

Abstract
Niemann-Pick type C (NPC) disease is a neurovisceral atypical lipid storage disorder involving the accumulation of cholesterol and other lipids in the late endocytic pathway. The pathogenic mechanism that links the accumulation of intracellular cholesterol with cell death in NPC disease in both the CNS and the liver is currently unknown. Oxidative stress has been observed in the livers and brains of NPC mice and in different NPC cellular models. Moreover, there is evidence of an elevation of oxidative stress markers in the serumof NPC patients. Recent evidence strongly suggests that mitochondrial dysfunction plays an important role in NPC pathogenesis and that mitochondria could be a significant source of oxidative stress in this disease. In this context, the accumulation of vitamin E in the late endosomal/lysosomal compartments in NPC could lead to a potential decrease of its bioavailability and could be another possible cause of oxidative damage. Another possible source of reactive species in NPC is the diminished activity of different antioxidant enzymes. Moreover, because NPC is mainly caused by the accumulation of free cholesterol, oxidized cholesterol derivatives produced by oxidative stress may contribute to the pathogenesis of the disease.
1. Introduction
Niemann-Pick type C disease (NPC) is a neurovisceral atypical lipid storage disorder that is mainly characterized by unesterified cholesterol accumulation in late endosomal/lysosomal (LE/Lys) compartments [1]. Several other kinds of lipids, such as lactosylceramide, glucosylceramide and GM2 and GM3 gangliosides, also accumulate in these compartments [2–5]. NPC is a fatal autosomal recessive disease that is caused by mutations in the Npc1 or Npc2 genes [6]. The Npc1 gene encodes a 1278-amino-acid lysosomal transmembrane protein with 13 transmembrane domains that have homology to the sterol-sensing domains (SSDs) of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase, SREBP cleavage-activating protein (SCAP), and Patched [7, 8]. NPC1 protein localizes to the Rab7-positive and mannose-6-P-receptor-negative late endosomal compartment [9–11]. The Npc2 gene encodes a soluble lysosomal protein that binds cholesterol [12] with a 1 : 1 stoichiometry and submicromolar affinity [13, 14]. Both proteins are involved in cholesterol trafficking from lysosomes. The current model for NPC1- and NPC2-mediated cholesterol efflux is as follows: after lysosomal hydrolysis of LDL-cholesteryl esters, cholesterol binds NPC2, which transfers it to NPC1 [15]; subsequently, NPC1 mediates the exit of cholesterol from lysosomes [16]. Mutations in the Npc1 gene account for approximately 95% of NPC cases, whereas mutations in the Npc2 gene explain the remaining 5% [17].
The disease is often diagnosed in early childhood, with patients typically displaying cerebellar ataxia, speaking and swallowing difficulty, and progressive dementia [1, 17]. The main neurological symptom of this disease is vertical supranuclear ophthalmoplegia, and its other prominent neurologic features include cataplexy, dysarthria, dysphagia, dystonia, and seizures [18, 19]. These symptoms are associated with damage to the central nervous system (CNS), especially in the cerebellum, where extensive and progressive neuronal death is observed [20].
Although neuronal damage is a major feature of NPC, most patients present considerable damage in the liver [2]. Indeed, NPC disease is recognized as a relatively common cause of liver disease in early life [21]. Approximately half of NPC patients suffer from liver disease, and NPC may be the most common metabolic disorder that is responsible for neonatal cholestasis [17].
The pathogenic mechanism linking the accumulation of intracellular cholesterol with cell death in NPC disease is currently unknown. However, increasing evidence indicating the presence of oxidative damage in NPC neurons and data connecting oxidative damage with fibrosis and apoptosis in the liver support the possibility that oxidative damage may induce these pathways in NPC disease. The question that remains unsolved is how oxidative damage is induced.
2. Cell Death and Oxidative Damage in NPC
The CNS is especially sensitive to oxidative stress damage [22]. This sensitivity can be explained by several features of the CNS: the high concentration of polyunsaturated fatty acids that are susceptible to lipid peroxidation, the relatively large amounts of oxygen consumed for energy production, and the fewer antioxidant defenses available to the CNS compared with other organs. In this sense, neurons are particularly vulnerable to oxidative stress because they have low levels of reduced glutathione (GSH) [23]. Oxidative stress has been observed in different NPC cellular models [24] as well as in the brains of NPC mice [25]; however, the functional relevance of oxidative stress to the disease process has not yet been established. In this context, we have reported increased levels of nitrotyrosinilated (N-Tyr) proteins in the cerebella of NPC mice by western blot analysis and an accumulation of N-Tyr-positive cells by immunofluorescence [26]. Furthermore, Smith et al. detected positive staining for N-Tyr in the thalami of NPC mice [25]. We have also found that, in a neuronal model of the disease, treatment with an antioxidant compound diminished c-Abl kinase activation and prevented cellular death and apoptosis [26]. This observation is particularly relevant because oxidative stress is a potent activator of the c-Abl/p73 proapoptotic pathway [27]. In fact, c-Abl/p73 activation kinetics correlate with the kinetics of the appearance of oxidative-stress markers in rat hippocampal neurons exposed to U18666A (U18), an inducer of cholesterol accumulation in lysosomes and a drug that has been widely used to induce the NPC phenotype in different cell types [28–30]. Highlighting the role of oxidative stress in NPC, we have shown that treatment with the antioxidant N-Acetyl Cysteine (NAC) prevented c-Abl/p73 activation and apoptosis in NPC-like neurons, suggesting that oxidative stress is the main upstream stimulus activating the c-Abl/p73 pathway in NPC neurons [26].
Livers of NPC mouse models present apoptosis, inflammation, and fibrosis. There is also functional damage, as evidenced by the large increases in the levels of liver disease markers such as plasmatic alanine and aspartate aminotransferases in NPC mice [31–33]. In support of the participation of oxidative stress in NPC mouse livers, we have recently found an increase in oxidative stress markers, such as protein carbonyls and oxidative-stress related genes, and a decrease in protective species, such as reduced glutathione [34].
Moreover, NPC patients present decreased antioxidant capacity (expressed as Trolox equivalents) and reduced Coenzyme Q10 in serum, which indicates a decrease in antioxidant defenses [35]. In addition, an increase in cholesterol oxidation products was detected in the plasma of NPC patients [36]. Additionally, microarray studies have shown that human NPC fibroblasts exhibit an activation of the apoptotic cascade and induction of genes related to oxidative stress [37, 38]. Oxidative damage is present in animal and cellular NPC models and in NPC patients; therefore, it may have a relevant role in the pathogenesis and progression of the disease.
3. Increase in Mitochondrial Cholesterol and Dysfunction in NPC Disease
One possible element associated with oxidative damage in NPC tissues could be mitochondrial dysfunction. In fact, mitochondrial dysfunction appears to be a key element in many neurodegenerative diseases and pathologies associated with liver and cardiac damage as a significant source of oxidative stress [39–44].
