-8 cells in end3∆ and sla2∆ cells: only PM and depolarized. A similar phenotype was observed in sla1∆ cells but to a lesser extent, with some internal punctate structures still visible and a polarized localization. In inp51∆ cells, Snc1-GFP localization was almost normal, with occasional depolarization. Of interest, erg3∆ and erg6∆ cells exhibited more internal Snc1-GFP localization compared with wild type, and erg3∆ cells had even less signal of Snc1-GFP at the PM than erg6∆. However, erg3∆ and erg6∆ cells still retained a polarized localization of Snc1-GFP, whereas erg2∆ cells were virtually indistinguishable from wild type. The two deletion strains defective in Bgl2 transport, sla2∆ and erg6∆, exhibited opposite Snc1-GFP localizations; wild type–like localization in erg6∆ cells, and PM only in sla2∆ cells. Steady-state Snc1-GFP localization is dependent on the rate of export from the Golgi membranes, the rate of endocytosis, and the rate of retrieval from endosomes. Clearly a decreased rate of endocytosis does not always lead to a PM-only localization of Snc1-GFP, as observed in inp51∆, erg2∆, erg3∆, and erg6∆ cells. Loss of function of proteins affecting multiple steps in the Snc1-GFP cycling (which is likely the case for Sla2, Erg6, and cofilin) leads to varying phenotypes.
Bgl2 rate of transport is decreased and accumulates in a late-Golgi compartment in cof1-8 cells at 30°C
To determine more precisely whether the rate of Bgl2 transport was affected in cof1-8 cells and to control for a potential increase in rate of Bgl2 synthesis in cof1-8 cells, we performed a pulse chase and immunoprecipitation of newly synthesized Bgl2 in intracellular versus extracellular fractions. Wild type and cof1-8 and cells were labeled at 30°C with [35S]methionine for 5 min and chased with 50 mM methionine for 0, 10, and 30 min (sec14-1 cells were labeled in the same way but at 37°C, with a 30-min pre-shift to this temperature). In wild-type cells Bgl2 is clearly secreted much faster when compared with cof1-8 and sec14-1 cells (Figure 3A). Results of three independent experiments were quantitated, and the percentage of internal Bgl2 compared with total (intracellular and extracellular) was plotted over time. At time 0 wild-type cells had 90.0% Bgl2 internal, compared with 95.3 and 94.1% for cof1-8 and sec14-1 cells, respectively, indicating that during the 5-min pulse period wild-type cells secreted ∼5% more than either mutant. By 10 min postchase, wild-type cells contained 46.3% internal Bgl2, compared with 66.9% in cof1-8 cells and 81.8% in sec14-1 cells, and by 30 min postchase, the difference in the rate of Bgl2 secretion was even more pronounced, with wild-type cells only retaining 12.1% of total Bgl2 internally, compared with 49.2 and 62.4% in cof1-8 and sec14-1 cells, respectively (Figure 3B). Therefore the accumulation of internal Bgl2 observed in cof1-8 cells is due to a decreased rate of transport and not to increased Bgl2 expression in cof1-8 cells compared with wild type.
FIGURE 3:

Bgl2 rate of transport is decreased and accumulates in a late Golgi compartment. (A) Bgl2 rate of transport was determined in wild-type and cof1-8 cells at 30°C and sec14-1 cells at 37°C by pulse-chase labeling of newly synthesized protein with [35S]methionine. Equal cell numbers were labeled with 100 μCi/time point, spheroplasts were generated, and Bgl2 was immunoprecipitated from intracellular and extracellular fractions at the indicated times and detected by SDS–PAGE/autoradiography. (B) Results of three independent Bgl2 pulse-chase and immunoprecipitations were quantitated, and the sum of internal and external fractions at a given time point was taken as 100%. The percentage internal Bgl2 was plotted vs. time using GraphPad Prism software. Error bars represent SEM. (C) Wild-type, cof1-8, and bgl2Δ cells were grown to logarithmic phase at 30°C. Cells were fixed in formaldehyde, spheroplasted, and permeabilized for indirect immunofluorescence staining with anti-Bgl2 antibody (green). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue), and the samples were slightly overexposed to clearly show the cells in wild type and bgl2Δ. Scale bar, 3 μm. (C) cof1-8 cells were grown at 30°C expressing the indicated Golgi membrane (Sec7-DsRed, trans-Golgi membrane; Anp1–red fluorescent protein [RFP], early Golgi membrane) and endosome (mCherry-Pep12 and mCherry-Tlg1) markers. Cells were treated as in B for immunofluorescence staining with anti-Bgl2 antibody (green) and anti-RFP (red) to detect the marker protein. Stacks of images were taken and treated with deconvolution software to reduce the background signal. Scale bar, 3 μm.
To determine the intracellular location of the Bgl2 in cof1-8 cells, we tested its location with respect to ER and Golgi membranes. Wild-type and cof1-8 cells grown at 30°C were spheroplasted and homogenized, and the membrane-bound compartments were fractionated to equilibrium on a continuous 30–60% sucrose gradient. The accumulation of Bgl2 in cof1-8 cells compared with wild type was readily reproducible in the fractioned membranes, and ER and Golgi membranes were separated by this method. The Bgl2-containing fractions were clearly excluded from the ER and followed a Golgi membrane–like distribution. The results of three independent gradients were compiled and plotted (Supplemental Figure S2). To further ascertain the location of Bgl2 in cof1-8 cells, we visualized endogenous Bgl2 by immunofluorescence microscopy using a purified anti-Bgl2 antibody. Cells were grown at 30°C and fixed in formaldehyde, and extracellular Bgl2 was removed by digestion of the cell wall before permeabilization and detection by immunofluorescence. In cof1-8 cells, Bgl2-containing punctate structures were easily observed. Such structures were only evident in wild-type cells with increased exposure, whereas bgl2∆ control cells contained no such elements (Figure 3C). Colocalization with Golgi membranes and endosome markers in cof1-8 cells revealed Bgl2 in a compartment closely apposed to the late Golgi membrane–specific marker Sec7-RFP, whereas no such proximity was observed with the early Golgi membrane marker Anp1-RFP or the endosome marker Pep12-RFP or Tlg1-RFP (Figure 3D). Together these data indicate that Bgl2 accumulates in cof1-8 cells in a late Golgi compartment.
Cargo-selective effect on secretion in cof1-8 cells
To determine whether cofilin affects protein secretion in general, we asked whether the cof1-8 allele affected total secretion of proteins into the medium at the permissive temperature of 30°C. Wild-type and cof1-8 cells were labeled with [35S]methionine for 10 min, and this pulse medium was replaced with chase medium containing 50 mM methionine to clearly see the rate of protein transport during 10-, 30-, and 60-min chase periods. Cells with a ts allele of the gene encoding N-ethylmaleimide (NEM)–sensitive factor, sec18-1, were cultured in the same way but shifted to the nonpermissive temperature of 37°C at 1 h before and during the pulse chase as a control to block secretion (Graham and Emr, 1991). There were no obvious changes in the total levels of proteins secreted, indicating that cof1-8 cells do not exhibit a general block in protein secretion. There were some minor differences between the proteins secreted into the medium from wild-type and cof1-8 cells at the 30-min time point, with three bands present in higher amounts (labeled +) and four bands present in lower amounts (labeled –); however, these differences were virtually undetectable at the 60-min time point, indicating there may be differences in the rate of secretion of some proteins but not in the overall amounts secreted (Figure 4A).
