

Abstract
The genetic adaptation of pathogens in host tissue plays a key role in the establishment of chronic infections. While whole genome sequencing has opened up the analysis of genetic changes occurring during long-term infections, the identification and characterization of adaptive traits is often obscured by a lack of knowledge of the underlying molecular processes. Our research addresses the role of Pseudomonas aeruginosa small colony variant (SCV) morphotypes in long-term infections. In the lungs of cystic fibrosis patients, the appearance of SCVs correlates with a prolonged persistence of infection and poor lung function. Formation of P. aeruginosa SCVs is linked to increased levels of the second messenger c-di-GMP. Our previous work identified the YfiBNR system as a key regulator of the SCV phenotype. The effector of this tripartite signaling module is the membrane bound diguanylate cyclase YfiN. Through a combination of genetic and biochemical analyses we first outline the mechanistic principles of YfiN regulation in detail. In particular, we identify a number of activating mutations in all three components of the Yfi regulatory system. YfiBNR is shown to function via tightly controlled competition between allosteric binding sites on the three Yfi proteins; a novel regulatory mechanism that is apparently widespread among periplasmic signaling systems in bacteria. We then show that during long-term lung infections of CF patients, activating mutations invade the population, driving SCV formation in vivo. The identification of mutational “scars” in the yfi genes of clinical isolates suggests that Yfi activity is both under positive and negative selection in vivo and that continuous adaptation of the c-di-GMP network contributes to the in vivo fitness of P. aeruginosa during chronic lung infections. These experiments uncover an important new principle of in vivo persistence, and identify the c-di-GMP network as a valid target for novel anti-infectives directed against chronic infections.
Author Summary
Here we investigate the molecular function of the important cyclic-di-GMP signaling system YfiBNR in the opportunistic pathogen Pseudomonas aeruginosa and demonstrate its importance for the evolution of persistent small colony variant (SCV) morphotypes in chronic cystic fibrosis (CF) lung infections. Previously we showed that YfiN is a membrane bound diguanylate cyclase, whose activity is controlled by the soluble periplasmic repressor YfiR and the outer-membrane peptidoglycan binding protein YfiB. In this study we use a combination of genetic and biochemical analyses to investigate the mechanistic principles of YfiN regulation. By examining a series of activating mutations throughout the yfi operon, we show that YfiBNR functions via tightly controlled competition between allosteric binding sites on the three Yfi proteins; a novel regulatory mechanism that is apparently widespread among periplasmic signaling systems in bacteria. We then show that during long-term CF lung infections, Yfi activating mutations invade the population, driving SCV formation in vivo. The identification of mutational “scars” in the yfi genes of clinical isolates further suggests that Yfi activity is both under positive and negative selection in vivo, with Yfi-mediated SCVs acting as an environmental pool for the generation of new smooth morphotypes.
Introduction
Pseudomonas aeruginosa is an opportunistic gram-negative pathogen that predominates in late stage cystic fibrosis (CF) lung infections [1]. Once established in the CF lung, P. aeruginosa is impossible to entirely eradicate, with repeated relapses of infection and the accompanying aggravation leading to progressive tissue degradation and eventually to death. Over the course of long-term chronic CF lung infections, P. aeruginosa undergoes phenotypic and genetic adaptation to the lung environment, resulting in both a progressive transition towards a persistent, low virulence state and a related diversification into a number of distinctive phenotypes [2], [3]. These include mucoid cells, which overproduce alginate and form distinctive slimy colonies [4], and small colony variants (SCVs), slow-growing isolates that show strong attachment to surfaces, auto-aggregation, enhanced exopolysaccharide production and biofilm formation [5], [6]. The appearance of SCVs correlates with a prolonged persistence of infection, poor lung function and increased antibiotic and serum resistance. Fatal systemic infections after lung transplantation and increased serum resistance have been associated with the recovery of SCVs of Burkholderia species [7], [8], [9]. P. aeruginosa SCVs also emerge in other situations that favor chronic infections including mechanically ventilated patients or patients suffering from chronic obstructive pulmonary disease [8], [10]. These studies suggest that persistent forms of P. aeruginosa represent genetic adaptations to the hostile milieu in the patient, with characteristics including resistance to phagocytosis [11], antimicrobial resistance due to slow growth or increased persister cell populations [7], [12], and reduced virulence [13] potentially contributing to selection. Consistent with this, our recent work [11] demonstrated a causal link between P. aeruginosa SCVs and persistence of infection in mice, supporting the hypothesis that under certain infection conditions the SCV phenotype confers a fitness advantage, and thus makes an important contribution to the pathogenesis of P. aeruginosa lung infections.
In recent years a strong link has emerged between enhanced levels of the second messenger cyclic di-GMP (c-di-GMP) [11], [14], [15], [16] and the SCV phenotype, via overproduction of exopolysaccharides [5], [16] or fimbrial adhesins [14], [17]. C-di-GMP is a ubiquitous and widespread signaling molecule that has been shown to influence a diverse range of cellular processes involved in the transition from a motile, single-cell lifestyle to sessile, surface attached consortia called biofilms [18], [19]. In P. aeruginosa c-di-GMP regulates multiple cellular processes, including exopolysaccharide production [20], [21], [22], exposure of fimbrial and proteinaceous adhesins [23], [24], rhamnolipid biosynthesis [25], siderophore production [11], and virulence and cytotoxicity systems [26], [27], [28], as well as assembly and function of pili [29], [30], [31] and flagella [32]. Since many of these cellular processes are subject to phenotypic adaptation during chronic P. aeruginosa lung infections, enzymes involved in c-di-GMP metabolism have emerged as possible targets of the underlying genotypic variation [2], [11], [14].
YfiBNR [11], [33] (also called AwsXRO [34], [35], [36], TpbB [37]) is a tripartite signaling system that modulates intracellular c-di-GMP levels in response to signals received in the periplasm [11]. The effector of the Yfi system, YfiN, is a membrane integral diguanylate cyclase consisting of a periplasmic PAS domain and cytoplasmic HAMP and catalytic GGDEF domains (Figure 1A). YfiN activity is repressed by the soluble periplasmic protein YfiR and stimulated by the outer membrane lipoprotein YfiB [11]. C-di-GMP produced by YfiN stimulates the production of the Pel and Psl exopolysaccharides, thereby promoting surface attachment in wild-type P. aeruginosa and generating an SCV phenotype when YfiN is activated or YfiR repression is released [11]. Homologs of the YfiBNR system are widespread, and have been shown to function similarly in Escherichia coli, Klebsiella pneumonia and Pseudomonas fluorescens SBW25, where they affect biofilm formation through cellulose production [33], [34] or Type 3 fimbriae expression [38].
Figure 1. Interactions between the YfiBNR proteins.
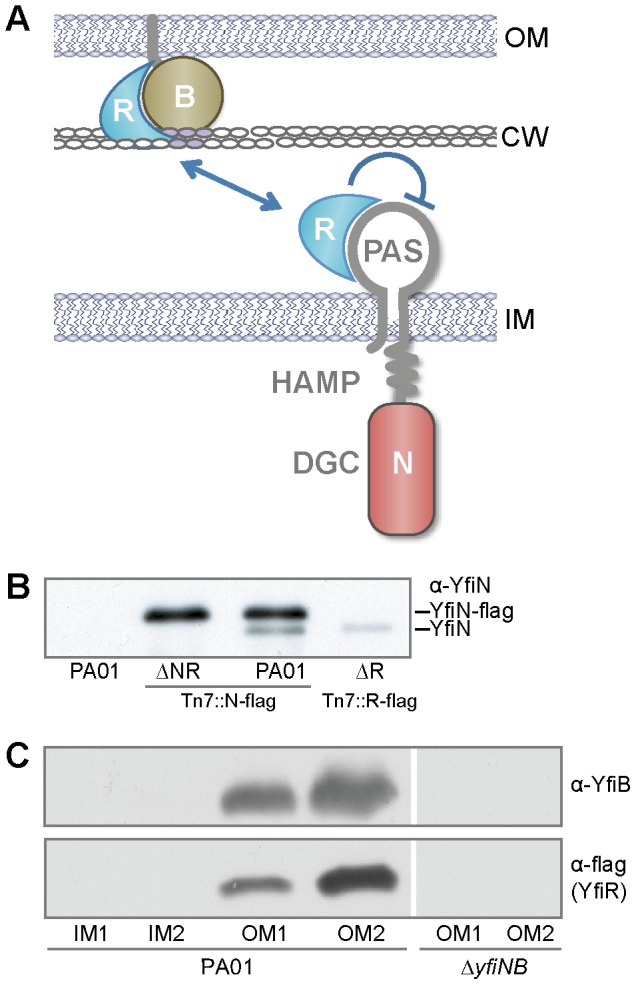
A) A model of yfiBNR interaction. YfiN is a membrane-localized DGC controlled by YfiR. YfiB, the outer-membrane bound Pal-like protein, activates YfiN by sequestering YfiR. B) Co-immunoprecipitation of YfiN with flag-tagged YfiN and YfiR. Immunoblot of boiled M2 resin samples with anti-YfiN antiserum shows YfiN (lower band) co-precipitating with YfiN-flag (upper band) or YfiR-flag. C) Membrane localization of YfiB and YfiR. Immunoblots of fractionated membrane samples with anti-YfiB (upper panel) and M2 antisera (lower 2 panels). The left two panels show membrane fractions for PA01 yfiR-M2, the right panel for ΔyfiBN yfiR-M2. IM1/2: inner membrane fractions, OM1/2: outer membrane fractions.
While the epistasis and mode of output of YfiBNR have been established [11], the mechanistic principles of YfiBNR control remain to be determined. Central unanswered questions include the nature of activating signals and the mechanism of YfiN activation. The periplasmic protein YfiR plays a key role in the signal transduction process as it bridges between the YfiN diguanylate cyclase in the inner membrane and the presumable YfiB sensor in the outer membrane. However, no structural or mechanistic information is available for any members of the YfiR family. In particular, it is unclear how YfiR interacts with the periplasmic PAS domain of YfiN. PAS are versatile domains that activate downstream signaling processes through a range of different mechanisms [39], [40], [41], [42] including ligand binding [41], light/oxygen-driven modification of bound flavin or heme groups [43], [44], homodimerization [45] and, in eukaryotes, heterodimerization [46], [47]. However, given the apparent ubiquity of PAS domains in bacterial signal transduction processes [48] additional activation mechanisms likely exist. Finally, the role and mode of action of YfiB have remained elusive. Epistasis experiments place YfiB upstream of YfiR, and suggest that YfiB activates YfiN by relieving YfiR-mediated repression [11]. YfiB is a structural homolog of Pal, a peptidoglycan binding protein and component of the Tol-Pal pathway required to maintain cell envelope integrity and function [49], [50], [51], [52], although it is unclear whether or not the two proteins are also functional analogs.
In this study we map several adaptive mutations in P. aeruginosa SCV isolates from CF patients to the c-di-GMP regulatory yfiBNR locus. Moreover, through the elucidation of the signal transduction mechanisms of the YfiBNR system we present a molecular rationale explaining how this system contributes to the evolution of distinct P. aeruginosa phenotypes and how the consecutive selection of gain- and loss-of-function yfiBNR mutations might contribute to P. aeruginosa adaptation in CF lungs. Firstly, a combination of genetic and biochemical analysis was used to produce a detailed molecular map of YfiBNR function. Through the isolation and characterization of “locked-on” and compensatory mutant alleles of all three components of the system, we provide evidence that YfiR inhibits YfiN allosterically, through a hydrophobic interaction between the C-terminus of YfiR and a conserved region of the periplasmic PAS domain of YfiN. Subsequent in silico analysis suggests that this YfiR-YfiN interaction represents a novel and widespread periplasmic signaling module, controlling diverse cytoplasmic outputs in a variety of species. YfiN repression is released through an YfiB-dependent sequestration of YfiR to the outer membrane fraction. The resulting structural rearrangements in YfiN are then propagated from the external PAS domain through the transmembrane helices and HAMP domain to activate the C-terminal catalytic GGDEF-domains. Our results suggest that the YfiB lipoprotein that spans the outer membrane and the peptidoglycan acts as a sensor of the YfiBNR system, and may be involved in transducing envelope stress into a rapid increase of c-di-GMP inside the cell and consequent biofilm formation through activation of the Pel and Psl exopolysaccharide systems.
In parallel, screening strategies with two libraries of clinical P. aeruginosa strains isolated from CF patients identified a number of SCVs with causal mutations throughout the yfiBNR locus. Activating substitutions were found in YfiN and a loss-of-function mutation was isolated in YfiR. In addition, several SCVs harbored mutations in the predicted yfiBNR promoter region. The observation that most of these mutations match the “locked-on” mutations that we isolated by in vitro genetics strongly argues that P. aeruginosa SCVs arise in patients' lungs through genetic alterations that activate the Yfi signaling system. Furthermore, the subsequent identification of clinical isolates containing both yfi activating mutations and loss-of-function mutations in yfiN suggests that the environment in the lung alternates over time between states that favor and disfavor the formation and fitness of SCVs. Thus, Yfi-mediated SCVs are under positive and negative selection in the dynamic environment of the CF lung thereby enabling P. aeruginosa to switch between slow growing and persistent SCVs and fast growing smooth morphotypes.