However, the role of mitochondrial dysfunction in the NPC disease phenotype remains undefined. Recent studies suggest that mitochondrial dysfunction and subsequent ATP deficiency may be responsible for the neuronal impairment in NPC disease [45]. These studies have demonstrated that NPC mouse brains contained smaller and more rounded mitochondria, with translucent matrices and irregular cisternae. Moreover, the levels of ATP in the brain, muscle, and livers of NPC mice are significantly decreased compared with those in WT mice [45]. Interestingly, several groups have shown that the mitochondria of NPC neurons and hepatocytes have a higher cholesterol content [45–48], and increased cholesterol has also been shown in the mitochondria of HeLa cells treated with U18 [49]. These data should be interpreted with caution, as it is difficult to obtain pure mitochondria uncontaminated by lysosomes because of the interaction between these organelles as a result of the increased autophagy in NPC cells [50, 51]. However, treatment with the cholesterol chelator cyclodextrin restores ATP synthesis and mitochondrial function [45], supporting the idea that the increase in mitochondrial cholesterol causes dysfunction per se. Therefore, a potential hypothesis is that the increase in mitochondrial cholesterol content and dysfunction may contribute to this damage (Figure 1).
Figure 1.
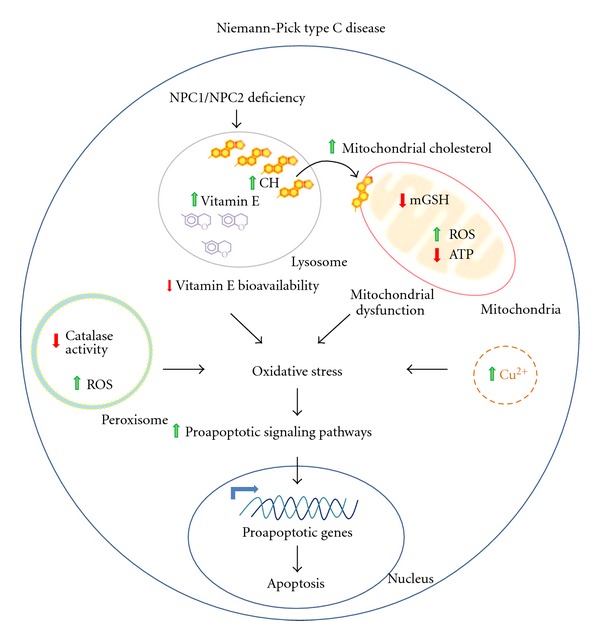
Oxidative stress as a pathogenic mechanism in Niemann-Pick C disease. NPC disease arises from deficiencies in one of two lysosomal proteins, NPC1 and NPC2, which are involved in the proper export of free cholesterol (CH) from lysosomes to different cellular compartments. Therefore, in NPC disease, free cholesterol accumulates inside the lysosomes and intracellular cholesterol transport is impaired. Remarkably, vitamin E also accumulates inside the lysosomes, possibly diminishing its bioavailability and decreasing the antioxidant capacity of the cell. Additionally, mitochondrial cholesterol is also increased, leading to a decrease in mGSH levels and diminishing mitochondria antioxidant capacity. In this scenario, there is an increase in mitochondrial ROS production and a decrease in ATP production. These two phenomena are well-known features of mitochondrial dysfunction. Furthermore, decreases of catalase activity and increases in ROS production inside the peroxisomes have been reported. In summary, in NPC disease, alterations in several different organelles, including lysosomes, mitocondria, and peroxisomes, along with disturbances in copper (Cu) transport, trigger oxidative stress damage, activating proapoptotic pathways and proapoptotic gene expression and inducing apoptotic cell death. Symbols: green up-arrows indicate increase or activation; red down-arrows indicate decrease or inhibition; bold black arrows represent multistep pathways; angled blue arrows indicate gene transcription activation.
Mitochondrial glutathione (mGSH) is the main line of defense against the reactive oxygen species (ROS) generated physiologically through the activity of the electron transport chain in the mitochondria and is therefore a key player in the maintenance of the appropriate mitochondrial redox environment, preventing mitochondrial dysfunction, and cell death. We have detected reduced GSH content in the livers of NPC mice [34], and Marí et al. reported a specific decrease of mGSH in NPC hepatocytes without changes in cytosolic GSH [46]. Interestingly, the increase in mitochondrial cholesterol seems to have a direct effect on the transport of GSH to the mitochondria, because the activity of its transporter depends on membrane fluidity [52, 53]. A decrease of membrane fluidity in mitochondria or an increased cholesterol : phospholipid molar ratio, as is observed in rat liver mitochondria after chronic alcohol intake, results in an impairment of mGSH transport through the 2-oxoglutarate carrier and, therefore, in a decrease in mGSH levels [54]. Supporting a connection between mitochondrial cholesterol content and reduced mGSH levels, evidence suggests that the hepatocytes of ob/ob mice or rats fed high-cholesterol diets have reduced mGSH levels and increased mitochondrial cholesterol content. Furthermore, hepatocytes from these mice and those obtained from NPC mice have decreased GSH content and increased sensitivity to TNF-α cell death signaling [46].
In summary, regarding the role of mitochondrial dysfunction in NPC disease, it has been observed in NPC models that important mitochondrial characteristics such as ATP synthesis, mitochondrial morphology, and the input of GSH to the mitochondrial matrix are altered. These alterations are correlated with increased mitochondrial cholesterol content and together result in a reduction in energy production, increased ROS production, and increased energy demand in NPC cells.
4. Peroxisomes Alterations in NPC Disease
Another relevant source of ROS are the peroxisomes [55]. In fact, peroxisomal catalase is one of the most important antioxidant enzymes that regulate intracellular H2O2 levels. Interestingly, different studies have shown early alterations of several peroxisomal enzymatic activities, such as catalase, and β-oxidation enzymes in the livers and cerebella of NPC mice (Figure 1) [56]. There is also one clinical case report of defective peroxisomal β-oxidation in an 18-month-old NPC patient [57]. Moreover, treatment of NPC mice with the peroxisomal inducers perfluorooctanoic acid or clofibrate rescued peroxisomal and lysosomal enzyme activities and decreased cholesterol content [58]. Furthermore, several groups have demonstrated that the activity and protein levels of catalase are regulated in a biphasic fashion by c-Abl kinase. At low H2O2 levels, catalase interacts with c-Abl, which stabilizes catalase by phosphorylating it at Tyr residues. Meanwhile, at high H2O2 levels or after sustained proapoptotic stimulus, c-Abl translocates to the nucleus to perform its proapoptotic function without phosphorylating catalase, which in turn is degraded [59–61]. Although it is still unknown where the interaction between intraperoxisomal catalase and c-Abl (which is located in cytosol, endoplasmic reticulum and the nucleus) occurs, this regulation has been demonstrated in several cell types, including fibroblasts and 293 cells [61]. As was mentioned above, we have demonstrated that in NPC neurons, the c-Abl kinase is activated and induces apoptosis [26, 62]. These antecedents suggest that reduced catalase activity in NPC cells could be related to alterations in c-Abl regulation mechanisms, contributing to the oxidative damage observed in the disease.
5. Intracellular Accumulation of Vitamin E in NPC Disease
Intracellular accumulation of vitamin E and decreased bioavailability of this antioxidant has emerged as one of the possible causes of oxidative damage in NPC. Vitamin E, and in particular α-tocopherol, its most important biological derivative, is one of the most relevant antioxidant defense molecules in addition to GSH. Both react directly with free radicals to form inactive, nonradical products [63–67].
