

Abstract
Since the identification of APOBEC3G (A3G) as a potent restriction factor of HIV-1, a tremendous amount of effort has led to a broadened understanding of both A3G and the APOBEC3 (A3) family to which it belongs. In spite of the fine-tuned viral counterattack to A3 activity, in the form of the HIV-1 Vif protein, enthusiasm for leveraging the Vif : A3G axis as a point of clinical intervention remains high. In an impressive explosion of information over the last decade, additional A3 family members have been identified as antiviral proteins, mechanistic details of the restrictive capacity of these proteins have been elucidated, structure-function studies have revealed important molecular details of the Vif : A3G interaction, and clinical cohorts have been scrutinized for correlations between A3 expression and function and viral pathogenesis. In the last year, novel and unexpected findings regarding the role of A3G in immunity have refocused efforts on exploring the potential of harnessing the natural power of this immune defense. These most recent reports allude to functions of the A3 proteins that extend beyond their well-characterized designation as restriction factors. The emerging story implicates the A3 family as not only defense proteins, but also as participants in the broader innate immune response.
1. Introduction
In 2002, the cloning of APOBEC3G (A3G; then called CEM15) and the identification of the protein product of this gene as the first cellular protein capable of restricting HIV-1 infection revealed a novel direction for chemotherapeutic intervention and ignited the search for additional defense proteins capable of counteracting viral invasion [1]. The report of this cloning solved a long-standing enigma in the field of HIV-1 pathogenesis. Early work examining and comparing the pathogenesis of wild-type and Vif-deficient HIV-1 had yielded conflicting results with some laboratories concluding that Vif was dispensable for productive infection while other groups maintained that Vif expression was essential [2–4]. Ultimately, it was decisively shown that the requirement for Vif was cell-type dependent; permissive cells supported the growth of HIV-1Δvif while nonpermissive cells limited such viral replication [5, 6]. Most interesting and relevant was the inability of Vif-deficient HIV to productively infect primary CD4+ T cells, one of the critical natural targets of HIV-1 infection [2, 3, 5, 7, 8]. The molecular explanation for the “Vif phenotype” remained unexplained for the subsequent decade. Proffered in this early work was the idea that permissive cells expressed a cellular factor that compensated for Vif. An equally valid suggestion was that nonpermissive cells harbored an inhibitory activity of HIV-1 that was itself overcome by the Vif protein. It was subsequently established, in a pair of elegant experiments utilizing heterokaryons formed from fusion of nonpermissive and permissive cell lines that, in fact, nonpermissive cells expressed an activity that suppressed HIV-1Δvif replication [9, 10]. The genetic relatedness of two T lymphocyte lines, one nonpermissive and the other permissive, was exploited in a classical subtractive hybridization experiment; A3G was identified as this described suppressive activity. It was found to be almost exclusively expressed by nonpermissive cells and its stable expression in permissive cells conveyed the ability to resist an HIV-1 challenge [1].
It was quickly appreciated that A3G was but one family member of a previously identified gene locus [11]. Subsequent investigation also revealed that A3G exhibited a potent DNA-mutating ability [12]. In humans, seven family members within the locus have been identified; rhesus macaques, the nonhuman primate that serves as the most important animal model for HIV treatment and vaccine testing, also have seven APOBEC3 genes, while the murine genome contains a single A3 gene [13–15]. In each of these organisms, the role the A3 genes play in counteracting viral invasion is critical. All seven A3 family members identified in humans exhibit powerful suppressive activity against a range of viruses while the homologous proteins in mice and primates appear to perform similar functions [16–18]. While A3 inhibitory activity is relatively broad, the most well-characterized and studied function is their striking ability to restrict retroviral infection [19]. In an evolutionary response to this restriction, the retroviruses have countered with a battery of genes exquisitely fine-tuned to overcome these endogenous defense proteins.
2. The Laboratory Setting
With one exception (A3C), each of the seven A3 family members in humans has been observed to be capable of combating HIV-1 [1, 17, 20–27]. Whether the antiviral activity observed is relevant during the course of a natural HIV-1 infection has not been unequivocally established for any of the family members and there are valid concerns raised in the interpretation of various data regarding levels of protein expression and potency. However, it is becoming increasingly clear that understanding the battle that is waged between the innate immune system and HIV-1 during acute infection is imperative and the A3 proteins are critical players in this initial encounter.
While the relative potencies of individual A3 family members in the setting of a natural infection have been difficult to assess, it has been convincingly established that, in the tissue culture setting, A3G exhibits the most potent activity against HIV-1. In a variety of cell types, both primary cells and established cells lines, and under varying experimental conditions, including both single-round infectivity assays and multiple-round replication assays, A3G suppresses the infectivity of HIV-1. HIV-1 Vif has evolved to counteract this impressive activity of A3G by preventing virion encapsidation of this host factor [28–35]. Vif acts as an adapter protein bridging A3G and a Cullin5-elongin B/C-Rbx ubiquitin ligase [36]. Within this complex A3G is ubiquitinylated and subsequently degraded in the 26S proteasome [36, 37]. Other modalities involving Vif prevention of A3G encapsidation have also been documented [28, 32, 34]. Interestingly, dominance of A3G over Vif has been noted under conditions of elevated and/or stabilized expression [1, 34, 36]. This ability to suppress HIV-1 even in the presence of Vif is noteworthy as it has distinct implications for the development of chemotherapeutics designed to interfere with the A3G : Vif axis.
The anti-HIV-1 functionality of A3G is multifaceted. Its most extensively characterized anti-HIV-1 function is its ability to catalyze cytidine deamination of HIV-1 DNA on the minus strand resulting in the detection of guanosine-to-adenosine transition mutations in reverse transcripts; upwards of 10% of guanosines may be mutated leading to the labeling of this A3G-mediated process as hypermutation [34, 38, 39]. The fate of such hypermutated transcripts is not well understood, but certainly this dramatic mutational burden effectively short-circuits viral infection.
