

Abstract
Mutations in the gene encoding β-III spectrin give rise to spinocerebellar ataxia type 5, a neurodegenerative disease characterized by progressive thinning of the molecular layer, loss of Purkinje cells and increasing motor deficits. A mouse lacking full-length β-III spectrin (β-III−/−) displays a similar phenotype. In vitro and in vivo analyses of Purkinje cells lacking β-III spectrin, reveal a critical role for β-III spectrin in Purkinje cell morphological development. Disruption of the normally well ordered dendritic arborization occurs in Purkinje cells from β-III−/− mice, specifically showing a loss of monoplanar organization, smaller average dendritic diameter and reduced densities of Purkinje cell spines and synapses. Early morphological defects appear to affect distribution of dendritic, but not axonal, proteins. This study confirms that thinning of the molecular layer associated with disease pathogenesis is a consequence of Purkinje cell dendritic degeneration, as Purkinje cells from 8-month-old β-III−/− mice have drastically reduced dendritic volumes, surface areas and total dendritic lengths compared with 5- to 6-week-old β-III−/− mice. These findings highlight a critical role of β-III spectrin in dendritic biology and are consistent with an early developmental defect in β-III−/− mice, with abnormal Purkinje cell dendritic morphology potentially underlying disease pathogenesis.
Introduction
Purkinje cells of the cerebellum have an elaborate, monoplanar dendritic tree with a high density of spines (Rall, 1977). The acquisition of this morphology, controlled by both intrinsic and extrinsic factors (Sotelo and Dusart, 2009), underlies important aspects of cerebellar function. For instance, it allows Purkinje cells, the sole output of the cerebellum, to integrate information from an array of synaptic inputs, with dendritic branching pattern and spine density determining the number and types of input the cell receives (Häusser et al., 2000). Dendritic morphological characteristics also influence how synaptic signals decay as they propagate toward the soma (Gulledge et al., 2005).
It has been well established that the assembly of a well ordered spectrin-actin filamentous network at the plasma membrane is required to maintain cellular morphology and physiological function (Bennett and Baines, 2001). For example, in erythrocytes spectrin is critical for mechanical support and maintenance of structural membrane integrity with β spectrin deficiency being associated with hemolytic anemias arising from the fragmentation of erythrocytes when placed under mechanical stress in the circulation (Greenquist et al., 1978; Lux et al., 1979; Agre et al., 1982, 1985). Studies using Caenorhabditis elegans have also demonstrated a role for spectrin in the maintenance of membrane integrity with loss of β spectrin in C. elegans resulting in axonal breakage (Hammarlund et al., 2000, 2007; Moorthy et al., 2000), while in Drosophila melanogaster synaptic retraction and consequently synapse elimination were observed at the neuromuscular junction when either α or β presynaptic spectrin was knocked down (Pielage et al., 2005, 2006).
Unlike invertebrates, vertebrates have two α (αI/αII)-subunits, four β (βI-βIV)-subunits, and a β-H subunit creating diversity and specialization of function, with β-III spectrin being expressed at high levels in the soma and dendrites of Purkinje cells (Ohara et al., 1998; Sakaguchi et al., 1998; Stankewich et al., 1998; Jackson et al., 2001). Mutations in the gene encoding β-III spectrin have been shown to underlie spinocerebellar ataxia type 5 (SCA5) (Ikeda et al., 2006) and we have previously reported the generation of a functional β-III spectrin knock-out mouse (β-III−/−) that showed characteristics of cerebellar ataxia, namely progressive motor deficits and age-related Purkinje cell loss (Perkins et al., 2010). Here, using both in vitro and in vivo morphometric analyses of Purkinje cells lacking β-III spectrin, we have investigated the role of β-III spectrin in dendritic development. The present study highlights a critical role for β-III spectrin in the monoplanar organization of the dendritic tree and shows loss of β-III spectrin to result in thinner dendrites and severe defects in dendritic spine development. Furthermore we identify defects in the distribution of dendritic, but not axonal, proteins.
Materials and Methods
Dissociated cerebellar cultures.
Cultures were prepared as previously described apart from cerebella were dissected at P0, digested in papain (Worthington) and dissociated cells were plated on poly-l-lysine-coated coverslips at cell density of 5 × 106/ml in 35 mm dishes (MatTek). Half the medium was also changed every 4 d (Linden, 1996; Furuya et al., 1998).
Immunofluorescence microscopy.
Dissociated cerebellar cultures were fixed with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4 for 15 min, and then incubated for 30 min at room temperature (RT) with blocking solution [10% goat serum with 0.2% Triton X-100 in 1× PBS]. Primary antibodies were applied overnight at 4°C [rabbit anti-β-III spectrin (1:1000), anti-EAAT4 (1:100), anti-GluRδ2 (1:2000; Frontier Science Japan), anti-Nav1.6 (1:100; Alomone Labs), anti-AnkG (1:50; Santa Cruz Biotechnology), guinea pig anti-Vglut1 (1:1000; Synaptic Systems), mouse anti-calbindin (1:5000; Swant)]. Cells were washed three times in PBS, incubated with secondary antibodies for 40 min at RT (goat anti-mouse IgG 488, anti-rabbit IgG 555, anti-guinea pig IgG 488 Invitrogen) followed by three rinses in PBS and coverslipping with Prolong Gold antifade reagent (Invitrogen). For paraffin sections brains were removed and immersion-fixed with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4 overnight at 4°C before embedding in paraffin. Sections (7 μm) were cut and mounted onto poly-l-lysine-coated slides and immunostained with rabbit anti-β-III spectrin (1:50), rabbit anti-Vglut1 (1:50: Invitrogen) and mouse anti-calbindin (1:50; Sigma). Secondary antibodies were cyanine 3 (Cy3)-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories) and FITC-conjugated goat anti-rabbit IgG (Cappel). Images were captured with a Zeiss inverted LSM510 confocal laser scanning microscope.
Golgi impregnation.
Both WT and β-III−/− mice were killed at P4, 8, 15, 30 and 90 and staining followed manufacturer's instructions (FD NeuroTechnologies) (male and female mice were used equally). Briefly after dissection brains and cerebella were incubated in premixed solution A and B for 2 weeks at RT in the dark before being transferred to solution C for a further 4 d in the dark but at 4°C. Solution A+B was only changed after the first 24 h whereas solution C was changed every day. Tissue was frozen and cut sections (120 μm) dried and mounted onto gelatin-coated slides overnight.
Electron microscopy.