Data from the literature and results from our lab show increased levels of α-tocopherol in the cerebella and a tendency to increase in the livers of NPC mice, and α-tocopherol accumulation was observed in several NPC in vitro models in a filipin-positive compartment (Figure 1) [68, 69]. Interestingly, one of the most damaged regions in NPC disease, the cerebellum, contains high levels of vitamin E [70]. Furthermore, mutations in the gene coding for the α-tocopherol transporter protein (α-Ttp) result in a neurologic syndrome of spinocerebellar ataxia called Ataxia with Vitamin E Deficiency or AVED. This condition is characterized by progressive ataxia, dysarthria, sensory loss, and severe damage of Purkinje cells [70, 71], sharing some symptoms with NPC patients. In addition, vitamin E supplementation has been demonstrated to protect against age-related deficits in Purkinje cell-β adrenergic receptor function and ethanol-induced Purkinje cell loss in rats [72, 73]. Moreover, interestingly, NPC1-like 1 (NPC1L1), which is the closest NPC1 homologue, mediates α-tocopherol transport in rat intestines [74].
Collectively, these data suggest that vitamin E transport through lysosomes could be mediated by NPC proteins and that it might therefore be altered in NPC cells. Because vitamin E is essential for health and increasing evidence suggest that antioxidants have a protective role in oxidative damage-associated diseases, studies on the accumulation and bioavailability of vitamin E in NPC cells are relevant for understanding the pathophysiology of this disease and the possible benefits of vitamin E therapy.
Interestingly, in addition to its antioxidant properties, α-tocopherol also induces signaling in cells [75]. It has been shown that α-tocopherol activates phosphatase 2A (PP2A), causing dephosphorylation and diminishing PKC alpha activity in various types of cells [75]. In addition, α-tocopherol inhibits TNF-α-induced ERK1/2 and p38 MAPKs activation [76]. These results suggest an anti-inflammatory action of α-tocopherol in addition to its antioxidant properties.
Our hypothesis is that vitamin E is trapped in LE/Lys and that it has a decreased bioavailability in NPC cells (Figure 1). However, we cannot disregard a different scenario in which the excess vitamin E compounds acting as prooxidants in NPC cells. In fact, α-TOH can play diverse roles in lipoprotein oxidation, displaying neutral, anti-, or, indeed, prooxidant activities under various conditions [77]. For example, in the presence of determinant Cu2+/LDL ratios, α-TOH acts as a mediator of LDL lipid peroxidation [78]. Therefore, an alternative hypothesis is that the lysosomal buildup of vitamin E found in NPC cells could actively contribute to the pathology, acting as a pro-oxidant molecule and increasing the levels of toxic cholesterol oxidation products.
It is therefore important to mention that a previous in vivo study [79] showed that treatment with vitamin E exerts a small but significant beneficial effect on locomotor performance in NPC mice. It may be assumed that the effects of this treatment were small because part of the vitamin E was trapped in the lysosomes and was not available as cell antioxidants, making it less likely that the small protective effect is due to its behavior as a pro-oxidant molecule. Furthermore, another study with vitamin C failed to obtain a significant benefit in NPC mice [25]; this failure might be related to the lack of neuroprotective properties of vitamin C in neurodegenerative diseases in which oxidative stress plays a key role and vitamin E serves as a neuroprotectant [80]. On the other hand, there is evidence for other potential NPC treatments that can be neuroprotective. Early treatment with the neurosteroid allopregnanolone also improved neurological symptoms and survival in NPC mice by correcting the neurosteroidogenic abnormalities [81]. Interestingly, allopregnanolone was demonstrated to work as a potent antioxidant in in vitro NPC models [82]. Treating NPC mice in vivo with curcumin, a potent activator of the antioxidant Nrf2 pathway [83], also improves the neurological symptoms and survival of NPC mice [5]. All these studies indicate a relevant role for oxidative stress in NPC-related neurodegeneration.
6. Glycosphingolipid Accumulation in NPC Disease
It is important to note that in NPC disease, there is not only cholesterol accumulation in the LE/Lys compartment but also an increased accumulation of other kinds of lipids. The improvement observed in patients treated with miglustat (Zavesca; Actelion Pharmaceuticals Ltd., Allschwil, Switzerland) can be attributed to its capacity to decrease the levels of glycosphingolipids. The accumulation of glycosphingolipids leads to defective intracellular calcium signaling, as has been demonstrated in several gangliosidoses [84]. Defective intracellular calcium could be detrimental for the cell because calcium plays vital roles in regulating a variety of cellular events, with impaired calcium homeostasis leading to endoplasmic reticulum (ER) stress, oxidative stress, and cell death. Moreover, Platt's group has demonstrated that sphingosine storage, an early event in NPC LE/Lys, causes a unique defect, inhibiting the filling of the LE/Lys calcium store [5]. Indeed, treatment of NPC1 CHO cells with thapsigargin elevated cytosolic calcium and corrected the NPC cellular phenotype, reducing glycosphingolipid and cholesterol accumulation. Additionally, the chelation of LE/Lys calcium content in healthy cells induces a set of cellular phenotypes identical to NPC, including defective endocytic transport and the subsequent storage of cholesterol, glycosphingolipids, and sphingomyelin [5, 85].
The depletion of glycosphingolipids by miglustat treatment reduces pathological lipid storage, improves endosomal uptake, and normalizes lipid trafficking in peripheral blood B lymphocytes [86]; however, the incomplete success of miglustat treatment in patients may be because it addresses only one aspect of the pathological cascade of NPC disease. Fu and colleagues [87] showed that miglustat therapy does not significantly improve oxidative stress in NPC patients. This result suggests that oxidative stress is an independent pathological process and that combination therapies that include an antioxidant may have an additional benefit in NPC patients.
7. Other Sources of Reactive Species in Oxidative Damage in NPC
Another possible source of reactive species in NPC is the diminished activity of different antioxidant enzymes. For example, catalase is one of the most important antioxidant enzymes participating in the regulation of intracellular H2O2, and, as mentioned previously, its levels are decreased in the livers and brains of NPC mice [56]. In addition, another antioxidant enzyme located in the cytosol and mitochondria, glutathione peroxidase 1, or GPx1, is regulated in the same way by the proapoptotic kinase c-Abl [61, 88], which is activated in NPC neurons. The activity of GPx1 is thought to be relevant to antioxidant defenses at the neuronal level, and GPx1−/− mice showed greater apoptosis-related sensitivity and damage in the brain when faced with extreme oxidative damage, such as that produced by ischemia-reperfusion [89].
Oxygen species are not the only inducers of oxidative damage to biomolecules. There is evidence for nitric-oxide- (NO-) mediated damage in NPC. Peroxynitrite binds a nitro group to tyrosines, inducing protein nitrotyrosination, which negates the physiological function of the proteins [90]. Interestingly, NO levels are elevated in neural stem cells (NSCs) from NPC mice and NSC self-renewal is decreased [91], suggesting that oxidative stress damage could be relevant in NPC neurodegeneration, even at the early stages of development [92]. Moreover, microarray analyses of human NPC fibroblasts have revealed an increase in NO synthase mRNA expression [37] and an increase in N-Tyr staining has been reported in fibroblasts [93].