FIGURE 4:
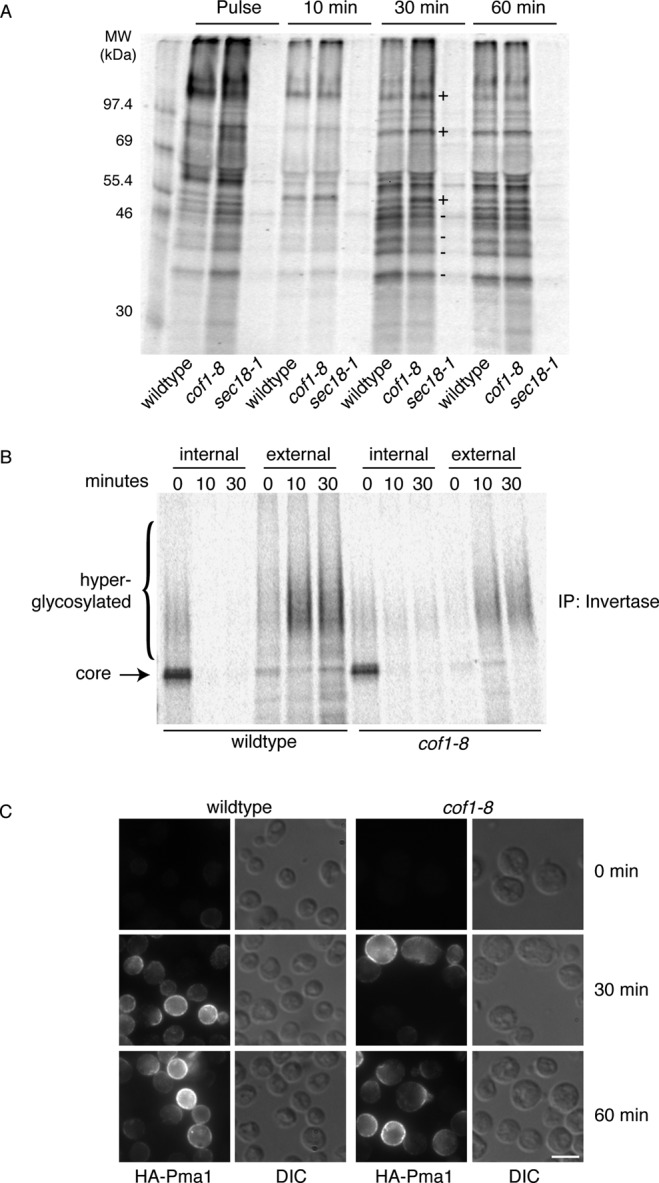
General secretion is not blocked in cof1-8 cells, but some cargoes are specifically affected. (A) Proteins secreted in the medium of wild-type and cof1-8 cells grown at 30°C and sec18-1 cells grown at 30°C and preshifted to 37°C for 1 h were detected by pulse-chase labeling of newly synthesized proteins. Cells were labeled with 150 μCi/ml [35S]methionine for 10 min. The pulse medium was removed and replaced with chase medium containing 50 mM methionine for 10, 30, and 60 min. Secreted proteins in the medium were precipitated with TCA and detected by SDS–PAGE/autoradiography. Bands labeled (+) are secreted at higher rates and bands labeled (–) are secreted at lower rates in cof1-8 cells. These differences were much less pronounced at 60 min. (B) Invertase rate of transport was determined in wild-type and cof1-8 cells at 30°C by pulse-chase labeling of newly synthesized protein with [35S]methionine. Cells were cultured in SC-methionine and 0.1% glucose for 15 min before (and during) labeling, equal cell numbers were labeled with 100 μCi/ time point, spheroplasts were generated, and invertase was immunoprecipitated from intracellular and extracellular fractions at the indicated times and detected by SDS–PAGE/autoradiography. (C) Transport of newly synthesized Pma1 at 30°C was determined in cells expressing Pma1-HA under the control of the MET25 methionine-repressible promoter. Synthesis was induced by washing cells free of methionine, and samples were fixed, spheroplasted, and permeabilized at the indicated time points for indirect immunofluorescence staining with anti-HA antibody. Scale bar, 5 μm.
To further determine cofilin's ability to affect export of proteins from the Golgi membranes, we tested a number of other cargoes directly that are transported by various routes from the Golgi apparatus. Invertase, a soluble enzyme of the periplasmic space, is secreted in response to glucose deprivation and known to be transported by HDSV (Harsay and Bretscher, 1995). The activity of invertase was measured in intact cells (extracellular pool) versus lysed cells (total) to determine the percentage of invertase secreted by cells starved of glucose for 2 h. Wild-type cells secreted 82% of invertase, whereas sec14-1 cells (grown and starved at nonpermissive temperature), known to block transport of this enzyme, exhibited 36% secretion (Bankaitis et al., 1989). However, cof1-8 cells exhibited a modest decrease in invertase secretion, to 69%, and this was only marginally statistically significant (p = 0.04; Supplemental Figure S3A). To determine more precisely the rate of transport of invertase, we performed pulse-chase and immunoprecipitation of newly synthesized invertase. After 15 min of glucose deprivation, cells were labeled with [35S]methionine for 5 min and then incubated with 50 mM methionine for 0, 10, and 30 min. Invertase was then immunoprecipitated from intracellular and extracellular fractions. Invertase is core glycosylated in the ER and is hyperglycosylated within the Golgi apparatus. In wild-type cells invertase was completely secreted in a hyperglycosylated form by 10 min postchase, with no detectable internal invertase. The cof1-8 cells exhibited a decrease in the amount of secreted invertase, and a very small amount of hyperglycosylated invertase was still present intracellularly at 10 and 30 min postchase (Figure 4B). This correlates well with the slight decrease we observed by measuring activity after 2 h of glucose deprivation and also indicates that this effect is due to a decrease in the rate of transport at the level of the Golgi apparatus.
The plasma membrane H+ ATPase Pma1 is transported from the TGN via LDSV (Harsay and Bretscher, 1995). We expressed Pma1-GFP in wild-type and cof1-8 cells and visualized its location by fluorescence microscopy. As expected, in wild-type cells Pma1-GFP localized uniformly in the plasma membrane. In cof1-8 cells grown at 30°C the localization of Pma1-GFP was unaffected, indicating that there is not a complete block in the LDSV pathway and no missorting of Pma1-GFP to the vacuole (Supplemental Figure S3B). However, as Pma1 is very stable at the PM, it is possible there are differences in the rate of transport not detectable by this method. Therefore we examined the localization of hemagglutinin (HA)-Pma1 under the control of MET25 promoter by immunofluorescence microscopy. HA-Pma1 reaches the PM after 90 min of derepression in methionine-free medium (Luo and Chang, 2000), and therefore we examined the localization after 30 and 60 min. Unfortunately, we were unable to detect intermediate steps (ER and Golgi membranes) of HA-Pma1 transport by this method, which was likely complicated by the fact that under repressing conditions a very low level of HA-Pma1 was detectable (even when double repressing amounts of methionine were used), and this was more pronounced in the wild-type cells (Figure 4E). Similarly, upon derepression, the overall expression level of HA-Pma1 was somewhat higher in wild-type cells compared with cof1-8 cells. Despite these slight differences in the expression of HA-Pma1, there was no major difference in the rate of transport of newly synthesized HA-Pma1, as both 30- and 60-min time points showed mostly PM staining in wild-type and cof1-8 cells, although we cannot exclude the possibility of a minor kinetic delay that could not be detected by this method (Figure 4C). Hxt1-CFP and Can1 are transmembrane proteins of the plasma membrane, and whereas the route for their transport from the TGN to the cell surface is not known, there was no defect in their trafficking in cof1-8 cells when compared with wild type (unpublished data). These findings indicate that the cof1-8 allele affects protein sorting at the Golgi apparatus, since the rate of transport of some cargoes leaving the Golgi membrane was affected without a general block in secretion or in the trafficking of a specific subset of Golgi membrane–derived vesicles destined to the cell surface.