Work from multiple groups has also uncovered deamination-independent anti-HIV effects of A3G that are seen during viral infection [22, 40–49]. The characterization of this editing-independent antiviral function has suggested a block to viral replication that occurs after entry but before integration. While the molecular details of this deaminase-independent function of A3G remain unclear, defective reverse transcription products are commonly observed, indicating that A3G likely acts during the process of reverse transcription. A more comprehensive understanding of this inhibition will be important. All members of the A3 family contain at least one conserved cytidine deaminase active site (CDA; family members A3B, A3D, A3F, and A3G contain two such domains) composed of the signature sequence His/Cys-X-Glu-X23–28-Pro-Cys-X2-Cys [11, 15]. Early structure-function analysis of A3G was performed by disrupting these suspected catalytic domains with site-directed mutagenesis [41]. The conserved histidine, glutamic acid, and cysteine resides in both the N-terminal and C-terminal domains of A3G were individually mutated and the resulting proteins were independently examined for their catalytic function as well as their ability to suppress HIV-1Δvif infection. The data clearly indicated that the C-terminal CDA domain was responsible for A3G enzymatic function. Unexpectedly the data also suggested that, under specific experimental conditions, significant anti-HIV-1 inhibition could be imparted in the absence of the characteristic mutagenic activity. Subsequent work in a range of experimental systems has supported these original observations. Controversy over these observations primarily stems from claims that these data have most often been cited in experimental settings using mutant A3G exhibiting elevated expression levels [41, 42, 50, 51]. In attempts to clarify the role of A3G expression levels a number of groups have compared A3G protein expression in transiently transfected cell lines and primary CD4+ T cells/macrophages, reporting that expression levels achieved during transient transfection exceed levels observed in primary cells. However, a few cautionary notes are warranted. A3G that is mutated, for instance, at the critical glutamic acid at residue 259 of the protein, has also been shown to have a more limited ability to block the process of reverse transcription thereby suggesting that distinguishing deamination-dependent and -independent activities may be challenging [16]. Additional support for a pleiotropic antiviral function of A3G is provided by observations in which the A3G phenotype is unaffected in cells that do not express uracil DNA glycosylase 2 or SMUG, enzymes responsible for the removal of uracils from single- or double-stranded DNA [52, 53]. As a significant suppressor of HIV-1, a multipronged ability of A3G to inhibit HIV-1 would have notable benefits to the invaded host.
Using a variety of cell lines and experimental conditions, the anti-HIV-1 activity of A3B, A3D, A3F, and A3H (haplotypes I, II, V, and VII) has also been conclusively demonstrated [17, 20, 21, 23–25, 27, 54, 55]. Hypermutation is often recorded as coincident with antiviral activity, although, in the case of A3B and A3F, as with A3G, there are observations of HIV-1 suppression in the absence of hypermutation [24, 42, 43]. Sensitivity to Vif regulation has been observed for A3D, A3F, A3G and A3H while A3B and A3H/Haplotype I resist Vif-mediated virion exclusion and thus exhibit detectable activity against wild-type HIV-1 virus. However, not all of these family members are equally likely to contribute to HIV-1 resistance during a natural infection; A3B is primarily expressed in B cells and makes it unlikely that this protein contributes appreciably to inhibition of HIV-1 [17, 20, 21, 23, 24, 56–58]. Similarly, the expression of the A3H/Haplotype I restricts wildtype HIV-1, but the protein is inherently unstable [20, 56]. A question with important clinical implications is whether this intrinsic instability may be overcome while harnessing the natural power to combat wild-type viral infection [58, 59].
Until recently, the role of A3A in HIV-1 inhibition was unappreciated outside of two significant observations: the first being a correlation between its expression in monocytes and the susceptibility of these cells to HIV-1 infection, and the second was that expression of A3A was confined to cells of the myeloid lineage and this expression was positively regulated by INF-α [60–62]. Berger et al. have now described a novel and critical role A3A plays in the early phase of HIV infection, specifically in myeloid cells [22]. When primary myeloid cells were infected with HIV-1 and the induction of expression at the A3 locus was examined, it was shown that these cells preferentially induced A3A, on both the mRNA and protein levels; induction of other A3 family members in these cells was not detected and A3A induction in peripheral blood lymphocytes was negligible. The induced A3A was protective upon HIV-1 challenge and depletion of A3A in primary macrophages and dendritic cells increased viral replication in both single-round infectivity assays and a spreading infection. Similar to other A3 proteins this viral restriction was primarily observed as a profound suppression in the accumulation of viral DNA suggesting interference with an early step of reverse transcription; limited editing of viral reverse transcripts was detectable, but the evidence suggested that enzymatic function was not the sole antiviral function. Notwithstanding its common role as an A3 family member involved in HIV-1 control, A3A exerts its antiviral function uniquely. It is not producer cell-derived A3A that impacts virus replication, but rather it is the pool of A3A present in the actual target cell itself that inhibits incoming HIV-1 particles. Data from independent laboratories strongly support these conclusions for this role of A3A in target myeloid cells [63–65].
Within cells of the myeloid lineage, A3A appears to be the critical suppressor, exerting its effect independently of its editing ability. In CD4+ T cells in the tissue culture model of infection, A3G activity dominates, and its inhibitory function is exerted utilizing both editing-dependent and -independent mechanisms. A3A functions in the target cell while A3G functions in the producer cell. Recent observations, however, have now suggested an unexpected and intricate antiviral role played by the A3G expressed in target cells [66]. Expression of either A3A or A3G activate the cellular DNA damage response (DDR) [67]. In the case of A3A, a G1/S-phase cell cycle arrest is also induced and its catalytic domain is implicated in the effect. While the relevance of these interesting observations in regard to HIV-1 infection is not immediately obvious (the A3A experiments were performed in human osteosarcoma cells) the role that the DDR response pathway plays in the innate immune response has only recently been explored and appreciated [68, 69]. Experimental observations support the idea that triggering the DDR pathway acts as an alerting mechanism for the innate immune system [66, 68, 70, 71]. In the emerging A3G story this certainly seems to be the case (Figure 1). Norman et al. examined expression of the critical Natural killer (NK) cell-activating ligand, NKG2D-L, in HIV-1-infected primary CD4+ T cells [66]. They compared expression of NKG2D-L under conditions of wildtype HIV-1 infection and HIV-1Δvif infection and found a surprising discrepancy: the combination of Vpr and A3G in the HIV-1Δvif infections activated the DDR ultimately leading to the upregulation of both A3G and NKG2D-L. Increased expression of NKG2D-L sensitized the HIV-1-infected cell to NK-mediated killing. In the presence of Vif this NK-mediated killing was blunted. The role of target cell-expressed A3G was further verified using shRNA's targeting A3G mRNA; loss of A3G in an HIV-1Δvif setting resulted in diminished NK-killing and increased (infected) cell survival. The authors suggest that, in a natural infection, the A3G-dependent sensitization of HIV-1-infected cells to NK-mediated killing is hindered by the loss of A3G through Vif-mediated degradation. It bears mentioning that infection of murine primary B cells with the transforming retrovirus Abelson murine leukemia virus (Ab-MuLV) also leads to the induction of activation-induced deaminase (AID) expression [72]. AID is a member of the larger APOBEC-AID family of cytidine deaminases (this grouping includes the founding member, APOBEC1, APOBEC2, APOBEC3A-H and AID). This induction of AID also results in the upregulation of an NKG2D ligand, rendering the infected cells susceptible to NK-mediated lysis. The in vivo effect is the profound containment of Ab-MuLV replication and the ability of the host animals to restrict the virus and survive this pathogenic encounter. This indirect effect of AID is also linked to the DDR-stimulated signaling pathways. Details on the mechanistic details of these antiviral functions have not yet been fully characterized. Particularly intriguing is whether the catalytic function of A3G and/or AID is necessary for these effects, and, if so, how is this enzymatic capacity utilized. With the description of the involvement of the DDR, it is suspected that the signature cytidine deaminase modality would be important but confirmation of such speculation is warranted. Based on these observations, therapeutic approaches that interfere with the process of Vif-regulated degradation of A3G could potentially strengthen not only a potent intracellular defense, but also impact the ability of NK cells to attack infected cells.