Considering other additional sources for oxidative stress, we previously reported copper accumulation (Figure 1) in the liver [34] and plasma, along with a decrease of copper excretion into the bile of Npc1−/− (NPC) mice (unpublished results). These data are in agreement with previous reports of copper increase and ceruloplasmin alterations in a cellular NPC model [94, 95]. Copper is an important micronutrient that plays an essential role in human physiology [96]. Increases in copper, iron, or zinc levels have been described as risk factors for oxidative stress damage in neurodegenerative pathologies, such as amyotrophic lateral sclerosis, Alzheimer's Disease, and Parkinson's Disease [97]. Furthermore, mutations in copper-binding proteins have been linked to those devastating disorders [98] and to Wilson disease and Menkes disease [99, 100]. Therefore, appropriate levels of copper are essential to avoid cellular damage by oxidative stress due to the rapid oxidation of copper, which causes damage to biomolecules and generates ROS, leading to cell death [101]. The liver plays a major role in this delicate maintenance of copper homeostasis, because most of the newly absorbed copper enters the liver after absorption from dietary sources in the small intestine. Additionally, the liver regulates the distribution of copper through its release into the plasma when bound to ceruloplasmin or by its excretion via bile [102]. Moreover, the expression of genes involved in metal homeostasis and transport, including iron, copper, and zinc, was reported to be altered in NPC fibroblast microarray studies [37, 38], suggesting alterations in metal levels in NPC disease.
As NPC is mainly caused by the accumulation of free cholesterol, it can be expected that oxidized cholesterol derivatives are produced in the presence of oxidative stress. In fact, an increase in cellular cholesterol oxidation products, such as 7-ketocholesterol (7-KC), 7β-hydroxycholesterol (7β-HC), and cholestane-3β,5α,6β-triol (3β,5α,6β-triol), has been described in NPC mouse tissues, plasma, and macrophages [36, 103, 104]. They are produced by the nonenzymatic oxidation of cholesterol with ROS and several of them have been reported to be cytotoxic in vitro [105]. Although the mechanism of oxysterol production in NPC is unclear, and most of the cholesterol should accumulate in lysosomes, the elevation of oxysterol levels in the disease has been consistently reproduced in mice and humans, and they have been proposed as biomarkers for NPC [36]. Moreover, 7-KC signaling has been shown to be modulated by vitamin E. It has been demonstrated that α-tocopherol impedes the cellular signaling of 7-KC, inhibiting its incorporation in lipid rafts [106]. As such, if there is less bioavailability of vitamin E in NPC cells, the proapoptotic effect of 7-KC, through its inhibition of the phosphatidylinositol 3-kinase/Akt survival pathway [107], could be further increased.
Remarkably, a study by Porter et al. [36] showed that cholesterol oxidation product levels correlated with the age of disease onset and disease severity in NPC mice and humans. Thus, cholesterol oxidation products have been proposed as blood-based biochemical markers for NPC disease that may prove informative for the diagnosis and treatment of this disorder and as outcome measures to monitor the response to therapy. These results suggest that oxidative damage is an important element in the pathology of NPC disease.
8. Putative Treatments for NPC Disease
Unfortunately, there is no fully effective treatment for this devastating and fatal disorder to date, only supportive measures for the relief of specific manifestations of the disease. Interventions to slow disease progression are the most promising therapies. Several experimental disease-specific therapies based on the molecular pathology of NPC have been tested in cell culture and animal models, including neurosteroids, cholesterol-binding agents (e.g., cyclodextrin), and molecules with antioxidant properties, such as curcumin and miglustat [108].
Among the cholesterol-binding agents, cyclodextrin seems to be promising [109], although data suggesting limited or no blood-brain barrier penetration of cyclodextrin following systemic administration have to be considered [110]. Treatment of NPC mice with cyclodextrin reduces the neurodegeneration and markedly extends the life span of NPC mice, suggesting a potential therapeutic approach for the treatment of individuals with NPC disease [111]. The mechanisms by which cyclodextrin mediates these beneficial effects are still unknown. The evidence shows that a single injection of cyclodextrin at p7 in NPC mice caused a marked increase in cholesteryl esters and the suppression of cholesterol synthesis in many organs, as well as changes in the expression of genes responsive to cholesterol levels [112], suggesting that cyclodextrin acutely reverses the lysosomal transport defect observed in NPC disease. However, although cyclodextrin has promising effects in the brain and liver, it has little or no effect at all on lung dysfunction, another important issue in NPC pathology [113]. Nonetheless, the positive data on cyclodextrin reported in model animals have encouraged its application as a potential treatment in NPC patients. In fact, last year, the European Medicines Agency (EMA) granted Hydroxypropyl-beta-cyclodextrin (HPbCD) orphan drug status and designated the compound as a potential treatment for NPC disease. In addition, the National Institutes of Health (NIH) in collaboration with the Therapeutics for Rare and Neglected Diseases Program (TRND) announced that they are developing a clinical trial utilizing cyclodextrin for NPC patients. This clinical trial is in the planning phase and is not yet officially approved by the FDA.
Regarding the use of antioxidant molecules, it has recently been reported that NPC patients showed significant decreases in the fractions of reduced coenzyme Q10 (CoQ10) [87]. This decrease in CoQ10 levels can cause changes similar to those reported in patients with deficiencies in CoQ10. CoQ10 deficiency is a rare human genetic condition that has been associated with a variety of clinical phenotypes: decreased activities of complex II + III, complex III and complex IV, reduced expression of the mitochondrial proteins involved in oxidative phosphorylation, decreased mitochondrial membrane potential, increased ROS activation of mitochondrial permeability transition (MPT), and reduced growth rates [114]. In fibroblasts from patients with CoQ deficiency, these abnormalities were partially restored by CoQ supplementation [114]. However, supplementation with CoQ10 does not correct the abnormal fraction of reduced CoQ10 found in NPC patients [87]. Although the treatment did not restore the levels of reduced CoQ10 in NPC patients, the authors did not analyze the effect of the treatment on mitochondrial function and ROS production.
Currently, there is an active clinical protocol to test the safety and efficacy of the antioxidant NAC in NPC patients at the National Institute of Health Clinical Center in the U.S.A. (http://clinicaltrials.gov/ct2/show/NCT00975689). NAC is a powerful antioxidant that acts by increasing cellular glutathione levels [115–117]. The advantage of using NAC lies in the fact that it has been approved by the FDA to treat acetaminophen poisoning or to reduce mucous stickiness in patients with cystic fibrosis. Plasma oxysterol analysis will be used as a biomarker in the treatment, because Ory's group has previously demonstrated an increase of oxysterols in the plasma of NPC mice [103]. Interestingly, NAC treatment shows beneficial effects not only in neurodegenerative diseases such as Alzheimer's but also in myocardial dysfunction [116]. In addition, we have reported a decrease of foamy cells on the livers of 7-week-old NPC mice after 2 weeks of treatment with NAC [34], and we also have shown a decrease in apoptosis in U18-treated neurons incubated with NAC [26]. Although the acute use of NAC has demonstrated clear beneficial effects [86], its chronic use for the treatment of diseases associated with oxidative damage is controversial [118], mainly because, to date, there have not been positive results reported in this clinical trial, so it is not clear if this treatment could be effective. A possible explanation for this lack of effectiveness is that the antioxidant effect of NAC results from its ability to restore the cytosolic levels of GSH, which is transported to mitochondria, where it exerts its detoxifying function. The mitochondrial transport of GSH is highly sensitive to membrane dynamics, and this transport is delayed in hepatocytes from NPC mice and is correlated with mitochondrial GSH (mGSH) depletion [46]. Therefore, NAC therapy would not be effective in NPC disease, because although the levels of cytosolic GSH are increased, GSH transport into mitochondria remains defective.