WT and β-III−/− mice at P15 and P120 were deeply anesthetized with 10% chloral hydrate and killed by transcardial perfusion with 4% paraformaldehyde/3% glutaraldehyde in 0.1 mm phosphate buffer, pH7.4, for 10–15 min (male and female mice were used equally). Brains and cerebella were dissected and postfixed in the same fixative for 4–16 h at 4°C. For ultrastructural analyses, 2 mm3 regions of cerebellum and hippocampus were dissected out, postfixed in OsO4, embedded in Epon and 1-μm-thick sections cut, stained with toluidine blue and viewed in a light microscope to select suitable areas for investigation. Ultrathin sections, 70-nm-thick were cut from selected areas and stained with uranyl acetate and lead citrate. Electron micrographs were acquired using Hitachi 7000.
Electrophysiology.
Mice were deeply anesthetized with halothane (Sigma) and decapitated under the United Kingdom Animals (Scientific Procedures) Act (1986). Cerebella were dissected out into ice-cold modified artificial CSF (ACSF) containing the following (in mm): 60 NaCl, 118 sucrose, 26 NaHCO3, 2.5 KCl, 11 glucose, 1.3 MgCl2, and 1 NaH2PO4 at pH 7.4 when bubbled with 95% O2/5% CO2. The cerebellar vermis was glued to the vibratome cutting platform (VT1200S, Leica Microsystems) with cyanoacrylate adhesive. Sagittal slices (200 μm thick) were cut and incubated for 30 min at 30°C in standard ACSF composed of the following (in mm): 119 NaCl, 2.5 CaCl2, 26 NaHCO3, 2.5 KCl, 11 glucose, 1.3 MgCl2, and 1 NaH2PO4 at pH 7.4 when bubbled with 95% O2/5% CO2. Slices were stored at room temperature until required for recording. Slices were transferred to a submerged recording chamber and superfused with standard ACSF (3–5 ml min−1) at room temperature. Purkinje cells were initially visualized with a 40× immersion objective and Normarski differential interference contrast (DIC) optics. Whole-cell recording were obtained from Purkinje cells using thick-walled borosilicate glass pipettes pulled to 3–6 MΩ. For Purkinje cell filling the internal solution contained the following (in mm): 0.2 Lucifer yellow (Sigma, L0144), 0.02 Alexa Fluor AR 568 hydrazide (Invitrogen, A-10441), 125 K-gluconate, 15 KCl, 10 HEPES, 5 EGTA, 2 MgCl2, 0.4 NaGTP, 2 NaATP, and 10 Na-phosphocreatine, adjusted to pH7.4 with KOH. Purkinje cells were voltage-clamped at −60 mV for 25–30 min and complete cell filling was monitored by Lucifer yellow fluorescence. Slices were then removed and fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, overnight at 4°C. Slices were washed twice in 0.1 m phosphate buffer, pH 7.4 and twice in dH2O then stored in Vectashield (Vector Laboratories) at 4°C. Slices were wet-mounted with Vectashield onto 0.13-mm-thick borosilicate glass and the Purkinje cell was imaged using the Alexa Fluor 568 dye and captured using a Zeiss inverted LSM510 confocal scanning laser microscope. For LTD induction the internal solution contained the following (in mm): 108 Cs-methanesulfonate, 9 NaCl, 9 HEPES, 1.8 EGTA, 1.8 MgCl2, 0.4 NaGTP, 2 MgATP, 63 sucrose, and 5 QX-314, adjusted to pH 7.4 with CsOH. Picrotoxin (50 μm) was added to the ACSF. Purkinje cells were voltage-clamped at −60 mV and parallel fiber (PF)-EPSCs were evoked by placing a patch-pipette filled with standard ACSF in the molecular layer and applying a square-pulse stimuli that evoked an EPSC of ∼500 pA in amplitude (lower stimulus required for β-III−/− animals). PF-EPSCs were recorded at a frequency of 0.1 Hz for 10 min to obtain a stable baseline. Induction of LTD was achieved with 30 single PF stimuli (at 0.1 Hz) together with a 200 ms depolarizing step to +20 mV as described by Kakegawa et al. (2008). Series resistances were <15 MΩ and were compensated for by 40–60%. Membrane currents were filtered at 5 kHz and sampled at 10 kHz. EPSCs were recorded using an Axopatch 200B amplifier (Molecular Devices) and in-house National Instruments software written by Tim O'Leary (O'Leary et al., 2010). Data were analyzed using IGOR Pro (WaveMetrics).
Image analysis and statistics.
Images of dissociated Purkinje cells (captured at 0.5 μm intervals) and filled Purkinje cells captured at Nyquist sampling rates and deconvolved using Huygens Deconvolution Software (Scientific Volume Imaging) were analyzed with filament tracer (Imaris 7.1) and NeuronStudio (Computational Neurobiology and Imaging Center). Colocalization analysis was performed with Imaris 7.1 Coloc mode. Spine analysis was performed using MetaMorph. Statistical analysis was performed using Student's t test, two sample assuming unequal variance, apart from analysis of filled Purkinje cells where Bonferroni-corrected Mann–Whitney tests were used after Kruskal–Wallis test.
Results
To investigate whether developmental defects can arise from loss of β-III spectrin we first looked at Purkinje cell morphology in dissociated cerebellar cultures. We initially confirmed that β-III spectrin displayed the same distribution in culture as in vivo by immunostaining cerebellar sections and dissociated cerebellar cultures with an antibody raised against a C-terminal epitope of β-III spectrin. This confirmed that in dissociated Purkinje cells β-III spectrin is found throughout the soma and dendritic tree but is not observed in the axon, similar to in vivo findings. Moreover this staining and that using an antibody against an N-terminal epitope of β-III spectrin revealed that β-III spectrin is located within Purkinje cell dendritic spines both in vivo and in vitro (Fig. 1).
Figure 1.

β-III spectrin localizes to Purkinje cell dendritic spines. A1, B1, Cerebellar sections (A1) and dissociated cerebellar cultures (B1) immunostained with C-terminal anti-β-III spectrin antibody show somatodendritic distribution of β-III spectrin in Purkinje cells (scale bar, 20 μm). Higher magnification reveals staining throughout dendritic spines with C-terminal (A2, B2) and N-terminal (A3, B3) anti-β-III spectrin antibody (bottom, scale bar, 10 μm).