Figure 1.
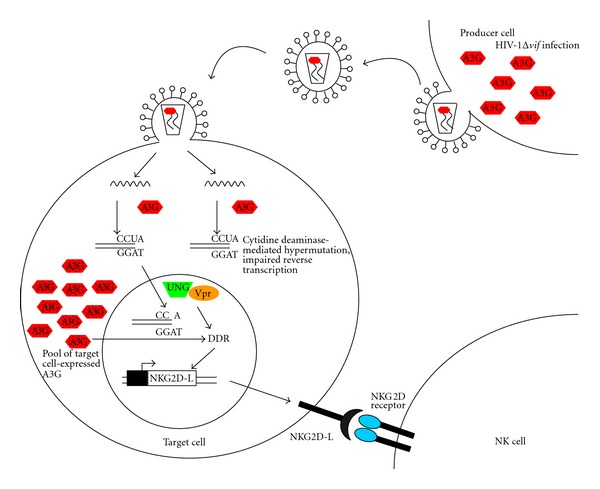
A3G can exert multiple antiviral effects against HIV-1 infection. Virion-packaged A3G restricts HIV-1Δvif replication via cytidine deaminase-mediated hypermutation as well as interfering with efficient reverse transcription. Additionally, the introduction of the uridines into the minus-strand DNA during reverse transcription triggers the DNA damage response (DDR). This induction of DDR involves other proteins, including the host protein, UNG, and the HIV-1 Vpr protein. Among other downstream effects, the DDR stimulates the transcriptional synthesis of NKG2D ligands. The subsequent expression of these proteins on the surface of the HIV-infected cell sensitizes it to NK cell lysis. It should also be noted that A3G expression within the target cell (designated as dotted symbols to distinguish it from the virion-packaged A3G). Also critically participates in the DDR activation.
3. The Picture in the Clinic
As astounding as our progress has been in understanding the molecular and mechanistic details of A3 proteins and their interaction with HIV-1, providing data for the in vivo relevance of A3 activity has been significantly more challenging. Experiments manipulating A3G in the laboratory have supported the proposition that elevated expression levels of this restriction factor can and do alter wildtype HIV-1 infectivity; clinical correlates of this in vitro observation have been more difficult to gather. With few exceptions, the clinical work to date has principally focused on A3G and the effect its fluctuating expression levels and catalytic activity can have on HIV-1 infection and progression. Clinical analyses do not often lend themselves to large sample sets, and the confounding combinatorial effects of host genetics and environment strain efforts of reproducibility. With these openly acknowledged limitations recognized, there remains an increasing amount of suggestive evidence that corroborates the idea that A3G expression and/or activity can modulate natural HIV-1 infection [59, 75, 77, 79–81] (Table 1).
Table 1.
Clinical studies correlating A3 family members and HIV-1 pathogenesis.
| A3 Family member | Correlation reported | Identification of cohort | Reference |
|---|---|---|---|
| A3B | Homozygous deletion of gene associated with higher: rates of HIV infection after exposure, viral set point, and rate of disease progression | 4216 HIV+ patients pooled from 5 longitudinal cohorts: ALIVE, MACS, SFCC, HGDS and MHCS [73] (US-based studies) | An et al. [74] |
|
| |||
| A3F and A3G | Level of detectable proviral hypermutations that exhibited A3F/A3G cytidine deaminase signatures associated with higher CD4+ cell count | 215 HIV+ female commercial sex workers plus 25 HIV+ women who were infected perinatally (Nairobi, Kenya) | Land et al. [75] |
|
| |||
| A3F and A3G | Elevated expression of A3F and A3G in PBMCs associated with establishment of lower viral set point | 30 women from a well-established [76] cohort of female commercial sex workers (Dakar, Senegal) | Ulenga et al. [77] |
|
| |||
| A3G | 186R polymorphism in African Americans associated with rapid progression to AIDS | 2430 HIV+ patients pooled from 5 longitudinal cohorts: ALIVE, MACS, SFCC, HGDS and MHCS [73] (US-based studies) | An et al. [78] |
|
| |||
| A3G | Elevated expression of A3G in CD14+ cells associated with resistance to HIV-1 infection after exposure | 30 HESN individuals (Florence, Italy) | Biasin et al. [79] |
|
| |||
| A3G | Elevated expression levels inversely associated with viral load in LTNPs | 6 uninfected volunteers; 17 HIV+ progressors; 8 HIV+ LTNPs | Jin et al. [80] |
|
| |||
| A3G | C40693T polymorphism, located within intronic sequences, associated with increased risk of infection | 122 HIV-exposed individuals; 69 sero converted after exposure, 53 retained seronegative status (Montreal, Canada) | Valcke et al. [73] |
|
| |||
| A3G | HESN individuals expressed elevated levels of A3G when compared to healthy controls; elevated levels of A3G associated with higher CD4+ cell count in HIV+ patients | 26 healthy controls, 37 HESN individuals, 45 HIV+ patients (Mexico City, Mexico) | Vázquez-Pérez et al. [81] |
|
| |||
| A3H | Haplotype I associated with protection from HIV-1 infection | 70 serodiscordant couples (Florence, Italy) | Cagliani et al. [59] |
HESN: highly exposed seronegative; LTNP: long-term nonprogressors.
