

Abstract
To reduce damage from toxic insults such as glutamate excitotoxicity and oxidative stresses, neurons may deploy an array of neuroprotective mechanisms. Recent reports show that progranulin (PGRN) gene null or missense mutations leading to inactive protein, are linked to frontotemporal lobar degeneration (FTLD), suggesting that survival of certain neuronal populations need full expression of functional PGRN. Here we show that extracellular PRGN stimulates phosphorylation/activation of the neuronal MEK/ERK/p90RSK and PI3K/Akt cell survival pathways and rescues cortical neurons from cell death induced by glutamate or oxidative stresses. Pharmacological inhibition of MEK/ERK/p90RSK signaling blocks the PGRN-induced phosphorylation and neuroprotection against glutamate toxicity whilst inhibition of either MEK/ERK/p90RSK or PI3K/Akt blocks PGRN protection against neurotoxin MPP+. Inhibition of both pathways had synergistic effects on PGRN-dependent neuroprotection against MPP+ toxicity suggesting both pathways contribute to the neuroprotective activities of PGRN. Extracellular PGRN is remarkably stable in neuronal cultures indicating neuroprotective activities are associated with full-length protein. Together, our data show that extracellular PRGN acts as a neuroprotective factor and support the hypothesis that in FTLD, reduction of functional brain PGRN results in reduced survival signaling and decreased neuronal protection against excitotoxicity and oxidative stress leading to accelerated neuronal cell death. That extracellular PGRN has neuroprotective functions against toxic insults suggests that in vitro preparations of this protein may be used therapeutically.
Keywords: progranulin, neuroprotection, neurodegeneration, frontotemporal lobar degeneration, excitotoxicity, oxidative stress, ERK, Akt
1. Introduction
Progranulin (PGRN), also identified in literature as PC cell-derived growth factor (PCDGF), acrogranin, or proepithelin, is a 593 residue polypeptide that contains a signal sequence and 7.5 homologous cysteine-rich granulin domains separated by linker sequences (Bateman, Bennett, 2009). Mature PGRN is secreted as a highly glycosylated protein of approximately 88–95 kDa that under certain conditions is processed in the linker regions by elastase or other proteases to produce biologically active peptides referred to as granulins or epithelins (Zhu et al., 2002; Kessenbrock et al., 2008; Butler et al., 2008). Processing of secreted PGRN is inhibited by the secretory leukocyte protease inhibitor (SLPI) (Zhu et al., 2002). PGRN and its derivatives, granulin peptides, are expressed in many tissues including epithelial cells, the gastrointestinal tract and hematopoietic cells (Daniel et al., 2000) and have been associated with multiple biological functions such as regulation of cell growth, cell cycle progression, embryonic development, and tissue repair (He, Bateman, 2003; Bateman, Bennett, 2009). Studies in non-neuronal systems indicate that PGRN activates cell signaling pathways including the extracellular regulated kinase (ERK1/2) and the phosphatidylinositol-3 kinase (PI3K)/Akt cell survival pathways (He et al., 2002; Lu, Serrero, 2001; Zanocco-Marani et al., 1999; Monami et al., 2006). During inflammation and wound healing, cells secrete the protease elastase that converts PGRN to granulin peptides which may have different, overlapping, or even opposite functions from the parent protein (Plowman et al., 1992; Tolkatchev et al., 2008; Zhu et al., 2002). For example, while full length PGRN acts as an anti-inflammatory agent, individual graunlin peptides have been shown to stimulate production of pro-inflammatory cytokines (Zhu et al., 2002). These observations suggest that a carefully maintained equilibrium between PGRN and granulin peptides may be important to tissue homeostasis.
In the central nervous system (CNS), PGRN is expressed in both neurons and microglia (Daniel et al., 2000; Matsuwaki et al., 2010; Ryan et al., 2009). Importantly, recent genetic studies showed that PGRN mutations are linked to frontotemporal lobar degeneration (FTLD), a form of dementia characterized by severe neuronal loss in the frontal and temporal brain regions of adult patients (Baker et al., 2006; Cruts et al., 2006). These findings renewed interest in the brain functions of this protein. To date, more than 70 FTLD-linked PGRN mutations have been detected with most of them causing functional null alleles (Sleegers et al., 2010). Although the autosomal dominant mode of inheritance of PGRN-linked familial FTLD might suggest gain of a toxic function, many PGRN mutants encode incomplete or inactive peptides indicating that PGRN haploinsufficiency can result in dominant transmission of neurodegeneration. Based on these findings, decreased plasma PGRN levels has been proposed as a biomarker for early diagnosis of this disorder (Sleegers et al., 2009; Finch et al., 2009; Ghidoni et al., 2008). Besides the null FTLD mutations that lead to decreased PGRN levels, there are at least 17 PGRN missense mutations linked to FTLD families (Gijselinck et al., 2008). The pathogenetic nature of these mutations is less clear than the PGRN null mutations, but it was recently reported that at least some missense mutations also cause a decrease in the levels of functional PGRN (Shankaran et al., 2008; Wang et al., 2010). Thus, there is strong evidence that a 50% reduction of functional PGRN (haploinsufficiency) leads to increased neuronal cell death in adult brains supporting the hypothesis that PGRN or its derivatives support neuronal survival.
Recently, it was reported that secreted PGRN binds sortilin and is subsequently delivered to lysosome through endocytosis (Hu et al., 2010). Additional studies suggest that PGRN functions as a neurotrophic agent that may promote survival of primary neuronal cultures under conditions of serum withdrawal (Van Damme et al., 2008; Gao et al., 2010; Ryan et al., 2009). Furthermore, PGRN knockdown decreased axonal outgrowth in zebrafish embryos and PGRN-deficient hippocampal slices are susceptible to glucose deprivation (Laird et al., 2010; Yin et al., 2010). Together, these reports indicate that PGRN function as a neurotrophic factor and may play important roles in neuronal physiology.
That reduction of PRGN leads to increased degeneration of cortical neurons in the CNS (Bateman, Bennett, 2009; Sleegers et al., 2010), raises the possibility this protein functions as a neuroprotctive factor. For example, PGRN may protect brain neurons from exposure to toxic insults such as glutamate-associated excitotoxicity and oxidative stress. Both processes are associated with neuronal activity and have been proposed to play important roles in the development neurodegenerative disorders (Fatokun et al., 2008; Lau, Tymianski, 2010). Here we show that extracellular PGRN activates neuronal ERK1/2 and Akt cell survival signaling and protects neurons from toxic insults associated with neurodegeneration.
2. Materials and Methods
All chemicals were purchased from Sigma except where indicated. All animal experiments were carried out in accordance with the rules and regulations at the Mount Sinai School of Medicine and the Biomedical Research Foundation of the Academy of Athens.