Results
The periplasmic regulator YfiR shuttles between inner and outer membrane
YfiN has previously been shown to function as a membrane bound diguanylate cyclase (DGC) whose activity is repressed by the soluble periplasmic protein YfiR [11], [35]. However, the mechanism of YfiR repression is currently unknown. The simplest potential repression mechanism is allosteric inhibition via direct binding of YfiR to the periplasmic PAS-like domain of YfiN. To test whether YfiR interacts with YfiN, co-immunoprecipitation experiments were carried out using an YfiR variant with a C-terminal flag tag. Wild-type YfiN was successfully pulled down by YfiR-flag (Figure 1B), indicating that these proteins interact and that YfiR might function by allosterically inhibiting YfiN activity. Furthermore, YfiN-flag was also shown to interact with YfiN wild type (Figure 1B), indicating that YfiN dimerizes in vivo, in common with other diguanylate cyclases [53].
YfiB is predicted to be an outer membrane lipoprotein on the basis of primary structure analysis and the fact that the protein is found exclusively in the insoluble fraction of lysed PA01 [11]. This was confirmed by membrane fractionation experiments that located YfiB exclusively in the outer membrane fractions (Figure 1C). Although co-immunoprecipitation experiments failed to show direct interaction between YfiB and YfiR (data not shown), YfiR-flag localization to the outer membrane fraction is strictly YfiB-dependent. In the absent of YfiB, YfiR-flag is stable (Figure S1B), but no longer associates with the membrane (Figure 1C, S1A).
Isolation of constitutive mutants delineates the mode of YfiN diguanylate cyclase activation
If YfiR represses YfiN activity through direct binding to its periplasmic PAS domain, it should be possible to isolate constitutively active YfiN variants that fail to bind YfiR. The positions of these activating residue substitutions would consequently provide insights into the mechanism of YfiN function and the binding interface of YfiN and YfiR. Previously, similar experiments have been successfully used to probe the structure-function relationship of the P. fluorescens DGC WspR [54], [55]. To identify YfiR-insensitive YfiN alleles, a screening system was designed in which yfiN and yfiR-flag are expressed from two separate plasmids in a ΔyfiNR background (see Materials and Methods). A pool of yfiN variants was produced by XL-1 red mutagenesis of the yfiN plasmid and screened for mutants that induced an SCV phenotype in the ΔyfiNR tester strain containing a plasmid-borne copy of yfiR. Sequencing identified the locations of twenty independent, activating yfiN mutations. Two residues were identified in the first transmembrane helix, ten were located towards the N-terminal end of the periplasmic PAS domain, four were found in the second transmembrane helix, and four towards the C-terminal end of the HAMP domain (Figure 2A). No mutations were found in the GGDEF domain. Since most of these mutations were isolated several times independently, we assume that the screen was approaching saturation (Table 1).
Figure 2. Activating mutations in YfiN.
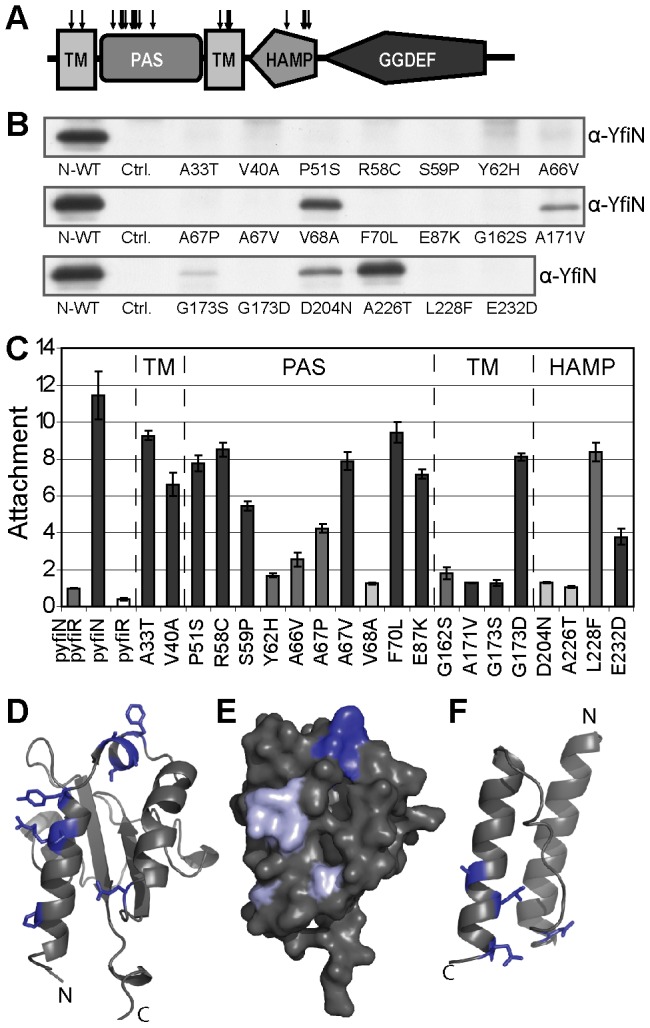
A) Locations of activating mutations in YfiN. Arrows indicate the positions of activating mutations throughout the structure of YfiN. TM: transmembrane helix. The cartoon is drawn to scale. B) Co-immunoprecipitation of active YfiN alleles with flag-tagged YfiR. Immunoblot of boiled M2 resin samples with anti-YfiN antiserum. N-WT shows the DyfiNR screening strain with both yfiN (WT) and yfiR-flag plasmids present. Ctrl. shows ΔyfiNR with pGm-yfiprom-N only. The point mutation present in YfiN is indicated for the remaining lanes, which show YfiN immunoprecipitated from ΔyfiNR strains containing both yfiR-flag and the mutated yfiN plasmids. C) Attachment of the active YfiN alleles is shown relative to ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag (p-yfiN p-yfiR). Controls containing the yfiN or yfiR-flag plasmid only are also shown. The point mutation present in YfiN is indicated for each bar. Light grey bars indicate mutants whose activity was compensated for by mutations at the C-terminus of YfiR. Mutants with mid grey bars were compensated for by mutations in the signal sequence, or by uncharacterized mutations, while those with dark grey bars were not compensated for during YfiR mutagenesis. The domain locations of mutants are indicated with TM, PAS etc. D) Cartoon showing the locations of activating substitutions (blue) on a homology model of the YfiN PAS domain (residues 44–154). The PAS model is based on the CitA periplasmic domain (see Materials and methods). E) Surface representation of the YfiN PAS model. The locations of activating mutants on the proposed homodimer interface are shown in light blue, those at the possible YfiR binding site are shown in dark blue. F) The locations of activating substitutions (blue) on a homology model of the YfiN HAMP domain (residues 183–236). N and C termini are marked in D) and F). The HAMP model is based on the Aer2 HAMP structure (see Materials and methods).
Table 1. YfiR-insensitive YfiN alleles.
| Location in YfiN | Residue change | Notes |
| First TM helix | A33T | |
| V40A | ||
| PAS, dimerization interface | P51S | |
| R58C | ||
| S59P | Equivalent mutation activates AwsX [35]. | |
| Y62H | ||
| A66V | ||
| A67P | ||
| A67V | ||
| V68A | ||
| F70L | ||
| E87K | Mutation present in Clin110. | |
| Second TM helix | G162S | |
| A171V | ||
| G173S | ||
| G173D | Mutation present in SCV20265. | |
| HAMP | D204N | Mutation at the equivalent position activates Tsr [59]. |
| A226T | ||
| L228F | ||
| E232D | Mutation at the equivalent position activates NarX [58]. |
PAS refers to the periplasmic PAS domain; HAMP refers to the HAMP domain.
Co-immunoprecipitation experiments showed that most of the activated YfiN alleles no longer bind to YfiR-flag (Figure 2B). In five cases (V68A, A171V, G173S, D204N, and A226T), residual YfiN-YfiR binding was still observed. In accordance with this, these five mutants produced the mildest phenotypes, with relatively low levels of surface attachment (Figure 2C) and partial SCV colony morphologies (Figure S2A). Expression of these five yfiN alleles in a ΔyfiNR strain produced a distinctive SCV phenotype (data not shown), indicating that the weaker SCV morphology seen with these alleles is likely due to partial inhibition by YfiR, rather than loss of YfiN function. The observation that activating mutations in YfiN abolished YfiR binding independently of their position within the protein (Figure 2B), suggested that the protein switches between discrete active and inactive states, and that the YfiR binding site is obscured in the active conformation.
A clearer suggestion of how YfiN functions was obtained when the positions of the YfiR-insensitive mutations were marked on homology models of the PAS and HAMP domains of YfiN. Activating mutations in the PAS domain appear to cluster into two regions of the 3-D structure modelled on the periplasmic domain of the sensor kinase CitA [41]. Assuming a similar dimerization mechanism for YfiN, the first group of substitutions would cluster at the interface between the two protein monomers (Figure 2D, 2E pale blue), where they might help to stabilize the active YfiN conformation. The remaining substitutions cluster on the domain surface most distal from the inner membrane. Three of these residues (Ala66, Ala67 and Val68) coordinate the position of the fourth (Phe70), which protrudes into the periplasmic space. These residues are predicted to form an exposed hydrophobic patch on the surface of the PAS domain, which we propose as a possible YfiR binding site (Figure 2E; dark blue). The four mutations in the HAMP domain lie close to one another and distal to the inner membrane (Figure 2F). Three mutations (positions 226, 228 and 232) are adjacent to residues required for helical bundle formation [56]. Specific substitutions at these positions may act to stabilise an active HAMP conformation relative to the inactive structure [56], [57]. In support of this, a substitution at the equivalent position to Glu232 in NarX renders the protein constitutively active [58]. The D204N mutation occupies an equivalent position in the helical linker region to the structurally important Leu237 residue of the E. coli serine-specific chemoreceptor Tsr. Mutations in this position have been shown to stabilise the activated form of Tsr [59], [60], suggesting a similar mechanism for the YfiN mutation.
Taken together, these results demonstrate that YfiN activation is crucially dependent on a number of key residues involved in intra-molecule signal transduction and on interfering with YfiR binding. We propose that the release of YfiR results in a conformational shift of the entire YfiN protein towards an active state, in which binding of the repressor is disfavored.
Compensatory mutations cluster in defined regions of the YfiR protein
To probe the YfiN-YfiR interaction in more detail, we undertook a screen for compensatory yfiR alleles, i.e. alleles that would restore wild-type (WT) colony morphology in the presence of some of the activated YfiN variants introduced above. Following PCR mutagenesis of yfiR, twenty-one alleles were isolated across eight yfiN mutant backgrounds (Table 2). Several yfiR alleles were independently isolated in several constitutive yfiN backgrounds, resulting in a total of fourteen unique, compensatory yfiR alleles, most of which cluster in the secretion signal sequence or in the C-terminal region of the protein (Table 2, Figure 3B). As the signal peptide is cleaved following export of YfiR into the periplasm, mutations in this region are not predicted to affect the final protein structure. Rather, these mutations might boost YfiR levels in the periplasm through increased translation or export of the protein. This was confirmed by immunoblot analysis demonstrating that signal peptide mutants indeed produced higher overall levels of YfiR than wild-type (Figure 3).
Table 2. Compensatory YfiR alleles.
| YfiN allele | No. | YfiR residue change(*) | Location in YfiR |
| Y62H | 12 | V21I | Signal peptide |
| 13 | D11N | Signal peptide | |
| A66V | 1 | S3L, D11Y, T76N | Signal peptide |
| A67P | 10 | V21I, Q187R | Signal peptide/proposed YfiN binding site |
| V68A | - | R93S, I169V | proposed YfiN binding site |
| - | E163G | proposed YfiN binding site | |
| - | Q187R | proposed YfiN binding site | |
| 3 | G29S, V112A | undefined | |
| 4 | S3T, D36E | Signal peptide | |
| 5 | V21I | Signal peptide | |
| 6 | L7S | Signal peptide | |
| G162S | - | Q125L | undefined |
| D204N | - | K63E | conserved region of YfiR |
| - | I169V | proposed YfiN binding site | |
| 11 | V21I | Signal peptide | |
| A226T | - | E163G | proposed YfiN binding site |
| 7 | L1M, Q10P, M86T, D102G, V135A | Signal peptide/improved start codon | |
| 8 | V21I | Signal peptide | |
| - | F151L | proposed YfiN binding site | |
| 9 | Q8H, I169V | proposed YfiN binding site | |
| L228F | 2 | Q8H, R60H | conserved region of YfiR |
(*): proposed activating mutations are highlighted in bold.
Figure 3. Compensatory YfiR alleles.