The role of Pmr1 in cofilin-dependent Bgl2 secretion
Pmr1 is the yeast orthologue of SPCA1, a Ca2+/Mn2+ ATPase localized to the Golgi apparatus (Antebi and Fink, 1992). Yeast cells lacking Pmr1 display various phenotypes associated with Golgi apparatus dysfunction, including secretion of heterologous yeast/mammalian proteins normally retained internally, secretion of CPY, and a defect in glycosylation (Rudolph et al., 1989; Antebi and Fink, 1992; Durr et al., 1998; Bonangelino et al., 2002). We previously identified SPCA1 as a downstream component of cofilin/actin–dependent protein sorting in human cells (von Blume et al., 2011). We therefore asked whether Pmr1 in yeast cells was also involved in cofilin-dependent cargo sorting from the late Golgi membranes. First we examined transport of CPY in pmr1∆ and cof1-8 cells. CPY is transported from the ER to the vacuole via the Golgi apparatus (Valls et al., 1987). Secretion of CPY extracellularly can occur if there is a block or delay in CPY transport from the late Golgi compartment to vacuole via endosomes, a phenotype previously reported for cof1-22 cells (Okreglak and Drubin, 2007) and pmr1∆ cells (Bonangelino et al., 2002). We tested whether cof1-8 cells grown at 30°C secreted CPY. Yeast harboring the indicated mutations and the wild-type cells were grown on solid medium on nitrocellulose filters for 48 h. Filters were removed and tested for the presence of secreted CPY by Western blotting. Both pmr1∆ and cof1-8 cells secreted CPY, similar to the vacuolar protein-sorting mutants vps4∆ and vps27∆, whereas wild-type cells did not (Figure 5A). CPY transport from the ER to Golgi apparatus results in the conversion from p1 to p2 form of CPY, and further transport to the vacuole leads to proteolytic processing of CPY to the mature form (Valls et al., 1987). The rate of CPY transport was assessed in cof1-8 and pmr1∆ cells grown at 30°C by pulse-chase analysis. Cells were labeled for 5 min with [35S]methionine and chased with 50 mM methionine for 0, 10, and 20 min, and CPY was immunoprecipitated from the lysates. Trafficking and processing of CPY occurs rapidly in wild-type cells, and by 10 min postchase all CPY was processed to the mature, vacuolar form. In cof1-8 cells, CPY trafficking was delayed, and at the 10-min time point all three forms were detected (Figure 5B). However, by 20 min postchase nearly all CPY was found in the mature form in cof1-8 cells. The decreased rate of CPY transport was even more pronounced in pmr1∆ cells, and at the 20-min time point a significant amount of CPY was still in the unprocessed form (Figure 5B). Therefore this decrease in the rate of CPY transport leads to its secretion from the Golgi membranes, further indicating a defect in protein sorting and not complete block of transport from the Golgi membranes in cof1-8 cells.
FIGURE 5:

cof1-8 and pmr1∆ cells have a synthetic sickness and share protein-sorting phenotypes. (A) Yeast cells were grown in YPD to logarithmic phase, and equal cell numbers were spotted on YPD solid medium and allowed to dry, and a nitrocellulose filter was overlaid for 48 h at 30°C. Secreted CPY was detected on the filter by Western blot against CPY. (B) CPY transport at 30°C was examined in wild-type, cof1-8, and pmr1∆ cells by pulse-chase labeling of newly synthesized protein with [35S]methionine. Equal cell numbers were labeled with 100 μCi/time point, and CPY was immunoprecipitated from lysates at the indicated times and detected by SDS–PAGE/autoradiography. (C) The indicated strains were grown to logarithmic phase at 25°C, and equal cell numbers were serially diluted and spotted on YPD medium and further incubated at 25 or 30°C for 48 h. (D) Yeast strains expressing the indicated vectors were grown to logarithmic phase, and equal cell numbers were serially diluted and spotted on selection medium to maintain plasmids and incubated 48 h at 30°C. (E, F) Cells were grown to logarithmic phase at 30°C, and equal cell numbers were treated to generate spheroplasts and washed to remove the cell wall; spheroplasts were analyzed by Western blot to determine the amount of intracellular Bgl2. Quantitation of Bgl2 was performed and normalized to Pgk1 loading control. Amounts relative to wild type were determined for three independent experiments, analyzed, and plotted using GraphPad Prism software. Error bars represent SEM.
Then we tested whether cofilin and PMR1 exhibit a genetic interaction. Double-mutant pmr1∆ cof1-8 strains were generated and tested by serial dilution for growth. There was a slight growth defect in the double mutant compared with single mutants at 25°C, and at 30°C a strong synthetic sickness was observed (Figure 5C). To test the specificity of the genetic interaction, double-mutant pmc1∆ cof1-8 strains were generated. Pmc1 is a homologue of Pmr1 that localizes to the vacuole (Cunningham and Fink, 1994). Pmc1 and Pmr1 can functionally complement one another, as either single deletion is viable, but a strain lacking both Pmr1 and Pmc1 is not viable (Cunningham and Fink, 1994). Cells with pmc1∆ cof1-8 did not exhibit any synthetic sickness as observed in pmr1∆ cof1-8 cells (Figure 5C). Furthermore, the growth defect in pmr1∆ cof1-8 cells at 30°C could be restored by expression of wild-type COF1 and/or PMR1 (Figure 5D).
The cofilin mutants and pmr1∆ cells secrete CPY. Therefore we examined secretion of Bgl2 and invertase in pmr1∆ cells, as described. Secretion of invertase was only mildly affected in cof1-8 cells, at 69 compared with 82% in wild-type cells (Figure 4B). Similarly, in pmr1∆ cells invertase was unaffected, secreting 79% (unpublished data). Bgl2 secretion was also affected in pmr1∆ cells, although not to the extent observed in cof1-8 cells (Figure 5E). However, statistical analysis of multiple experiments revealed that the increased intracellular Bgl2 accumulation observed in pmr1∆ cells was slightly significant (Figure 5E). In addition, in the cof1-8 pmr1∆ cells the accumulated Bgl2 was increased, and this was also statistically significant compared with cof1-8 cells alone (Figure 5E). pmr1∆ cells display a compensatory increase in PMC1 expression, and Pmc1 localizes not only to the vacuole, but also to Golgi membranes, where it aids in maintaining calcium homeostasis in the absence of Pmr1 (Marchi et al., 1999). If Bgl2 transport is dependent on proper calcium levels in the Golgi apparatus and these are only partially perturbed in pmr1∆ due to Pmc1 activity, then this could account for the milder accumulation of Bgl2 in pmr1∆ cells compared with cof1-8. The increased accumulation of Bgl2 observed in cof1-8 pmr1∆ cells indicates that cofilin still functions to regulate calcium homeostasis of the Golgi apparatus in the absence of Pmr1, and this is likely through the relocalized Pmc1. In addition, we found that overexpression of exogenous PMR1 in cof1-8 cells could restore the transport of Bgl2 (Figure 5F), further supporting that Pmr1 acts downstream of cofilin in regulating transport of Bgl2.