Reduced dendritic surface area and spine density in Purkinje cells lacking β-III spectrin in vitro
Dissociated cultures from WT and β-III−/− spectrin mice were fixed and immunostained for calbindin after 8, 15, and 22 d in vitro (DIV) and the area of the soma and dendritic arborization quantified (Fig. 2A,B). This revealed that at 8 DIV the dendritic area of Purkinje cells lacking β-III spectrin is significantly smaller than WT Purkinje neurons (∼52% of WT cells) but the area of somas is slightly larger (∼110% of WT cells). However, by 15 and 22 DIV, although the dendritic area is still smaller in Purkinje cells lacking β-III spectrin, the difference, while still significant, is not as great (∼88 and ∼77% of WT cells, respectively) and there is no longer any difference in the area of Purkinje cell somas. Importantly, quantification of spine density (also based on calbindin immunostaining) revealed significantly lower spine densities in Purkinje cells lacking β-III spectrin at all time points compared with WT cells (Fig. 2C,D).
Figure 2.

Reduced dendritic surface area and spine density in cultured β-III−/− Purkinje cells. A, Dissociated cerebellar cultures from WT and β-III−/− mice stained for calbindin after 8, 15, and 22 DIV (scale bar, 100 μm). B, Quantification reveals smaller dendritic area in β-III−/− mice at all time points and larger somatic area at 8 DIV only (WT, N = 44–50; β-III−/−, N = 43–54). C, High-magnification images of dissociated Purkinje cells immunostained for calbindin (scale bar, 10 μm). D, Reduction in dendritic spine density in dissociated Purkinje cells from β-III−/− mice at all time points (N = 10–11 for both genotypes). All data are means ± SEM. *p < 0.05; ***p < 0.001.
β-III spectrin loss specifically affects Purkinje cell spine density
Given the morphological defects observed in dissociated Purkinje cell cultures we went on to examine whether similar deficits could be observed in vivo. For this we undertook Golgi impregnation and looked at the effect loss of β-III spectrin had on Purkinje cell spine density. Similar to the in vitro findings a >80% reduction in spine density was observed in β-III−/− Purkinje cells from as early as P8 (Fig. 3A,B). To ascertain whether this dramatic effect of β-III spectrin on spine development/maintenance was specific to Purkinje neurons, or a more ubiquitous phenomenon, we looked at the effect of β-III spectrin loss on spine density in hippocampal CA1 pyramidal neurons (Fig. 3C,D), since after the cerebellum the highest level of β-III spectrin expression is within the hippocampus (Jackson et al., 2001). We observed no difference in spine density of pyramidal neurons in β-III−/− mice at P8, 15, 30 and 90 (Fig. 3D). Similarly transmission electron microscopy revealed a reduction in number of synapses within the cerebellar molecular layer of β-III−/− mice compared with WT but no difference in the hippocampus (Fig. 4A–D). Together the in vivo data demonstrate a specific effect of β-III spectrin's absence on Purkinje cell spine development and synapse formation onto these cells.
Figure 3.

Loss of β-III spectrin alters Purkinje cell spine density in vivo. A, High-magnification images of Golgi impregnated Purkinje cells from WT and β-III−/− mice at P8, 15, 30 and 90 (scale bar, 10 μm). B, Reduced Purkinje cell dendritic spine density from P8 (WT, N = 2–3, n = 18–36; β-III−/−, N = 2–3, n = 13–39). C, High-magnification images of Golgi impregnated CA1 pyramidal neurons from WT and β-III−/− mice at P8, 15, 30 and 90 (scale bar, 10 μm). D, No difference in hippocampal pyramidal dendritic spine density in β-III−/− mice compared with WT animals at any age (WT, N = 2–3, n = 25–40; β-III−/−, N = 2–3, n = 20–45). All data are means ± SEM; ***p < 0.001.
Figure 4.
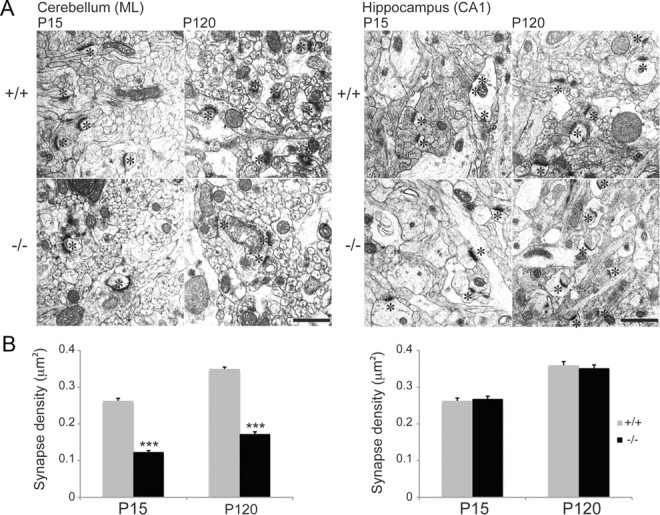
Reduction in parallel fiber-Purkinje cell synapses in β-III−/− mice. A, Representative electron micrographs from cerebellum and hippocampus of WT and β-III−/− mice at P15 and P120. Asterisks denote postsynaptic densities (scale bar, 1 μm). B, Quantification of synapse density shows an early loss of PF-PC synapses but no loss of hippocampal synapses in β-III−/− mice [WT, N = 2, n = 97–124; β-III−/−, N = 2, n = 125–144 fields of view (28 μm2)]. All data are means ± SEM; ***p < 0.001.
β-III spectrin required for wider diameter dendrites and monoplanar dendritic tree in vivo
Golgi impregnation was insufficient for morphometric analyses of entire Purkinje cell dendritic trees and therefore individual Purkinje cells in acute cerebellar sagittal slices from young (5- to 6-week-old) and old (8 months of age) mice were filled with Alexa Fluor 568 by diffusion from a whole-cell patch pipette and visualized by confocal microscopy. Serial stacks of the confocal fluorescent images were used for three-dimensional reconstruction of the entire dendritic arbors (Fig. 5A–C). Quantification showed that total dendritic surface area and actual dendritic volume of Purkinje cells from 5- to 6-week-old β-III−/− mice were significantly smaller than WT Purkinje cells but total dendritic length was the same (Fig. 5D–F). However, average distal dendrite diameter was narrower in young β-III−/− mice compared with WT mice (Fig. 5G). Analysis of cells from 8-month-old animals confirmed our previous assumption that the observed thinning of the molecular layer in old β-III−/− mice was due to degeneration of the Purkinje cell dendritic tree (Perkins et al., 2010) as Purkinje cells from 8-month-old β-III−/− mice were found to be substantially smaller than age-matched WT cells in all morphometric parameters (total dendritic surface area, volume and length; Fig. 5D–F). In contrast there was no difference in basal diameter of the primary dendrite (Fig. 5H) or cell body diameter (Fig. 5I) between genotypes at either age. Finally, loss of β-III spectrin resulted in abnormal branching of higher order dendrites (Fig. 5J,K) and disruption to the monoplanar dendritic arborization of Purkinje neurons in young mice, visualized by much greater dendritic protrusion in the z (coronal)-plane (Fig. 5L,M).