In infected individuals, hypermutated HIV-1 proviral genomes and elevated A3G expression levels have been correlated with both lower viral loads and increased CD4+ T cells counts [75, 80–83]. In a relatively large study, Land et al. noted the significant association between proviral hypermutation and increased peripheral blood CD4+ T cell count. A3G expression was not directly quantified and the detected hypermutation was used as a surrogate for catalytic function of A3G.
More direct analysis of A3G expression in the setting of a natural HIV-1 infection has also yielded tantalizing hints of A3G control. Working with a small cohort of women, one group recently reported an interesting correlation between individuals expressing higher levels of A3G before HIV-1 infection with the establishment of a lower viral set point after infection [77]. Perhaps the most interesting cohorts in which to examine A3G expression levels and the importance of these levels during viral infection in vivo are long-term nonprogressors (LTNPs), elite suppressors (ESs), and highly exposed seronegative (HESN) individuals. To date, there has been no reporting of A3G expression (or activity) as an explanation for the innate ability of an ES to completely control the HIV-1 virus. However, there has been an observation that elevated A3G levels do correlate with higher CD4+ T cell counts and lower viremia within a group of identified LTNPs, suggesting that, under certain conditions, overexpression of A3G may be protective [80]. Two independent studies, examining approximately 67 individuals who have been repeatedly exposed to HIV-1, yet retain their seronegative status, have also presented evidence that elevated A3G expression levels correlate with viral restriction [79, 81]. Amongst these two cohorts a variety of cell types were studied, including PBMCs, CD4+ T cells, CD8+ T cells, CD14+ monocytes, and cervical cells. These cells were assayed for the level of A3G expression primarily determined at the transcriptional level; in a small number of instances, protein expression was also determined. Calculated levels of mRNA and protein in HESN individuals were then compared to both HIV+ individuals and healthy controls and, in both experimental groups, HESN expressed statistically higher levels of A3G expression. One study carried the results further and was also able to show that PBMCs isolated from HESN individuals were able to more effectively limit a wildtype HIV-1 challenge [79]. Interestingly, both PBMCs and CD14+ cells, isolated from these HESN individuals, appeared to exhibit higher responsiveness to IFN-α treatment as measured by the induction of A3G expression.
Finally, a recent experiment utilizing the SIV/macaque model for HIV-1 infection also suggests that investigating and understanding the consequences of increased A3G (and A3F, in this case) expression levels may elucidate the protective role these defense proteins can play in vivo [84]. Infected macaques were separated by clinical stage (chronic versus AIDS stage of infection) and compared to uninfected controls. In isolated PBMCs, CD4+ T cells, and peripheral lymph nodes there was a demonstrated negative correlation between A3F/G mRNA expression and viral loads. In addition, the difference in A3F/G expression between control and infected animals was even more pronounced when individuals whose disease course mimicked that of HIV-1/LTNPs were specifically compared. One of the novel aspects of this reporting was the kinetic observation of the in vivo regulation of A3G gene expression after SIV challenge. Seven days after infection A3G expression levels began to rise and this expression induction peaked on Day 10 after infection. Peak viremia was measured on Day 14. The concomitant rise of A3G levels, leading the rise of replicating virus levels, suggests that the struggle for control between this intracellular restriction factor and the invading pathogen occurs early, during acute infection. This supports previous reports noting the HIV-1-induction of A3G expression and the critical importance this early encounter may play on establishing viral set point [22, 79, 81, 84–86].
In spite of the meticulous analyses and important work accomplished, the current clinical understanding of how and whether A3 family members modulate HIV-1 infection is limited and somewhat unsatisfactory. A consensus has not yet emerged and such agreement will likely require a more collaborative and coordinated effort, across cohorts and experimental approaches. The details of designing such experiments are themselves still fraught with unknown parameters; for instance, which cell types and/or tissues should be examined? Is an examination of proviral hypermutation or viral genome editing enough to serve as a marker for A3G antiviral function? Is a measurement of A3G mRNA sufficient to draw conclusions regarding expression of the protein and resultant antiviral activity? At least two groups have noted a disconcerting disconnect between A3G mRNA and protein expression in PBMCs [60, 79]. Do other A3 family members play distinct roles at discrete stages of viral infection? In spite of this minefield of questions and the intrinsic limits placed on a data set as soon as a cohort of study is chosen, ventures into the clinical realm are paramount and it is only this data that can ultimately reveal the role of the A3 family in potentially containing HIV-1 infection.
4. The Murine Story
In contrast to the undetermined impact human A3 proteins have in limiting natural HIV-1 infection, systematic and directed experiments in mice have conclusively shown that murine A3 (mA3) is essential in containing and restricting several murine retroviruses: MMTV, a betaretrovirus (mouse mammary tumor virus), F-MuLV (Friend murine leukemia virus), a gammaretrovirus, as well as FV (Friend virus) [86–88]. Other murine gammaretroviruses, such as MLV (murine leukemia virus), are resistant to mA3 restriction [89–91]. Unlike the complex APOBEC3 locus in humans, which contains a tandem array of seven genes, the murine genome encodes a single APOBEC3 gene, mA3 [11, 92]. The knockout of mA3 was achieved quickly after the identification and cloning of A3G [93]. While a preliminary examination of the mice was relatively uninteresting, detailed characterization of the response of these animals to specific viral challenge was both illuminating and exciting.
In a series of informative and elegant in vivo experiments, it was shown that MMTV spreads more rapidly and is disseminated more extensively in mice lacking a functional mA3 gene as compared to wildtype mice. The mA3 knockout mice exhibited higher initial viral loads and a shorter time to the development of mammary tumors [86]. It was interesting to note that the protection afforded by mA3 was not absolute; mA3 blunted, but did not completely inhibit, MMTV infection, suggesting even partial protection has a significant role in in vivo pathogenesis. The molecular mechanism of this mA3-dependent repression of infection remains unidentified, although it does appear that this antiviral function is exerted independently of any detectable hypermutation or viral genome editing. In this setting mA3-mediated containment of MMTV bears a striking resemblance to A3A-dependent control of HIV-1 in myeloid cells: neither inhibition requires a detectable hypermutation function, although the block to viral infection traces to an early post-entry step, and antiviral function is exerted by protein expressed in target cells [22, 86]. In the case of mA3, antiviral function was a combinatorial effect of both virion-packaged and endogenously expressed protein. In terms of potentially harnessing the innate power of the A3 proteins, the most intriguing observation was the reporting that pre-treatment of wildtype mice with either LPS or INF-α upregulated mA3 expression in dendritic cells, the first cells infected during MMTV exposure. This early elevation of mA3 expression directly correlated with increased resistance to MMTV. Mice lacking mA3 were unable to restrict viral infection despite either treatment [94]. This result speaks directly to some of the underlying concerns regarding the detrimental consequences of manipulating the expression of A3G and certainly bolsters the hypothesis that increased expression of this protein could ameliorate restriction of HIV-1 infection.