DISCUSSION
Our findings reveal the requirement of cofilin for the secretion of the cell wall–specific protein Bgl2 from the late Golgi compartment. Mutant cofilin (cof1-8 allele) also affected the sorting of CPY from the Golgi membranes, similar to the finding in mammalian cells, in which cofilin knockdown led to cathepsin D secretion (von Blume et al., 2009). Many other proteins were exported from the late Golgi membranes normally in cof1-8 cells, although some differences in the rate of transport were observed, again mimicking the observed phenotype in mammalian cells (von Blume et al., 2009). As cofilin is essential, we must work with mutant alleles, which obviously retain some function. This may account for the fact that we do not see more protein-sorting events affected in cof1-8 cells. The situation is further complicated in yeast, as it is known that they can reroute a number of cargoes between different secretory pathways when one is not available. For example, invertase, which normally follows the HDSV-clathrin–dependent pathway, can be transported by the LDSV when necessary, and similarly, when CPY cannot be transported to the vacuole by the HDSV pathway, it is secreted by the LDSV pathway (Gurunathan et al., 2002). We observed a mild defect in the secretion of invertase in cof1-8 cells, which may be a reflection of this rerouting, as CPY transport was also affected in cof1-8 cells. The following data suggest a direct role for cofilin in the selective sorting/export of the secretory cargo from the late Golgi compartment:
Bgl2 and CPY trafficking is affected in the cof1-8 mutant even at the permissive temperature of 30°C. At this temperature, there is a milder defect in endocytosis, since FM4-64 was endocytosed at a normal rate in cof1-8 cells, although the steady-state localization of the v-SNARE Snc1-GFP was altered and mostly visible at the plasma membrane.
Many of the endocytic mutants have no effect on the trafficking from the Golgi apparatus to the cell surface and display varying Snc1-GFP localizations, indicating that defective endocytosis cannot account for the sorting defects in cof1-8 mutants. The two proteins that show a clear phenotype for transport of Bgl2 are Sla2 and Erg6. Sla2 and the mammalian orthologue Hip1R have been implicated in export of cargo at the TGN (Mulholland et al., 1997; Carreno et al., 2004). Sterols are important for trafficking out of the Golgi membranes in both mammalian and yeast cells, and the requirement of Erg6 in Bgl2 export is further proof of this requirement in selective cargo export from the Golgi apparatus (Proszynski et al., 2005).
Bgl2 and Pma1 are transported in the same class of vesicles from the Golgi apparatus to the cell surface (Harsay and Bretscher, 1995). The fact that cof1-8 does not affect trafficking of Pma1 strongly suggests that the defect is in the sorting and/or packing of Bgl2 and not in the budding or the departure of the specific vesicles from the late Golgi compartment.
Bgl2 rate of transport is decreased, and it is retained in a late Golgi compartment, which further supports our proposal that cofilin is required for the sorting of secretory cargo in the late Golgi membranes.
Cofilin and Pmr1 function in the same sorting pathway at the Golgi apparatus
Pmr1 was first identified in a screen for “supersecreting” mutants based on elevated secretion levels of heterologously expressed proteins (Smith et al., 1985). The protein was then characterized as a Ca2+ ATPase localized to the Golgi apparatus and identified as orthologue of human SPCA1 (Antebi and Fink, 1992; Durr et al., 1998; Ton et al., 2002). Defects associated with loss of Pmr1 in yeast can be complemented by expression of human SPCA1 in yeast, indicating a conserved function in eukaryotic cells for the Ca2+/Mn2+ ATPases of the Golgi membranes (Sorin et al., 1997; Ton et al., 2002). In addition, we recently showed SPCA1 functions in a cofilin/actin–dependent sorting mechanism of secretory cargo at the TGN in mammalian cells (von Blume et al., 2011).
Pmr1 was shown to affect vacuolar protein sorting, and loss of PMR1 and cof1-8 show synthetic sickness, which affirms the role of these two proteins in a similar pathway. Moreover, pmr1∆ cof1-8 double mutants show a more severe effect on the secretion of Bgl2 from the late Golgi membranes compared with that observed in either of the single mutants alone. Taken alone, these findings are confusing, as we would not expect an increase Bgl2 accumulation if Pmr1 acts downstream of cofilin. Similarly, we would expect at least the same defect in Bgl2 transport in pmr1∆ cells as is observed for cof1-8. The vacuolar Ca2+ ATPase Pmc1 forms an essential pair with Pmr1, indicating that loss of both calcium pumps leads to lethal mis-maintenance of overall calcium homeostasis (Cunningham and Fink, 1994; Marchi et al., 1999). Of interest, it has been shown that a portion of Pmc1 is retained in the Golgi membranes when Pmr1 is not present, and this could account for the mild effect in Bgl2 transport observed in pmr1∆ cells. cof1-8 and pmc1∆ do not show a synthetic genetic interaction as observed in cof1-8 pmr1∆ cells, although Pmc1 can aid in maintaining Golgi membrane calcium homeostasis and may be able to function downstream of cofilin in regulating sorting of Bgl2 from the TGN, as cof1-8 pmr1∆ cells showed an increased accumulation of Bgl2 compared with either individual mutant. The fact that overexpression of PMR1 in cof1-8 cells rescued the Bgl2 secretion defect strongly indicates the involvement of Pmr1 downstream of the cofilin-dependent cargo sorting in the Golgi membranes.
It is noteworthy that Pma1 and Bgl2, which are transported in the same carriers from Golgi membranes to the cell surface, have different requirements for their sorting: the sorting of Pma1 is pH dependent, whereas Bgl2 export depends on Ca2+ (Huang and Chang, 2011). This highlights the importance of ion homeostasis in the sorting of specific secretory cargo in the late Golgi compartments.
Taken together, these new findings suggest that the function of cofilin/actin and the Ca2+ ATPase Pmr1/SPCA1 in secretory cargo sorting in the late Golgi membranes is conserved in eukaryotic cells. The obvious next challenge is to identify events that are triggered by the cofilin- and Pmr1/SPCA1-dependent Ca2+ import into the late Golgi compartments. Does binding of Ca2+ change the properties of specific cargoes and enable their packing into transport carriers destined to the cell surface? Is there a specific protein (receptor) that binds specific cargoes in a Ca2+-dependent manner and packs them into the budding transport carriers? What is the precise role of actin in this process? Is it required for the activation of Pmr1 or generation of a specific domain that sequesters the secretory cargo sorting machinery? Yeast is a good system to search for proteins involved specifically in the export of Bgl2 and could reveal the identity of components of the Ca2+-dependent cargo sorting in the late Golgi membranes.
MATERIALS AND METHODS
Media
Yeast cells were grown in rich YPD (1% yeast extract, 2% peptone, and 2% glucose) or synthetic complete (SC) media (0.67% yeast nitrogen base, 2% glucose supplemented with amino acid drop-out mix from Sigma-Aldrich [St. Louis, MO; lacking uracil, histidine, leucine and tryptophan] or Clontech [Mountain View, CA; lacking methionine]), with required amino acids for auxotrophies and/or plasmid selection added as required.
Yeast strains and plasmid construction
Cofilin mutant strains were acquired from David Drubin (University of California, Berkeley) and sequenced to confirm the mutation. The cof1-8 mutant was not correct upon sequencing, and to maintain isogenic strains a cof1-8 strain in the background of BY4741 was obtained from Charlie Boone (University of Toronto, Toronto, Canada) and used for all analyses (see Table 1). Strains ACY4, ACY18, and ACY19 were generated by homologous recombination of two PCR-generated fragments, one containing 80 base pairs upstream of the cof1-8 allele to the STOP plus the beginning of the NatNT2 cassette from vector pYM17 (Janke et al., 2004) and a fragment containing the NatNT2 cassette plus 45 base pairs downstream of COF1 locus. Fragment 1 was amplified from genomic DNA from the cof1-8, KanMx4 strain using primers 5′-GGAAACAAGAAAAGACTGGTTAGCAACTAC-3′ and 5′-TAATTAACCCGGGGATCCGTCGACCTGCAGCGTACGTTAATGAGAACCAGCGCCTCTGCTGACTCT-3′ (COF1 sequence in bold). Fragment 2 was amplified using pYM17 as template and primers 5′-CGTACGCTGCAGGTCGACGGATCC-3′ and 5′-TTTCATTTTTCTTGAAGATTGTTGTCATTTGTGAAATCATTTACCATCGATGAATTCGAGCTCGATTAC-3′ (downstream COF1 sequence in bold). Strains were assessed for temperature sensitivity and confirmed by sequencing of the COF1 gene. To construct the COF1 plasmid, wild-type COF1 was amplified from genomic DNA from START to STOP using primers 5′-CTAGCTACTAGTATGTCTAGATCTGGGTATGC-3′ and 5′-CTAGCTCTCGAGTTAATGAGAACCAGCGCCTCTG-3′ (COF1 sequences in bold) and cloned into a pRS416 vector containing the ADH1 promoter and CYC1 terminator (provided by Sebastion Leon, Université Paris Diderot, Paris, France) using SpeI and XhoI restriction enzymes (New England BioLabs, Ipswich, MA).