Figure 5.

Aberrant Purkinje cell dendritic morphology in β-III−/− mice. A, Representative images of Purkinje cells filled with Alexa Fluor 568 from WT and β-III−/− mice at 5–6 weeks and 8-months of age (scale bar, 50 μm). B, C, Examples of dendritic reconstruction (B) and dendritic tracing (C) using Imaris software. D–F, Quantification of morphological parameters measured from reconstructed images shows reduction in total dendritic surface area (D) and volume (E) but no difference in total dendritic length (F) in young β-III−/− mice compared with age-matched WT animals. Significant reductions in all parameters in old β-III−/− mice (D–F). G, Narrower distal dendrite diameters in β-III−/− mice. H, No difference in basal diameter of primary dendrite between genotypes. I, Purkinje cell body diameter not altered in β-III−/− mice. J, Graphic representation of higher order dendrites in young WT and β-III−/− Purkinje cell. K, Larger number of branch intersections in Purkinje cell from young β-III−/− mice. L, Filled Purkinje cells viewed in coronal plane (scale bar, 50 μm). M, Quantification of coronal dendritic protrusion in young and old WT and β-III−/− mice. All data are medians with 25th and 75th percentiles indicated (WT, N = 3–5, n = 9–10; β-III−/−, N = 3–4, n = 8; Kruskal–Wallis test, *p < 0.05; **p < 0.01 ***p < 0.001).
Normal parallel fiber abundance in β-III−/− mice
To determine whether the morphological changes were a consequence of reduced parallel fiber abundance we looked at expression of Vglut1, a presynaptic marker selective for parallel fiber terminals. Using confocal immunofluorescence and Western blot analysis we observed no difference in overall expression levels between β-III−/− and WT animals at 3-weeks of age but the staining appeared more diffuse with fewer bright puncta (Fig. 6A,B). The early morphological defects therefore appear not to be due to loss of parallel fiber terminals. Similarly in dissociated cultures there was wide spread staining of Vglut1 on Purkinje cells lacking β-III spectrin and quantification revealed that the degree of colocalization of Vglut1 with GluRδ2, a parallel fiber-Purkinje cell postsynaptic marker, was lower in β-III−/− Purkinje cells (Fig. 6C), highlighting the redistribution at the membrane of a postsynaptic protein thought to interact with β-III spectrin (Hirai et al., 1999). More GluRδ2 protein was observed within the cell body of β-III−/− Purkinje cells compared with WT from 8 DIV (Fig. 6D) and the GluRδ2 located at the dendritic plasma membrane in β-III−/− Purkinje cells, instead of being located within spines was distributed over a large area of the dendrites (Fig. 6E). To determine whether there was any physiological effect of this mislocalization we examined whether long term depression (LTD), thought to be the cellular basis of motor learning (Ito, 1989, 2001; Hansel et al., 2001), was normal in β-III−/− mice, since GluRδ2 is essential for induction of LTD (Hirano et al., 1994; Jeromin et al., 1996). This revealed that the downstream signaling of GluRδ2 was unaffected in β-III−/− mice as there was no impairment in cerebellar LTD following conjunctive stimulation, which consisted of 30 single PF stimuli together with a 200 ms depolarization of the Purkinje cell (Fig. 6F). The amplitude of PF-EPSCs 25–30 min after conjunctive stimulation was 41 ± 0.4% (N = 5, n = 7) of baseline responses, similar to that of WT cells (42 ± 0.3% of baseline, N = 5, n = 8).
Figure 6.
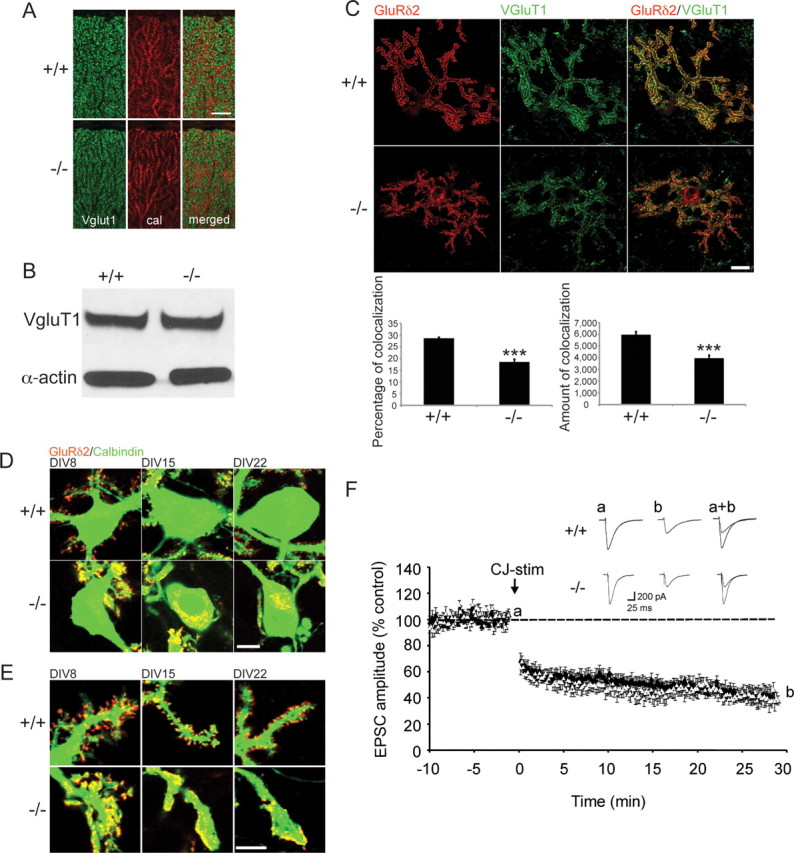
No change in parallel-fiber afferents in β-III−/− mice but GluRδ2 is differently distributed. A, Parasagittal cerebellar sections immunostained for Vglut1 show more diffuse staining but similar levels of reactivity throughout molecular layer of 3-week-old β-III−/− mice (scale bar, 20 μm). B, Western blot analysis of total cerebellar homogenates from 3-week-old mice shows no loss of Vglut1 in β-III−/− mice. C, Reduced colocalization of Vglut1 and GluRδ2 in dissociated Purkinje cells (n = 29–34; scale bar, 20 μm). Data are means ± SEM; ***p < 0.001. D, Presence of GluRδ2 reactivity in Purkinje cell soma of β-III−/− mice (scale bar, 10 μm). E, GluRδ2 displays different dendritic distribution in β-III−/− mice (scale bar, 10 μm). F, Averaged data of cerebellar LTD recorded from 5- to 6-week-old WT (closed triangle) and β-III−/− mice (open triangle). The inset traces show PF-EPSC just before (a) and 30 min after (b) conjunctive-stimulation (CJ-stim) and their superimposition (a+b).