Finally, it is interesting to note that in addition to reducing MMTV replication, virion-incorporated mA3 has also been shown to be able to markedly reduce the transmission of virus [95]. MMTV, as a number of other retroviruses, including HIV-1, is transmitted vertically through breastfeeding. In an investigation examining the route of transmission, Okeoma et al. report that not only was mA3 mRNA readily detectable in mammary epithelial tissue but that this packaged mA3 significantly decreased MMTV transmission. In an effort to extend these observations to HIV-1 infection, this group examined expression of the A3 genes in primary human mammary tissue and found significant levels of both A3F and A3G mRNA. Whether this expression translates into protection from the vertical transmission of HIV-1 is not yet clear. However, the trajectory of this study is interesting taken in the context of HIV-1 infection in which breastfeeding accounts for approximately 40% of vertical transmission [96].
FV causes immunosuppression and leukemia in mice. Interestingly, mice strains are differentially susceptible to FV, and a number of genes have been implicated in the resistance to this disease [97]. Both cell-mediated and humoral responses appear necessary for recovery and, naturally enough, the major histocompatibility complex (MHC) locus has been identified as important. However, an essential non-MHC gene, Recovery from Friend virus gene 3 (Rfv3), has also been implicated [98]. Mice strains resistant to FV (e.g., C57BL/6), possess Rfv3 resistance alleles, develop high concentrations of protective neutralizing antibodies, and recover from viremia. Mice strains susceptible to FV infection (e.g., BALB/c) fail to mount the protective humoral response, develop splenomegaly and erythroleukemia, and succumb to viral infection. In a revealing study, passive immunization of susceptible mice decreased mortality dramatically, suggesting that the Rfv3 locus critically influences the production of the protective neutralizing antibodies [99].
The first reporting of the genetic region encompassing Rfv3 was in 1979 [98]. It was to be 30 years before two groups simultaneously identified Rfv3 as mA3 [87, 88]. Using a range of both in vivo and in vitro experiments they convincingly showed that mA3 expression was critical to the restriction of FV infection and resulted in the suppression of virus particle infectivity. This inhibition to viral replication occurred after entry, but before integration, presumably affecting an early stage of FV infection (potentially reverse transcription). The description of the restrictive capacity of mA3 was reminiscent of the extensive data characterizing the A3G anti-HIV-1 function. It should also be noted that, in the FV system, mA3 function was exerted independently of any detectable cytidine deamination activity. While the observations supported the identity of mA3 as the suppressive factor, a consensus on what distinguished a resistant mA3 allele from a susceptible allele was less discernable. Preliminary data implicated the influence of mA3 polymorphisms on expression level, essentially suggesting the resistant mA3 alleles were more highly expressed [87, 88, 100]. In addition, there was also suggestion of an important role for a coinherited B-cell-activating factor receptor (BAFF-R) allele [101].
Recent work probing the resistant versus susceptible mA3 alleles has supported previous suggestion that an mA3 splice variant lacking exon 5 may be more potent than a full-length isoform [89, 102]. This latest work suggests that the mA3Δexon 5 variant is more efficiently translated and the overall combinatorial effect of elevated mRNA levels and preferential translation of the mA3Δexon 5 account for significantly higher levels of mA3 protein capable of potently restricting FV infection [102]. A small number of genetic variants within the A3 family and their respective relationship to HIV-1 disease acquisition and progression have been described: the H186R variant of A3G is associated with rapid progression in African American populations, the C40693T variant of A3G, as well as the homozygous loss of A3B, may be associated with increased infection susceptibility, and Haplotype I of A3H may provide resistance to infection [59, 73, 74, 78]. To date, a molecular understanding of how these variants modify (or fail to modify) HIV-1 disease is sorely lacking. Details of the defining characteristics of the resistant mA3 alleles are of significant interest upon contemplation that such differences, when identified, could be thoroughly dissected in a relevant in vivo setting, perhaps providing valuable insight into mechanistic detail. Such details may expand our understanding of the human versions of the A3 family and the critical polymorphisms.
What is also underscored in these reports is the importance of characterizing both expression levels and allelic differences of individual A3 genes within this family. Fluctuations of A3G mRNA levels, in which A3G gene expression is upregulated, have been reported across the immature-to-mature differentiation transition in dendritic cells (DCs) [65, 103]. The ability of mature DC's to resist HIV-1 infection is well documented, and this correlative observation is intriguing [104, 105]. An observation reported in the MMTV system is also suggestive: the DC's of mice stimulated with LPS 24 hours prior to a viral challenge exhibited a modest (3-4-fold) increase in mA3 mRNA levels, but displayed a significantly increased restriction of MMTV [94]. Finally, a recent paper examining a novel role for A3G in the sensitization of infected cells to NK-mediated lysis suggests that small fluctuations in A3G expression levels may have profound functional consequences [66]. These studies are interesting for their suggestion that modest elevations of mA3 and A3G gene expression can lead to impressive increases in viral restriction.
5. Concluding Remarks
The unfolding story of the multifunctional characteristics of the A3 family is fascinating. When the identification and characterization of A3G as a potent restriction factor emerged, the field raised numerous important questions and formulated strategies for capitalizing on this natural innate defense. Over several years, the identity of the entire A3 family of proteins as important innate restriction factors has been established. The ability of A3G to inhibit HIV-1Δvif infection has been analyzed by a significant number of laboratories, but the full complement of molecular details on how it exerts its antiviral function has not yet been gathered. Cytidine deamination undoubtedly occurs in the setting of a natural viral infection, but it is not entirely clear whether this enzymatic function is the only modality through which A3G can obstruct HIV-1 in vivo. An improved understanding of the details of how antiviral functions are exerted is needed. In addition, the important, and likely critical contribution of additional A3 family members in vivo, remains largely uncharacterized, although recent work using a tissue-culture model would suggest that a collaborative effort amongst family members is essential [17]. Utilization of both the MMTV/mA3 and FV/mA3 murine systems may be particularly useful. They are the only in vivo models of A3 restriction that currently exist. Alteration of the murine genome is relatively tractable and there is a single A3 gene in the rodent genome; potentially, mA3 genetic variants may be assessed in this setting. Other outstanding questions include the determination of whether any of the A3 proteins require cofactors or post-translational modifications to function effectively. An important co-factor for APOBEC1 has been delineated and while there is a preliminary suggestion that A3F/3G antiviral activity requires a co-factor, no specific proteins have been identified to date [106, 107].