TABLE 1:
Yeast strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3∆1 leu2∆0 ura3∆0 met15∆0 | EUROSCARF |
| BY4742 | MATα his3∆1 leu2∆0 ura3∆0 lys2∆0 | EUROSCARF |
| cof1-8 | BY4741 cof1-8, KanMx4 | Charlie Boone |
| Wild type | MATα cof1-4 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| cof1-4 | MATα cof1-4, LEU2 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| cof1-5 | MATα cof1-5, LEU2 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| cof1-22 | MATα cof1-22, LEU2 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| cof1-18 | MATα cof1-18, LEU2 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| cof1-19 | MATα cof1-19, LEU2 ura-52 his3Δ200 leu2-3, 112 lys2-801 ade2-101 | David Drubin |
| sla1∆ | BY4742 sla1∆::KanMx4 | EUROSCARF |
| sla2∆ | BY4742 sla2∆::KanMx4 | EUROSCARF |
| inp51∆ | BY4742 inp51∆::KanMx4 | EUROSCARF |
| end3∆ | BY4742 end3∆::KanMx4 | EUROSCARF |
| erg2∆ | BY4742 erg2∆::KanMx4 | EUROSCARF |
| erg3∆ | BY4742 erg3∆::KanMx4 | EUROSCARF |
| erg6∆ | BY4742 erg6∆::KanMx4 | EUROSCARF |
| Anp1-RFP | MATα ANP1-RFP, KanMx6 his3∆1 leu2∆0 ura3∆0 lys2∆0 | Erin O'shea (Harvard Cambridge, MA) |
| ACY4 | MATα ANP1-RFP, KanMx6 cof1-8, natNT2 his3∆1 leu2∆0 ura3∆0 lys2∆0 | This study |
| pmr1∆ | BY4742 pmr1∆::KanMx4 | EUROSCARF |
| pmc1∆ | BY4742 pmc1∆::KanMx4 | EUROSCARF |
| ACY18 | BY4742 pmr1∆::KanMx4 cof1-8, natNT2 | This study |
| ACY19 | BY4742 pmc1∆::KanMx4 cof1-8, natNT2 | This study |
| SF282-1D | MATa sec18-1 mal mel gal2 CUP1 SUC2 | Randy Schekman |
| CMY505 | BY4741 sec14-1, NatMx4 | Chris McMaster (Dalhousie University, Halifax, Canada) |
EUROSCARF, European Saccharomyces cerevisiae Archive for Functional Analysis, Institute for Molecular Biosciences, Johann Wolfgang Goethe-University Frankfurt, Frankfurt, Germany.
Bgl2 accumulation
Bgl2 accumulation was performed as described with minor modifications (Kozminski et al., 2006). Cells were cultured in YPD or SC to select for various plasmids at 30°C to logarithmic phase, and equal cell numbers were harvested by centrifugation at 2000 × g for 5 min. Cell pellets were resuspended in 10 mM NaN3/NaF solution and incubated on ice for 10 min, harvested at 10, 000 × g for 1 min, resuspended in fresh pre-spheroplasting buffer (100 mM Tris-H2SO4, pH 9.4; 50 mM β-mercaptoethanol; 10 mM NaN3; 10 mM NaF) and further incubated on ice for 15 min. Cells were harvested as before, washed once in spheroplast buffer without zymolyase (50 mM KH2PO4-KOH, pH 7.4; 1.4 M sorbitol; 10 mM NaN3), resuspended in spheroplast buffer containing 167 μg/ml zymolyase 100T (Seikagaku Biobusiness, Tokyo, Japan), and incubated with gentle mixing for 30 min at 30°C. Spheroplasts were harvested by centrifugation at 5000 × g for 10 min and resuspended in 1× SDS sample buffer before separation by SDS–PAGE and Western blotting to detect Bgl2 (a gift from Randy Schekman, University of California, Berkeley) and Pgk1 loading control (Invitrogen).
Invertase secretion
Invertase secretion was performed using standard methods (Goldstein and Lampen, 1975) with minor modifications. Yeast cells were grown to logarithmic phase at 30°C in YPD, and equal cell numbers were collected by centrifugation at 2000 × g for 5 min. Cell pellets were washed twice, resuspended in 0.1% glucose YPD, and cultured for 2 h further at 30°C to induce SUC2 gene expression. Cells were harvested as before, washed twice, and resuspended in cold 10 mM NaN3. An equal cell number was transferred to two new tubes—one containing 10 mM NaN3 to keep cells intact (secreted fraction) and the other containing 10 mM NaN3 plus 0.2% Triton X-100 to lyse cells (total internal and secreted fractions). In triplicate, equal cell numbers of secreted and total samples were used to assay invertase activity in 0.1 M sodium acetate (pH 5.1). Tubes were equilibrated at 30°C, and 2.5 mol of sucrose (invertase substrate) was added in timed intervals to start the reaction. Exactly 30 min later the reaction was terminated by the addition of 0.2 M sodium phosphate (pH 7.0) with 10 mM NEM and boiling for 3 min. The glucose produced by the invertase was measured by standard glucose oxidase reaction. Tubes were equilibrated at 30°C, and fresh glucostat reagent (0.1 M potassium phosphate, pH 7.0, 4.34 U glucose oxidase, 2.5 μg/μl, 0.1 mM NEM, 150 μg/ml O-dianisidine, and 1 mg/ml horseradish peroxidase) was added in timed intervals. Samples were incubated at 30°C for a maximum of 30 min (less, if color change was observed quickly), and the reaction was terminated with the addition of 12 N H2SO4 before reading absorbance at 540 nm.
[35S]methionine labeling of secreted proteins
Cells were grown SC-methionine to logarithmic phase at 30°C (and shifted to 37°C for 1 h in the case of sec18-1 control), labeled for 15 min with 150 μCi/ml [35S]methionine, and chased with 50 mM methionine for 30 min. Secreted proteins in the medium were precipitated with trichloroacetic acid (TCA) and detected by SDS–PAGE/autoradiography.
CPY secretion filter assay
Yeast cells were grown in YPD to logarithmic phase, and equal cell numbers (5 μl of 1 OD) were spotted on YPD solid medium; a nitrocellulose filter was overlaid for 48 h. Secreted CPY was detected on the filter by Western blot against CPY (Invitrogen, Carlsbad, CA).