Following this line of evidence, it has recently been shown that a curcumin derivative, J147, has the ability to prevent memory deficits in AD transgenic mice, and this effect is correlated with the reduction of markers for oxidative stress damage and inflammation [119]. Furthermore, it has previously been reported that curcumin treatment can normalize cytosolic calcium levels, prolonging the survival of NPC mice [5]. However, this explanation might not be the only one for the curcumin protective effect, as it is also an antioxidant molecule.
Another putative drug that may be useful in ameliorating NPC symptomatology is imatinib, a specific c-Abl kinase inhibitor. Imatinib treatment increased the quality of life of NPC mice by delaying body weight loss and neurological symptoms, decreasing cerebellar apoptosis and inflammation, and increasing Purkinje cell survival. However, the inhibition of c-Abl with imatinib did not affect oxidative stress levels, suggesting that c-Abl/p73 activation in NPC is downstream of oxidative stress. Nonetheless, inhibition of the c-Abl/p73 module is still an interesting therapeutic target in NPC and perhaps in other neurological disorders as well [62]. Interestingly, imatinib (Gleevec, STI571) is a U.S. Food and Drug Administration-approved drug used in patients with chronic myelogenous leukemia, in which the therapeutic target is the aberrant oncogenic fusion protein Bcr-Abl [120, 121].
Finally, it seems that the path to an effective therapy for NPC disease could be a multidrug approach that combines antioxidant and anti-inflammatory compounds with other drugs that improve lipid metabolism. In this sense, it is important to note that the first and only approved therapy for patients with NPC, miglustat [122–124], is a reversible inhibitor of glucosylceramide synthase, which catalyzes the first committed step of glycosphingolipid synthesis. Miglustat is currently approved in the European Union (EU), USA, Canada, Brazil, Australia, Turkey, Israel, Switzerland, South Korea, and New Zealand for the treatment of patients with Gaucher disease. In January 2009, the EU Commission extended miglustat's indication to include the treatment of progressive neurological manifestations in adult pediatric patients with NPC. This extension was followed by authorization in Brazil and South Korea [108, 125].
9. Concluding Remarks
NPC disease is a fatal disorder without an efficient treatment available. Several approaches have attempted to at least delay the disease progress. A better understanding of the mechanisms involved in the pathogenesis of NPC is necessary to design appropriate and effective therapeutic approaches. Increasing evidence indicates that oxidative stress damage has an important role in the pathophysiology of NPC disease. Therefore, drugs that can decrease or ameliorate oxidative stress damage and apoptosis must be used in combination with other pharmacological strategies that restore the proper metabolism and transport of lipids, especially cholesterol, in NPC patients.
Acknowledgments
The authors acknowledge the involvement of many colleagues who have participated in the studies and contributed with helpful discussions. This work is supported by Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT, Grants no. 3100026 (to M. C. Vázquez), no. 1080221 (to A. Alvarez), and no. 1110310 (to S. Zanlungo) and Fondo Nacional de Desarrollo de Areas Prioritarias, FONDAP, Project no. 15090007, Center for Genome Regulation (CGR) to SZ.
References
- 1.Patterson MC, Vanier MT, Suzuki K, et al. Niemann-Pick disease type C. A lipid trafficking disorder. In: Scriver CR, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. Vol. 2. New York, NY, USA: Mulencer Hill; 2001. [Google Scholar]
- 2.Liscum L, Klansek JJ. Niemann-Pick disease type C. Current Opinion in Lipidology. 1998;9(2):131–135. doi: 10.1097/00041433-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. Journal of Cell Biology. 1989;108(5):1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liscum L, Sturley SL. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochimica et Biophysica Acta. 2004;1685(1–3):22–27. doi: 10.1016/j.bbalip.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Lloyd-Evans E, Morgan AJ, He X, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature Medicine. 2008;14(11):1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 6.Vanier MT, Duthel S, Rodriguez-Lafrasse C, Pentchev P, Carstea ED. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. American Journal of Human Genetics. 1996;58(1):118–125. [PMC free article] [PubMed] [Google Scholar]
- 7.Carstea ED, Morris JA, Coleman KG, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 8.Davies JP, Ioannou YA. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. The Journal of Biological Chemistry. 2000;275(32):24367–24374. doi: 10.1074/jbc.M002184200. [DOI] [PubMed] [Google Scholar]
- 9.Frolov A, Srivastava K, Daphna-Iken D, Traub LM, Schaffer JE, Ory DS. Cholesterol overload promotes morphogenesis of a Niemann-Pick C (NPC)-like compartment independent of inhibition of NPC1 or HE1/NPC2 function. The Journal of Biological Chemistry. 2001;276(49):46414–46421. doi: 10.1074/jbc.M108099200. [DOI] [PubMed] [Google Scholar]
- 10.Neufeld EB, Wastney M, Patel S, et al. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. The Journal of Biological Chemistry. 1999;274(14):9627–9635. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- 11.Zhang M, Dwyer NK, Neufeld EB, et al. Sterol-modulated glycolipid sorting occurs in Niemann-Pick C1 late endosomes. The Journal of Biological Chemistry. 2001;276(5):3417–3425. doi: 10.1074/jbc.M005393200. [DOI] [PubMed] [Google Scholar]
- 12.Naureckiene S, Sleat DE, Lacklan H, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 13.Friedland N, Liou HL, Lobel P, Stock AM. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2512–2517. doi: 10.1073/pnas.0437840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ory DS. The Niemann-Pick disease genes: regulators of cellular cholesterol homeostasis. Trends in Cardiovascular Medicine. 2004;14(2):66–72. doi: 10.1016/j.tcm.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Kwon HJ, Abi-Mosleh L, Wang ML, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du X, Kumar J, Ferguson C, et al. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. Journal of Cell Biology. 2011;192(1):121–135. doi: 10.1083/jcb.201004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wraith JE, Baumgartner MR, Bembi B, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Molecular Genetics and Metabolism. 2009;98(1-2):152–165. doi: 10.1016/j.ymgme.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Pentchev PGV, Vanier MT, Suzuki K, Patterson MC. Niemann-Pick disease type C: a cellular cholesterol lipidosis. In: Scriver CRB, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY, USA: McGraw Hill; 1995. pp. 2625–2639. [Google Scholar]
- 19.Ory DS. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochimica et Biophysica Acta. 2000;1529(1–3):331–339. doi: 10.1016/s1388-1981(00)00158-x. [DOI] [PubMed] [Google Scholar]