Early defects in distribution of dendritic but not axonal proteins
In another mutant mouse with disrupted β-III spectrin expression (Spnb3−/−) EAAT4, a Purkinje cell protein known to interact with β-III spectrin (Jackson et al., 2001) was reported to accumulate in the cell soma and dendritic shafts of Purkinje cells from aged mice (Stankewich et al., 2010). Therefore, the localization of EAAT4 was examined in dissociated Purkinje cells from β-III−/− mice. This revealed large accumulations of EAAT4 in the cell body and dendrites after only 8 DIV, indicating early defects in protein distribution (Fig. 7A,B). In contrast no defects, either in vivo or in vitro, were observed in the development or the localization of proteins to the axon initial segment (AIS). Ankyrin G, an AIS-restricted protein (Davis et al., 1996), was still targeted to this region in β-III−/− Purkinje cells (Fig. 7C). Furthermore, the AnkG immunoreactivity revealed no difference in the length or width of AISs in β-III−/− Purkinje cells compared with WT animals (Length, WT 12.8 ± 0.4; β-III−/− 12.1 ± 0.6 μm; p = 0.378: Width, WT 0.9 ± 0.04; β-III−/− 1.02 ± 0.02; p = 0.113; N = 2, n = 13 for both genotypes). Similarly, a normal localization of Nav1.6 was observed at the AIS (Fig. 7D) indicating the loss of β-III spectrin has a specific effect on the distribution of dendritic, but not axonal proteins.
Figure 7.

No disruption to AIS or mislocalization of axonal proteins in β-III−/− mice. A, B, Accumulations of EAAT4 protein observed in cell bodies (A) and dendrites (B) of dissociated β-III−/− Purkinje cells after 8 DIV (scale bar, 10 μm). C, D, High levels of AnkG (C) and Nav1.6 (D) reactivity localized to AIS with no aberrant accumulation in Purkinje cell bodies of β-III−/− mice (scale bar, 20 μm).
Discussion
In this study we show that β-III spectrin is critical for the correct development and maintenance of Purkinje cell dendritic structure. In young β-III−/− mice the dendritic tree is no longer well ordered and planar, and although total dendritic length is unchanged the dendrites are thinner and have very reduced spine density, resulting in alterations to dendritic protein distribution. In addition, in old β-III−/− mice there is substantial loss of total dendritic length, surface area, and volume. To our knowledge, this is the first full morphometric study of a SCA mouse model, yielding important findings concerning the mechanisms of planar dendritic organization, spine formation and development of the Purkinje cell dendritic tree.
Spectrin and membrane integrity
Studies looking at the structure of the erythrocyte membrane have shown that spectrins are required for both mechanical resilience and elasticity. These features have been shown to arise through the formation of flexible rod-like spectrin heterodimers, which self-associate into tetramers (Ungewickell and Gratzer, 1978; Shotton et al., 1979) and subsequently interact with ankyrin, protein 4.1 and actin giving rise to stable membrane skeletons (Bennett and Stenbuck, 1980; Speicher et al., 1982; Cohen, 1983; Bennett, 1985; Cianci et al., 1988; Kennedy et al., 1991). Here we show that in Purkinje cells the loss of β-III spectrin function appears to disrupt the formation of a normal supportive membrane skeleton as the dendrites are thinner in its absence, resulting in a loss in dendritic surface area, analogous to the reduced erythrocytic surface area observed in β-I spectrin deficient patients with hereditary spherocytosis (HS) (Chasis et al., 1988). It is relevant to note, however, that although β-I spectrin deficiency is associated with HS, the disease is mainly a consequence of null mutations within ankyrin R (Eber et al., 1996; Randon et al., 1997; Hayette et al., 1998). Therefore, it is possible that Purkinje cell structural defects may arise in SCA5 if the disease-causing mutations result in conformational changes in β-III spectrin that not only disrupt membrane skeleton stability by hindering formation of spectrin dimers/tetramers, but also reduce or weaken interaction of spectrin with associated proteins, such as ankyrin.
Molecular processes for Purkinje cell development
It is known that intrinsic properties, rather than the presence of presynaptic partners, governs Purkinje cell spine formation, as spine development is normal in mice lacking granule cell afferents (Rakic and Sidman, 1973; Sotelo, 1975; Hirano et al., 1977; O'Brien and Unwin, 2006). Similarly, here we show that spine formation is abnormal in the absence of any change in granule cell afferent terminals as indicated by Vglut1 staining, indicating the importance of Purkinje cell intrinsic properties. Although intracellular calcium concentrations are thought to shape spine morphology (Segal et al., 2000; Vecellio et al., 2000), little is known about how the three-dimensional arrangement of spines around Purkinje cell dendritic shafts is achieved. It has been suggested that the presence of a regularly spaced filamentous cytoskeletal protein lattice could form the basis for spine distribution (O'Brien and Unwin, 2006). Here we show that loss of one such protein, β-III spectrin, does lead to severe defects in both spine formation and three-dimensional organization of dendrites, resulting in disrupted planar organization of the dendritic tree. β-III spectrin would therefore appear to be one important factor in governing the regular patterning of Purkinje cell dendrites and spine development. The identification of other key proteins that interact with β-III spectrin will be instrumental in unraveling the complex mechanism of dendritic development and spine morphogenesis.