CPY labeling and immunoprecipitation
Cells were grown in SC-methionine at 30°C, and equal cell numbers (∼1 OD per time point) were labeled with 100 μCi/time point of [35S]methionine and chased with 50 mM methionine for the indicated times. Cell lysates were generated in 200 μl of 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, and protease inhibitors by glass bead disruption. SDS was added to 0.5%, and lysates were boiled 5 min before addition of 800 μl of IP buffer (30 mM Tris-HCl, pH 7.5, 120 mM NaCl, 5 mM EDTA, 1% Triton X-100). Immunoprecipitates (IPs) were cleared by centrifugation at 10,000 × g for 10 min, and 35 μl of Protein A/G PLUS Agarose (Qiagen, Valencia, CA) and 2 μl of CPY monoclonal antibody (Invitrogen) were added and incubated overnight at 4°C. IPs were washed extensively, first in IP buffer with 0.1% SDS, then in IP buffer plus 2 M urea, followed again by IP buffer with 0.1% SDS three more times. Samples were resuspended in SDS sample buffer and detected by SDS–PAGE/autoradiography.
Bgl2 and invertase labeling and immunoprecipitation
These were performed as described in Graham (2001). Briefly, cells were grown in SC-methionine at 30°C (or 37°C as indicated for ts controls), and equal cell numbers (∼1 OD per time point for Bgl2 and 3 OD per time point for invertase) were labeled with 100 μCi/time point of [35S]methionine and chased with 50 mM methionine for the indicated times. In the case of invertase, cells were cultured in SC-methionine containing 0.1% glucose for 15 min before and during the labeling. At the indicated times cells were removed and added to 2× stop/spheroplasting buffer (2 M sorbitol, 50 mM Tris-HCl, pH 7.4, 40 mM NaN3, 40 mM NaF, 1 mg/ml bovine serum albumin). After 10 min on ice, 1 μl of β-mercaptoethanol and 30 μg of zymolyase were added, and spheroplasts were generated at 37°C for 20 min with gentle shaking. Cells were separated from cell walls by centrifugation at 5000 × g for 5 min, and cell wall proteins were TCA precipitated. Cell pellets and TCA pellets were resuspended in 100 μl of SDS/urea buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% SDS, 6 M urea), incubated at 100°C for 4 min, and 900 μl of IP buffer (50 mM Tris-HCl, 0.1 mM EDTA, 150 mM NaCl, 1% NP40) was added. Cell lysates were cleared by centrifugation at 16,000 × g for 10 min at 4°C. Immunoprecipitations were performed overnight at 4°C with 30 μl of Protein A/G PLUS Agarose and 1 μl of Bgl2 (from Randy Schekman) or invertase (from Howard Riezman, University of Geneva, Geneva, Switzerland) antibody. Immunoprecipitations were washed as described, resuspended in SDS sample buffer, and detected by SDS–PAGE/autoradiography.
Subcellular fractionation
Approximately 150 OD600 of wild-type and cof1-8 cells cultured in YPDA at 30°C were harvested by centrifugation at 3000 × g, washed once with ice-cold 10 mM NaN3/NaF, and further incubated on ice in NaN3/NaF. Cells were resuspended at 20 OD/ml in pre-spheroplasting buffer (10 mM NaN3, 10 mM NaF, 100 mM Tris-H2SO4, pH 9.4, 0.36 μl/ml β-mercaptoethanol), and incubated for 20 min at 25°C. Cells were collected and spheroplasted at 50 OD600/ml in spheroplasting buffer (40 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–NaOH, pH 7.5, 1.4 M sorbitol, 1 μl/ml β-mercaptoethanol) by treatment with 50 U/OD600mn zymolyase 100T for 45–60 min at 37°C. Efficiency of spheroplasting was monitored by measuring OD600 of a 1:1000 dilution in H2O versus spheroplast buffer, with a 10-fold reduction in OD600 indicating complete spheroplasting. Spheroplasts were harvested, resuspended in 1.5 ml of lysis buffer (10 mM HEPES-NaOH, 1 mM EDTA, 0.3 M sorbitol, protease inhibitors), and lysed by 30 strokes of a Dounce homogenizer. Lysates were cleared twice by centrifugation (600 × g for 3 min). Equal protein concentrations were loaded on top of a continuous sucrose gradient (10 ml of 30–60% sucrose in 10 mM HEPES-NaOH, 1 mM EDTA) and centrifuged for 18 h at 100,000 × g. Fractions of 1 ml were collected from the top, and proteins were separated by SDS–PAGE and analyzed by Western blotting using antibodies against Mnn9 (a gift from Yoichi Noda, University of Tokyo, Tokyo, Japan) and Kar2 (a gift from Mark Rose, Princeton University, Princeton, NJ). Protein bands were quantitated with the Odyssey 2.1 software for three independent experiments, and the averages were plotted using Prism software (GraphPad Software, La Jolla, CA).
Fluorescence microscopy
Cells were imaged with a Leica DMI6000B microscope (Leica, Wetzlar, Germany) equipped with a DFC 360 FX camera using an HCX Pl APO 100× 1.4 objective. Images were taken using Leica LAS AF software. Snc1-GFP and Pma1-GFP were expressed exogenously from their own promoters and visualized in live cells cultured at 30°C in SC-uracil. The plasmid expressing Snc1-GFP was a gift from Hugh Pelham (MRC Laboratory of Molecular Biology, Cambridge, UK), and Pma1-GFP was kindly provided by Annick Breton (CNRS [Centre national de la recherche scientifique], Bordeaux, France).
FM4-64 uptake and trafficking were determined as described previously (Vida and Emr, 1995) with slight modifications. Cells were grown in YPD at 30°C and directly labeled with FM4-64 or shifted to 37°C for 1 h before labeling with 40 μM FM4-64 (Invitrogen) for 15 min (at 30 or 37°C, respectively). Cells were washed and resuspended in fresh, prewarmed YPD and further incubated at 30 or 37°C for 45 min, and live cells were immediately visualized.
Actin was visualized by staining with phalloidin linked to Alexa 488 (Invitrogen). Cells were grown at 30°C in YPD and directly fixed or shifted to 37°C for 1 h. Cells were fixed in 3.7% formaldehyde for 30 min and washed in phosphate-buffered saline (PBS). Fixed cells were permeabilized for 15 min with 0.1% Triton X-100 in PBS with 2% bovine serum albumin (BSA), washed in PBS, and incubated with 0.2 U/ml Alexa 488–phalloidin in PBS with 2% BSA for 1 h at room temperature. Cells were washed again in PBS and visualized immediately or stored at 4°C for visualization later.
Immunocolocalization of Bgl2
The immunocolocalization experiments were performed on wild-type and cof1-8 cells containing either genomic Anp1-RFP or expressing exogenously Sec7-DsRed (kindly provided by Scott Emr, Cornell University, Ithaca, NY), mCherry-Tlg1, or mCherry-Pep12 (kindly provided by David Katzmann, Mayo Clinic, Rochester, MN). Cells were grown in SC medium to logarithmic phase at 30°C and fixed for 30 min in 3.7% formaldehyde. Immunofluorescence was performed as described (Pringle et al., 1989), with slight modifications. Cells were washed twice in 0.1 M potassium phosphate (pH 7.4) and 1.2 M sorbitol (phos/sorb) and resuspended in 1 ml of phos/sorb. Cells were treated with 50 mg/ml zymolyase 10 T and 2 ml of β-mercaptoethanol at 30°C for 30 min. Cells were harvested gently by centrifugation at 3000 rpm for 5 min, washed twice, and resuspended in 200–500 ml of phos/sorb. Polylysine (molecular weight, >300,000; Sigma-Aldrich)-coated slides were prepared in advance by adding of 10 ml polylysine (1 mg/ml) to the well of a Teflon-printed microscope slide (Immuno-Cell, Mechelen, Belgium), incubating it at room temperature for 10 min, washing it with distilled water, and allowing it to air dry. Spheroplasted cells (∼20 ml) were spotted onto the coated slides and incubated at room temperature for 20 min. Cells were permeabilized by immersing the slide in ice-cold methanol for 6 min and ice-cold acetone for 30 s. Cells were immediately covered in PBS with 3% BSA for rehydration and blocking and incubated for 30 min at room temperature. Cells were treated with primary antibody in a humid chamber at 4°C overnight, washed three times for 10 min in PBS, and treated with secondary antibody for 2 h in the dark at room temperature. Cells were washed again three times for 10 min in PBS before mounting and visualization. A purified Bgl2 antibody (a gift from Patrick Brennwald, University of North Carolina at Chapel Hill, Chapel Hill, NC) was used at 1:1000, and anti-RFP mouse antibody (Abcam) was used at 1:500. Secondary antibodies were anti–mouse Alexa 594 and anti–rabbit Alexa 488 (Invitrogen), each used at 1:5000. Images were taken in three-dimensional stacks with 10–12 slices of 0.1–0.3 mm and subjected to Leica deconvolution software.