Manipulation of the Vif : A3G interaction is also a viable point of chemotherapeutic intervention. To date only one compound specifically targeting Vif and thereby liberating functional A3G from Vif regulation has been reported; rapidly evolving technology may soon identify others [108]. A more comprehensive understanding of the interface involved in this viral and cellular protein association could identify new target sequences. For instance, recent identification of the transcription factor CBF-β as a member of the ubiquitin-ligase complex recruited by Vif to degrade A3G may prove interesting when considering novel drug targets [109]. Liberating A3G from Vif-mediated control has been shown to impact HIV-1 replication in vitro and suggests elevated levels of A3G can have a significant impact on the kinetics of viral replication, but whether expression levels of A3 genes can be modulated in vivo remains to be determined. A better fundamental understanding of gene regulation and the important regulatory elements within this family is also essential. To date only one of the promoters within the A3 family has been identified and characterized [85].
A more collaborative and concerted effort in the examination of various cohorts is more likely to reveal whether there exist meaningful associations between A3 genes and the ability to completely resist or partially restrict HIV-1. In light of the recent data being produced in the murine systems, an examination of rapid progressors and various A3 genetic variants is warranted. Additionally, data sets analyzing A3G genetic variants, while relevant and useful, may have missed important information about other family members; the recent findings involving A3A would suggest that this gene would also be important to examine in a number of cohorts.
Expanded roles for members of the A3 family have also been reported. These reports attribute an importance to A3 proteins that extends beyond the relatively simple arena of restriction factor. A3G's participation in marking cells for NK-mediated lysis would expand the reach of the A3 family into induced innate immunity, a series of cellular interactions important in bridging the innate and adaptive responses. Further describing and characterizing this observation will be important as it has potentially important implications for treatment during acute infection and vaccine design. In ten years the field has exploded, from the recognition of a single potential restriction factor (A3G) to an impressive understanding of a family of proteins that influence, modulate, and enhance the innate immune response. It begs the tantalizing question: what will the next decade bring?
References
- 1.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 2.Fisher AG, Ensoli B, Ivanoff L. The sor gene of HIV-1 is required for efficient virus transmission in vitro . Science. 1987;237(4817):888–893. doi: 10.1126/science.3497453. [DOI] [PubMed] [Google Scholar]
- 3.Strebel K, Daugherty D, Clouse K. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328(6132):728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 4.Sodroski J, Goh WC, Rosen C. Replicative and cytopathic potential of HTLV-III/LAV with sor gene deletions. Science. 1986;231(4745):1549–1553. doi: 10.1126/science.3006244. [DOI] [PubMed] [Google Scholar]
- 5.Gabuzda DH, Lawrence K, Langhoff E, et al. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. Journal of Virology. 1992;66(11):6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakai H, Shibata R, Sakuragi JI, Sakuragi S, Kawamura M, Adachi A. Cell-dependent requirement of human immunodeficiency virus type 1 vif protein for maturation of virus particles. Journal of Virology. 1993;67(3):1663–1666. doi: 10.1128/jvi.67.3.1663-1666.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon JHM, Miller DL, Fouchier RAM, Soares MA, Peden KWC, Malim MH. The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. The EMBO Journal. 1998;17(5):1259–1267. doi: 10.1093/emboj/17.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Schwedler U, Song J, Aiken C, Trono D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. Journal of Virology. 1993;67(8):4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madani N, Kabat D. An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. Journal of Virology. 1998;72(12):10251–10255. doi: 10.1128/jvi.72.12.10251-10255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon JHM, Gaddis NC, Fouchier RAM, Malim MH. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nature Medicine. 1998;4(12):1397–1400. doi: 10.1038/3987. [DOI] [PubMed] [Google Scholar]
- 11.Jarmuz A, Chester A, Bayliss J, et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79(3):285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 12.Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Molecular Cell. 2002;10(5):1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 13.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biology. 2008;9(6, article no. 229) doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt K, Guo K, Algaier M, et al. Differential virus restriction patterns of rhesus macaque and human APOBEC3A: implications for lentivirus evolution. Virology. 2011;419(1):24–42. doi: 10.1016/j.virol.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wedekind JE, Dance GSC, Sowden MP, Smith HC. Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends in Genetics. 2003;19(4):207–216. doi: 10.1016/S0168-9525(03)00054-4. [DOI] [PubMed] [Google Scholar]
- 16.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philosophical Transactions of the Royal Society B. 2009;364(1517):675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hultquist JF, Lengyel J, Refsland EW, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-Deficient HIV-1. Journal of Virology. 2011;85(21):11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross SR. Are viruses inhibited by APOBEC3 molecules from their host species. PLoS Pathogens. 2009;5(4) doi: 10.1371/journal.ppat.1000347. Article ID e1000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annual Review of Immunology. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 20.OhAinle M, Kerns JA, Li MMH, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host and Microbe. 2008;4(3):249–259. doi: 10.1016/j.chom.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339(2):281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Berger G, Durand S, Fargier G, et al. APOBEC3a is a specific inhibitor of the early phases of hiv-1 infection in myeloid cells. PLoS Pathogens. 2011;7(9) doi: 10.1371/journal.ppat.1002221. Article ID e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. Journal of Virology. 2006;80(21):10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Current Biology. 2004;14(15):1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 25.Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. Journal of Virology. 2004;78(11):6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. The EMBO Journal. 2004;23(12):2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang Y, Lai MS, Wang X, Han Y, Lampen R, Zheng YH. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. The Journal of Biological Chemistry. 2008;283(17):11606–11614. doi: 10.1074/jbc.M707586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Molecular Cell. 2003;12(3):591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 29.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nature Medicine. 2003;9(11):1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 30.Conticello SG, Harris RS, Neuberger MS. The Vif Protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Current Biology. 2003;13(22):2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 31.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nature Medicine. 2003;9(11):1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 32.Kao S, Miyagi E, Khan MA, et al. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology. 2004;1, article 27 doi: 10.1186/1742-4690-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. The Journal of Biological Chemistry. 2004;279(9):7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 34.Mariani R, Chen D, Schröfelbauer B, et al. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114(1):21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Potash MJ, Volsky DJ. Functional domains of APOBEC3G required for antiviral activity. Journal of Cellular Biochemistry. 2004;92(3):560–572. doi: 10.1002/jcb.20082. [DOI] [PubMed] [Google Scholar]