GluRδ2 has been shown to interact with β spectrin (Hirai and Matsuda, 1999) and the N terminus of GluRδ2 has been shown to be involved in the formation and stabilization of parallel fiber-Purkinje cell synapses (Kurihara et al., 1997; Lalouette et al., 2001). However, the fact that total spine density is normal in the GluRδ2 knock-out mouse (Kurihara et al., 1997) demonstrates that GluRδ2 is not an intrinsic factor required for spine formation. Therefore, the absence of spines in β-III−/− Purkinje cells is unlikely to be due to the mislocalization of GluRδ2, but alterations in the membrane distribution of GluRδ2 could play a part in the loss of parallel fiber-Purkinje cell synapses. However, it seems that despite the observed redistribution of GluRδ2 in β-III−/− Purkinje cells induction of LTD in β-III−/− mice was similar to WT demonstrating normal signaling via the C terminus of GluRδ2, the region essential for LTD induction (Kohda et al., 2007; Kakegawa et al., 2008). The lack of LTD impairment in young β-III−/− mice does correlate with our finding that 3-week-old β-III−/− mice display signs of motor learning, shown by their ability to improve performance on less demanding motor tasks (Perkins et al., 2010). Nevertheless the accumulation of dendritic proteins within the perikaryon of β-III−/− Purkinje cells correlates with studies reporting a role for β-III spectrin in protein trafficking via dynein-mediated vesicular transport (Clarkson et al., 2010; Holleran et al., 2001; Lorenzo et al., 2010; Stankewich et al., 2010).
Physiological consequences of β-III spectrin loss
Determining what in fact the physiological consequences of these morphological defects are will be crucial to understanding the cellular mechanisms leading to Purkinje cell dysfunction and death in SCA5. One possibility is that since Purkinje cell density is not altered in young β-III−/− mice (Perkins et al., 2010) the protrusion of dendrites beyond their normal dendritic fields would lead to overlap with adjacent fields, and hence the potential for multiple climbing fiber innervation. In the majority of mouse models that exhibit motor impairments Purkinje cell innervation by multiple climbing fibers (CFs) has been found in adulthood (Crepel and Mariani, 1976; Mariani et al., 1977; Crepel et al., 1980; Mariani and Changeux, 1980; Aiba et al., 1994; Chen et al., 1995; Kano et al., 1995, 1997, 1998; Kashiwabuchi et al., 1995; Offermanns et al., 1997; Watase et al., 1998). However, to our knowledge no three-dimensional morphological studies have been performed with any of these mutant mice and so it may be that overlapping dendritic trees could underlie some of the observed defects in climbing fiber innervation. One plausible mechanism for such effects could be an increased potential for multiple CF innervation by transverse CF branches (Miyazaki and Watanabe, 2011) as a result of the interdigitation of the dendritic trees from neighboring Purkinje cells.
The current morphological study has provided further insight into the mechanism underlying previous observations of enhanced parallel fiber-mediated EPSCs in young β-III−/− mice (Perkins et al., 2010). The current data reveal this is not a consequence of increased Purkinje cell spine density, as in fact this appears to be reduced in β-III−/− mice. Instead the discovery that, in the absence of β-III spectrin, dendrites are thinner suggests that increased PF-EPSCs may arise from larger changes in membrane potential upon stimulation, a consequence of smaller dendritic diameters (Rall, 1977), and subsequently earlier activation of low-voltage gated channels. Over time this hyperexcitable state of Purkinje cells lacking β-III spectrin may result in them being more prone to damage through excitotoxicity and explain the observed degeneration of dendritic structures in old β-III−/− mice. Further evidence supporting this interpretation comes from the SCA1 transgenic mouse, for which it was proposed that Purkinje cells would be closer to firing threshold due to smaller somata and reduced dendritic arborization (Inoue et al., 2001). Alternatively, or in combination, the larger EPSCs may be a consequence of the formation of dendritic shaft synapses in the absence of spines, since shaft synapses are thought to produce larger synaptic currents resulting in neurons being more vulnerable to cell death (Fishbein and Segal, 2007). Another possibility is that altered transmembrane ion channel expression in the absence of β-III spectrin and/or changes to the composition/distribution of AMPA receptor subunits could underlie the increased conductance.
In conclusion, these morphometric analyses reveal a critical role for β-III spectrin in the development of the well ordered monoplanar dendritic arborization of Purkinje neurons and have identified β-III spectrin as an important intrinsic factor in spine morphogenesis. Further analysis of proteins whose cellular trafficking or stability are impaired either due to the loss-of or presence of mutant forms of β-III spectrin will provide a greater understanding of cellular mechanisms that underlie SCA pathogenesis. Future investigations also need to address whether or not the late-onset dendritic degeneration and cell loss observed in SCA5 patients are in fact downstream consequences of a developmental defect, and, consequently, whether developmental defects might be the crux of various progressive neurodegenerative diseases.
Footnotes
This work was supported by grants from the National Institutes of Health (NS056158) and The Wellcome Trust (077946). We thank Mohamed Farah for help with electron microscopy, Trudi Gillespie at the University of Edinburgh for technical assistance with microscopy, and David Wyllie, David Sterratt, Laura Ranum, David Linden, Lyle Ostrow, and Rita Sattler for useful discussions.
References
- Agre P, Orringer EP, Bennett V. Deficient red-cell spectrin in severe, recessively inherited spherocytosis. N Engl J Med. 1982;306:1155–1161. doi: 10.1056/NEJM198205133061906. [DOI] [PubMed] [Google Scholar]
- Agre P, Casella JF, Zinkham WH, McMillan C, Bennett V. Partial deficiency of erythrocyte spectrin in hereditary spherocytosis. Nature. 1985;314:380–383. doi: 10.1038/314380a0. [DOI] [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Bennett V. The membrane skeleton of human erythrocytes and its implications for more complex cells. Annu Rev Biochem. 1985;54:273–304. doi: 10.1146/annurev.bi.54.070185.001421. [DOI] [PubMed] [Google Scholar]
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- Bennett V, Stenbuck PJ. Human erythrocyte ankyrin. Purification and properties. J Biol Chem. 1980;255:2540–2548. [PubMed] [Google Scholar]