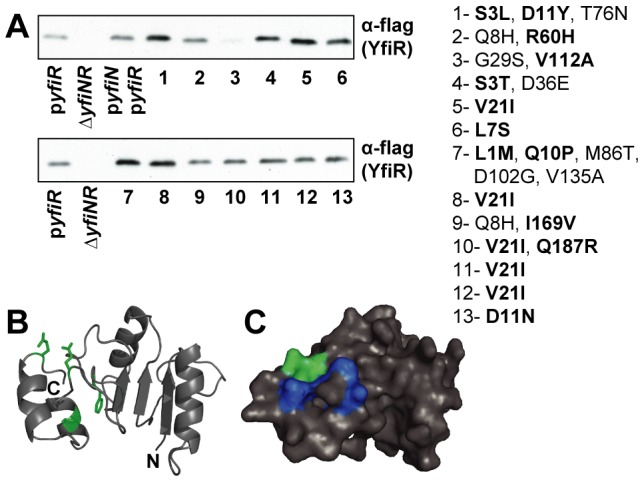
A) Immunoblots with M2 antiserum, showing levels of compensatory YfiR-flag variants in whole cell lysates. pyfiR: ΔyfiNR pMR-yfiR-flag, ΔyfiNR: strain without vector, pyfiN/pyfiR: ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag, lanes 1–13: ΔyfiNR pGm-yfiprom-N with compensatory pMR-yfiR-flag plasmids, proposed activating mutations are highlighted in bold. Mutants in lane 1, 4–8, 10–13 harbor mutations in the signal peptide that enhance expression, see also Table 2. B) Cartoon showing the locations of activating substitutions (green) on a homology model of YfiR (comprising residues 68–190). N and C termini are marked. The YfiR model is based on multiple structures (see Materials and methods). C) Surface representation of the YfiR model. The locations of activating mutants are shown in green, hydrophobic residues forming the possible YfiN binding surface are shown in dark blue.
Compensatory mutations in the C-terminus of YfiR were isolated in three distinct yfiN mutant backgrounds, all of which produced mild SCV phenotypes and showed residual binding of YfiR (Table 1, Figure 2). This suggests that mutations in this region of YfiR enhance binding to YfiN, but cannot compensate for a total loss of protein-protein interaction. In a cross-complementation experiment, all four C-terminal YfiR mutants (F151L, E163G, I169V and Q187R) were able to suppress the SCV phenotype of all three weak YfiN mutants (Figure S2B), indicating that the enhanced effectiveness of these mutants was the result of an overall increase in YfiR binding affinity, rather than through complementation of specific YfiN mutations. When plotted onto a homology model of YfiR, the four C-terminal mutations surround a hydrophobic region on the surface of the model (Figure 3B) that presents a possible candidate for the YfiN binding surface. These residues are highly conserved among YfiR homologs, especially the central Phe residue at position 162. Likewise, despite little overall conservation of the YfiN PAS domain, the predicted YfiR binding site on its surface (‘AAVVF’ motif) is highly conserved, but absent in the PAS domain of CitA [41], [61] (see below).
A plausible model for YfiN-YfiR interaction arises from these observations, in which the exposed phenylalanine on the surface of YfiN is hidden from the aqueous environment by hydrophobic interaction with the C-terminus of YfiR. Together, these data demonstrate that activating yfiN alleles can be overcome by compensatory mutations in yfiR, again lending support for a direct repression of YfiN by the periplasmic protein YfiR.
Genetic dissection of the YfiB outer membrane sensor
YfiB is predicted to be an outer-membrane lipoprotein with a PAL-like peptidoglycan (PG) binding domain. Overproduction of YfiB leads to YfiN-dependent SCV formation [11]. How this effect is exerted on YfiN is not clear and no detailed model for YfiB function in P. aeruginosa exists. To investigate the function of YfiB, a screen was conducted for activating mutants that induced an SCV phenotype in PA01 without overproduction of the protein. A total of 20 yfiB alleles were isolated, each containing one or more amino acid substitutions. All activating yfiB alleles caused increased surface attachment and biofilm formation (Figure 4A). Strikingly, while mutations were distributed throughout the sequence of yfiB, at least one substitution was found between residues 35 and 55 in all cases. These affected a total of seven positions, five of which were also isolated as single activating substitutions (Figure 4A). Alleles with single mutations in the YfiB N-terminus generally had strong effects that were weakened by the presence of secondary mutations (for example, YfiB L43P produces higher attachment than L43P alleles containing additional mutations). Together this argued that the majority of mutations leading to YfiBNR activation cluster in this region of the YfiB protein. Some of these variants had very strong activating effects despite showing severely reduced stability (Figure 4A). Three further, similar substitutions (I40F, V42M, and E45G) were found in conjunction with other singly isolated activating mutations. While neither the V42M nor the E45G substitutions contributed positively to YfiB activity, the YfiB-I40F-F48L allele produced a far stronger phenotype than F48L alone, suggesting that the I40F mutation also contributes to YfiB activation (Figure 4A). When the locations of the activating mutations were plotted onto a 3-D homology model of YfiB, they clustered around the first helix of the PAL domain (Figure 4B). Interestingly, activating residues in YfiB surround a predicted surface-exposed hydrophobic region, similar to that seen in the model of YfiR (Figure 3C). This hydrophobic patch is highly conserved in YfiB homologs, but absent in the YfiB structural homolog OprL [61], [62]. Most notably, W55, predicted to form the core of the hydrophobic binding site, is very highly conserved, only replaced in a small minority of cases with phenylalanine (note that the YfiB W55L mutation forms a weak SCV in PA01).
Figure 4. Mutational analysis of YfiB.

A) The effect of activating yfiB mutants, expressed from pME6032 in ΔyfiBNR Tn7::yfiNR, on attachment is shown relative to PA01 (PA01 ctrl.). ‘YfiB WT’ indicates pME6032-yfiB. The point mutants in YfiB are indicated for the remaining lanes. Those mutations thought to contribute to YfiB activation are marked in bold. The immunoblot shows the levels of YfiB protein present in each strain. B) Left: Cartoon showing the locations of activating substitutions (red) on a homology model of YfiB (comprising residues 27–168). The YfiB model is based on the Omp/Pal structure (see Materials and methods for details). The N terminus and peptidoglycan binding site are marked. Right: Surface representation of the YfiB model. The locations of activating mutants are shown in red, hydrophobic residues forming the possible YfiR binding surface are shown in dark blue. C) Co-localization of YfiB and YfiR at the outer membrane. Immunoblots of fractionated soluble and membrane samples with anti-YfiB and M2 antisera as shown. ‘YfiB WT’ indicates ΔyfiBNR Tn7::yfiR-flag containing pME6032-yfiB. ‘PG-’ indicates the same background strain containing pME-yfiB-PG- (YfiB containing the D102A and G105A substitutions), while ‘F48S’ contains the hyperactive yfiB F48S plasmid. D) The effect of different yfiB mutants, expressed from pME6032 in ΔyfiBNR Tn7::yfiNR, on attachment is shown relative to pME6032 only (ctrl.). ‘PG-’ mutants ± F48S or L43P mutations as indicated. In ‘LA-’ mutants, the lipid anchor is missing, the signal peptide has been replaced with that from YfiR. ‘ΔBNR LA-’ indicates the yfiB LA- mutant in the ΔyfiBNR strain background. The immunoblot shows the levels of YfiB protein present in each strain. E) Colony morphologies on LB Congo-red agar upon over-expression of yfiB mutants.
To determine whether binding to PG is important for YfiB function, mutants were constructed with two critical peptidoglycan-binding residues [63] replaced with alanine. Expression of the resulting gene, yfiB-D102A-G105A (PG-) did not induce an SCV colony morphology and had no effect on attachment, despite producing wild type-like levels of protein (Figure 4D, E). To assess the role of the YfiB OM lipid anchor (LA), the YfiB signal peptide was replaced with that from YfiR, which lacks the Cys lipid acceptor residue. The resulting mutant (LA-) slightly increased attachment upon induction in an YfiN-dependent manner (Figure 4D). This residual activity was dependent on peptidoglycan binding, as a PG-/LA- mutant was fully inactive (Figure 4D). These data suggest that peptidoglycan binding and, to a lesser extent, membrane anchoring are required for YfiB activity. When the mutations disrupting PG binding were combined with activating mutations in yfiB (F48S or L43P; see above), very low YfiB protein levels were detected, possibly as a result of reduced protein stability (Figure 4D). Importantly, the resulting alleles still led to increased attachment and generated an SCV phenotype (Figure 4D, E). This suggested that the activating mutations are dominant over the loss of peptidoglycan binding, and that they are able to fix YfiB in its active conformation independent of PG binding.
YfiB sequesters YfiR at the outer membrane (Figure 1). To examine the relationship between this activity and YfiB function, membrane fractionation was carried out for strains expressing PG- and hyperactive (F48S) YfiB variants alongside an yfiR-flag allele. While wild-type YfiB and the constitutive mutant F48S were able to sequester YfiR to the outer membrane fraction, no localization was seen for the PG binding mutant of YfiB (Figure 4C). Together, these data suggest that YfiB is able to release YfiN repression by sequestering YfiR at the outer membrane and that this activity requires both YfiB peptidoglycan binding and anchoring in the outer membrane. Since we were unable to co-immunoprecipitate YfiB and YfiR, it is unclear if YfiB sequesters YfiR through direct protein-protein contact or if additional component(s) mediate this interaction.
The finding that both lipid anchor and peptidoglycan binding are required for full activity of YfiB, together with the mapping of gain-of-function mutations close to the YfiB N-terminus, prompted us to examine the role of the N-terminal linker region in YfiB mediated signaling. The need for YfiB anchoring in cell wall and outer membrane suggests that YfiR sequestration might respond to the distance spanned by the protein. To test this, we modulated the length of the 13-amino acid long linker connecting the lipid acceptor cysteine with the PAL domain (Figure 5A). While extending the linker by 9 amino acids produced only a modest attachment effect upon induction (Figure 5C), shortening the linker by 5 amino acids produced a strong constitutive phenotype with respect to YfiR sequestration to the outer membrane (Figure 5B), surface attachment (Figure 5C), and SCV morphology (Figure 5D). This suggests that the linker region plays an important role in YfiB-mediated signaling and that YfiR sequestration might critically depend on the ‘wingspread’ of the YfiB connector between the two outermost layers of the cell.
Figure 5. Effect of YfiB linker mutants.

A) Sequence of the N-terminal YfiB ‘linker’, between the outer membrane (lipid anchor on the highlighted cysteine) and the Pal-like domain. The first activating mutant position (Figure 4) is shown in red. ‘short-linker’ indicates a deletion of 5 amino acids, leaving the lipobox intact. ‘long-linker’ indicates an insertion/duplication of 9 amino acids (dashed line). B) Co-localization of YfiB and YfiR at the outer membrane. Immunoblots of fractionated soluble and membrane samples with anti-YfiB and M2 antisera as shown. ‘YfiB WT’ indicates ΔyfiBNR Tn7::yfiNR containing pME6032-yfiB. ‘short’/‘long’ indicate the strains containing pME6032-yfiB linker mutants. C) The effect of different yfiB mutants, expressed from pME6032 in ΔyfiBNR Tn7::yfiNR, on attachment is shown relative to pME6032 only (ctrl.). The immunoblot shows the levels of YfiB protein present in each strain. D) Colony morphologies on LB Congo-red agar upon over-expression of yfiB mutants.
YfiBNR as a potential periplasmic stress sensor system
The above experiments, together with the observation that the YfiB homolog Pal is involved in cell envelope homeostasis and function [64] argued that YfiB might induce an up-regulation of c-di-GMP levels in response to cell envelope stress. Consistent with this, the YfiBNR system plays a role in the cellular response to elevated salt concentrations (high osmolarity) and exposure to outer-membrane disturbing detergents like SDS (Figure S3). However, while such a mechanism is consistent with our experiments suggesting that YfiB envelope anchoring is important for its regulatory role, attempts to link YfiB to distinct forms of envelope stress have so far remained unsuccessful.
In silico analysis of available bacterial genome sequences revealed that the Yfi signaling family is widespread in gram-negative bacteria and that several of its members lacked the respective YfiB component (see below). This argued for an additional level of signal input within this regulatory network. In a previous screen for mutants leading to an SCV phenotype [11], we identified transposon insertions in PA5489 that codes for the periplasmic thiol:disulfide interchange protein DsbA. This protein catalyzes cysteine crosslinking, and is responsible for the correct folding of periplasmic proteins [65]. Given that YfiR contains four highly conserved cysteines, we hypothesised that disruption of DsbA may result in YfiR misfolding and consequent release of YfiN repression (YfiN contains no periplasmic cysteines). To test if the dsbA SCV phenotype was dependent on the Yfi system, dsbA was deleted in PA01 wild type and in different yfi mutant strains. Deletion of dsbA produced a strong SCV phenotype in PA01 and in the yfiB mutant, but not in the ΔyfiBNR strain (Figure 6A). Other phenotypes associated with dsbA deletion such as a reduction in secreted protease levels were unaffected in the ΔyfiBNR background (data not shown). Also, disruption of the wspR gene encoding another major P. aeruginosa diguanylate cyclase [15], did not abrogate the SCV phenotype of the ΔdsbA background (Figure 6A). Consistent with a lower YfiR stability resulting from protein misfolding, YfiR levels, but not YfiN levels, were markedly reduced in the ΔdsbA background as compared to wild-type PA01 (Figure 6B). Finally, the ΔdsbA SCV phenotype could be abolished by over-expression of yfiR-flag in trans (Figure 6A,B). Altogether, this suggested that in the absence of DsbA, misfolding of YfiR leads to specific activation of the YfiN diguanylate cyclase.