- 20.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochimica et Biophysica Acta. 2004;1685(1–3):48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Vanier MT. Niemann-Pick disease type C. Orphanet Journal of Rare Diseases. 2010;5(1):p. 16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Sub-Cellular Biochemistry. 2008;49:241–268. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dringen R. Metabolism and functions of glutathione in brain. Progress in Neurobiology. 2000;62(6):649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 24.Zampieri S, Mellon SH, Butters TD, et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. Journal of Cellular and Molecular Medicine. 2009;13(9 B):3786–3796. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith D, Wallom KL, Williams IM, Jeyakumar M, Platt FM. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiology of Disease. 2009;36(2):242–251. doi: 10.1016/j.nbd.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Klein A, Maldonado C, Vargas LM, et al. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiology of Disease. 2011;41(1):209–218. doi: 10.1016/j.nbd.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c-Abl tyrosine kinase by amyloid-β-peptide and reactive oxygen species. Neurobiology of Disease. 2004;17(2):326–336. doi: 10.1016/j.nbd.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one. The Journal of Biological Chemistry. 1989;264(20):11796–11806. [PubMed] [Google Scholar]
- 29.Sparrow SM, Carter JM, Ridgway ND, Cook HW, Byers DM. U18666A inhibits intracellular cholesterol transport and neurotransmitter release in human neuroblastoma cells. Neurochemical Research. 1999;24(1):69–77. doi: 10.1023/a:1020932130753. [DOI] [PubMed] [Google Scholar]
- 30.Koh CHV, Whiteman M, Li QX, et al. Chronic exposure to U18666A is associated with oxidative stress in cultured murine cortical neurons. Journal of Neurochemistry. 2006;98(4):1278–1289. doi: 10.1111/j.1471-4159.2006.03958.x. [DOI] [PubMed] [Google Scholar]
- 31.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 2005;42(4):886–893. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- 32.Beltroy EP, Liu B, Dietschy JM, Turley SD. Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease. Journal of Lipid Research. 2007;48(4):869–881. doi: 10.1194/jlr.M600488-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Garver WS, Jelinek D, Oyarzo JN, et al. Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. Journal of Cellular Biochemistry. 2007;101(2):498–516. doi: 10.1002/jcb.21200. [DOI] [PubMed] [Google Scholar]
- 34.Vázquez MC, del Pozo T, Robledo FA, et al. Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028777. Article ID e28777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann-Pick disease, type C. Molecular Genetics and Metabolism. 2010;101(2-3):214–218. doi: 10.1016/j.ymgme.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter FD, Scherrer DE, Lanier MH, et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Science Translational Medicine. 2010;2(56) doi: 10.1126/scitranslmed.3001417. Article ID 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy JV, Ganley IG, Pfeffer SR. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS ONE. 2006;1(1, article e19) doi: 10.1371/journal.pone.0000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Windt A, Rai M, Kytömäki L, et al. Gene set enrichment analyses revealed several affected pathways in Niemann-Pick disease type C fibroblasts. DNA and Cell Biology. 2007;26(9):665–671. doi: 10.1089/dna.2006.0570. [DOI] [PubMed] [Google Scholar]
- 39.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovascular Research. 2009;81(3):449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 40.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radical Biology and Medicine. 2008;44(7):1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urtasun R, de la Rosa LC, Nieto N. Oxidative and nitrosative stress and fibrogenic response. Clinics in Liver Disease. 2008;12(4):769–790. doi: 10.1016/j.cld.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy PH. Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectrums. 2009;14(8, supplement 7):8–13. doi: 10.1017/s1092852900024901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England Journal of Medicine. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 44.Yu H, Liu J, Li J, Zang T, Luo G, Shen J. Protection of mitochondrial integrity from oxidative stress by selenium-containing glutathione transferase. Applied Biochemistry and Biotechnology. 2005;127(2):133–142. doi: 10.1385/abab:127:2:133. [DOI] [PubMed] [Google Scholar]
- 45.Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. The Journal of Biological Chemistry. 2005;280(12):11731–11739. doi: 10.1074/jbc.M412898200. [DOI] [PubMed] [Google Scholar]
- 46.Marí M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metabolism. 2006;4(3):185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Charman M, Kennedy BE, Osborne N, Karten B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. Journal of Lipid Research. 2010;51(5):1023–1034. doi: 10.1194/jlr.M002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernández A, Llacuna L, Fernández-Checa JC, Colell A. Mitochondrial cholesterol loading exacerbates amyloid β peptide-induced inflammation and neurotoxicity. Journal of Neuroscience. 2009;29(20):6394–6405. doi: 10.1523/JNEUROSCI.4909-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death and Differentiation. 2008;15(3):484–493. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- 50.Pacheco CD, Lieberman AP. The pathogenesis of Niemann-Pick type C disease: a role for autophagy? Expert Reviews in Molecular Medicine. 2008;10:p. e26. doi: 10.1017/S146239940800080X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishibashi S, Yamazaki T, Okamoto K. Association of autophagy with cholesterol-accumulated compartments in Niemann-Pick disease type C cells. Journal of Clinical Neuroscience. 2009;16(7):954–959. doi: 10.1016/j.jocn.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 52.Colell A, García-Ruiz C, Morales A, et al. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: effect of membrane physical properties and S- adenosyl-L-methionine. Hepatology. 1997;26(3):699–708. doi: 10.1002/hep.510260323. [DOI] [PubMed] [Google Scholar]
- 53.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicology and Applied Pharmacology. 2005;204(3):263–273. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxidants and Redox Signaling. 2009;11(11):2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. BioFactors. 2009;35(4):346–355. doi: 10.1002/biof.48. [DOI] [PubMed] [Google Scholar]
- 56.Schedin S, Sindelar PJ, Pentchev P, Brunk U, Dallner G. Peroxisomal impairment in Niemann-Pick type C disease. The Journal of Biological Chemistry. 1997;272(10):6245–6251. doi: 10.1074/jbc.272.10.6245. [DOI] [PubMed] [Google Scholar]
- 57.Sequeira JSS, Vellodi A, Vanier MT, Clayton PT. Niemann-Pick disease type C and defective peroxisomal β-oxidation of branched-chain substrates. Journal of Inherited Metabolic Disease. 1998;21(2):149–154. doi: 10.1023/a:1005395709826. [DOI] [PubMed] [Google Scholar]
- 58.Schedin S, Pentchev P, Dallner G. Reduced cholesterol accumulation and improved deficient peroxisomal functions in a murine model of Niemann-Pick type C disease upon treatment with peroxisomal proliferators. Biochemical Pharmacology. 1998;56(9):1195–1199. doi: 10.1016/s0006-2952(98)00234-2. [DOI] [PubMed] [Google Scholar]