- Chasis JA, Agre P, Mohandas N. Decreased membrane mechanical stability and in vivo loss of surface area reflect spectrin deficiencies in hereditary spherocytosis. J Clin Invest. 1988;82:617–623. doi: 10.1172/JCI113640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Cianci CD, Giorgi M, Morrow JS. Phosphorylation of ankyrin down-regulates its cooperative interaction with spectrin and protein 3. J Cell Biochem. 1988;37:301–315. doi: 10.1002/jcb.240370305. [DOI] [PubMed] [Google Scholar]
- Clarkson YL, Gillespie T, Perkins EM, Lyndon AR, Jackson M. Beta-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum Mol Genet. 2010;19:3634–3641. doi: 10.1093/hmg/ddq279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CM. The molecular organization of the red cell membrane skeleton. Semin Hematol. 1983;20:141–158. [PubMed] [Google Scholar]
- Crepel F, Mariani J. Multiple innervation of Purkinje cells by climbing fibers in the cerebellum of the Weaver Mutant Mouse. J Neurobiol. 1976;7:579–582. doi: 10.1002/neu.480070610. [DOI] [PubMed] [Google Scholar]
- Crepel F, Delhaye-Bouchaud N, Guastavino JM, Sampaio I. Multiple innervation of cerebellar Purkinje cells by climbing fibres in staggerer mutant mouse. Nature. 1980;283:483–484. doi: 10.1038/283483a0. [DOI] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain−) and NrCAM at nodal axon segments. J Cell Biol. 1996;135:1355–1367. doi: 10.1083/jcb.135.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eber SW, Gonzalez JM, Lux ML, Scarpa AL, Tse WT, Dornwell M, Herbers J, Kugler W, Ozcan R, Pekrun A, Gallagher PG, Schröter W, Forget BG, Lux SE. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996;13:214–218. doi: 10.1038/ng0696-214. [DOI] [PubMed] [Google Scholar]
- Fishbein I, Segal M. Miniature synaptic currents become neurotoxic to chronically silenced neurons. Cereb Cortex. 2007;17:1292–1306. doi: 10.1093/cercor/bhl037. [DOI] [PubMed] [Google Scholar]
- Furuya S, Makino A, Hirabayashi Y. An improved method for culturing cerebellar Purkinje cells with differentiated dendrites under a mixed monolayer setting. Brain Res Brain Res Protoc. 1998;3:192–198. doi: 10.1016/s1385-299x(98)00040-3. [DOI] [PubMed] [Google Scholar]
- Greenquist AC, Shohet SB, Bernstein SE. Marked reduction of spectrin in hereditary spherocytosis in the common house mouse. Blood. 1978;51:1149–1155. [PubMed] [Google Scholar]
- Gulledge AT, Kampa BM, Stuart GJ. Synaptic integration in dendritic trees. J Neurobiol. 2005;64:75–90. doi: 10.1002/neu.20144. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Davis WS, Jorgensen EM. Mutations in beta-spectrin disrupt axon outgrowth and sarcomere structure. J Cell Biol. 2000;149:931–942. doi: 10.1083/jcb.149.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, Bastiani MJ. Axons break in animals lacking beta-spectrin. J Cell Biol. 2007;176:269–275. doi: 10.1083/jcb.200611117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Hayette S, Carré G, Bozon M, Alloisio N, Maillet P, Wilmotte R, Pascal O, Reynaud J, Reman O, Stéphan JL, Morlé L, Delaunay J. Two distinct truncated variants of ankyrin associated with hereditary spherocytosis. Am J Hematol. 1998;58:36–41. doi: 10.1002/(sici)1096-8652(199805)58:1<36::aid-ajh7>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Hirai H, Matsuda S. Interaction of the C-terminal domain of delta glutamate receptor with spectrin in the dendritic spines of cultured Purkinje cells. Neurosci Res. 1999;34:281–287. doi: 10.1016/s0168-0102(99)00061-9. [DOI] [PubMed] [Google Scholar]
- Hirano A, Dembitzer HM, Yoon CH. Development of Purkinje cell somatic spines in the weaver mouse. Acta Neuropathol. 1977;40:85–90. doi: 10.1007/BF00688577. [DOI] [PubMed] [Google Scholar]
- Hirano T, Kasono K, Araki K, Shinozuka K, Mishina M. Involvement of the glutamate receptor delta 2 subunit in the long-term depression of glutamate responsiveness in cultured rat Purkinje cells. Neurosci Lett. 1994;182:172–176. doi: 10.1016/0304-3940(94)90790-0. [DOI] [PubMed] [Google Scholar]
- Holleran EA, Ligon LA, Tokito M, Stankewich MC, Morrow JS, Holzbaur EL. beta III spectrin binds to the Arp1 subunit of dynactin. J Biol Chem. 2001;276:36598–36605. doi: 10.1074/jbc.M104838200. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Dürr A, Zühlke C, Bürk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- Inoue T, Lin X, Kohlmeier KA, Orr HT, Zoghbi HY, Ross WN. Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J Neurophysiol. 2001;85:1750–1760. doi: 10.1152/jn.2001.85.4.1750. [DOI] [PubMed] [Google Scholar]
- Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Jackson M, Song W, Liu MY, Jin L, Dykes-Hoberg M, Lin CI, Bowers WJ, Federoff HJ, Sternweis PC, Rothstein JD. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature. 2001;410:89–93. doi: 10.1038/35065091. [DOI] [PubMed] [Google Scholar]
- Jeromin A, Huganir RL, Linden DJ. Suppression of the glutamate receptor delta 2 subunit produces a specific impairment in cerebellar long-term depression. J Neurophysiol. 1996;76:3578–3583. doi: 10.1152/jn.1996.76.5.3578. [DOI] [PubMed] [Google Scholar]
- Kakegawa W, Miyazaki T, Emi K, Matsuda K, Kohda K, Motohashi J, Mishina M, Kawahara S, Watanabe M, Yuzaki M. Differential regulation of synaptic plasticity and cerebellar motor learning by the C-terminal PDZ-binding motif of GluRdelta2. J Neurosci. 2008;28:1460–1468. doi: 10.1523/JNEUROSCI.2553-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–79. doi: 10.1016/s0896-6273(01)80047-7. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Watanabe M, Kurihara H, Offermanns S, Jiang H, Wu Y, Jun K, Shin HS, Inoue Y, Simon MI, Wu D. Phospholipase cbeta4 is specifically involved in climbing fiber synapse elimination in the developing cerebellum. Proc Natl Acad Sci U S A. 1998;95:15724–15729. doi: 10.1073/pnas.95.26.15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Kennedy SP, Warren SL, Forget BG, Morrow JS. Ankyrin binds to the 15th repetitive unit of erythroid and nonerythroid beta-spectrin. J Cell Biol. 1991;115:267–277. doi: 10.1083/jcb.115.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda K, Kakegawa W, Matsuda S, Nakagami R, Kakiya N, Yuzaki M. The extreme C-terminus of GluRdelta2 is essential for induction of long-term depression in cerebellar slices. Eur J Neurosci. 2007;25:1357–1362. doi: 10.1111/j.1460-9568.2007.05412.x. [DOI] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M. Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta2 subunit. J Neurosci. 1997;17:9613–9623. doi: 10.1523/JNEUROSCI.17-24-09613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalouette A, Lohof A, Sotelo C, Guénet J, Mariani J. Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the delta-2 ionotropic glutamate receptor. Neuroscience. 2001;105:443–455. doi: 10.1016/s0306-4522(01)00193-2. [DOI] [PubMed] [Google Scholar]