Figure 6. Reducing effects on yfiBNR activity.

A) Colony morphologies of yfiBNR/dsbA single and double mutants on LB Congo-red agar. Indication of the following abbreviations: ΔBNR – ΔyfiBNR, ΔB – ΔyfiBNR Tn7::yfiNR, ΔR – ΔyfiR, pR-flag – pMR-yfiR-flag, pdsbA – pME6032ara-dsbA, pME – pME6032ara. B) Immunoblots of PA01 and ΔdsbA with M2 and anti-YfiN antisera, showing levels of YfiR-flag and YfiN in whole cell lysates. C) The effect of increasing concentrations of DTT on attachment (bars), of the ΔBNR and ΔB mutants is shown relative to wild-type PA01. Curves represent relative optical density with standard errors.
The observation that YfiR activity requires DsbA suggested that reducing conditions in the periplasm might block YfiR and lead to YfiN activation. To test this, growth and surface attachment were scored for various PA01 mutants with increasing concentrations of the reducing agent DTT. As shown in Figure 6C, deleting yfiBNR markedly increased the sensitivity of PA01 to reducing conditions. While the yfi + strain showed attachment up to 20 mM DTT, the yfi mutant showed no attachment above 2.5 mM DTT. Importantly, deletion of yfiB alone had no appreciable effect on PA01 growth or attachment in DTT, strongly suggesting that the activation of YfiN under reducing conditions is a consequence of YfiR misfolding rather than signaling through YfiB (Figure 6C). Together, these data suggest that YfiR constitutes an YfiB-independent sensing device that is able to activate the YfiN diguanylate cyclase in response to the redox status of the periplasm.
The YfiBNR system is under positive and negative selection in CF lungs
“In vitro” mutagenesis of the yfiBNR operon has revealed several routes to Yfi-mediated SCV formation. Disruption of yfiR induces a strong SCV phenotype [11], and activating mutations can arise in yfiN or yfiB (Figure 2, 4). To determine whether mutations in the yfi genes were responsible for the SCV phenotypes of clinical P. aeruginosa isolates from CF patients, we sequenced the yfiBNR operon of clinically derived SCVs and analyzed the contribution of candidate SNPs. Firstly, 45 clinical SCV strains were pooled and their genomes sequenced (S. Häussler and A. Dötsch, unpublished data). The yfiBNR operon sequence of each pool (representing a consensus sequence drawn from 15 individual SCV genome sequences) was compared with that of PA01, the positions of SNPs were identified (Table S3), and non-synonymous SNPs reproduced and introduced into the SCV screening strains described above (for details see Materials and Methods). From a total of 11 mutations identified, seven showed no noticeable effect. However, several mutations were identified in yfiN (G173D, D223N, L227M) or yfiR (C26R) that induced an SCV morphology and greatly enhanced attachment in the screening strain (Figure 2C, 7A, D). The amino acid residues altered in YfiN either matched (G173) or were located in the immediate vicinity of positions (D223, L227) identified by in vitro mutagenesis (Figure 2). These data demonstrate that SCVs arise in the airways of CF patients through the selection of clones carrying mutations in yfi genes that lead to de-repression the Yfi pathway.
Figure 7. Clinical YfiBNR mutants.

A) Colony morphologies of ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag strains with the point mutants indicated present in either YfiR or YfiN. ‘WT’: both plasmids contain wild-type yfiN/yfiR alleles. All colony morphologies are on LB Congo-red agar. B) Colony morphologies of Clin110 and Clin163 strains. pR indicates that the strain contains pME-araC-yfiR, the asterisk indicates induction. C) Colony morphologies of ΔyfiBNR strains complemented with the yfiBNR operon inserted into the att-Tn7 site. Mutations in the complementing copy of yfiN are indicated in each case. ‘WT’: wild type yfiN. D) Attachment relative to ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag (pNpR) of clinical yfiN/yfiR mutants. ‘pR’ and ‘pN’ contain pMR-yfiR-flag or pGm-yfiprom-N only. E) Attachment of Clin110 ± pME-araC-yfiR and Clin163 relative to PA01.
Next, we analyzed two clinical SCV strains in more detail. First, the SCV morphology and strong surface attachment of strain Clin110 were suppressed by overexpression of yfiR in trans (Figure 7B, E). Sequence analysis revealed that Clin110 YfiN contained the activating mutation E87K (Figure 2; Tables 1, S3). Interestingly, a second isolate (Clin163) was recovered 18 months later from the sputum of the same patient and identified as a descendent of Clin110 by comparison of synonymous SNPs throughout the yfiBNR operon. Clin163 displayed a smooth colony morphology and low surface attachment (Figure 7B, E), in spite of the fact that it contained the yfiN E87K mutation. Clin163 harbored an additional mutation (G329C) in the GGDEF active site motif of the diguanylate cyclase domain. As such active site mutations are known to destroy enzyme activity [54], this strongly suggested that the recovery of smooth colony morphology in Clin163 was due to YfiN inactivation (Figure 7B). In support of this, the c-di-GMP level in Clin110 (1517±280 pmol/mg protein) was measured, and shown to be ∼30 times higher than in PA01 (51.5±20.7). The level in Clin163 in contrast, was much lower (114±41.6). Finally, expression of a phosphodiesterase in trans markedly reduced Clin110 attachment (Figure S4). Second, we analyzed strain SCV20265, a clinical CF isolate whose SCV morphology is abolished by transposon insertions in yfiN or yfiB [14]. Sequence analysis of the yfiBNR operon from SCV20265 revealed two yfiN mutations; the activating mutation G173D (Figure 2; Table 1) and an in-frame deletion of codons 255–257 within the GGDEF diguanylate cyclase domain (Table S3). To determine their individual contributions, yfiBNR operons containing one or both yfiN mutations present were introduced into the att::Tn7 site of a ΔyfiBNR strain. G173D alone induced a robust SCV morphology in PA01, while the Δ255–257 deletion abolished the SCV phenotype in combination with G173D (Figure 7C). Furthermore, YfiN alleles containing only the Δ255–257 deletion were completely inactive on the basis of attachment assays with yfiN expression in trans (data not shown). While an SCV-inducing mutation was found in SCV20265 YfiN, the final protein is inactive. Clearly, additional SCV-inducing mutations must have arisen to complement the loss of YfiN activity, the nature of which is the subject of active investigation.
Together these data argue that the YfiBNR system is under both positive and negative selection in P. aeruginosa colonizing the lung of CF patients. Since SCV isolates have high reproductive costs [11], it is possible that alternating selection for rapid growth and persistence acts on the c-di-GMP network, thereby accumulating gain- and loss-of-function alleles in key components like YfiN.
Yfi defines a widespread and highly modular bacterial signaling system
Homologs of the YfiB, YfiN, and YfiR proteins were determined and plotted on a 16S rRNA-based phylogenetic tree to represent the taxonomic spread of the system (Figure 8A), for more details see Materials and Methods. 144 genomes were found to contain complete or partial yfiBNR operons. Genera containing complete, conserved yfiBNR operons (total 99) were found in the alpha-, beta- and gamma-proteobacteria, with most examples clustering in the gamma and beta classes. Two types of degenerate yfi operons were also identified. Firstly, operons containing yfiN and yfiB homologs in synteny, but no yfiR, were found in ten predominantly gamma-proteobacterial genomes (Table S4). In most cases, the transmembrane helices and GGDEF output domain of YfiN, as well as the PAL domain of YfiB were conserved. However, no homology was observed for the periplasmic domain of YfiN, and none of the proposed YfiR binding residues were conserved (Figure 8C, D), indicating that these systems function in a markedly different manner to P. aeruginosa YfiBNR. Secondly, 35 genomes were identified containing all three yfi homologs, but with only two of them in synteny (Table S4). Closer examination suggested that the genes in synteny were almost always yfiN and yfiR. The γ-proteobacteria Salmonella and Dickeya contain fully conserved yfiR and yfiN genes but lack yfiB. Interestingly, with growing evolutionary distance from Pseudomonas sp., the general divergence of the Yfi systems increased. Almost all of these operons lacked an yfiB homolog, and in many cases the GGDEF output domain of YfiN was replaced with GGDEF-EAL pairs, histidine-phosphotransfer domains (HPT) or sensor histidine kinases (Table S4). Variation was also seen at the level of signal input. In Thauera sp. MZ1T a system was identified with two YfiR homologs and a single YfiN with a hybrid sensor kinase output. The genomic location of the yfiN-yfiR homologs was highly variable, with some systems part of operons and others found alone. Nonetheless, alignment of PA01 YfiR and YfiN with some of the most divergent homologs revealed that the hydrophobic residues identified above as important for function were conserved throughout (Figure S5), suggesting that the basic concept of YfiR regulation of YfiN remains unchanged even in these highly divergent systems.
Figure 8. In silico analysis of the YfiBNR system.

A) Lineage tree illustrating the distribution of the yfiBNR genes along bacterial lineages. Individual branches indicate separate genera. Genera marked in large, bold type contain species with complete yfiBNR operons. Those marked with an asterisk (*) contain species with yfiB and yfiN only, conserved and in synteny. The remaining genera contain species with two yfi genes conserved and in synteny (usually yfiN and yfiR) and a third yfi homolog elsewhere in the genome. Bacterial classes are indicated with coloured shading. Alpha, Beta etc. refer to the respective proteobacterial class. B-D) Weblogo representations of YfiBNR residue conservation. The height of the letter in each case indicates the degree of conservation at that position. Hydrophobic residues are coloured blue, hydrophilic residues red. Asterisks (*) indicate the sites of activating mutations, while blue underlining indicates those residues suggested to contribute to a hydrophobic protein binding site. B) YfiB residues 33–72. C) YfiN PAS-domain residues 48–87, YfiN HAMP-domain residues 198–237. D) YfiR residues 122–191.
Altogether, these analyses suggest that the YfiR-YfiN[PAS] core represents a widespread, highly modular and versatile signaling system in bacteria that during evolution has integrated additional signal inputs through the acquisition of YfiB and distinct activities through the combination of different output domains.
Discussion
During long-term colonization of CF lungs, Pseudomonas aeruginosa clones undergo specific genotypic adaptation to the host environment. As a result, the genotypes of strains present in advanced CF infections differ substantially from strains that originally invade the respiratory tract of these patients [2]. During adaptation to chronic behavior P. aeruginosa experiences a widespread loss of virulence factors and concomitant ‘induction’ of persistence traits. However, little is known about the exact nature of these traits or about the adaptive processes that lead to their expression. In this study we investigate specific genetic adaptations of the P. aeruginosa c-di-GMP signaling network and their role in the establishment of chronic infections of CF lungs. We demonstrate that during long-term lung infections, mutations affecting the activity of key regulators of c-di-GMP production invade the bacterial population in the lung, driving the formation of persistent SCV morphotypes in vivo. Our studies concentrate on the YfiBNR system, one of the previously identified clinically relevant systems for c-di-GMP production in P. aeruginosa [11]. The finding that the YfiBNR proteins are subject to both positive and negative selection allows us to make specific predictions regarding the adaptive processes occurring during long-term chronic infections of CF lungs, i.e. that the fitness landscape of the lung is heterogeneous and dynamic, providing selective pressure for both smooth and SCV phenotypes in different regions of the lung, or periods during infection. The recognition of genetic changes occurring in the patient was facilitated by a comprehensive analysis of Yfi-mediated regulation in P. aeruginosa, emphasizing the importance of a thorough molecular and mechanistic understanding of physiological and regulatory processes for the analysis of adaptive behavior in vivo.
While the epistasis and physiological significance of the YfiBNR system has been described previously [11], [35], its mechanism of action has remained largely unknown. We provide biochemical and genetic evidence that the activity of membrane-integral diguanylate cyclase YfiN is suppressed via interaction between its extracellular PAS domain and the periplasmic repressor protein YfiR. Release of repression, either by YfiR disruption or by YfiB-mediated sequestration to the outer layer of the cell envelope leads to a conformational shift in YfiN that propagates through the PAS and transmembrane domains to switch the cytoplasmic HAMP domain from an inactive to an active conformation. This activating conformational change is subsequently transmitted to the C-terminus, where DGC activity is likely determined by the formation of a properly aligned GGDEF homodimer [53], [66], [67]. Thus, YfiN appears to function by switching between discrete inactive and active functional states depending on the presence or absence of bound YfiR. In this respect, YfiN is similar to a number of other well-characterized transmembrane signaling proteins, including histidine kinases and methyl-accepting chemotaxis proteins [56] in which ligand binding provokes activating conformational changes that propagate throughout the protein structure.