- 36.Yu X, Yu Y, Liu B, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. The Journal of Biological Chemistry. 2005;280(19):18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- 38.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424(6944):94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thielen BK, Klein KC, Walker LW, et al. T cells contain an RNase-insensitive inhibitor of APOBEC3G deaminase activity. PLoS Pathogens. 2007;3(9):1320–1334. doi: 10.1371/journal.ppat.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman ENC, Holmes RK, Craig HM, et al. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Current Biology. 2005;15(2):166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 42.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. The Journal of Biological Chemistry. 2007;282(4):2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 43.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends in Biochemical Sciences. 2007;32(3):118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 44.Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. Journal of Virology. 2006;80(17):8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 46.Luo K, Wang T, Liu B, et al. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. Journal of Virology. 2007;81(13):7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of tRNA3Lys-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. Journal of Virology. 2006;80(23):11710–11722. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. The Journal of Biological Chemistry. 2007;282(44):32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- 49.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathogens. 2008;4(12) doi: 10.1371/journal.ppat.1000231. Article ID e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyagi E, Opi S, Takeuchi H, et al. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. Journal of Virology. 2007;81(24):13346–13353. doi: 10.1128/JVI.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schumacher AJ, Haché G, MacDuff DA, Brown WL, Harris RS. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. Journal of Virology. 2008;82(6):2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langlois MA, Neuberger MS. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. Journal of Virology. 2008;82(9):4660–4664. doi: 10.1128/JVI.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase APOBEC3G. Journal of Virology. 2006;80(2):875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bogerd HP, Wiegand HL, Doehle BP, Cullen BR. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology. 2007;364(2):486–493. doi: 10.1016/j.virol.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan L, Sarkis PTN, Wang T, Tian C, Yu XF. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. The FASEB Journal. 2009;23(1):279–287. doi: 10.1096/fj.07-088781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li MMH, Emerman M. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. Journal of Virology. 2011;85(16):8197–8207. doi: 10.1128/JVI.00624-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. Journal of Virology. 2011;85(7):3142–3152. doi: 10.1128/JVI.02049-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gourraud PA, Karaouni A, Woo JM, et al. APOBEC3H haplotypes and HIV-1 pro-viral vif DNA sequence diversity in early untreated human immunodeficiency virus-1 infection. Human Immunology. 2011;72(3):207–212. doi: 10.1016/j.humimm.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cagliani R, Riva S, Fumagalli M, et al. A positively selected APOBEC3H haplotype is associated with natural resistance to HIV-1 infection. Evolution. 2011;65:3311–3322. doi: 10.1111/j.1558-5646.2011.01368.x. [DOI] [PubMed] [Google Scholar]
- 60.Koning FA, Newman ENC, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. Journal of Virology. 2009;83(18):9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peng G, Ke JL, Jin W, Greenwell-Wild T, Wahl SM. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. Journal of Experimental Medicine. 2006;203(1):41–46. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Research. 2010;38(13):4274–4284. doi: 10.1093/nar/gkq174. Article ID gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koning FA, Goujon C, Bauby H, Malim MH. Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. Journal of Virology. 2011;85(24):13448–13452. doi: 10.1128/JVI.00775-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thielen BK, McNevin JP, McElrath MJ, Hunt BVS, Klein KC, Lingappa JR. Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. The Journal of Biological Chemistry. 2010;285(36):27753–27766. doi: 10.1074/jbc.M110.102822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pion M, Granelli-Piperno A, Mangeat B, et al. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. Journal of Experimental Medicine. 2006;203(13):2887–2893. doi: 10.1084/jem.20061519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norman JM, Mashiba M, McNamara LA, et al. The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nature Immunology. 2011;12(10):975–983. doi: 10.1038/ni.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Reports. 2011;12(5):444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gasser S, Raulet DH. The DNA damage response arouses the immune system. Cancer Research. 2006;66(8):3959–3962. doi: 10.1158/0008-5472.CAN-05-4603. [DOI] [PubMed] [Google Scholar]
- 69.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436(7054):1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17(1):19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 71.Croxford JL, Gasser S. Damage control: how HIV survives the editor APOBEC3G. Nature Immunology. 2011;12(10):925–927. doi: 10.1038/ni.2115. [DOI] [PubMed] [Google Scholar]
- 72.Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24(6):779–786. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 73.Valcke HS, Bernard NF, Bruneau J, Alary M, Tsoukas CM, Roger M. APOBEC3G genetic variants and their association with risk of HIV infection in highly exposed Caucasians. AIDS. 2006;20(15):1984–1986. doi: 10.1097/01.aids.0000247124.35129.e1. [DOI] [PubMed] [Google Scholar]
- 74.An P, Johnson R, Phair J, et al. APOBEC3B deletion and risk of HIV-1 acquisition. Journal of Infectious Diseases. 2009;200(7):1054–1058. doi: 10.1086/605644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Land AM, Ball TB, Luo M, et al. Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. Journal of Virology. 2008;82(16):8172–8182. doi: 10.1128/JVI.01115-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanki PJ, M’Boup S, Marlink R, et al. Prevalence and risk determinants of human immunodeficiency virus type 2 (HIV-2) and human immunodeficiency virus type 1 (HIV-1) in West African female prostitutes. American Journal of Epidemiology. 1992;136(7):895–907. doi: 10.1093/aje/136.7.895. [DOI] [PubMed] [Google Scholar]