Immunolocalization of HA-Pma1
The pMET25-HA-PMA1 vector was kindly provided by Amy Chang (University of Michigan), and cells were treated as described, with minor modification (Luo and Chang, 2000). Cells were grown in repressing conditions to logarithmic phase, washed in distilled water, and grown in methionine-free medium to derepress the MET25 promoter to allow HA-PMA1 expression. Normally, 600 μM methionine is sufficient to repress the MET25 promoter, but concentrations up to 2 mM were used to avoid the low-level of expression observed in wild-type cells. Cells were fixed for 2 h in 0.1 M KPO4, pH 6.6, washed in the same buffer, and treated for immunofluorescence as described. The HA epitope was detected with a monoclonal HA antibody (Covance, Berkeley, CA) used at a concentration of 1:1000 and the secondary anti–mouse Alexa 488 (Invitrogen).
Serial dilutions to assess growth
Cells were grown to logarithmic phase at 25°C, set to an OD600 of 0.1, and serially diluted three times. Cells were plated to appropriate medium (YPD or SC lacking uracil or leucine to select plasmids as required) using a 96-well replica plate and incubated at the indicated temperatures for 48–72 h. The PMR1–expressing vector was provided by Kyle Cunningham (Johns Hopkins University, Baltimore, MD) and contains PMR1 under the control of its own promoter cloned in a URA3-marked 2μ vector.
Supplementary Material
Acknowledgments
We thank all members of the Malhotra lab for helpful discussion. We also thank Randy Schekman and Edina Harsay for reagents and technical advice. Vivek Malhotra is an Institució Catalana de Recerca i Estudis Avançats Professor at the Center for Genomic Regulation, and the work in his lab is funded by grants from the Plan Nacional (BFU2008-00414), Consolider (CSD2009-00016), AGAUR SGR2009-1488 Grups de Recerca Emergents (AGAUR-Catalan Government), and European Research Council (268692). The project has received research funding from the European Union. This article reflects only the author's views. The Union is not liable for any use that may be made of the information contained therein.
Abbreviations used:
- CPY
carboxypeptidase Y
- HDSV
high-density secretory vesicle
- LDSV
low-density secretory vesicle
- PIP2
phosphatidylinositol 4,5-bisphosphate
- SPCA
secretory pathway calcium ATPase
- TGN
trans-Golgi network
- ts
temperature sensitive
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-09-0826) on May 2, 2012.
REFERENCES
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- Antebi A, Fink GR. The yeast Ca(2+)-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol Biol Cell. 1992;3:633–654. doi: 10.1091/mbc.3.6.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthington BA, Bennett LG, Skatrud PL, Guynn CJ, Barbuch RJ, Ulbright CE, Bard M. Cloning, disruption and sequence of the gene encoding yeast C-5 sterol desaturase. Gene. 1991;102:39–44. doi: 10.1016/0378-1119(91)90535-j. [DOI] [PubMed] [Google Scholar]
- Ashman WH, Barbuch RJ, Ulbright CE, Jarrett HW, Bard M. Cloning and disruption of the yeast C-8 sterol isomerase gene. Lipids. 1991;26:628–632. doi: 10.1007/BF02536427. [DOI] [PubMed] [Google Scholar]
- Bankaitis VA, Malehorn DE, Emr SD, Greene R. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranski TJ, Faust PL, Kornfeld S. Generation of a lysosomal enzyme targeting signal in the secretory protein pepsinogen. Cell. 1990;63:281–291. doi: 10.1016/0092-8674(90)90161-7. [DOI] [PubMed] [Google Scholar]
- Bonangelino CJ, Chavez EM, Bonifacino JS. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2486–2501. doi: 10.1091/mbc.02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno S, Engqvist-Goldstein AE, Zhang CX, McDonald KL, Drubin DG. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J Cell Biol. 2004;165:781–788. doi: 10.1083/jcb.200403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MG, Amberg DC. Biochemical and genetic analyses provide insight into the structural and mechanistic properties of actin filament disassembly by the Aip1p cofilin complex in Saccharomyces cerevisiae. Genetics. 2007;176:1527–1539. doi: 10.1534/genetics.107.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Stevens TH. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J Cell Biol. 1996;133:529–541. doi: 10.1083/jcb.133.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KW, Fink GR. Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J Cell Biol. 1994;124:351–363. doi: 10.1083/jcb.124.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curwin AJ, Fairn GD, McMaster CR. Phospholipid transfer protein Sec14 is required for trafficking from endosomes and regulates distinct trans-Golgi export pathways. J Biol Chem. 2009;284:7364–7375. doi: 10.1074/jbc.M808732200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D, Sundarababu S, Gerst JE. Involvement of long chain fatty acid elongation in the trafficking of secretory vesicles in yeast. J Cell Biol. 1998;143:1167–1182. doi: 10.1083/jcb.143.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr G, Strayle J, Plemper R, Elbs S, Klee SK, Catty P, Wolf DH, Rudolph HK. The medial-Golgi ion pump Pmr1 supplies the yeast secretory pathway with Ca2+ and Mn2+ required for glycosylation, sorting, and endoplasmic reticulum-associated protein degradation. Mol Biol Cell. 1998;9:1149–1162. doi: 10.1091/mbc.9.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- Goldstein A, Lampen JO. Beta-D-fructofuranoside fructohydrolase from yeast. Methods Enzymol. 1975;42:504–511. doi: 10.1016/0076-6879(75)42159-0. [DOI] [PubMed] [Google Scholar]
- Graham TR. Curr Protoc Cell Biol Chapter 7, Unit 7.6. 2001. Metabolic labeling and immunoprecipitation of yeast proteins. [DOI] [PubMed] [Google Scholar]
- Graham TR, Emr SD. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurunathan S, David D, Gerst JE. Dynamin and clathrin are required for the biogenesis of a distinct class of secretory vesicles in yeast. EMBO J. 2002;21:602–614. doi: 10.1093/emboj/21.4.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsay E, Bretscher A. Parallel secretory pathways to the cell surface in yeast. J Cell Biol. 1995;131:297–310. doi: 10.1083/jcb.131.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsay E, Schekman R. A subset of yeast vacuolar protein sorting mutants is blocked in one branch of the exocytic pathway. J Cell Biol. 2002;156:271–285. doi: 10.1083/jcb.200109077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chang A. pH-dependent cargo sorting from the Golgi. J Biol Chem. 2011;286:10058–10065. doi: 10.1074/jbc.M110.197889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Karpova TS, Reck-Peterson SL, Elkind NB, Mooseker MS, Novick PJ, Cooper JA. Role of actin and Myo2p in polarized secretion and growth of Saccharomyces cerevisiae. Mol Biol Cell. 2000;11:1727–1737. doi: 10.1091/mbc.11.