- Linden DJ. A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron. 1996;17:483–490. doi: 10.1016/s0896-6273(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Lorenzo DN, Li MG, Mische SE, Armbrust KR, Ranum LP, Hays TS. Spectrin mutations that cause spinocerebellar ataxia type 5 impair axonal transport and induce neurodegeneration in Drosophila. J Cell Biol. 2010;189:143–158. doi: 10.1083/jcb.200905158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux SE, Pease B, Tomaselli MB, John KM, Bernstein SE. Hemolytic anemias associated with deficient or dysfunctional spectrin. Prog Clin Biol Res. 1979;30:463–469. [PubMed] [Google Scholar]
- Mariani J, Changeux JP. Multiple innervation of Purkinje cells by climbing fibers in the cerebellum of the adult staggerer mutant mouse. J Neurobiol. 1980;11:41–50. doi: 10.1002/neu.480110106. [DOI] [PubMed] [Google Scholar]
- Mariani J, Crepel F, Mikoshiba K, Changeux JP, Sotelo C. Anatomical, physiological and biochemical studies of the cerebellum from Reeler mutant mouse. Philos Trans R Soc Lond B Biol Sci. 1977;281:1–28. doi: 10.1098/rstb.1977.0121. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Watanabe M. Development of an anatomical technique for visualizing the mode of climbing fiber innervation in Purkinje cells and its application to mutant mice lacking GluRdelta2 and Ca(v)2.1. Anat Sci Int. 2011;86:10–18. doi: 10.1007/s12565-010-0095-1. [DOI] [PubMed] [Google Scholar]
- Moorthy S, Chen L, Bennett V. Caenorhabditis elegans beta-G spectrin is dispensable for establishment of epithelial polarity, but essential for muscular and neuronal function. J Cell Biol. 2000;149:915–930. doi: 10.1083/jcb.149.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J, Unwin N. Organization of spines on the dendrites of Purkinje cells. Proc Natl Acad Sci U S A. 2006;103:1575–1580. doi: 10.1073/pnas.0507884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc Natl Acad Sci U S A. 1997;94:14089–14094. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara O, Ohara R, Yamakawa H, Nakajima D, Nakayama M. Characterization of a new beta-spectrin gene which is predominantly expressed in brain. Brain Res Mol Brain Res. 1998;57:181–192. doi: 10.1016/s0169-328x(98)00068-0. [DOI] [PubMed] [Google Scholar]
- O'Leary T, van Rossum MC, Wyllie DJ. Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J Physiol. 2010;588:157–170. doi: 10.1113/jphysiol.2009.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJ, Dutia MB, Jackson M. Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J Neurosci. 2010;30:4857–4867. doi: 10.1523/JNEUROSCI.6065-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielage J, Fetter RD, Davis GW. Presynaptic spectrin is essential for synapse stabilization. Curr Biol. 2005;15:918–928. doi: 10.1016/j.cub.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Pielage J, Fetter RD, Davis GW. A postsynaptic spectrin scaffold defines active zone size, spacing, and efficacy at the Drosophila neuromuscular junction. J Cell Biol. 2006;175:491–503. doi: 10.1083/jcb.200607036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Sidman RL. Organization of cerebellar cortex secondary to deficit of granule cells in weaver mutant mice. J Comp Neurol. 1973;152:133–161. doi: 10.1002/cne.901520203. [DOI] [PubMed] [Google Scholar]
- Rall W. Core conductor theory and cable properties of neurons. In: Kandel ER, editor. Handbook of Physiology: the nervous system I. Bethesda: American Physiological Society; 1977. pp. 39–97. [Google Scholar]
- Randon J, Miraglia del Giudice E, Bozon M, Perrotta S, De Vivo M, Iolascon A, Delaunay J, Morle L. Frequent de novo mutations of the ANK1 gene mimic a recessive mode of transmission in hereditary spherocytosis: three new ANK1 variants: ankyrins Bari, Napoli II and Anzio. Br J Haematol. 1997;96:500–506. doi: 10.1046/j.1365-2141.1997.d01-2074.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi G, Orita S, Naito A, Maeda M, Igarashi H, Sasaki T, Takai Y. A novel brain-specific isoform of beta spectrin: isolation and its interaction with Munc13. Biochem Biophys Res Commun. 1998;248:846–851. doi: 10.1006/bbrc.1998.9067. [DOI] [PubMed] [Google Scholar]
- Segal I, Korkotian I, Murphy DD. Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci. 2000;23:53–57. doi: 10.1016/s0166-2236(99)01499-x. [DOI] [PubMed] [Google Scholar]
- Shotton DM, Burke BE, Branton D. The molecular structure of human erythrocyte spectrin. Biophysical and electron microscopic studies. J Mol Biol. 1979;131:303–329. doi: 10.1016/0022-2836(79)90078-0. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Anatomical, physiological and biochemical studies of the cerebellum from mutant mice. II. Morphological study of cerebellar cortical neurons and circuits in the weaver mouse. Brain Res. 1975;94:19–44. doi: 10.1016/0006-8993(75)90874-4. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Dusart I. Intrinsic versus extrinsic determinants during the development of Purkinje cell dendrites. Neuroscience. 2009;162:589–600. doi: 10.1016/j.neuroscience.2008.12.035. [DOI] [PubMed] [Google Scholar]
- Speicher DW, Morrow JS, Knowles WJ, Marchesi VT. A structural model of human erythrocyte spectrin. Alignment of chemical and functional domains. J Biol Chem. 1982;257:9093–9101. [PubMed] [Google Scholar]
- Stankewich MC, Tse WT, Peters LL, Ch'ng Y, John KM, Stabach PR, Devarajan P, Morrow JS, Lux SE. A widely expressed betaIII spectrin associated with Golgi and cytoplasmic vesicles. Proc Natl Acad Sci U S A. 1998;95:14158–14163. doi: 10.1073/pnas.95.24.14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankewich MC, Gwynn B, Ardito T, Ji L, Kim J, Robledo RF, Lux SE, Peters LL, Morrow JS. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc Natl Acad Sci U S A. 2010;107:6022–6027. doi: 10.1073/pnas.1001522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E, Gratzer W. Self-association of human spectrin. A thermodynamic and kinetic study. Eur J Biochem. 1978;88:379–385. doi: 10.1111/j.1432-1033.1978.tb12459.x. [DOI] [PubMed] [Google Scholar]
- Vecellio M, Schwaller B, Meyer M, Hunziker W, Celio MR. Alterations in Purkinje cell spines of calbindin D-28 k and parvalbumin knock-out mice. Eur J Neurosci. 2000;12:945–954. doi: 10.1046/j.1460-9568.2000.00986.x. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]