Our analyses place YfiR at the center of the Yfi signaling pathway with two potential interaction partners, YfiN and YfiB, localized in the inner and outer membrane, respectively. Specific partner selection of YfiR would then determine YfiN signaling activity. YfiR appears to be unique, without functional homologs in other signaling systems. Nonetheless, the combination of mutagenesis, structural modeling and analysis of sequence conservation applied in this study allowed us to propose that YfiR may bind to YfiN via a hydrophobic binding site composed of residues in the C-terminal region of the protein. Sequence conservation indicates that hydrophobic amino acids (Val160, Phe162, Leu183) on the surface of the modeled YfiR structure may be especially important in this regard. Consistent with such a mode of action, compensatory mutations in YfiR that are able to suppress activated forms of YfiN are positioned in the immediate vicinity of this hydrophobic patch. Likewise, our genetic studies indicate that YfiR binds to a highly conserved stretch of five amino acids (AAVVF) on the surface of the YfiN PAS domain. Even conservative substitutions in this region abolish the YfiR binding surface and lead to YfiN activation. It is possible that the valine and alanine residues coordinate the central phenylalanine to protrude into the periplasm thereby providing a hydrophobic binding site for YfiR. To our knowledge, this is the first recorded example of a prokaryotic PAS domain that signals via protein-protein interaction. Structural studies are underway to clarify the molecular details of this interaction.
YfiB spans the peptidoglycan and the outer membrane and appears to function by sequestering YfiR away from the inner membrane and YfiN. While direct YfiR-YfiB interaction could not be demonstrated, YfiR localization to the outer membrane fraction requires the presence and activity of YfiB. The location of activating mutants in YfiB provided insight into the mechanism of YfiB action, leading us to propose that activation may result from the exposure of a patch of hydrophobic residues on the YfiB surface, particularly the highly conserved tryptophan at position 55 (Figure 8). According to our model of YfiB structure and topology, these hydrophobic residues form an YfiR binding site immediately proximal to the insertion site of the protein into the outer membrane. Since both peptidoglycan binding and outer membrane anchoring are essential for YfiB function, the protein might sense envelope integrity or the distance between the two outermost layers of the cell, a mechanism that has also been suggested for the outer membrane protein Pal [51]. Changes in the relationship between the outer membrane and peptidoglycan would expose or obscure the hydrophobic YfiR binding site, controlling the amount of YfiR bound to YfiB and hence the level of YfiN activation. Such a model is supported by the observation that shortening of the linker between the Pal-like domain of YfiB and its anchoring site in the outer membrane leads to YfiR recruitment and activation of YfiN, while a longer linker retains overall YfiB activity but does not lead to enhanced activation. A corollary of this model is that the YfiR-YfiN interaction would need to exist in a state of dynamic equilibrium, with some unbound YfiR and YfiN present at all times. Rather than directly challenging the YfiR-YfiN interaction, YfiB would act as a ‘sink’ for unbound YfiR, removing it from the periplasm and thus shifting the equilibrium towards unbound, active YfiN.
The model for YfiBNR function proposed above implies a biological role for the Yfi system in sensing physical stresses affecting the outer membrane and/or the peptidoglycan, and triggering c-di-GMP-dependent outputs as a response to that stress. The nature of these output systems is the subject of ongoing research, but is currently thought to focus on the control of exopolysaccharide production at the transcriptional and post transcriptional levels [11]. Our experiments failed to directly demonstrate a role for YfiB in the response to envelope stress. Although the Yfi system as a whole is required for full biofilm induction at high osmolarity or in the presence of SDS, two stressors that are known to affect the integrity of the cell envelope, we could not demonstrate a specific role for YfiB in this process. In addition to YfiB-mediated activation, YfiN can be activated by YfiR disruption. This is most visible in a genetic yfiR mutant, but may also occur when YfiR does not fold correctly due to incorrect/incomplete cysteine crosslink formation. The four cysteine residues in YfiR are strictly conserved amongst complete YfiBNR systems, supporting their importance for YfiR function. In response to external reducing agents P. aeruginosa stages a biofilm response that fully depends on the presence of YfiN and YfiR, but not YfiB, and which greatly enhances PA01 tolerance under these conditions in comparison to an yfiBNR mutant strain. YfiR misfolding thus potentially represents a second mode of YfiBNR sensory input, and suggests a role for the system in the response to reducing environments. For example, it is conceivable that the Yfi system is able to sense anoxic conditions through the redox sensitive cysteines of YfiR and by that induce biofilm in the absence of oxygen. The option to employ YfiR directly as a signal input domain, either as a cysteine-dependent redox sensor or through another as-yet-undiscovered mechanism, predicts the existence of Yfi-like signaling systems in which YfiB is missing. This is indeed the case. While complete yfiBNR systems are widespread in the gamma- and beta-proteobacteria (with the alpha-proteobacteria Sphingopyxis representing the only genus external to these two classes) many YfiNR pairs lacking an YfiB component are found in species both closely related and widely divergent from P. aeruginosa. In particular, species in the Salmonella and Dickeya genera have conserved YfiN and YfiR homologs, but lack YfiB. Given the positions of Salmonella and Dickeya in the YfiBNR lineage tree it seems likely that these genera may once have had functional YfiB homologs, but have since lost them. Evolution has not only modulated the input side of the YfiBNR system, the output domains of more divergent Yfi homologs are also variable. Careful in silico analysis identified histidine kinases, GGDEF/EAL pairs and MCP domains in place of the YfiN GGDEF domain. Despite this diversity of signaling outputs, the residues predicted for YfiN-YfiR interaction are well conserved. This led us to hypothesize that this YfiR-YfiN [PAS/HAMP] core may be a more ancient signaling module, which in evolution has been connected in a modular fashion to different output and additional input domains. This core module, characterized by the binding of a periplasmic repressor to the PAS-like domain of a transmembrane signal transduction system, represents a novel and apparently widespread signaling motif in bacteria. The YfiN-YfiR interaction shares some striking characteristics with the LapD-LapG system of Pseudomonas fluorescens [68], [69], [70] and Pseudomonas putida [71]. The unique periplasmic domain of the c-di-GMP signaling protein LapD sequesters the periplasmic protease LapG when c-di-GMP is present, preventing it from cleaving the adhesin LapA. Periplasmic protein interaction systems such as LapAG and YfiBNR may well be representatives of a broader regulatory principle in bacterial signaling.
The mutational analysis described in this study demonstrated several possible genetic routes to yfi-mediated SCV formation. In addition to loss-of-function mutations in yfiR, mutations that mimic an active conformation may arise at specific locations in the PAS, HAMP or transmembrane regions of YfiN, or towards the N-terminus of YfiB. When analyzing the yfi operons of clinical SCVs we identified several that contained either an inactivating mutation in yfiR or an activating mutation in yfiN, arguing that the Yfi signal transduction system is under strong positive selection in the lung environment. Apparently, one route to generate SCVs in vivo is by an increase of c-di-GMP through mutations that activate or de-repress a diguanylate cyclase. Consistent with this, the methylesterase wspF, whose disruption induces c-di-GMP dependent SCV formation in vitro [72], was shown to be mutated in a P. aeruginosa strain following adaptation to the CF lung [2]. Similarly, Rau et al. have identified a persistent CF isolate in which the GGDEF/EAL protein MorA is strongly upregulated [73]. Furthermore, we find that in a large fraction of P. aeruginosa SCVs isolated from CF lungs the characteristic morphotype can be alleviated or reversed to a more rapidly growing smooth phenotype by the expression of a phosphodiesterase (Jaeger, Bos and Jenal, unpublished). Of the five SCV-inducing mutations isolated in the yfi system, four were gain of function mutations in YfiN, with only one loss-of-function mutation found in YfiR. This is surprising, as one expects loss-of-function mutations in yfiR to occur more frequently than activating mutations in yfiN. These findings seem to suggest that the strongest Yfi-mediated SCVs, produced when YfiR is absent and YfiN is fully active, are disfavored in the CF lung compared with weaker morphotypes produced by activated YfiN alleles. It is possible that YfiR has a supplementary role in cell signaling and that loss-of-function mutations in the yfiR gene incur an unforeseen fitness cost in the human host. Alternatively, activating mutations in yfiN might be favored because they allow the system to retain some degree of flexibility, where a secondary genetic or regulatory increase in YfiR activity could override the activating effect of the original mutation in yfiN. Careful analysis of selected clinical SCVs indeed provided evidence for consecutive rounds of mutations affecting the yfi system. The finding that some clinical strains contained both gain- and loss-of function mutations in yfiN strongly suggested that SCVs can function as an environmental pool for the generation of new smooth morphotypes. Apparently, SCVs do not represent a dead end in the dynamic and heterogeneous environment of chronically infected CF airways and may be continuously favored and disfavored over time and/or different lung environments. In this way the behavior of clinical P. aeruginosa SCVs reflects that of P. fluorescens cultured in vitro [36], where sequential gain and loss of function mutations arose in c-di-GMP related systems (including Yfi/Aws), in response to environmental changes. The variable fitness of SCVs might well be explained by the characteristic slow growth of these morphotypes [11], which may contribute substantially to persistence under very stringent in vivo conditions but pose dramatic costs under conditions that favor rapid growth. E.g. antibiotic treatment regimens or other insults that alter the fragile homeostasis between airway pathogens and local host defenses [74] might favor selection for persistence traits, while in the absence of such conditions rapidly growing sub-types might have a considerable advantage. Continuous adaptation of the c-di-GMP network could facilitate these physiological adjustments and thereby contribute to the in vivo fitness of P. aeruginosa during chronic lung infections.
CF patients are permanently colonized by P. aeruginosa with the same bacterial lineage persisting continuously in the lungs for years or even decades and without being eradicated by chemotherapy. The development of effective treatments rests on a detailed understanding of how bacteria adapt to the airway and resist host defenses and antibiotics. Several reports have indicated a striking correlation between mutagenesis and persistence of P. aeruginosa in CF lungs [2], [75], [76], [77]. Smith et al. [2] have reported that during chronic infection P. aeruginosa adapts by loss-of-function mutations that remove virulence factors required for the acute phase of infection. But while this study outlines factors that are disadvantageous in chronic infection, it does not provide information about what is needed to enhance fitness in CF airways and sustain the clonal expansion of P. aeruginosa during chronic infection. Alleles causing (often subtle) positive changes in protein function are more difficult to identify and rationalize than loss-of-function mutations and often require experimental input to be validated. E.g., the identification and elucidation of the yfi gain-of-function alleles in clinical isolates was critically dependent on the mechanistic understanding that could only be provided by a thorough molecular analysis of the pathway. Based on these studies we propose that rapid evolution of diguanylate cyclases and possibly other components of the c-di-GMP network contributes to the remarkable flexibility and degree of persistence of P. aeruginosa cells in CF lungs. A thorough understanding of the underlying physiological processes is mandatory to predict, delineate and interfere with these in vivo selection processes.
Materials and Methods
Ethics statement
The clinical Pseudomonas aeruginosa strains used for this research were cultured from patient samples collected for routine microbiological testing at the University Children's Hospital, Basel or the Medical Microbiological Department of the Hannover Medical School. Subculturing and analysis of bacteria was performed anonymously. No additional procedures were carried out on the patients. Cultures were sampled following regular procedures with written informed consent, in agreement with the guidelines of the “Ethikkommission beider Basel EKBB” or with approval by the Ethical Board of the Hannover Medical School.
Strains and growth conditions
Strains and plasmids used in this study are listed in Table S1. Primers are listed in Table S2. Unless otherwise stated, all P. aeruginosa and E. coli strains were grown at 37°C in Luria Bertani (LB) medium [78], solidified with 1.3% agar where appropriate. For P. aeruginosa, gentamycin was used at 30 µg/ml (E. coli 20 µg/ml), carbenicillin at 100 µg/ml and tetracycline at 50/100 µg/ml (E. coli 12.5 µg/ml). For E. coli, ampicillin was used at 100 µg/ml. Congo Red dye was added to a final concentration 0.04%. For inducible plasmids, IPTG was added to a final concentration 0.2 or 1 mM and arabinose to 0.2% as appropriate.
Molecular biology procedures
Cloning was carried out in accordance with standard molecular biology techniques. To produce a chromosomal flag-tagged copy of yfiN E. coli DY330 [79] was transformed with plasmid pMR20-yfiBNR [11]. The PCR fragment amplified with primers A and B from plasmid pSUB11 [80] were then used to produce a C-terminal yfiN fusion in pMR20 by the method described by Yu et al. [79]. The resulting yfiN-flag fragment was then ligated between the HindIII and BamHI sites of pUC18T-mini-Tn7T-Gm [81].
The yfiR screening plasmid pMR-yfiR-flag was constructed by replacement of the lac-promoter/lacI fragment of pMR20-yfiR-M2 [11] with a chloramphenicol cassette amplified using primers C and D, according to the method of Yu et al. [79]. The chloramphenicol gene was then excised by Flp-mediated excision using the plasmid pFLP2 [82]. To produce the yfiN screening plasmid pGm-yfiprom-N, the tetracycline resistance cassette was excised from pME6032 [83] between BamHI and XbaI, and replaced with the aacCI gentamycin resistance cassette, amplified with primers E and F from pBBR-MCS5 [84]. The lac-promoter/lacI fragment was then excised between EcoRI and BamHI and the EcoRI site was blunted with klenow polymerase. The yfiN fragment was amplified with primers G and H from PA01 ΔyfiR [11] and ligated between BamHI and the blunt site.