2.1 Primary neuronal cultures
cortical neuronal cultures from embryonic brains of E18.5 Wistar rats were prepared as described (Vogiatzi et al., 2008; Xu et al., 2009). Dissociated cells were plated onto poly–D-lysine-coated plates at a density of approximately 60,000/cm2. Cells were maintained in Neurobasal medium supplemented with 1% B27 (Invitrogen), L-glutamine (0.5 mM) and penicillin/streptomycin (1% v/v) and used at 8 to 12 days in vitro (DIV). Under these conditions post-mitotic neurons represent more than 98% of cultured cells (Paxinou et al., 2001).
2.2 Production and purification of PRGN
HEK293T cells stably transfected with vector pcDNA3.1/V5-His-TOPO expressing human PGRN (provided by Dr. Bateman, McGill University) were grown in DMEM supplemented with 10% FBS and 0.5 mg/ml G418. At 100% cell confluency, the growth medium was replaced with fresh DMEM and condition media were collected 48 hrs later. An approximately 90 KDa protein corresponding to glycosylated full-length PGRN was present in conditioned media of transfected, but not control, cultures (see supplemental Fig. 1). His-tagged PGRN was bound to Ni-NTA agarose beads (Qiagen) overnight and collected beads were washed with PBS containing 10mM imidazole. PGRN was then eluted with PBS containing 200mM imidazole. Silver-stained SDS gels showed purified full-length PGRN protein with minimal degradation (supplemental Fig. 1). For PGRN stability experiments, PGRN was added to neuronal cultures for various times as indicated in the text. Elastase alone (0.3U/ml) or elastase pre-incubated with protease inhibitors for 30 min, was added to the media together with PGRN. At the end of reaction, media were retrieved, denatured and analyzed by Western Blot using anti-V5 tag antibody (Invitrogen).
2.3 Cell survival assays
three independent assays were used to measure PGRN-dependent neuroprotection against glutamate or H2O2 toxicity as indicated in Results. Hoechst staining assay of neuronal viability (Arndt-Jovin, Jovin, 1977) was determined according to manufacturer’s instructions (Sigma). Briefly, neurons on poly-D-lysine-coated 24-well plates were treated with glutamate for 3 hrs, fixed in 4% paraformaldehyde for 20 min at room temperature and stained with Hoechst 33342 for 10 min. Neurons were then observed under a fluorescence microscope on ultraviolet illumination. Numbers of viable neurons were counted in ten fields per well with at least 20 neurons per field. Results are expressed as percent of control value. MTT cell viability assay based on the cleavage of yellow tetrazolium salt MTT to purple formazan (Denizot, Lang, 1986) was performed according to manufacturer’s instructions (Sigma). In summary, neurons at 8–12 DIV grown on poly-D-lysine-coated 96-well plates were treated with toxic agents for 3 hrs and MTT solution (1 mg/ml) was added to each well. Plates were incubated at 37 °C for 2 hrs and the reaction was terminated by 0.1N HCl in isopropanol for 1 hr. Absorbance was then measured at 560 nm by spectrophotometric microplate reader (Thermo Scientific) with background subtraction at 620 nm. Data are expressed as a percent of control value. Lactate dehydrogenase (LDH) release assays (Koh, Choi, 1987), were performed using the Cytotoxicity Detection Kit Plus (Roche) according to manufacturer’s instruction. Results are expressed as the percentage of LDH release by non-treated cells.
2.4 Assessment of MPP+ toxicity
rat cortical neurons cultured on poly-D-lysine-coated 12-well plates for five days were treated with 35 nM progranulin or BSA (as control) for 24 hrs prior to addition of 40 μM MPP+. Twenty four hours after MPP+ addition, culture media were removed and cells were lysed in a detergent containing solution that enables the quantification of viable cells by counting the number of intact nuclei in a haemacytometer as described (Stefanis et al., 1999; Farinelli et al., 1998). Cell counts were performed in triplicate and are reported as mean ± SE. In experiments with inhibitors of ERK and Akt (Fig 6), MPP+ treatment was modified in order to achieve comparable death rates in sorter time periods. This was crucial because inhibitors are unstable in culture and may exert non-specific toxic effects when used for longer times. Titration experiments with or without the inhibitors showed that 4 hours treatments with the compounds, was the optimal regiment. Axonal degeneration and loss of neuritic processes were observed using phase contrast microscopy.
Figure 6. ERK1/2 and Akt signaling mediate the neuroprotective activities of PGRN against MPP+ toxicity.
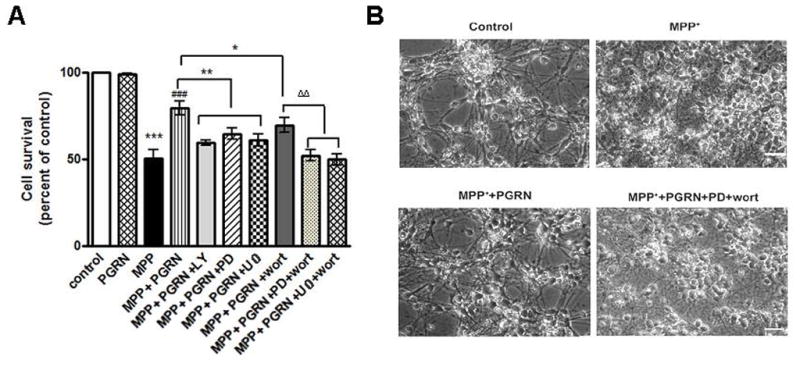
(A) Rat cortical cultures as in Fig. 1E were pre-incubated with PGRN (15nM) for 30 min with or without inhibitors of MEK/ERK (U0126 or PD98059) or PI3K/Akt (wortmannin or LY-294002) signaling as indicated and then treated with 40μM MPP+ for 4 hrs. Following MPP+ treatment, cells were lysed with a nuclear sparing buffer. Intact nuclei were counted in a haemacytometer. Data are presented as mean ± SE, obtained from three separate experiments (one way ANOVA followed by the Student-Newman-Keuls’ test, ***p<0.001, comparing between cultures treated with MPP+ and untreated (control) or PGRN alone treated cultures; ###p<0.001, comparing between MPP+ and MPP++PGRN treated cultures; *p<0.05, **p<0.01, comparing between cultures treated with MPP++PGRN in the presence and absence of MEK/ERK1/2 and PI3K/Akt inhibitors; ΔΔp<0.01, comparing between cultures treated with MPP++PGRN+wortmannin in the presence and absence of PD98059 or U0126). (B) Representative photomicrographs of cortical neurons treated as in A. MPP+ treatment of neuronal cultures caused retraction of neuritic processes and eventually neuronal death (MPP+). PGRN preserves the neuritic processes of MPP+-treated cultures (MPP+PGRN) and this PGRN effect is abolished in the presence of MEK/ERK and PI3K/Akt inhibitors PD98059 and wortmannin, respectively. Magnification is X20.