- 59.Cao C, Leng Y, Kufe D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. The Journal of Biological Chemistry. 2003;278(32):29667–29675. doi: 10.1074/jbc.M301292200. [DOI] [PubMed] [Google Scholar]
- 60.Cao C, Leng Y, Liu X, Yi Y, Li P, Kufe D. Catalase is regulated by ubiquitination and proteosomal degradation. Role of the c-Abl and Arg tyrosine kinases. Biochemistry. 2003;42(35):10348–10353. doi: 10.1021/bi035023f. [DOI] [PubMed] [Google Scholar]
- 61.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxidants and Redox Signaling. 2005;7(5-6):619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 62.Alvarez AR, Klein A, Castro J, et al. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. The FASEB Journal. 2008;22(10):3617–3627. doi: 10.1096/fj.07-102715. [DOI] [PubMed] [Google Scholar]
- 63.Burton GW, Traber MG. Vitamin E: antioxidant activity, biokinetics, and bioavailability. Annual Review of Nutrition. 1990;10:357–382. doi: 10.1146/annurev.nu.10.070190.002041. [DOI] [PubMed] [Google Scholar]
- 64.Burton GW, Ingold KU. Vitamin E as an in vitro and in vivo antioxidant. Annals of the New York Academy of Sciences. 1989;570:7–22. doi: 10.1111/j.1749-6632.1989.tb14904.x. [DOI] [PubMed] [Google Scholar]
- 65.Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Annals of the New York Academy of Sciences. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 66.Rigotti A. Absorption, transport, and tissue delivery of vitamin E. Molecular Aspects of Medicine. 2007;28(5-6):423–436. doi: 10.1016/j.mam.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Research Reviews. 2005;4(2):288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 68.Yévenes LF, Klein A, Castro JF, et al. Lysosomal vitamin E accumulation in Niemann-Pick type C disease. Biochimica et Biophysica. 2011;1822(2):150–160. doi: 10.1016/j.bbadis.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 69.Ulatowski L, Parker R, Davidson C, et al. Altered vitamin E status in Niemann-Pick type C disease. Journal of Lipid Research. 2011;52(7):1400–1410. doi: 10.1194/jlr.M015560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Serra SA, Ill-Raga G, Coma M, et al. The role of vitamin E on intracellular signaling pathways in brain: molecular basis for the treatment of neurodegenerative processes. In: Matthew WB, editor. Vitamin E: New Research. New York, NY, USA: Nova Science; 2006. [Google Scholar]
- 71.Copp RP, Wisniewski T, Hentati F, Larnaout A, Ben Hamida M, Kayden HJ. Localization of α-tocopherol transfer protein in the brains of patients with ataxia with vitamin E deficiency and other oxidative stress related neurodegenerative disorders. Brain Research. 1999;822(1-2):80–87. doi: 10.1016/s0006-8993(99)01090-2. [DOI] [PubMed] [Google Scholar]
- 72.Gould TJ, Chadman K, Bickford PC. Antioxidant protection of cerebellar β-adrenergic receptor function in aged F344 rats. Neuroscience Letters. 1998;250(3):165–168. doi: 10.1016/s0304-3940(98)00477-7. [DOI] [PubMed] [Google Scholar]
- 73.Heaton MB, Mitchell JJ, Paiva M. Amelioration of ethanol-induced neurotoxicity in the neonatal rat central nervous system by antioxidant therapy. Alcoholism. 2000;24(4):512–518. [PubMed] [Google Scholar]
- 74.Narushima K, Takada T, Yamanashi Y, Suzuki H. Niemann-Pick C1-like 1 mediates α-tocopherol transport. Molecular Pharmacology. 2008;74(1):42–49. doi: 10.1124/mol.107.043034. [DOI] [PubMed] [Google Scholar]
- 75.Azzi A, Ricciarelli R, Zingg JM. Non-antioxidant molecular functions of α-tocopherol (vitamin E) FEBS Letters. 2002;519(1–3):8–10. doi: 10.1016/s0014-5793(02)02706-0. [DOI] [PubMed] [Google Scholar]
- 76.Ekstrand-Hammarström B, Österlund C, Lilliehöök B, Bucht A. Vitamin E down-modulates mitogen-activated protein kinases, nuclear factor-κB and inflammatory responses in lung epithelial cells. Clinical and Experimental Immunology. 2007;147(2):359–369. doi: 10.1111/j.1365-2249.2006.03285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mustacich DJ, Leonard SW, Patel NK, Traber MG. α-tocopherol β-oxidation localized to rat liver mitochondria. Free Radical Biology and Medicine. 2010;48(1):73–81. doi: 10.1016/j.freeradbiomed.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Upston JM, Terentis AC, Stocker R. Tocopherol-mediated peroxidation of lipoproteins: implications for vitamin E as a potential antiatherogenic supplement. The FASEB Journal. 1999;13(9):977–994. doi: 10.1096/fasebj.13.9.977. [DOI] [PubMed] [Google Scholar]
- 79.Bascuñan-Castillo EC, Erickson RP, Howison CM, et al. Tamoxifen and vitamin E treatments delay symptoms in the mouse model of Niemann-Pick C. Journal of Applied Genetics. 2004;45(4):461–467. [PubMed] [Google Scholar]
- 80.Quintanilla RA, Muñoz FJ, Metcalfe MJ, et al. Trolox and 17β-estradiol protect against amyloid β-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. The Journal of Biological Chemistry. 2005;280(12):11615–11625. doi: 10.1074/jbc.M411936200. [DOI] [PubMed] [Google Scholar]
- 81.Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nature Medicine. 2004;10(7):704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 82.Zampieri S, Mellon SH, Butters TD, et al. Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. Journal of Cellular and Molecular Medicine. 2009;13(9 B):3786–3796. doi: 10.1111/j.1582-4934.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balogun E, Hoque M, Gong P, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochemical Journal. 2003;371(part 3):887–895. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. The Journal of Biological Chemistry. 2010;285(27):20423–20427. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lloyd-Evans E, Platt FM. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic. 2010;11(4):419–428. doi: 10.1111/j.1600-0854.2010.01032.x. [DOI] [PubMed] [Google Scholar]
- 86.Lachmann RH, te Vruchte D, Lloyd-Evans E, et al. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiology of Disease. 2004;16(3):654–658. doi: 10.1016/j.nbd.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 87.Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann-Pick disease, type C. Molecular Genetics and Metabolism. 2010;101(2-3):214–218. doi: 10.1016/j.ymgme.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cao C, Leng Y, Huang W, Liu X, Kufe D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. The Journal of Biological Chemistry. 2003;278(41):39609–39614. doi: 10.1074/jbc.M305770200. [DOI] [PubMed] [Google Scholar]
- 89.Crack PJ, Taylor JM, Flentjar NJ, et al. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. Journal of Neurochemistry. 2001;78(6):1389–1399. doi: 10.1046/j.1471-4159.2001.00535.x. [DOI] [PubMed] [Google Scholar]
- 90.Guix FX, Ill-Raga G, Bravo R, et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain. 2009;132(5):1335–1345. doi: 10.1093/brain/awp023. [DOI] [PubMed] [Google Scholar]
- 91.Kim SJ, Lim MS, Kang SK, Lee YS, Kang KS. Impaired functions of neural stem cells by abnormal nitric oxide-mediated signaling in an in vitro model of Niemann-Pick type C disease. Cell Research. 2008;18(6):686–694. doi: 10.1038/cr.2008.48. [DOI] [PubMed] [Google Scholar]