The three pME-yfiB-PG- variants were constructed by ligation of yfiB fragments (amplified with primers I and U, from the appropriate pME6032-yfiB gain-of-function plasmids) between the EcoRI and AscI sites of pME6032-yfiB [11]. To make the yfiB-LA- constructs, yfiR was amplified with primers V and P, and yfiB with primers W and J from genomic DNA (or pME-yfiB-PG- as appropriate). The MfeI-PstI yfiR fragment and the PstI-BamHI yfiB fragments were then ligated between the EcoRI and BglII sites of pME6032. The long/short yfiB inserts were produced by SOE-PCR using primers AH/AI together with primers X/Y or Z/AA, and ligated into pME6032 between EcoRI and BglII. To produce pME-araC-dsbA, the dsbA fragment amplified with primers K and L from PA01 genomic DNA was ligated between the EcoRI-KpnI sites of pME-araC [11], [83]. To make pME-araC-yfiR the PCR product amplified with primers V and P was cut with MfeI-BamHI and ligated between the EcoRI-BglII sites of pME-araC.
To produce pTn7-yfiBN20265-R, the yfiBNR operon of SCV20265 was amplified using primers G and J and ligated between the HindIII and BamHI sites of pUC18T-mini-Tn7T-Gm. The G173D and Δ255-7 variants were produced by digesting yfiBNR PCR products from SCV20265 and WT PA01 templates at a PstI site midway between the two mutations, then ligating both inserts between BamHI and HindIII as above. Clinical yfiN/yfiR mutant alleles were produced by SOE PCR using primers AR/AS and AJ-AQ for yfiN, and AZ/BA and AT-AY for yfiR, respectively. Inserts were then ligated into NcoI-BamHI-cut pME6032-Gm or HindIII-BamHI-cut pMR-yfiR-flag respectively.
XL1-red mutagenesis
Mutator strain E. coli XL1-red (Stratagene) was transformed with the plasmid pGm-yfiprom-N according to the method of Chung et al. [85] and plated onto LB gentamycin plates. After overnight growth at 37°C the transformant colonies were pooled and plasmid DNA extracted. This pooled DNA was then used to transform the screening strain ΔyfiNR pMR-yfiR-flag. Samples were plated onto LB plates containing gentamycin, tetracycline and Congo Red and transformants exhibiting an SCV phenotype were selected as potentially carrying activated YfiN alleles. These SCVs were restreaked onto fresh plates and the yfiN gene amplified in each case via colony PCR, using primers G and H. Mutations in yfiN were then identified by sequencing.
Screening by error-prone PCR
Plasmids pME6032-yfiB [11] and pMR-yfiR-flag were used as templates for error-prone PCR reactions following the method described in [86], and consisting of 2 min 94°C, then 25 cycles of 45 sec 94°C, 45 sec 55°C and 90 sec 72°C, before a final extension for 10 min at 72°C. Each 50 µl reaction contained 0.2 mM dNTPs, Taq polymerase buffer, 5U Taq polymerase, 0.3 µM forward and reverse primers (I/J and G/P), 1.0 µl template DNA and 0.5–1.0 µl mutagenesis buffer (4 mM dTTP, 4 mM dCTP, 27.5 mM MgCl2, and 2.5 mM MnCl2). Pools of mutated yfiR and yfiB fragments were then ligated into their parent vectors between HindIII - BamHI, and EcoRI - BglII respectively. To screen for yfiB mutants, ΔyfiBNR Tn7::yfiNR [11] was transformed with pooled pME6032-yfiB and SCV transformants were isolated from selective LB plates. Mutations in yfiB were identified by sequencing. For yfiR mutants, ΔyfiNR was transformed with pooled pMR-yfiR-flag and the resulting transformant colonies used to make electrocompetent cells. Plasmids carrying the 20 activated yfiN alleles were extracted and purified, then used to transform the ΔyfiNR pMR-yfiR-flag pooled cells. Transformants were screened on selective LB plates for smooth colonies containing potential dominant YfiR alleles. To eliminate suppressor mutations in the screening strain, pMR-yfiR-flag DNA was extracted from smooth strains and used to transform ΔyfiNR containing the relevant activated-yfiN plasmid. The yfiR alleles present in those strains that displayed a smooth morphotype at this stage were analyzed by sequencing.
Transduction with phage E79tv2
The E79tv2 [87] transducing lysate for the Tn5::wspR strain was prepared and used according to the protocol described in our previous work [11].
Deletion of the dsbA gene
The strains PA01 ΔdsbA and ΔyfiBNR ΔdsbA were constructed via an adaptation of the protocol described elsewhere [88]. Briefly, the dsbA deletion construct was produced by SOE-PCR using primers Q-T, and contained homologous flanking regions to the target gene. This construct was ligated into pME3087 between HindIII-BamHI. The resulting vector was then used to delete dsbA by two-step allelic exchange. Following transformation into the target strain, single crossovers were selected on tetracycline and restreaked. Cultures from single crossovers were grown overnight in LB medium, and diluted 1∶100 into fresh medium. After 2 hours, 20 µg/ml tetracycline was added to inhibit the growth of cells that had lost the tetracycline cassette. After a further hour of growth, 2000 µg/ml carbenicillin was added to select against growing bacteria. Cultures were grown for a further 4–6 hours, before cells were harvested by centrifugation, washed once in LB and used to inoculate an overnight culture. This counter-selection was done twice, before plating of a dilution series of the final samples onto LB agar. Individual colonies were patched onto LB plates ± tetracycline, tetracycline sensitive colonies were tested for deletion of dsbA by colony PCR.
Attachment assays
Assays to test the different alleles of the yfiBNR system were performed as described in [11], [89]. Briefly, 96 well plates containing 150/200 µl LB medium/well were inoculated with single colonies using sterile toothpicks, and incubated overnight at 37°C without shaking. Plates were washed three times with distilled water. The attached cell material was then stained with 0.1% Crystal Violet solution (5% methanol, 5% isopropanol) before further washing to remove excess dye. Crystal Violet was re-dissolved in 20% acetic acid solution and absorbance measured at 600 nm. Assays were performed with 6 wells/strain and repeated independently for each experiment.
The attachment assays for phenotypic testing of stress conditions were performed with the following modifications. Strains were grown on LB plates O/N at 37°C then scraped from the plates and diluted in the appropriate medium used for the experiment. 96 well plates containing 200 µl medium/well were inoculated with the diluted bacterial suspension to a final OD600 of 0.001, plates were incubated overnight at 37°C without shaking. Envelope stress was tested on M9 minimal medium (47.6 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 18.6 mM NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.03 mM Fe-III-citrate) containing 10 mM Na-succinate as a carbon and energy source and 0.01–0.25% sodium dodecyl sulfate (SDS) [90] and with osmotic stress minimal medium M63 (100 mM KH2PO4, 15 mM (NH4)2SO4, 2 µM FeSO4, 1 mM MgSO4, 0.001 mg/ml Thiamin) [78] with 20 mM Na-succinate, 0.2% Glucose and 0.01–0.4 M NaCl [91]. Reducing conditions were tested on MHB (Mueller-Hinton broth, cation adjusted, Becton Dickinson) containing 1.25–20 mM Dithiothreitol (DTT). Assays were performed at least 3 times independently with 12 wells/condition tested, shown is the mean of all experiments.
Immunoblot analysis
Samples were separated on 15% Tris-HCl gels and blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were incubated overnight in blocking solution (1× PBS pH 7.4, 0.01% Tween20, 5% milk powder), after which proteins were detected with 1/5000 (α-M2, α-YfiB) or 1/2000 (α-YfiN) specific antiserum and 1/10,000 rabbit anti-mouse (α-M2) or swine anti-rabbit (α-YfiB, α-YfiN) secondary antibody (DakoCytomation). Bound antibodies were visualized using ECL chemiluminescent detection reagent (Perkin-Elmer). Samples were normalized following comparison of optical density for the initial cell samples in each case.
Co-immunoprecipitation
PA01 overnight cultures (or colonies scraped from agar plates, in the case of SCV strains) were pelleted by centrifugation and re-suspended in ice-cold IP buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 1 mM EDTA, 1.0% v/v Triton X-100, protease inhibitor), then incubated at 4°C with end-over-end agitation for 6 hours. All subsequent steps were carried out on ice. Samples were centrifuged (15,000 g, 20 min, 4°C), and the supernatant was removed and incubated with 20 µg/ml protein-A agarose beads at 4°C with end-over-end agitation for 30 min to remove non-specifically binding proteins. The samples were then centrifuged (3,000 g, 1 min 4°C) to pellet the beads, an aliquot of the supernatant was taken for analysis, and the remaining supernatant was incubated overnight with 20 µg/ml ANTI-flag M2 affinity gel (Sigma) at 4°C with end-over-end agitation. Samples were pelleted by centrifugation (3,000 g, 1 min 4°C), the supernatant was discarded and the beads re-suspended in 1.0 ml ice-cold IP buffer. This wash step was repeated between 3 and 5 times. The beads were then re-suspended in SDS sample buffer, boiled for 10 minutes at 95°C and pelleted by centrifugation. Interacting proteins in the supernatant were detected by immunoblotting.
Membrane fractionation
The fractionation protocol was adapted from the method described in [92]. 50 ml overnight cultures of PA01 strains were pelleted by centrifugation, washed once with Tris buffer (30 mM Tris pH 8.0), and re-suspended in 6.0 ml Tris buffer containing 20% w/v sucrose, 1 mg DNase, 1 mg RNase, and 2 mg lysozyme. Samples were incubated for 20 minutes at room temperature, then lysed by French Press, diluted to 10% sucrose with Tris buffer and centrifuged to remove cell debris (3,500 g, 15 min, 4°C). Protease inhibitors were added at this stage as a precaution. The soluble and membrane fractions were then separated by ultracentrifugation (160,000 g, 105 min, 4°C) on a Tris buffered 15%/70% sucrose gradient. The membrane fraction was recovered from the 15%–70% interface and diluted to about 30% with Tris buffer, before loading onto a Tris buffered, sucrose step-gradient (steps contained 70, 64, 58 and 52% sucrose). The samples were then ultracentrifuged for 18 hours (160,000 g, 4°C) to separate inner and outer membranes. The four bands visible after ultracentrifugation were recovered, diluted in 1–2 volumes Tris buffer and pelleted (15,000 g, 35 min, 4°C), before being re-suspended in Tris buffer and analyzed by immunoblotting. Western blot samples were normalized according to the total protein content of the different fractions, determined with a Protein assay kit (BioRad).
Quantitation of cyclic-di-GMP levels
C-di-GMP levels of clinical strains were measured by liquid chromatography-tandem mass spectrometry after [93]. The protein content of each pellet (post-extraction) was determined with a Protein Assay kit (BioRad). Measurements were in triplicate and values expressed as pmol c-di-GMP/mg protein.
Sequence analysis of clinical SCV strains
In total 45 auto-aggregative SCV strains isolated from the sputum of CF patients were grouped in three pools of 15 strains. Each strain was cultivated individually in 20 ml LB medium at 37°C and shaking at 180 rpm to an OD600 of 1.0. 1 ml of each culture was harvested by centrifugation (8000 rpm, 1 minute, 4°C) and the cell pellet was washed once with 1 ml sterile deionized water. Pellets were resuspended in sterile deionized water, pooled at equal amounts and DNA was isolated from the pooled cell suspensions using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions.
DNA samples were further prepared and sequenced using 76 bp paired end sequencing on a Genome Analyzer II-x (Illumina). Libraries of 250 bp prepared according the manufacturer's instructions “Preparing Samples for Paired-End-Sequencing”. Cluster generation was performed using the Illumina cluster station, sequencing for read 1 and read 2 on the Genome Analyzer followed a standard protocol. The fluorescent images were processed to sequences using the Genome Analyzer Pipeline Analysis software 1.6 (Illumina).
The sequence reads were quality-trimmed using the perl script Trim.pl by Nik Joshi (obtained from http://wiki.bioinformatics.ucdavis.edu/index.php/Trim.pl) in the “windowed adaptive trimming” mode, removing any sequence with a quality score <10. Detection of single nucleotide polymorphisms (SNPs) was performed with the MAQ software [Li et al, 2008, Genome Res 18∶1851] using a perl script that calculates the frequencies of SNPs in pooled sequences [Holt et al., 2009, Bioinformatics 25∶2074].
Identification of YfiBNR homologs in other systems
An initial set of genomes displaying synteny conservation among homologs to PA1119 (GenBank:15596316), PA1120 (GenBank:15596317) and PA1121 (GenBank:15596318) from Pseudomonas aeruginosa PAO1 (GenBank:AE004091) was manually defined. Synteny conservation was assessed for complete microbial genomes of the RefSeq database on the MAGE database server [94]. The three sets of protein sequences corresponding to each homologous group were aligned with CLUSTAL W (default settings) [95]. Hidden Markov Models were then computed with HMMER.3 (http://hmmer.org/). Models were calibrated and searched against a local copy of the microbial complete genome database (NCBI) with HMMER.3. A series of Perl scripts were used to sort the outputs and to count occurrences of complete or partial systems as described in Figure 8. Occurrences of homologs (E-value<10−4) were then reported on an illustrative phylogenetic tree based on 16S rRNA sequences from the ribosomal database project (RDP, http://rdp.cme.msu.edu/index.jsp) [96]. All sequences were >1200 nucleotides and tagged as “good quality” according to RDP. Type strains and isolated samples were preferred. At least two sequences/genus were downloaded as alignment files from RDP. Consensus was inferred using EMBOSS (http://www.sanger.ac.uk/ Software/EMBOSS). Genus consensuses were aligned with CLUSTAL W (default settings) and phylogenetic analyses were conducted in MEGA4 [97]. Evolutionary history was inferred using the UPGMA method [98], and evolutionary distances were computed using the Maximum Composite Likelihood method. All positions containing gaps and missing data were eliminated (Complete deletion option), leaving a total of 1143 positions in the final dataset.