2.5 Western Blot analysis
Western blots analysis was performed as described (Vogiatzi et al., 2008; Xu et al., 2009). Briefly, neurons were washed with cold PBS and solubilized in lysis buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, phosphatase and protease inhibitor cocktail. Cell lysates were centrifuged at 16,000 × g for 40 min and 30μg of supernatant protein was loaded onto each well of 10% SDS-PAGE. The following antibodies were used for blotting analysis: anti-PGRN (Zymed), anti-His (Qiagen), anti-V5 (Invitrogen), anti-ERK1/2 and pERK Thr202/Tyr204 (Cell Signaling), anti-Akt and pAkt Ser473 (Cell Signaling), anti-p90RSK and pRSK Thr359/Ser363 as well as pRSK Ser380 (Cell signaling).
2.6 Statistical analysis
all data are expressed as mean ± SE. All data were normalized to the control (100%) and levene’s test (embedded in SPSS) was used to assess the homogeneity of variance. Accordingly, statistical significance of differences was evaluated either with paired t-test or with one-way ANOVA followed by the Student-Newman-Keuls’ test as post-hoc multiple comparisons depending on the significance of levene’s test. p values < 0.05 were considered significant.
3. Results
3.1 Extracellular PGRN promotes neuronal survival against toxic insults
Decreased expression of PRGN is associated with increased degeneration of cortical neurons in the CNS (Bateman, Bennett, 2009; Sleegers et al., 2010), raising the possibility this protein functions as a neuroprotective factor. For example, PGRN may protect brain neuronal populations from exposure to toxic insults such as glutamate-associated excitotoxity and oxidative stress. To better understand the effects of PGRN on neuronal survival, we generated adequate amounts of purified protein using recombinant DNA technology (see Materials and Methods) and asked whether exogenous PGRN is able to protect rat cortical neuronal cultures from glutamate toxicity. Treatment of our cultures with 50 μM glutamate reduced neuronal cell viability to 53±4%, determined by Hoechst staining, a commonly used protocol that evaluates cell survival by counting intact cell nuclei (see Materials and Methods). Pretreatment of the neuronal cultures with recombinant PGRN however, significantly decreased the glutamate-induced neuronal cell death (73±5% survival) (Fig. 1A). The neuroprotective effect of PGRN against glutamate excitotoxicity was verified by employing the MTT as well as the LDH assays, both of which are commonly used to evaluate cell toxicity and survival (Figs. 1B and 1C, respectively). Glutamate toxicity, measured by the amount of LDH released to the culture medium, was reduced by PGRN proportionally to its concentration and the effect of PGRN at 35 nM was comparable to the neuroprotective effect induced by neurotrophin BDNF used as a positive control (Fig. 1C).
Figure 1. Neuroprotective functions of extracellular PGRN.
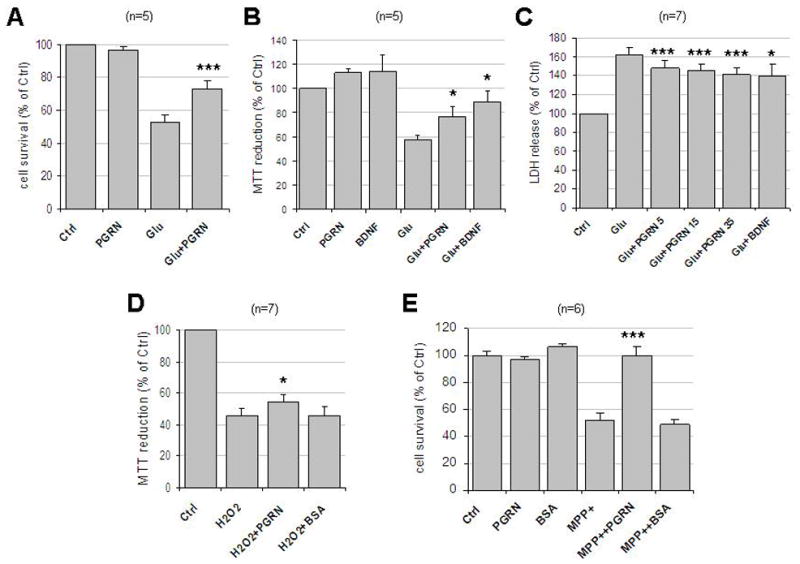
(A) Rat cortical neurons were cultured in 24-well plates in Neurobasal Media plus B27 supplement. At 8–12 DIV, neurons were pre-treated with 35nM PGRN for 30 min followed by 50μM glutamate incubation for 3 hrs. Cells were then fixed with 4% paraformaldehyde and stained with Hoechst 33342. Five pictures were taken from each well and each condition represented the average of four wells. Cell survival was measured by counting the number of cells with normal nuclear morphology. Results (mean ± SE) were calculated from five independent experiments. ***, p<0.005 comparing between cultures treated with glutamate in the presence or absence of PGRN (paired t-test). (B) Cortical neurons as above cultured in 96-well plates were pre-treated with 35nM PGRN or 50ng/ml BDNF for 18 hrs followed by 50μM glutamate for 3 hrs. Cell viability was evaluated by MTT assay and normalized to control (ctrl). Results (mean ± SE) were summarized from five independent experiments and in each experiment each condition is the average of six identical wells. *, p<0.05 comparing between cultures treated with glutamate in the presence or absence of PGRN or BDNF (paired t-test). (C) Neuronal cultures as above were pretreated with different concentrations (5nM, 15nM or 35nM) of PGRN or 50ng/ml BDNF for 2hrs followed by 50μM of glutamate treatment for 24 hrs. The cell-free culture supernatant was collected and LDH release was determined as per Manufacturer’s instructions. Results (mean ± SE) were summarized from seven independent experiments. *, p<0.05; ***, p<0.005 comparing between cultures treated with glutamate in the presence and absence of PGRN or BNDF (paired t-test). Numbers next to PGRN indicate concentrations in nM. (D) Rat cortical neuronal cultures as in (B) were pre-treated with either 35nM PGRN or BSA for 24 hrs followed by 25μM H2O2 treatment overnight. Cell viability was evaluated as in (B) and normalized to control. Results (mean ± SE) were summarized from seven independent experiments and in each experiment each condition is the average of six identical wells. *, p<0.05 comparing glutamate-treated cultures in the presence or absence of PGRN (paired t-test). (E) Neurons in 12-well plates were pre-treated with either 35 nM PGRN or BSA for 24 hrs followed by 40 μM MPP+ treatment for 24 hrs. Cell survival was evaluated by counting the number of intact nuclei in a haemacytometer after lysis (see Materials and Methods). Results (mean ± SE) were summarized from six independent experiments. ***, p<0.005 comparing between cultures treated with glutamate in the presence and absence of PGRN (paired t-test). Ctrl, no treatment; Glu, glutamate.