- 92.German DC, Quintero EM, Liang CL, Xie C, Dietschy JM. Degeneration of neurons and glia in the Niemann-Pick C mouse is unrelated to the low-density lipoprotein receptor. Neuroscience. 2001;105(4):999–1005. doi: 10.1016/s0306-4522(01)00230-5. [DOI] [PubMed] [Google Scholar]
- 93.Mani K, Cheng F, Fransson LÅ. Constitutive and vitamin C-induced, NO-catalyzed release of heparan sulfate from recycling glypican-1 in late endosomes. Glycobiology. 2006;16(12):1251–1261. doi: 10.1093/glycob/cwl045. [DOI] [PubMed] [Google Scholar]
- 94.Yanagimoto C, Harada M, Kumemura H, et al. Copper incorporation into ceruloplasmin is regulated by Niemann-Pick C1 protein. Hepatology Research. 2011;41(5):484–491. doi: 10.1111/j.1872-034X.2011.00788.x. [DOI] [PubMed] [Google Scholar]
- 95.Yanagimoto C, Harada M, Kumemura H, et al. Niemann-Pick C1 protein transports copper to the secretory compartment from late endosomes where ATP7B resides. Experimental Cell Research. 2009;315(2):119–126. doi: 10.1016/j.yexcr.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 96.Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. American Journal of Clinical Nutrition. 1998;67(supplement 5):952S–959S. doi: 10.1093/ajcn/67.5.952S. [DOI] [PubMed] [Google Scholar]
- 97.Barnham KJ, Bush AI. Metals in Alzheimer’s and Parkinson’s diseases. Current Opinion in Chemical Biology. 2008;12(2):222–228. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 98.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis) Chemical Reviews. 2006;106(6):1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 99.Kaler SG. Menkes disease. Advances in pediatrics. 1994;41:263–304. [PubMed] [Google Scholar]
- 100.Langner C, Denk H. Wilson disease. Virchows Archiv. 2004;445(2):111–118. doi: 10.1007/s00428-004-1047-8. [DOI] [PubMed] [Google Scholar]
- 101.Linder MC, editor. Biochemistry of Copper. New York, NY, USA: Plenum Press; 1991. [Google Scholar]
- 102.Mercer JFB, Llanos RM. Molecular and cellular aspects of copper transport in developing mammals. Journal of Nutrition. 2003;133(5, supplement 1):1481S–1484S. doi: 10.1093/jn/133.5.1481S. [DOI] [PubMed] [Google Scholar]
- 103.Zhang JR, Coleman T, Langmade SJ, et al. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. Journal of Clinical Investigation. 2008;118(6):2281–2290. doi: 10.1172/JCI32561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tint GS, Pentchev P, Xu G, et al. Cholesterol and oxygenated cholesterol concentrations are markedly elevated in peripheral tissue but not in brain from mice with the Niemann-Pick type C phenotype. Journal of Inherited Metabolic Disease. 1998;21(8):853–863. doi: 10.1023/a:1005474803278. [DOI] [PubMed] [Google Scholar]
- 105.Lund EG, Björkhem I. Role of oxysterols in the regulation of cholesterol homeostasis: a critical evaluation 1. Accounts of Chemical Research. 1995;28(6):241–249. [Google Scholar]
- 106.Royer MC, Lemaine-Ewing S, Desrumaux C, et al. 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched (Lipid Raft) domains is impaired by vitamin E. A specific role for α-tocopherol with consequences on cell death. The Journal of Biological Chemistry. 2009;284(23):15826–15834. doi: 10.1074/jbc.M808641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vejux A, Guyot S, Montange T, Riedinger JM, Kahn E, Lizard G. Phospholipidosis and down-regulation of the PI3-K/PDK-1/Akt signalling pathway are vitamin E inhibitable events associated with 7-ketocholesterol-induced apoptosis. Journal of Nutritional Biochemistry. 2009;20(1):45–61. doi: 10.1016/j.jnutbio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 108.Pérez-Poyato MS, Pineda M. New agents and approaches to treatment in Niemann-Pick type C disease. Current Pharmaceutical Biotechnology. 2011;12(6):897–901. doi: 10.2174/138920111795542697. [DOI] [PubMed] [Google Scholar]
- 109.Sturleya SL, Pattersonb MC, Pentchevc P. Unraveling the sterol-trafficking defect in Niemann-Pick C disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2093–2094. doi: 10.1073/pnas.0812934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang MS, Boddapati S, Sierks MR. Cyclodextrins promote protein aggregation posing risks for therapeutic applications. Biochemical and Biophysical Research Communications. 2009;386(3):526–531. doi: 10.1016/j.bbrc.2009.06.077. [DOI] [PubMed] [Google Scholar]
- 111.Vance JE, Peake KB. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Current Opinion in Lipidology. 2011;22(3):204–209. doi: 10.1097/MOL.0b013e3283453e69. [DOI] [PubMed] [Google Scholar]
- 112.Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1 −/− mouse. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muralidhar A, Borbon IA, Esharif DM, et al. Pulmonary function and pathology in hydroxypropyl-beta-cyclodextin-treated and untreated npc1 −/− mice. Molecular Genetics and Metabolism. 2011;103(2):142–147. doi: 10.1016/j.ymgme.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodriguez-Hernandez A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5(1):19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- 115.Arakawa M, Ito Y. N-acetylcysteine and neurodegenerative diseases: basic and clinical pharmacology. Cerebellum. 2007;6(4):308–314. doi: 10.1080/14734220601142878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine-a safe antidote for cysteine/glutathione deficiency. Current Opinion in Pharmacology. 2007;7(4):355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opinion on Biological Therapy. 2008;8(12):1955–1962. doi: 10.1517/14728220802517901. [DOI] [PubMed] [Google Scholar]
- 118.Aitio ML. N-acetylcysteine—passe-partout or much ado about nothing? British Journal of Clinical Pharmacology. 2006;61(1):5–15. doi: 10.1111/j.1365-2125.2005.02523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen Q, Prior M, Dargusch R, et al. A novel neurotrophic drug for cognitive enhancement and Alzheimer's disease. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0027865. Article ID e27865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (ST1571, imatinib), a rationally developed, targeted anticancer drug. Nature Reviews Drug Discovery. 2002;1(7):493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 121.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Ab1 tyrosine kinase on the growth of Bcr-Ab1 positive cells. Nature Medicine. 1996;2(5):561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 122.Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. The Lancet Neurology. 2007;6(9):765–772. doi: 10.1016/S1474-4422(07)70194-1. [DOI] [PubMed] [Google Scholar]
- 123.Galanaud D, Tourbah A, Lehéricy S, et al. 24 month-treatment with miglustat of three patients with Niemann-Pick disease type C: follow up using brain spectroscopy. Molecular Genetics and Metabolism. 2009;96(2):55–58. doi: 10.1016/j.ymgme.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 124.Wraith JE, Vecchio D, Jacklin E, et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Molecular Genetics and Metabolism. 2010;99(4):351–357. doi: 10.1016/j.ymgme.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 125.Wraith JE, Imrie J. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. Therapeutics and Clinical Risk Management. 2009;5(1):877–887. doi: 10.2147/tcrm.s5777. [DOI] [PMC free article] [PubMed] [Google Scholar]