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RW, et al. Segregation of sphingolipids and sterols during formation of secretory vesicles at the trans-Golgi network. J Cell Biol. 2009;185:601–612. doi: 10.1083/jcb.200901145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Alfaro G, Dighe S, Beh CT. Homologues of oxysterol-binding proteins affect Cdc42p- and Rho1p-mediated cell polarization in Saccharomyces cerevisiae. Traffic. 2006;7:1224–1242. doi: 10.1111/j.1600-0854.2006.00467.x. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Fedorov EV, Fedorov AA, Almo SC, Drubin DG. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 1997;16:5520–5530. doi: 10.1093/emboj/16.18.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- Lees ND, Skaggs B, Kirsch DR, Bard M. Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae—a review. Lipids. 1995;30:221–226. doi: 10.1007/BF02537824. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Nichols BJ, Prescianotto-Baschong C, Riezman H, Pelham HR. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol Biol Cell. 2000;11:23–38. doi: 10.1091/mbc.11.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR. A human homologue of the yeast HDEL receptor. Nature. 1990;348:162–163. doi: 10.1038/348162a0. [DOI] [PubMed] [Google Scholar]
- Lin MC, Galletta BJ, Sept D, Cooper JA. Overlapping and distinct functions for cofilin, coronin and Aip1 in actin dynamics in vivo. J Cell Sci. 2010;123:1329–1342. doi: 10.1242/jcs.065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Chang A. An endosome-to-plasma membrane pathway involved in trafficking of a mutant plasma membrane ATPase in yeast. Mol Biol Cell. 2000;11:579–592. doi: 10.1091/mbc.11.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi V, Sorin A, Wei Y, Rao R. Induction of vacuolar Ca2+-ATPase and H+/Ca2+ exchange activity in yeast mutants lacking Pmr1, the Golgi Ca2+-ATPase. FEBS Lett. 1999;454:181–186. doi: 10.1016/s0014-5793(99)00803-0. [DOI] [PubMed] [Google Scholar]
- Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland J, Wesp A, Riezman H, Botstein D. Yeast actin cytoskeleton mutants accumulate a new class of Golgi-derived secretary vesicle. Mol Biol Cell. 1997;8:1481–1499. doi: 10.1091/mbc.8.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Ojala PJ, Paavilainen V, Lappalainen P. Identification of yeast cofilin residues specific for actin monomer and PIP2 binding. Biochemistry. 2001;40:15562–15569. doi: 10.1021/bi0117697. [DOI] [PubMed] [Google Scholar]
- Okreglak V, Drubin DG. Cofilin recruitment and function during actin-mediated endocytosis dictated by actin nucleotide state. J Cell Biol. 2007;178:1251–1264. doi: 10.1083/jcb.200703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks LW, Smith SJ, Crowley JH. Biochemical and physiological effects of sterol alterations in yeast—a review. Lipids. 1995;30:227–230. doi: 10.1007/BF02537825. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Preston RA, Adams AE, Stearns T, Drubin DG, Haarer BK, Jones EW. Fluorescence microscopy methods for yeast. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- Proszynski TJ, et al. A genome-wide visual screen reveals a role for sphingolipids and ergosterol in cell surface delivery in yeast. Proc Natl Acad Sci USA. 2005;102:17981–17986. doi: 10.1073/pnas.0509107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M, Poon PP, Schindler C, Murray LE, Kama R, Gabriely G, Singer RA, Spang A, Johnston GC, Gerst JE. The Gcs1 Arf-GAP mediates Snc1,2 v-SNARE retrieval to the Golgi in yeast. Mol Biol Cell. 2006;17:1845–1858. doi: 10.1091/mbc.E05-09-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph HK, Antebi A, Fink GR, Buckley CM, Dorman TE, LeVitre J, Davidow LS, Mao JI, Moir DT. The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell. 1989;58:133–145. doi: 10.1016/0092-8674(89)90410-8. [DOI] [PubMed] [Google Scholar]
- Singer-Kruger B, Nemoto Y, Daniell L, Ferro-Novick S, De Camilli P. Synaptojanin family members are implicated in endocytic membrane traffic in yeast. J Cell Sci. 1998;111:3347–3356. doi: 10.1242/jcs.111.22.3347. [DOI] [PubMed] [Google Scholar]
- Smith RA, Duncan MJ, Moir DT. Heterologous protein secretion from yeast. Science. 1985;229:1219–1224. doi: 10.1126/science.3939723. [DOI] [PubMed] [Google Scholar]
- Sorin A, Rosas G, Rao R. PMR1, a Ca2+-ATPase in yeast Golgi, has properties distinct from sarco/endoplasmic reticulum and plasma membrane calcium pumps. J Biol Chem. 1997;272:9895–9901. doi: 10.1074/jbc.272.15.9895. [DOI] [PubMed] [Google Scholar]
- Tang HY, Munn A, Cai M. EH domain proteins Pan1p and End3p are components of a complex that plays a dual role in organization of the cortical actin cytoskeleton and endocytosis in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:4294–4304. doi: 10.1128/mcb.17.8.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HY, Xu J, Cai M. Pan1p, End3p, and S1a1p, three yeast proteins required for normal cortical actin cytoskeleton organization, associate with each other and play essential roles in cell wall morphogenesis. Mol Cell Biol. 2000;20:12–25. doi: 10.1128/mcb.20.1.12-25.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton VK, Mandal D, Vahadji C, Rao R. Functional expression in yeast of the human secretory pathway Ca(2+), Mn(2+)-ATPase defective in Hailey-Hailey disease. J Biol Chem. 2002;277:6422–6427. doi: 10.1074/jbc.M110612200. [DOI] [PubMed] [Google Scholar]
- Valls LA, Hunter CP, Rothman JH, Stevens TH. Protein sorting in yeast: the localization determinant of yeast vacuolar carboxypeptidase Y resides in the propeptide. Cell. 1987;48:887–897. doi: 10.1016/0092-8674(87)90085-7. [DOI] [PubMed] [Google Scholar]
- Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Blume J, Alleaume AM, Cantero-Recasens G, Curwin A, Carreras-Sureda A, Zimmermann T, van Galen J, Wakana Y, Valverde MA, Malhotra V. ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev Cell. 2011;20:652–662. doi: 10.1016/j.devcel.2011.03.014. [DOI] [PubMed] [Google Scholar]
- von Blume J, Duran JM, Forlanelli E, Alleaume AM, Egorov M, Polishchuk R, Molina H, Malhotra V. Actin remodeling by ADF/cofilin is required for cargo sorting at the trans-Golgi network. J Cell Biol. 2009;187:1055–1069. doi: 10.1083/jcb.200908040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren DT, Andrews PD, Gourlay CW, Ayscough KR. Sla1p couples the yeast endocytic machinery to proteins regulating actin dynamics. J Cell Sci. 2002;115:1703–1715. doi: 10.1242/jcs.115.8.1703. [DOI] [PubMed] [Google Scholar]
- Wesp A, Hicke L, Palecek J, Lombardi R, Aust T, Munn AL, Riezman H. End4p/Sla2p interacts with actin-associated proteins for endocytosis in Saccharomyces cerevisiae. Mol Biol Cell. 1997;8:2291–2306. doi: 10.1091/mbc.8.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JR, Munro S. A yeast homolog of the mammalian mannose 6-phosphate receptors contributes to the sorting of vacuolar hydrolases. Curr Biol. 2001;11:1074–1078. doi: 10.1016/s0960-9822(01)00273-1. [DOI] [PubMed] [Google Scholar]
- Yang S, Cope MJ, Drubin DG. Sla2p is associated with the yeast cortical actin cytoskeleton via redundant localization signals. Mol Biol Cell. 1999;10:2265–2283. doi: 10.1091/mbc.10.7.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.