To explore whether PGRN is able to protect neurons from oxidative stress, we treated rat cortical neuronal cultures with hydrogen peroxide (H2O2), a commonly used reagent for induction of oxidative stress. Addition of 25 μM H2O2 reduced neuronal viability, measured using the MTT, to about 46±4% of control while pre-incubation with 35 nM PGRN significantly protected neurons from oxidative damage increasing cell survival to approximately 54±5% of control. BSA used at concentrations similar to PGRN had no effect on neuronal cell viability (Fig. 1D). MPP+, the active derivative of MPTP, is known to exert neuronal toxicity, in part, by releasing reactive oxygen species (ROS) from the mitochondria, as a result of complex I inhibition (Przedborski, Vila, 2003). To study the PGRN effects against MPP+ toxicity, we exposed cortical neurons to 40 μM MPP+ for 24 hrs and cell survival was evaluated by counting intact nuclei. MPP+ treatment decreased neuronal survival to 47±7% of control (Fig. 1E), whilst pre-treatment of these cultures with 35 nM of PGRN restored survival to nearly control levels (Fig. 1E). In contrast, pre-treatment with BSA had no effect on the MPP+-induced neuronal death (Fig. 1E). Together, these results indicate that PRGN has a strong neuroprotective effect against MPP+ neurotoxicity. Furthermore, our data suggest that the neuroprotective effect of PGRN depends on the toxic agent. Thus, there is a significance difference between PGRN and Glu+PGRN (p=0.016; 1A) but no significance between PGRN and MPP++ PGRN (p=0.516; 1E) suggesting PGRN almost completely reverses the toxic effects of MPP+ but not those of glutamate.
Depending on culture conditions and cell type, secreted PGRN can be processed by extracellular proteases, including elastase and proteinase 3, to produce granulin peptides that have been proposed to have different functions from the parent protein (Zhu et al., 2002; He, Bateman, 2003; Kessenbrock et al., 2008). To explore whether neurons secrete proteases able to process PGRN, purified protein was added to the medium of rat cortical neuronal cultures, and non-degradated PGRN was recovered and analyzed. Figure 2 shows that PGRN is remarkably stable in the culture media of primary neurons even following overnight incubation (lanes 2, 3, 6 and 9). In contrast, PGRN was quickly degraded in the presence of exogenous elastate (lanes 4 and 7), a process inhibited by inhibitors of elastase (lanes 5 and 8). These data show that exogenous PGRN is not metabolized to any significant extent by primary neuronal cultures in vitro and suggest that it is unlikely that this protein is processed to granulins by cortical neurons in vivo. Furthermore, our observations suggest that the neuroprotective properties of exogenous PGRN are most probably due to full-length PRGN protein rather than granulin peptides.
Figure 2. Stability of extracellular PGRN in neuronal culture media.
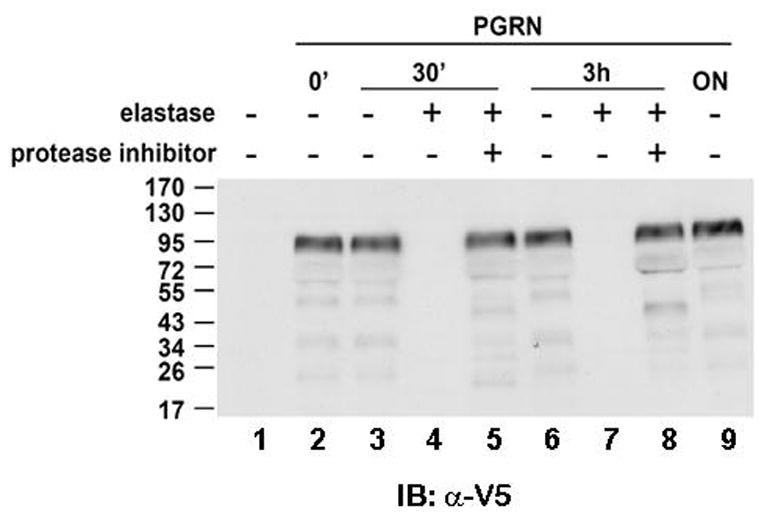
35 nM of recombinant PGRN (see Methods) was added to neuronal cultures for various time periods as indicated. Elastase (0.3 U/ml) or mixture of elastase with protease inhibitor cocktail was added together with PGRN at the same time. Protease inhibitors were pre-incubated with elastase for 30 min before adding into culture media. At the end of reaction, 5 μl of media were retrieved, denatured and analyzed by Western Blot with anti-V5 tag antibody. Numbers to the left of the blots indicate the position and size (kDa) of molecular mass markers. IB, immunoblot.
3.2 Extracellular PGRN activates ERK and Akt signaling in cortical neurons
To elucidate the mechanism by which PGRN exerts its neuroprotective function, we examined its effects on cell signaling pathways including those of ERK1/2 and Akt kinases. Both pathways have been reported to be activated in response to PGRN in non-neuronal cell lines (He et al., 2002; Lu, Serrero, 2001; Zanocco-Marani et al., 1999; Monami et al., 2006) and we reasoned that these may also be activated in primary neurons. ERK1/2 and Akt kinases are involved in cell survival and activation of the corresponding signaling pathways is indicated by phosphorylation of ERK1/2 at residues Thr202/Tyr204 and of Akt at Ser473 (Fayard et al., 2005; Payne et al., 1991). Figure 3A shows that PGRN treatment leads to a rapid increase in the phosphorylation of both ERK1/2 and Akt kinases at residues Thr202/Tyr204 and Ser473, respectively. Interestingly, ERK1/2 phosphorylation peaked earlier than Akt phosphorylation (Fig. 3A) and since there is no evidence that Akt is downstream of ERK, these PGRN-induced phosphorylation events may be independent of each other suggesting PGRN independently activates both survival pathways. To confirm the specificity of ERK and Akt activation by PGRN, we employed pharmacological agents that specifically target MEK/ERK1/2 and PI3K/Akt kinases. Pre-incubation of our cultures with MEK/ERK1/2 inhibitor U0126 decreased phosphorylation of ERK1/2 while pre-treatment with PI3K/Akt inhibitor LY-294002 abolished PGRN-induced Akt phosphorylation. MEK/ERK1/2 inhibitor PD98059 and PI3K/Akt inhibitor wortmannin gave results similar to those obtained with U0126 and LY-294002 respectively (Figs. 3B and C).
Figure 3. Extracellular PGRN activates ERK and Akt signaling pathways of cortical neurons.