Molecular models of the YfiBNR proteins were produced using HHPRED (http://toolkit.tuebingen.mpg.de/hhpred) and visualized with PYMOL (http://pymol.org/). Models were based on the following structures: YfiN PAS: 1p0z_A, YfiN HAMP: 3lnr_A, YfiR: 2iss_D, 2ywj_A, 3lft_A, 3dfu_A, 2abw_A, 2ioj_A, and 1yqg_A, YfiB: 2kls_A and 2ldt_A. Sequence conservation plots were visualized using WebLogo 3 [99].
Supporting Information
Localization and stability of YfiR. A) Outer Membrane localization of YfiR-flag. Immunoblot of fractionated membrane samples stained with M2 antisera. The panel shows the soluble and the outer membrane fractions for ΔyfiBNR, ΔyfiNR and ΔyfiBNR Tn7::yfiNR harboring pMR-yfiR-flag (p-yfiR). B) Stability of YfiR-flag in whole cell lysate. Immunoblot of ΔyfiR, ΔyfiNR and ΔyfiBNR with yfiR-flag inserted into the att-Tn7 site, and one time point with the strains over expressing YfiR (ΔyfiBNR, ΔyfiNR and ΔyfiBNR Tn7::yfiNR harboring pMR-yfiR-flag (p-yfiR)). ‘t’ indicates different time points (hours post inoculation); ‘O/N’ overnight incubation.
(PDF)
Colony morphologies with activated YfiN alleles. A) Colony morphologies of ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag strains with the point mutants indicated present in YfiN. ‘pyfiR pyfiN’ denotes the wild-type control; ‘pyfiN’ denotes a strain containing pGm-yfiprom-N only. B) Colony morphologies for cross-complementation strains. The YfiN (bold) and YfiR (italic) mutant alleles present are shown in each case.
(TIF)
Attachment of Yfi mutants in response to the presence of SDS or changes in osmolarity. A) Attachment on M9 medium with 20 mM Na-succinate and increasing concentrations of SDS is shown relative to PA01. B) Attachment with low and high osmolarity, on M63 medium with 20 mM Na-succinate, 0.2% glucose, and increasing concentrations of NaCl is shown relative to PA01. 50% and 75% indicate dilutions of the medium with distilled water. ΔBNR indicates ΔyfiBNR and ΔB indicates ΔyfiBNR Tn7::yfiNR. Bars represent the level of absolute attachment and curves represent optical density (OD) of total cells with standard errors.
(PDF)
Attachment of clinical iosolates in the presence of a phosphodiesterase. The effect of a PDE, in Clin110 and Clin163, on attachment is shown relative to strains expressing an active site mutant (PDE*). The PDE (PA5295) and the active site mutant are expressed from a vanillate inducible vector (pBV-PA5295 and pBV-PA5295E328A respectively).
(PDF)
Sequence conservation across distant YfiN/YfiR homologs. Sequence alignments for YfiR (residues 55–190) and the YfiN PAS domain (residues 61–115), across PA01 and five distant homologs. The five species whose yfiNR genes were compared were: Opitutus terrae PB90-1, Acidobacteria bacterium Ellin345, Geobacter sp. M21, Rhodothermus marinus DSM 4252, and Desulfobacterium autotrophicum HRM2. Fully conserved residues are marked with an asterisk (*), similarities are marked with (:) or (.). Putative hydrophobic binding site residues are enclosed in black boxes. Residues are colored according to the chemical nature of their side chains.
(PDF)
Bacterial strains and plasmids used in this study.
(PDF)
Primers used in this study.
(PDF)
Single nucleotide polymorphism (SNP) positions in the sequences of clinical SCVs, compared to P. aeruginosa PA01.
(PDF)
Identification of YfiBNR homologs in other systems.
(PDF)
Acknowledgments
Clinical P. aeruginosa isolates were kindly provided by Prof. Dr. Jürgen Hammer and Dr. Daniel Trachsel from the Universitäts-Kinderspital Basel (UKBB) and Dr. Reno Frei from the Universität Spital Basel. Cyclic-di-GMP measurements were provided by Prof. Volkhard Kaever at the Hannover Medical School. The authors would like to thank Dr. Alex Boehm for insightful conversations about this work.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by SNF research grants 31-108186 and 31003A_130469 to UJ (SNF, http://www.snf.ch/E/Pages/default.aspx), the Mukoviszidose e.V (S07/05, http://muko.info/), the Bundesministerium für Bildung und Forschung (01FP09104A, BMBF, http://www.bmbf.de/) and an ERC starter grant (http://erc.europa.eu/) and an ERC starter grant to SH (RESISTOME 1500092). AB was supported by the International Research Training Group 1273 funded by the German Research Foundation (DFG, http://www.dfg.de/index.jsp). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology. 2007;153:917–923. doi: 10.1099/mic.0.2006/004077-0. [DOI] [PubMed] [Google Scholar]
- 2.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huse HK, Kwon T, Zlosnik JE, Speert DP, Marcotte EM, et al. Parallel Evolution in Pseudomonas aeruginosa over 39,000 Generations In Vivo. MBio. 2010;1:e00199–10. doi: 10.1128/mBio.00199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Govan JRDV. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirisits MJ, Prost L, Starkey M, Parsek MR. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2005;71:4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haussler S, Ziegler I, Lottel A, von Gotz F, Rohde M, et al. Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J Med Microbiol. 2003;52:295–301. doi: 10.1099/jmm.0.05069-0. [DOI] [PubMed] [Google Scholar]
- 7.Haussler S, Tummler B, Weissbrodt H, Rohde M, Steinmetz I. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis. 1999;29:621–625. doi: 10.1086/598644. [DOI] [PubMed] [Google Scholar]
- 8.Reinhardt A, Kohler T, Wood P, Rohner P, Dumas JL, et al. Development and persistence of antimicrobial resistance in Pseudomonas aeruginosa: a longitudinal observation in mechanically ventilated patients. Antimicrob Agents Chemother. 2007;51:1341–1350. doi: 10.1128/AAC.01278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haussler S, Lehmann C, Breselge C, Rohde M, Classen M, et al. Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complex. Eur J Clin Microbiol Infect Dis. 2003;22:249–253. doi: 10.1007/s10096-003-0901-y. [DOI] [PubMed] [Google Scholar]
- 10.Murphy TF, Brauer AL, Sethi S, Kilian M, Cai X, et al. Haemophilus haemolyticus: a human respiratory tract commensal to be distinguished from Haemophilus influenzae. J Infect Dis. 2007;195:81–89. doi: 10.1086/509824. [DOI] [PubMed] [Google Scholar]
- 11.Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, et al. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog. 2010;6:e1000804. doi: 10.1371/journal.ppat.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spoering AL, Lewis K. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol. 2001;183:6746–6751. doi: 10.1128/JB.183.23.6746-6751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke V, Robinson JO, Richardson CJ, Bundell CS. Longitudinal studies of virulence factors of Pseudomonas aeruginosa in cystic fibrosis. Pathology. 1991;23:145–148. doi: 10.3109/00313029109060814. [DOI] [PubMed] [Google Scholar]
- 14.Meissner A, Wild V, Simm R, Rohde M, Erck C, et al. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ Microbiol. 2007;9:2475–2485. doi: 10.1111/j.1462-2920.2007.01366.x. [DOI] [PubMed] [Google Scholar]
- 15.Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. 2009;191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haussler S. Biofilm formation by the small colony variant phenotype of Pseudomonas aeruginosa. Environ Microbiol. 2004;6:546–551. doi: 10.1111/j.1462-2920.2004.00618.x. [DOI] [PubMed] [Google Scholar]
- 18.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 19.Jenal U, Malone J. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu Rev Genet. 2006;40:385–407. doi: 10.1146/annurev.genet.40.110405.090423. [DOI] [PubMed] [Google Scholar]
- 20.Merighi M, Lee VT, Hyodo M, Hayakawa Y, Lory S. The second messenger bis-(3′–5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol Microbiol. 2007;65:876–895. doi: 10.1111/j.1365-2958.2007.05817.x. [DOI] [PubMed] [Google Scholar]
- 21.Lee VT, Matewish JM, Kessler JL, Hyodo M, Kayakawa Y, et al. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol. 2007;65:1474–1484. doi: 10.1111/j.1365-2958.2007.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hickman JW, Harwood CS. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol. 2008;69:376–389. doi: 10.1111/j.1365-2958.2008.06281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulasekara HD, Ventre I, Kulasekara BR, Lazdunski A, Filloux A, et al. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol. 2005;55:368–380. doi: 10.1111/j.1365-2958.2004.04402.x. [DOI] [PubMed] [Google Scholar]
- 24.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, et al. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol. 2010;75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An S, Wu J, Zhang LH. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-Di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl Environ Microbiol. 2010;76:8160–8173. doi: 10.1128/AEM.01233-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′–5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A. 2006;103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuchma SL, Connolly JP, O'Toole GA. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol. 2005;187:1441–1454. doi: 10.1128/JB.187.4.1441-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moscoso JA, Mikkelsen H, Heeb S, Williams P, Filloux A. The Pseudomonas aeruginosa sensor RetS switches Type III and Type VI secretion via c-di-GMP signalling. Environ Microbiol. 2011;13:3128–38. doi: 10.1111/j.1462-2920.2011.02595.x. [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Whitchurch CB, Mattick JS. FimX, a multidomain protein connecting environmental signals to twitching motility in Pseudomonas aeruginosa. J Bacteriol. 2003;185:7068–7076. doi: 10.1128/JB.185.24.7068-7076.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kazmierczak BI, Lebron MB, Murray TS. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol. 2006;60:1026–1043. doi: 10.1111/j.1365-2958.2006.05156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alm RA, Bodero AJ, Free PD, Mattick JS. Identification of a novel gene, pilZ, essential for type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J Bacteriol. 1996;178:46–53. doi: 10.1128/jb.178.1.46-53.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merritt JH, Ha DG, Cowles KN, Lu W, Morales DK, et al. Specific Control of Pseudomonas aeruginosa Surface-Associated Behaviors by Two c-di-GMP Diguanylate Cyclases. MBio. 2010;1:e00183–10. doi: 10.1128/mBio.00183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girgis HS, Liu Y, Ryu WS, Tavazoie S. A comprehensive genetic characterization of bacterial motility. PLoS Genet. 2007;3:1644–1660. doi: 10.1371/journal.pgen.0030154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giddens SR, Jackson RW, Moon CD, Jacobs MA, Zhang XX, et al. Mutational activation of niche-specific genes provides insight into regulatory networks and bacterial function in a complex environment. Proc Natl Acad Sci U S A. 2007;104:18247–18252. doi: 10.1073/pnas.0706739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald MJ, Gehrig SM, Meintjes PL, Zhang XX, Rainey PB. Adaptive divergence in experimental populations of Pseudomonas fluorescens. IV. Genetic constraints guide evolutionary trajectories in a parallel adaptive radiation. Genetics. 2009;183:1041–1053. doi: 10.1534/genetics.109.107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beaumont HJ, Gallie J, Kost C, Ferguson GC, Rainey PB. Experimental evolution of bet hedging. Nature. 2009;462:90–93. doi: 10.1038/nature08504. [DOI] [PubMed] [Google Scholar]
- 37.Ueda A, Wood TK. Connecting quorum sensing, c-di-GMP, pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog. 2009;5:e1000483. doi: 10.1371/journal.ppat.1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilksch JJ, Yang J, Clements A, Gabbe JL, Short KR, et al. MrkH, a novel c-di-GMP-dependent transcriptional activator, controls klebsiella pneumoniae biofilm formation by regulating type 3 fimbriae expression. PLoS Pathog. 2011;7:e1002204. doi: 10.1371/journal.ppat.1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pappalardo L, Janausch IG, Vijayan V, Zientz E, Junker J, et al. The NMR structure of the sensory domain of the membranous two-component fumarate sensor (histidine protein kinase) DcuS of Escherichia coli. J Biol Chem. 2003;278:39185–39188. doi: 10.1074/jbc.C300344200. [DOI] [PubMed] [Google Scholar]
- 40.Reinelt S, Hofmann E, Gerharz T, Bott M, Madden DR. The structure of the periplasmic ligand-binding domain of the sensor kinase CitA reveals the first extracellular PAS domain. J Biol Chem. 2003;278:39189–39196. doi: 10.1074/jbc.M305864200. [DOI] [PubMed] [Google Scholar]
- 41.Sevvana M, Vijayan V, Zweckstetter M, Reinelt S, Madden DR, et al. A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J Mol Biol. 2008;377:512–523. doi: 10.1016/j.jmb.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 42.Moglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009;17:1282–1294. doi: 10.1016/j.str.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Key J, Moffat K. Crystal structures of deoxy and CO-bound bjFixLH reveal details of ligand recognition and signaling. Biochemistry. 2005;44:4627–4635. doi: 10.1021/bi047942r. [DOI] [PubMed] [Google Scholar]