- 77.Ulenga NK, Sarr AD, Thakore-Meloni S, Sankalé JL, Eisen G, Kanki PJ. Relationship between human immunodeficiency type 1 infection and expression of human APOBEC3G and APOBEC3F. Journal of Infectious Diseases. 2008;198(4):486–492. doi: 10.1086/590212. [DOI] [PubMed] [Google Scholar]
- 78.An P, Bleiber G, Duggal P, et al. APOBEC3G genetic variants and their influence on the progression to AIDS. Journal of Virology. 2004;78(20):11070–11076. doi: 10.1128/JVI.78.20.11070-11076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biasin M, Piacentini L, Lo Caputo S, et al. Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G: a possible role in the resistance to HIV of HIV-exposed seronegative individuals. Journal of Infectious Diseases. 2007;195(7):960–964. doi: 10.1086/511988. [DOI] [PubMed] [Google Scholar]
- 80.Jin X, Brooks A, Chen H, Bennett R, Reichman R, Smith H. APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. Journal of Virology. 2005;79(17):11513–11516. doi: 10.1128/JVI.79.17.11513-11516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vázquez-Pérez JA, Ormsby CE, Hernández-Juan R, Torres KJ, Reyes-Terán G. APOBEC3G mRNA expression in exposed seronegative and early stage HIV infected individuals decreases with removal of exposure and with disease progression. Retrovirology. 2009;6, article no. 23 doi: 10.1186/1742-4690-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pace C, Keller J, Nolan D, et al. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. Journal of Virology. 2006;80(18):9259–9269. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo . Journal of Virology. 2005;79(3):1975–1980. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mußil B, Sauermann U, Motzkus D, Stahl-Hennig C, Sopper S. Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology. 2011;8(article 77) doi: 10.1186/1742-4690-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farrow MA, Kim EY, Wolinsky SM, Sheehy AM. NFAT and IRF proteins regulate transcription of the anti-HIV gene, APOBEC3G. The Journal of Biological Chemistry. 2011;286(4):2567–2577. doi: 10.1074/jbc.M110.154377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo . Nature. 2007;445(7130):927–930. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- 87.Santiago ML, Montano M, Benitez R, et al. APOBEC3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science. 2008;321(5894):1343–1346. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takeda E, Tsuji-Kawahara S, Sakamoto M, et al. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo . Journal of Virology. 2008;82(22):10998–11008. doi: 10.1128/JVI.01311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abudu A, Takaori-Kondo A, Izumi T, et al. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Current Biology. 2006;16(15):1565–1570. doi: 10.1016/j.cub.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 90.Browne EP, Littman DR. Species-specific restriction of APOBEC3-mediated hypermutation. Journal of Virology. 2008;82(3):1305–1313. doi: 10.1128/JVI.01371-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doehle BP, Schäfer A, Wiegand HL, Bogerd HP, Cullen BR. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. Journal of Virology. 2005;79(13):8201–8207. doi: 10.1128/JVI.79.13.8201-8207.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conticello SG, Thomas CJF, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Molecular Biology and Evolution. 2005;22(2):367–377. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- 93.Mikl MC, Watt IN, Lu M, et al. Mice deficient in APOBEC2 and APOBEC3. Molecular and Cellular Biology. 2005;25(16):7270–7277. doi: 10.1128/MCB.25.16.7270-7277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Okeoma CM, Low A, Bailis W, Fan HY, Peterlin BM, Ross SR. Induction of APOBEC3 in vivo causes increased restriction of retrovirus infection. Journal of Virology. 2009;83(8):3486–3495. doi: 10.1128/JVI.02347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Okeoma CM, Huegel AL, Lingappa J, Feldman MD, Ross SR. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host and Microbe. 2010;8(6):534–543. doi: 10.1016/j.chom.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coovadia HM, Rollins NC, Bland RM, et al. Mother-to-child transmission of HIV-1 infection during exclusive breastfeeding in the first 6 months of life: an intervention cohort study. The Lancet. 2007;369(9567):1107–1116. doi: 10.1016/S0140-6736(07)60283-9. [DOI] [PubMed] [Google Scholar]
- 97.Hasenkrug KJ, Chesebro B. Immunity to retroviral infection: the friend virus model. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(15):7811–7816. doi: 10.1073/pnas.94.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chesebro B, Wehrly K. Identification of a non-H-2 gene (Rfv-3) influencing recovery from viremia and leukemia induced by Friend virus complex. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(1):425–429. doi: 10.1073/pnas.76.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hasenkrug KJ, Brooks DM, Chesebro B. Passive immunotherapy for retroviral disease: influence of major histocompatibility complex type and T-cell responsiveness. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(23):10492–10495. doi: 10.1073/pnas.92.23.10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Santiago ML, Smith DS, Barrett BS, et al. Persistent friend virus replication and disease in APOBEC3-deficient mice expressing functional B-cell-activating factor receptor. Journal of Virology. 2011;85(1):189–199. doi: 10.1128/JVI.01838-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsuji-Kawahara S, Chikaishi T, Takeda E, et al. Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic APOBEC3 and BAFF-R loci in friend virus-infected mice. Journal of Virology. 2010;84(12):6082–6095. doi: 10.1128/JVI.02516-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li J, Hakata Y, Takeda E, et al. Two genetic determinants acquired late in Mus evolution regulate the inclusion of exon 5, which alters mouse APOBEC3 translation efficiency. PLoS Pathogens. 2012;8(1) doi: 10.1371/journal.ppat.1002478. Article ID e1002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. The Journal of Biological Chemistry. 2007;282(6):3539–3546. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- 104.Piguet V, Blauvelt A. Essential roles for dendritic cells in the pathogenesis and potential treatment of HIV disease. Journal of Investigative Dermatology. 2002;119(2):365–369. doi: 10.1046/j.1523-1747.2002.01840.x. [DOI] [PubMed] [Google Scholar]
- 105.Steinman RM, Granelli-Piperno A, Pope M, et al. The interaction of immunodeficiency viruses with dendritic cells. Current Topics in Microbiology and Immunology. 2003;276:1–30. doi: 10.1007/978-3-662-06508-2_1. [DOI] [PubMed] [Google Scholar]
- 106.Han Y, Wang X, Dang Y, Zheng YH. APOBEC3G and APOBEC3F require an endogenous cofactor to block HIV-1 replication. PLoS Pathogens. 2008;4(7) doi: 10.1371/journal.ppat.1000095. Article ID e1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Henderson JO, Blanc V, Davidson NO. Isolation, characterization and developmental regulation of the human apobec-1 complementation factor (ACF) gene. Biochimica et Biophysica Acta. 2001;1522(1):22–30. doi: 10.1016/s0167-4781(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 108.Nathans R, Cao H, Sharova N, et al. Small-molecule inhibition of HIV-1 Vif. Nature Biotechnology. 2008;26(10):1187–1192. doi: 10.1038/nbt.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jäger S, Kim DY, Hultquist JF, et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012;481(7381):371–375. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]