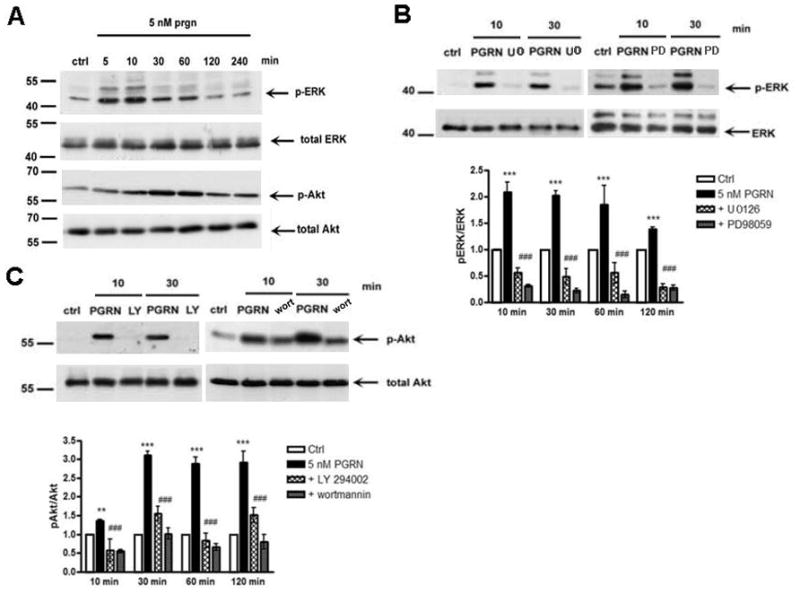
(A) Rat cortical neurons in 12-well plates were treated at 8DIV with 5nM PGRN for the indicated time periods. Untreated cultures were used as controls (ctrl). Following incubation, cells were collected and assayed on Western blots for the proteins indicated to the right of the blots. A representative blot out of three independent experiments is shown (B) ERK inhibitors U0126 and PD98059 (25μM each) blocked PGRN-induced ERK1/2 phosphorylation. Inhibitors were added to cultures for 30 min prior to addition of 5nM PGRN. Neurons were subsequently collected at indicated times and subjected to SDS-PAGE and Western blot as above. Densitometric analysis of the amounts of p-ERK in the presence of inhibitors expressed as pERK to ERK ratio that was set as 1 for control (white bar). Other bars represent phosphoprotein to protein ratios relative to control. (C) PI3K/Akt inhibitors wortmannin (wort, 1μM) or LY-294002 (25μM) blocks PGRN-induced Akt phosphorylation. Inhibitors and PGRN were added to cultures as in (B) and neurons were collected at indicated times and subjected to SDS-PAGE and Western blot as above. Densitometric analysis of the amounts of p-Akt in the presence of inhibitors is expressed as p-Akt to Akt ratio as above. Data were obtained from three separate experiments (**p<0.01, ***p<0.001, one way ANOVA followed by the Student-Newman-Keuls’ test, comparing between cultures treated with PGRN and without treatment (ctrl); ###p<0.001, comparing between cultures treated with PGRN in the presence or absence of inhibitors). Signal variability is indicated by error bars.
An important downstream effector of ERK1/2 kinase is the p90 ribosomal S6 kinase (p90RSK) which is activated upon phosphorylation at residues Thr359/Ser363 and Ser380. Following activation p90RSK travels to the nucleus where it regulates gene expression (Anjum, Blenis, 2008). Figure 4 shows that treatment of neuronal cultures with PGRN, induced a rapid increase in the phosphorylation of p90RSK residues Thr359/Ser363 and Ser380. Furthermore, the PGRN-induced phosphorylation of p90RSK was blocked by MEK/ERK1/2 inhibitor U0126 indicating that ERK1/2 mediated the PGRN-induced phosphorylation of p90RSK. Taken together, these results show that PGRN specifically activates the ERK1/2 and Akt cell survival pathways in neuronal populations and suggest a mechanism by which PGRN protects neurons from toxic insults.
Figure 4. PRGN stimulates phosphorylation of p90 ribosomal S6 kinase (p90RSK) in primary cortical neuronal cultures.
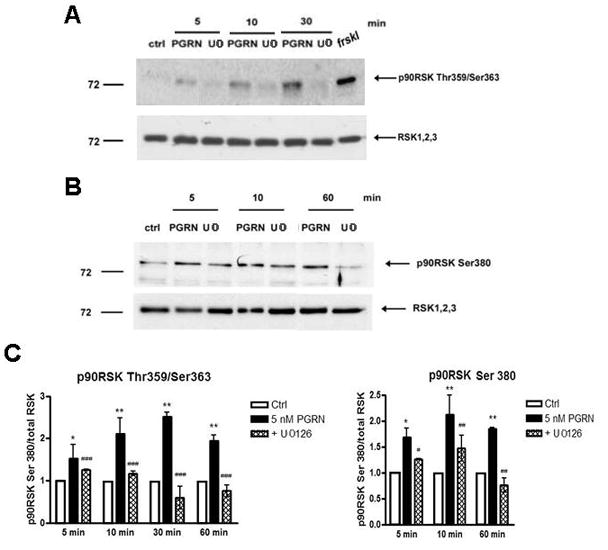
(A) Rat neuronal cultures prepared as above were treated with 5 nM PGRN for the indicated time periods, in the absence or presence of MEK/ERK inhibitor U0126 (25 μM) and at the end of treatment, neurons were lysed and assayed by Western blotting for levels p90RSK phosphorylated at Thr359/Ser363 and total RSK 1,2,3 proteins. (B) Cortical neurons were treated as in A for the indicated periods and levels of phoshorylated p90RSK at Ser380 and total RSK 1,2,3 proteins were assayed as above. (C) Densitometric analysis of p90RSK Thr359/Ser363 and p90RSK Ser380 in neuronal cultures treated as above was performed and analysed as described in Fig. 3. Data were obtained from three separate experiments (*p<0.05, **p<0.01, one way ANOVA followed by the Student-Newman-Keuls’ test, comparing between cultures treated with PGRN and without treatment (ctrl); #p<0.05, ##p<0.01, ###p<0.001, comparing between cultures treated with PGRN in the presence and absence of U0126).
3.3 Inhibition of ERK1/2 and Akt signaling abolishes the neuroprotective effects of extracellular PGRN
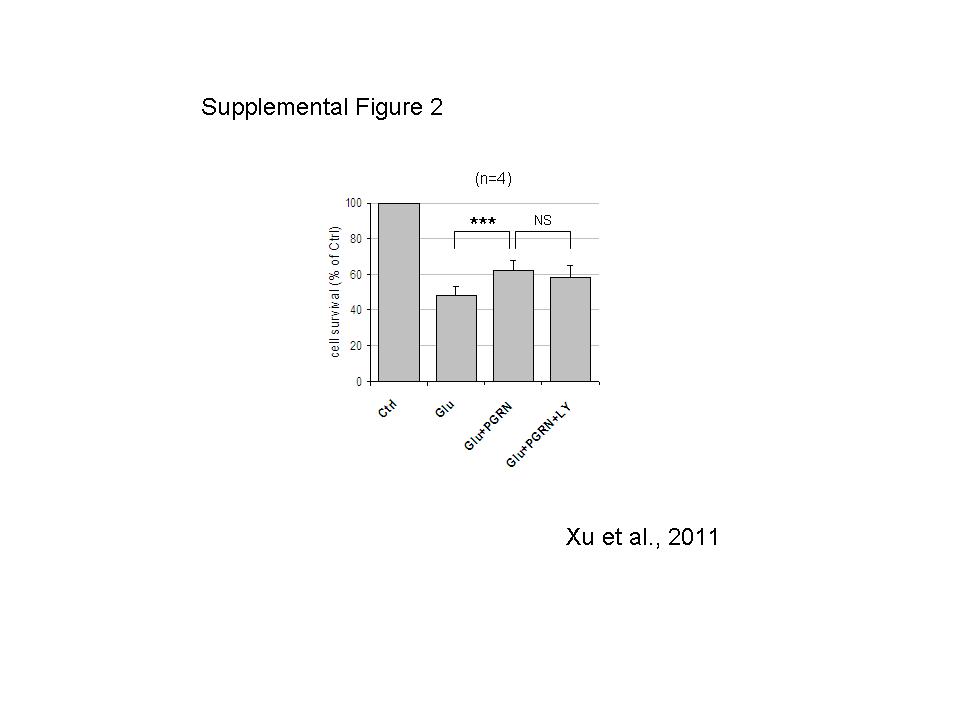
To examine whether the PGRN-dependent neuroprotection is indeed mediated by ERK1/2 signaling, we used MEK/ERK1/2 inhibitor U0126. Figure 5A shows that the PGRN-dependent neuronal survival of glutamate-treated cultures is inhibited by U0126. Consistent with these results obtained with the cell counting assay, U0126 also blocked the PGRN-dependent decrease of LDH in the media of glutamate-treated neuronal cultures (Fig. 5B). U0126 alone had no effect on cell survival or LDH release (Fig. 5). Despite the PRGN-induced increase in Akt phosphorylation however, Akt inhibitors, including LY-294002, had no effect on the PGRN-dependent neuroprotection (supplemental Fig. 2), suggesting that the Akt signaling has little or no effect on the neuroprotective activities of PGRN against glutamate toxicity and that MEK/ERK1/2 signaling may be the main pathway mediating the PRGN neuroprotection against glutamate excitotoxicity.