- 44.Crosson S, Rajagopal S, Moffat K. The LOV domain family: photoresponsive signaling modules coupled to diverse output domains. Biochemistry. 2003;42:2–10. doi: 10.1021/bi026978l. [DOI] [PubMed] [Google Scholar]
- 45.Lee J, Tomchick DR, Brautigam CA, Machius M, Kort R, et al. Changes at the KinA PAS-A dimerization interface influence histidine kinase function. Biochemistry. 2008;47:4051–4064. doi: 10.1021/bi7021156. [DOI] [PubMed] [Google Scholar]
- 46.Razeto A, Ramakrishnan V, Litterst CM, Giller K, Griesinger C, et al. Structure of the NCoA-1/SRC-1 PAS-B domain bound to the LXXLL motif of the STAT6 transactivation domain. J Mol Biol. 2004;336:319–329. doi: 10.1016/j.jmb.2003.12.057. [DOI] [PubMed] [Google Scholar]
- 47.Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang C, Tesar C, Gu M, Babnigg G, Joachimiak A, et al. Extracytoplasmic PAS-like domains are common in signal transduction proteins. J Bacteriol. 2009;192:1156–1159. doi: 10.1128/JB.01508-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godlewska R, Wisniewska K, Pietras Z, Jagusztyn-Krynicka EK. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett. 2009;298:1–11. doi: 10.1111/j.1574-6968.2009.01659.x. [DOI] [PubMed] [Google Scholar]
- 50.Yeh YC, Comolli LR, Downing KH, Shapiro L, McAdams HH. The caulobacter Tol-Pal complex is essential for outer membrane integrity and the positioning of a polar localization factor. J Bacteriol. 2010;192:4847–4858. doi: 10.1128/JB.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonsor DA, Hecht O, Vankemmelbeke M, Sharma A, Krachler AM, et al. Allosteric beta-propeller signalling in TolB and its manipulation by translocating colicins. EMBO J. 2009;28:2846–2857. doi: 10.1038/emboj.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, et al. Structure of BeF3- -modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure. 2007;15:915–927. doi: 10.1016/j.str.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 54.Malone JG, Williams R, Christen M, Jenal U, Spiers AJ, et al. The structure-function relationship of WspR, a Pseudomonas fluorescens response regulator with a GGDEF output domain. Microbiology. 2007;153:980–994. doi: 10.1099/mic.0.2006/002824-0. [DOI] [PubMed] [Google Scholar]
- 55.Goymer P, Kahn SG, Malone JG, Gehrig SM, Spiers AJ, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. II. Role of the GGDEF regulator WspR in evolution and development of the wrinkly spreader phenotype. Genetics. 2006;173:515–526. doi: 10.1534/genetics.106.055863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parkinson JS. Signaling mechanisms of HAMP domains in chemoreceptors and sensor kinases. Annu Rev Microbiol. 2010;64:101–122. doi: 10.1146/annurev.micro.112408.134215. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Q, Ames P, Parkinson JS. Mutational analyses of HAMP helices suggest a dynamic bundle model of input-output signalling in chemoreceptors. Mol Microbiol. 2009;73:801–814. doi: 10.1111/j.1365-2958.2009.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collins LA, Egan SM, Stewart V. Mutational analysis reveals functional similarity between NARX, a nitrate sensor in Escherichia coli K-12, and the methyl-accepting chemotaxis proteins. J Bacteriol. 1992;174:3667–3675. doi: 10.1128/jb.174.11.3667-3675.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ames P, Zhou Q, Parkinson JS. Mutational analysis of the connector segment in the HAMP domain of Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 2008;190:6676–6685. doi: 10.1128/JB.00750-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manson MD. The tie that binds the dynamic duo: the connector between AS1 and AS2 in the HAMP domain of the Escherichia coli Tsr chemoreceptor. J Bacteriol. 2008;190:6544–6547. doi: 10.1128/JB.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 62.Lim A, Jr, De Vos D, Brauns M, Mossialos D, Gaballa A, et al. Molecular and immunological characterization of OprL, the 18 kDa outer-membrane peptidoglycan-associated lipoprotein (PAL) of Pseudomonas aeruginosa. Microbiology. 1997;143(Pt 5):1709–1716. doi: 10.1099/00221287-143-5-1709. [DOI] [PubMed] [Google Scholar]
- 63.Parsons LM, Lin F, Orban J. Peptidoglycan recognition by Pal, an outer membrane lipoprotein. Biochemistry. 2006;45:2122–2128. doi: 10.1021/bi052227i. [DOI] [PubMed] [Google Scholar]
- 64.Godlewska R, Wisniewska K, Pietras Z, Jagusztyn-Krynicka EK. Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett. 2009;298:1–11. doi: 10.1111/j.1574-6968.2009.01659.x. [DOI] [PubMed] [Google Scholar]
- 65.Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 66.Chan C, Paul R, Samoray D, Amiot NC, Giese B, et al. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci U S A. 2004;101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, et al. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 2008;6:e67. doi: 10.1371/journal.pbio.0060067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newell PD, Monds RD, O'Toole GA. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci U S A. 2009;106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Newell PD, Boyd CD, Sondermann H, O'Toole GA. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol. 2011;9:e1000587. doi: 10.1371/journal.pbio.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gjermansen M, Nilsson M, Yang L, Tolker-Nielsen T. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms: genetic elements and molecular mechanisms. Mol Microbiol. 2010;75:815–826. doi: 10.1111/j.1365-2958.2009.06793.x. [DOI] [PubMed] [Google Scholar]
- 72.D'Argenio DA, Calfee MW, Rainey PB, Pesci EC. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol. 2002;184:6481–6489. doi: 10.1128/JB.184.23.6481-6489.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, et al. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol. 2010;12:1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x. [DOI] [PubMed] [Google Scholar]
- 74.Rosenfeld M, Emerson J, Williams-Warren J, Pepe M, Smith A, et al. Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr. 2001;139:359–365. doi: 10.1067/mpd.2001.117288. [DOI] [PubMed] [Google Scholar]
- 75.Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 76.Kresse AU, Dinesh SD, Larbig K, Romling U. Impact of large chromosomal inversions on the adaptation and evolution of Pseudomonas aeruginosa chronically colonizing cystic fibrosis lungs. Mol Microbiol. 2003;47:145–158. doi: 10.1046/j.1365-2958.2003.03261.x. [DOI] [PubMed] [Google Scholar]
- 77.Bianconi I, Milani A, Cigana C, Paroni M, Levesque RC, et al. Positive signature-tagged mutagenesis in Pseudomonas aeruginosa: tracking patho-adaptive mutations promoting airways chronic infection. PLoS Pathog. 2011;7:e1001270. doi: 10.1371/journal.ppat.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller JH. Experiments in molecular genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1972. pp. 352–355. [Google Scholar]
- 79.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A. 2001;98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 82.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 83.Heeb S, Itoh Y, Nishijyo T, Schnider U, Keel C, et al. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol Plant Microbe Interact. 2000;13:232–237. doi: 10.1094/MPMI.2000.13.2.232. [DOI] [PubMed] [Google Scholar]
- 84.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 85.Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rasila TS, Pajunen MI, Savilahti H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal Biochem. 2009;388:71–80. doi: 10.1016/j.ab.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 87.Morgan AF. Transduction of Pseudomonas aeruginosa with a mutant of bacteriophage E79. J Bacteriol. 1979;139:137–140. doi: 10.1128/jb.139.1.137-140.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Voisard C, Bull CT, Keel C, Laville J, Maurhofer M, et al. Biocontrol of root diseases by Pseudomonas fluorescens CHA0: current concepts and experimental approaches. In: O'Gara F, Dowling DN, Boesten B, editors. Molecular Ecology of Rhizosphere Microorganisms. Weinheim, Germany: Wiley-VCH; 1994. pp. 67–89. [Google Scholar]
- 89.Merritt JH, Kadouri DE, O'Toole GA. Growing and analyzing static biofilms. Curr Protoc Microbiol Chapter. 2005;1:Unit 1B 1. doi: 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klebensberger J, Rui O, Fritz E, Schink B, Philipp B. Cell aggregation of Pseudomonas aeruginosa strain PAO1 as an energy-dependent stress response during growth with sodium dodecyl sulfate. Arch Microbiol. 2006;185:417–427. doi: 10.1007/s00203-006-0111-y. [DOI] [PubMed] [Google Scholar]
- 91.Diab F, Bernard T, Bazire A, Haras D, Blanco C, et al. Succinate-mediated catabolite repression control on the production of glycine betaine catabolic enzymes in Pseudomonas aeruginosa PAO1 under low and elevated salinities. Microbiology. 2006;152:1395–1406. doi: 10.1099/mic.0.28652-0. [DOI] [PubMed] [Google Scholar]
- 92.Hancock RE, Nikaido H. Outer membranes of gram-negative bacteria. XIX. Isolation from Pseudomonas aeruginosa PAO1 and use in reconstitution and definition of the permeability barrier. J Bacteriol. 1978;136:381–390. doi: 10.1128/jb.136.1.381-390.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spangler C, Bohm A, Jenal U, Seifert R, Kaever V. A liquid chromatography-coupled tandem mass spectrometry method for quantitation of cyclic di-guanosine monophosphate. J Microbiol Methods. 2010;81:226–231. doi: 10.1016/j.mimet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 94.Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, et al. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006;34:53–65. doi: 10.1093/nar/gkj406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 98.Sneath PH, Sokal RR. Numerical taxonomy. Nature. 1962;193:855–860. doi: 10.1038/193855a0. [DOI] [PubMed] [Google Scholar]
- 99.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Localization and stability of YfiR. A) Outer Membrane localization of YfiR-flag. Immunoblot of fractionated membrane samples stained with M2 antisera. The panel shows the soluble and the outer membrane fractions for ΔyfiBNR, ΔyfiNR and ΔyfiBNR Tn7::yfiNR harboring pMR-yfiR-flag (p-yfiR). B) Stability of YfiR-flag in whole cell lysate. Immunoblot of ΔyfiR, ΔyfiNR and ΔyfiBNR with yfiR-flag inserted into the att-Tn7 site, and one time point with the strains over expressing YfiR (ΔyfiBNR, ΔyfiNR and ΔyfiBNR Tn7::yfiNR harboring pMR-yfiR-flag (p-yfiR)). ‘t’ indicates different time points (hours post inoculation); ‘O/N’ overnight incubation.
(PDF)
Colony morphologies with activated YfiN alleles. A) Colony morphologies of ΔyfiNR pGm-yfiprom-N, pMR-yfiR-flag strains with the point mutants indicated present in YfiN. ‘pyfiR pyfiN’ denotes the wild-type control; ‘pyfiN’ denotes a strain containing pGm-yfiprom-N only. B) Colony morphologies for cross-complementation strains. The YfiN (bold) and YfiR (italic) mutant alleles present are shown in each case.
(TIF)
Attachment of Yfi mutants in response to the presence of SDS or changes in osmolarity. A) Attachment on M9 medium with 20 mM Na-succinate and increasing concentrations of SDS is shown relative to PA01. B) Attachment with low and high osmolarity, on M63 medium with 20 mM Na-succinate, 0.2% glucose, and increasing concentrations of NaCl is shown relative to PA01. 50% and 75% indicate dilutions of the medium with distilled water. ΔBNR indicates ΔyfiBNR and ΔB indicates ΔyfiBNR Tn7::yfiNR. Bars represent the level of absolute attachment and curves represent optical density (OD) of total cells with standard errors.
(PDF)
Attachment of clinical iosolates in the presence of a phosphodiesterase. The effect of a PDE, in Clin110 and Clin163, on attachment is shown relative to strains expressing an active site mutant (PDE*). The PDE (PA5295) and the active site mutant are expressed from a vanillate inducible vector (pBV-PA5295 and pBV-PA5295E328A respectively).
(PDF)
Sequence conservation across distant YfiN/YfiR homologs. Sequence alignments for YfiR (residues 55–190) and the YfiN PAS domain (residues 61–115), across PA01 and five distant homologs. The five species whose yfiNR genes were compared were: Opitutus terrae PB90-1, Acidobacteria bacterium Ellin345, Geobacter sp. M21, Rhodothermus marinus DSM 4252, and Desulfobacterium autotrophicum HRM2. Fully conserved residues are marked with an asterisk (*), similarities are marked with (:) or (.). Putative hydrophobic binding site residues are enclosed in black boxes. Residues are colored according to the chemical nature of their side chains.
(PDF)
Bacterial strains and plasmids used in this study.
(PDF)
Primers used in this study.
(PDF)
Single nucleotide polymorphism (SNP) positions in the sequences of clinical SCVs, compared to P. aeruginosa PA01.
(PDF)
Identification of YfiBNR homologs in other systems.
(PDF)