Figure 5. Inhibition of ERK1/2 kinase blocks the neuroprotective function of extracellular PGRN against glutamate toxicity.
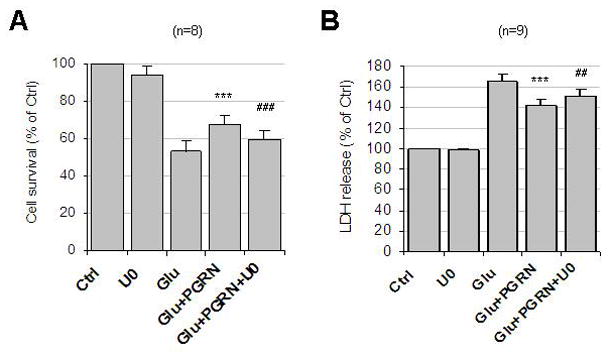
(A) Rat cortical cultures of 8–12 DIV were pretreated for 1 hour with U0126 (10μM) and then with 35nM PGRN for 2 hrs, followed by 50 μM glutamate for 24 hrs. Cells were then fixed, stained and analyzed as described in the legend of Fig. 2A. Results (mean ± SE) were summarized from eight independent experiments. ***, p<0.001 comparing cultures treated with glutamate in the presence and absence of PGRN (paired t-test); ###, p<0.001 between cultures treated with PGRN/glutamate in the presence and absence of U0126 (paired t-test). (B) Conditioned media of neuronal cultures prepared as above was replaced with fresh media and neurons were treated with U0126, PGRN and glutamate as described above. Cell-free supernatants were then collected and LDH release was determined as described in the legend to Fig. 2C. Results (mean ± SE) were summarized from nine independent experiments. ***, p<0.001 comparing between cultures treated with glutamate in the presence and absence of PGRN; ##, p<0.01 comparing between cultures treated with PGRN/glutamate in the presence and absence of U0126 (paired t-test).
We then employed pharmacological inhibitors to ask whether the ERK1/2 and Akt signaling pathways are involved in the neuroprotective function of PGRN against MPP+-induced toxicity. Figure 6A shows that PGRN significantly inhibited the neuronal cell death induced by toxin MPP+ and this neuroprotective effect was blocked by MEK/ERK1/2 inhibitors including U0126 and PD98059. In contrast to the glutamate-induced toxicity however, the neuroprotective effect of PGRN against MPP+ toxicity was partially blocked by inhibitors of the PI3K/Akt cell survival signaling like LY-294002 and wortmannin (Fig. 6A). These data suggest that both the ERK1/2 and Akt signaling pathways contribute to the neuroprotective activities of PGRN against MPP+ toxicity. This suggestion is further supported by data that inhibition of both MEK/ERK1/2 and PI3K/Akt pathways act synergistically suppressing further the PGRN-dependent neuronal survival of MPP+-treated cultures (Fig. 6A). Furthermore, our data indicate that these pathways contribute independently to the neuroprotective effects of PGRN against MPP+ toxicity. Morphological examination of MPP+-treated neuronal cultures showed that PGRN decreases degeneration of neuritic processes caused by exposure to MPP+ and this protective process is blocked in the presence of inhibitors against both MEK/ERK1/2 and PI3K/Akt kinases (Fig. 6B). Together, our results reveal a novel neuroprotective function of PGRN against toxic agents, such as neurotoxin MPP+, and suggest that this neuroprotective function of PGRN involves activation of both ERK1/2 and Akt cell survival signaling pathways.
4. Discussion
Progranulin is a secreted protein that has been shown to play important roles in many biological processes including inflammation, wound repair, tumorgenesis and embryonic development (Bateman, Bennett, 2009). Genetic studies show that PGRN mutations leading to reduced levels of functional protein (haploinsufficiency) associate with specific neurodegeneration in the frontotemporal region of the brain (Baker et al., 2006; Cruts et al., 2006) suggesting that PRGN functions in neuronal physiology and survival, and that specific cortical neuronal populations may need full protein expression for sustained survival. Indeed, recent studies indicate that PGRN has neurotrophic activities and promotes neuronal survival under conditions of serum or trophic factor withdrawal (Van Damme et al., 2008; Gao et al., 2010; Ryan et al., 2009). Chronic exposure of brain neurons to toxic insults such as glutamate excitotoxicity and oxidative stress however, has been proposed as an important factor contributing to neurodegenerative disorders characterized by progressive loss of cortical neurons (Fatokun et al., 2008; Lau, Tymianski, 2010). In addition, these neurotoxic mechanisms may operate in acute conditions like stroke where production of oxygen free radicals or hyperactivity of glutamate receptors may compound neuronal damage and death. We reason that to avoid or minimize severe neuronal damage inflicted by toxic insults, neurons may deploy an array of neuroprotective mechanisms and that PGRN may be part of this neuronal defense against toxic insults. Neurons deprived of the full protection of PGRN, like in FTLD-linked PGRN mutations, may then be more vulnerable to toxic conditions than neurons that express normal level of PGRN. Over the years this chronic vulnerability may translate into accelerated neuronal cell loss and dementia.
PRGN is secreted as a glycosylated polypeptide indicating that it exerts at least some of its biological functions in the extracellular space. To explore the neuroprotective properties of extracellular PGRN and to preserve possible modifications specific to mammalian cells, we produced and purified PGRN protein from the culture media of human embryonic kidney (HEK293) cells overexpressing human PGRN. Purified protein was then tested in primary neuronal cultures to ask whether PGRN protects neurons from specific toxic insults such as glutamate-associated excitotoxicity and oxidative stresses evoked by H2O2 and MPP+. Our results show that extracellular PGRN has potent neuroprotective functions mediated, at least in part, by the activation of neuronal MEK/ERK and PI3K/Akt signal transduction pathways both of which are stimulated by extracellular PGRN.
Under certain conditions including inflammation, PGRN is processed by specific proteases, such as elastase, to granulin peptides proposed to mediate some of the functions of the parent protein (Plowman et al., 1992; Tolkatchev et al., 2008; Zhu et al., 2002). We thus examined the extent to which extracellular PGRN is processed in the medium of our neuronal cultures. Our data show that PGRN is remarkably stable in the media of neuronal cell cultures as we failed to detect any significant degradation of PGRN even after overnight incubation. Media PRGN was completely degraded by exogenous elastase suggesting neurons secrete little or no elastase or other PRGN processing enzymes to the medium (Fig. 2). Our observations support the hypothesis that the detected neuroprotective effects of exogenous PGRN are due to the full-length protein rather than its processing products granulins, although they do not exclude similar or parallel neuronal functions of these peptides. Our finding also suggests that secreted neuronal PRGN may act as an autocrine signaling factor to stimulate neuroprotection. Recent reports identify sortilin as a cell surface binding partner of PGRN indicating that this protein is internalized through sortilin-mediated endocytosis (Hu et al., 2010). It is thus important to explore whether sortilin-mediated endocytosis of PGRN is involved in neuroprotection. Although our data show no decrease in the levels of extracellular PGRN, we cannot exclude the possibility that small and undetectable fractions of PGRN are endocytosed by neuronal cell surface sortilin. Alternatively, sortilin may be merely a regulator/controller of extracellular PGRN levels by endocytosis destined for lysosomal degradation, and the putative receptor for neuronal survival is yet to be found.
Exogenous PGRN rescued cell death in neuronal cultures subjected to toxic insults such as glutamate, H2O2 and MPP+, but did not significantly alter the survival rate of cultures in the absence of toxic insults. Interestingly, the neuroprotective effects of PGRN, measured by different methodologies, were most pronounced against MPP+-induced oxidative insult. MPP+ is the active derivative of MPTP, a mitochondrial toxin that induces Parkinsonism in humans and experimental animals (Przedborski, Vila, 2003). The primary event in MPP+-induced cell death is the production of ROS with subsequent mitochondrial dysfunction and apoptotic death. On the other hand, glutamate-induced excitotoxicity is receptor-mediated and primarily involves Ca2+ overload followed by ROS generation and necrotic death. The differential effect of PGRN on neuronal survival of glutamate- or MPP+-treated cultures suggests that the neuroprotective effect of PGRN may depend on the specific cell death mechanism involved in each condition and that PRGN may be more potent against apoptotic than necrotic death. Furthermore, the potent protective effect of PGRN against the MPP+-induced neuronal cell death raises the possibility that PGRN reduces the release of reactive oxygen species through the inhibition of complex I in the mitochondria and may affect neurodegenerative mechanisms specific to Parkinson’s disease including degeneration of substantial nigra neurons. In this respect, it is of interest that PGRN has been proposed to be involved in Parkinson’s disease (Brouwers et al., 2007; Sleegers et al., 2010).
Haploinsufficiency of PGRN in FTLD seems to necessitate a delicate mechanism to strictly maintain the extracellular levels of PGRN and recent reports suggest that PGRN-deficient hippocampal slices starved for glucose and oxygen show greater cell death compared to wild-type tissues (Yin et al., 2010). Our data however, reveal specific neuronal signaling pathways regulated by PGRN. Furthermore, that increasing extracellular PRGN increases neuroprotection indicates that reduction of extracellular PRGN (like in FTLD) will decrease protection from toxic insults. In addition, our results reveal neuroprotective signaling stimulated by increased PRGN and this signaling should be less effective in conditions of reduced PGRN. Taken together, PGRN is not simply neurotrophic, but also neuroprotective against various noxious insults, a property that may be important to the survival of non-mitotic neuronal cells throughout life. Accordingly, compared to wild type neurons, neurons deficient in PGRN may be more vulnerable to chronic insults such as increased activity of the glutamatergic system or oxidative stress. Similar mechanism may apply to other neurodegenerative diseases as PGRN has been implicated in Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) (Sleegers et al., 2010).
Treatment of neuronal cultures with PGRN increased phosphorylation of Akt and ERK kinases at epitopes associated with kinase activation and these phosphorylation events were sensitive to specific inhibitors of the MEK/ERK1/2 and PI3K/Akt signaling pathways (Fig. 3). Together, these data indicate that exogenous PRGN activates both the MEK/ERK1/2 and PI3K/Akt cell survival pathways of neuronal cells and suggest that these pathways may be involved in the PGRN-dependent neuroprotection against toxic insults. A recent report indicates that treatment of PGRN null (PGRN −/−) neurons with recombinant PGRN failed to stimulate phosphorylation of ERK1/2 (Kleinberger et al., 2010). Presently it is unclear whether this discrepancy is due to differences in the activities of the PGRN preparations used or to methodological differences in the protocols employed.
An important downstream effector of ERK1/2 is protein p90 ribosomal S6 kinase (p90RSK) which is phosphorylated and activated by ERK in the cytoplasm. Following activation, p90RSK translocates to the nucleus where it activates the serum response factor (SRF). Treatment of neuronal cultures with PGRN leads to the phosphorylation of specific p90RSK residues, a process inhibited by MEK/ERK inhibitor U0126 (Fig. 4). Since this inhibitor targets MEK, the upstream activator of ERK, our results indicate that exogenous PGRN stimulates the neuronal MEK/ERK/p90RSK cell signaling cascade and suggest that stimulation of this pathway contributes to the neuroprotective properties of PGRN. Indeed, inhibition of this cascade blocks the neuroprotective activity of PGRN against glutamate excitotoxicity (Fig. 5) further supporting the hypothesis that PGRN-induced activation of the MEK/ERK/p90RSK pathway plays important roles in the neuroprotective functions of this protein. Pharmacological inhibition of the MEK/ERK/p90RSK pathway also blocked the PGRN-dependent neuroprotection against neurotoxic agent MPP+ (Fig. 6). In addition, inhibition of the PI3K/Akt signaling pathway using LY-294002 or wortmannin also blocked the PGRN-dependent neuroprotection against MPP+. Interestingly, combined pharmacological inhibition of both pathways had a synergistic effect suppressing neuronal survival further than did inhibition of either pathway alone, suggesting that both pathways contribute to the neuroprotective activity of PGRN against MPP+. Notably, the PGRN-conferred neuroprotection against MPP+ was also characterized morphologically by sparing of neuronal processes (Fig. 6B). This PGRN effect is noteworthy especially in a post-mitotic system of primary cortical neuronal cultures older than 7 DIV (Baki et al., 2008). Since MPP+-induced death is characterized by axonal degeneration which proceeds soma demise (Przedborski, Vila, 2001), our results suggest that PGRN not only induces neurite outgrowth (Van Damme et al., 2008; Gao et al., 2010; Ryan et al., 2009) but also preserves integrity of neuronal processes (Fig. 6B). Together, our results support the hypothesis that reduction of functional PGRN in FTDL patients with PGRN mutations results in reduced activity of neuronal cell survival pathways such as MEK/ERK/p90RSK and PI3K/Akt thus decreasing neuronal protection against chronic toxic insults and leading to increased rates of neuronal cell death. That PGRN added to growth media acts as a neuroprotective factor against toxic insults suggests that cellular expression of PGRN may not be necessary for neuroprotection and that in vitro preparations of this protein may be used therapeutically if appropriately delivered to brain tissue, a hypothesis that needs experimental verification.
FTLD patients with PGRN mutations are often characterized by the presence of misfolded, polyubiquitinated and abnormally phosphorylated C-terminal fragments of TAR DNA-binding protein 43 (TDP-43) in tau-negative and ubiquitin-positive neuronal inclusion bodies (Neumann et al., 2006; Sleegers et al., 2010). Recent reports suggest that neurons derived from PGRN-deficient mice accumulate phosphorylated TDP-43 fragments (Yin et al., 2010; Kleinberger et al., 2010) while suppressing PGRN induces a caspase-dependent cleavage of TDP-43 a process that may be relevant to accumulation of TDP-43 fragments (Zhang et al., 2007; Kleinberger et al., 2010). Thus, it would be important to explore whether the MEK/ERK/p90RSK and PI3K/Akt signaling pathways are involved in the molecular modifications that promote formation and translocation of TDP-43 aggregates. Deciphering the cellular signaling pathways that mediate the neuroprotective effects of PGRN and their involvement in the accumulation of abnormal aggregates common to the disease may lead to the development of new therapeutic interventions for the treatment of FTLD and related disorders.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. A. Bateman, McGill University, Canada, for human PGRN cDNA. This work was supported by NIH grants AG-17926, AG-08200, NS 047229 and Alzheimer’s Association grant IIRG-10-174237.
Footnotes
6. Conflicts of interest statement: The authors declare that there is no conflict of interest associated with this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- Arndt-Jovin DJ, Jovin TM. Analysis and sorting of living cells according to deoxyribonucleic acid content. J Histochem Cytochem. 1977;25:585–589. doi: 10.1177/25.7.70450. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28:483–490. doi: 10.1523/JNEUROSCI.4067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- Brouwers N, Nuytemans K, van der ZJ, Gijselinck I, Engelborghs S, Theuns J, Kumar-Singh S, Pickut BA, Pals P, Dermaut B, Bogaerts V, De Pooter T, Serneels S, Van den BM, Cuijt I, Mattheijssens M, Peeters K, Sciot R, Martin JJ, Cras P, Santens P, Vandenberghe R, De Deyn PP, Cruts M, Van Broeckhoven C, Sleegers K. Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol. 2007;64:1436–1446. doi: 10.1001/archneur.64.10.1436. [DOI] [PubMed] [Google Scholar]
- Butler GS, Dean RA, Tam EM, Overall CM. Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: Dynamics of matrix metalloproteinase-14 (MT1-MMP) mediated membrane protein shedding. Mol Cell Biol. 2008;28:4896–4914. doi: 10.1128/MCB.01775-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der ZJ, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den BM, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA, Friedman WJ. Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci. 1998;18:5112–5123. doi: 10.1523/JNEUROSCI.18-14-05112.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun AA, Stone TW, Smith RA. Oxidative stress in neurodegeneration and available means of protection. Front Biosci. 2008;13:3288–3311. doi: 10.2741/2926. [DOI] [PubMed] [Google Scholar]
- Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Joselin AP, Wang L, Kar A, Ray P, Bateman A, Goate AM, Wu JY. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3beta. Protein Cell. 2010;1:552–562. doi: 10.1007/s13238-010-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: An update. Hum Mutat. 2008;29:1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- He Z, Ismail A, Kriazhev L, Sadvakassova G, Bateman A. Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res. 2002;62:5590–5596. [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Frohlich L, Sixt M, Lammermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fassler R, Jenne DE. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Wils H, Ponsaerts P, Joris G, Timmermans JP, Van Broeckhoven C, Kumar-Singh S. Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J Neurochem. 2010;115:735–747. doi: 10.1111/j.1471-4159.2010.06961.x. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Laird AS, Van Hoecke A, De Muynck L, Timmers M, Van Den BL, Van Damme P, Robberecht W. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One. 2010;5:e13368. doi: 10.1371/journal.pone.0013368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lu R, Serrero G. Mediation of estrogen mitogenic effect in human breast cancer MCF-7 cells by PC-cell-derived growth factor (PCDGF/granulin precursor) Proc Natl Acad Sci U S A. 2001;98:142–147. doi: 10.1073/pnas.011525198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuwaki T, Asakura R, Suzuki M, Yamanouchi K, Nishihara M. Age-Dependent Changes in Progranulin Expression in the Mouse Brain. J Reprod Dev. 2010 doi: 10.1262/jrd.10-116s. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Monami G, Gonzalez EM, Hellman M, Gomella LG, Baffa R, Iozzo RV, Morrione A. Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex. Cancer Res. 2006;66:7103–7110. doi: 10.1158/0008-5472.CAN-06-0633. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. 2001;21:8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase) EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowman GD, Green JM, Neubauer MG, Buckley SD, McDonald VL, Todaro GJ, Shoyab M. The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J Biol Chem. 1992;267:13073–13078. [PubMed] [Google Scholar]
- Przedborski S, Vila M. The last decade in Parkinson’s disease research. Basic sciences. Adv Neurol. 2001;86:177–186. [PubMed] [Google Scholar]
- Przedborski S, Vila M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci. 2003;991:189–198. [PubMed] [Google Scholar]
- Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der ZJ, Peeters K, Mattheijssens M, Cruts M, Vandenberghe R, De Deyn PP, Robberecht W, Van Broeckhoven C. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–609. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- Sleegers K, Cruts M, Van Broeckhoven C. Molecular Pathways of Frontotemporal Lobar Degeneration. Annu Rev Neurosci. 2010;33:71–88. doi: 10.1146/annurev-neuro-060909-153144. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkatchev D, Malik S, Vinogradova A, Wang P, Chen Z, Xu P, Bennett HP, Bateman A, Ni F. Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 2008;17:711–724. doi: 10.1110/ps.073295308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den BL, Robberecht W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo-Martinez M, Wang L, Wu JY, Robberecht W, Goate A. Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem. 2010;112:1305–1315. doi: 10.1111/j.1471-4159.2009.06546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Litterst C, Georgakopoulos A, Zaganas I, Robakis NK. Peptide EphB2/CTF2 generated by the {gamma}-secretase processing of EphB2 receptor, promotes tyrosine phosphorylation and cell surface localization of NMDA receptors. J Biol Chem. 2009;284:27220–27228. doi: 10.1074/jbc.M109.048728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanocco-Marani T, Bateman A, Romano G, Valentinis B, He ZH, Baserga R. Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res. 1999;59:5331–5340. [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.